

Abstract
The attachment of ubiquitin (Ub) to lysines on substrates or itself by ubiquitin-conjugating (E2) and ubiquitin ligase (E3) enzymes results in protein ubiquitination. Lysine selection is important for generating diverse substrate-Ub structures and targeting proteins to different fates; however, the mechanisms of lysine selection are not clearly understood. The positioning of lysine(s) toward the E2/E3 active site and residues proximal to lysines are critical in their selection. We investigated determinants of lysine specificity of the ubiquitin-conjugating enzyme Cdc34, toward substrate and Ub lysines. Evaluation of the relative importance of different residues positioned −2, −1, +1 and +2 toward ubiquitination of its substrate, Sic1, on lysine 50 showed that charged residues in the −1 and −2 positions negatively impact on ubiquitination. Modeling suggests that charged residues at these positions alter the native salt-bridge interactions in Ub and Cdc34, resulting in misplacement of Sic1 lysine 50 in the Cdc34 catalytic cleft. During polyubiquitination, Cdc34 showed a strong preference for Ub lysine 48 (K48), with lower activity towards lysine 11 (K11) and lysine 63 (K63). Mutating the −2, −1, +1 and +2 sites surrounding K11 and K63 to mimic those surrounding K48 did not improve their ubiquitination, indicating that further determinants are important for Ub K48 specificity. Modeling the ternary structure of acceptor Ub with the Cdc34~Ub complex as well as in vitro ubiquitination assays unveiled the importance of K6 and Q62 of acceptor Ub for Ub K48 polyubiquitination. These findings provide molecular and structural insight into substrate lysine and Ub K48 specificity by Cdc34.
Keywords: ubiquitin, cell cycle, Cdc34, lysine, Sic1, SCF
Introduction
Protein ubiquitination involves the covalent attachment of the 8 kDa Ub to proteins and is important in regulating nearly all aspects of cellular function.1 Protein ubiquitination requires a cascade of three classes of enzymes.1 First, the C-terminal glycine of (Ub) forms a thioester bond with the catalytic cysteine of the E1 Ub-activating enzyme in an adenosine-5-triphosphate (ATP)-dependent manner. Ub is then transferred from the E1 to the active-site cysteine of an E2 Ub-conjugating enzyme in a transesterification reaction. Finally, E2s in conjunction with a substrate-binding E3 ligase, transfers Ub to a substrate lysine (K) to form an isopeptide bond. Two major families of E3s exist. The RING (really interesting new gene) finger E3s, which lack catalytic activity, recruit the substrate and E2~Ub conjugate into a single complex to facilitate ubiquitination on substrate lysine.2 Conversely, catalytic HECT (homologous to E6-AP C terminus) E3s accept Ub from the E2 via a catalytic cysteine in a transesterification reaction, then transfer Ub to a substrate lysine.1
Protein ubiquitination is a very versatile process, due to the ability of Ub to be conjugated to substrate lysines, its own lysines or its N terminus, which generates a diverse range of structures.3 Attachment of one Ub to substrate results in monoubiquitination, while conjugation of two or more Ubs to substrate lysines results in multiple monoubiquitination.4 Furthermore, once attached to substrate, Ub itself can act as an acceptor for conjugation of further Ubs, resulting in polyubiquitin chain extension. Ub contains seven lysines at positions 6, 11, 27, 29, 33, 48 and 63, all of which can serve as acceptors to generate polyubiquitin chains.3,5,6 Alternatively, linear head-to-tail polyubiquitin chains can be formed where the C-terminal glycine of Ub is attached to the α-amino group of a methionine from another Ub by the HOIL/HOIP/SHARPIN linear ubiquitin chain assembly E3 ligase complex (LUBAC).7-10
The generation of different substrate ubiquitin structures is important for obtaining structural diversity, which, in turn, allows proteins to be targeted to different fates. For example, in response to DNA damage the E2 Rad6 in association with the RING E3, Rad18, promotes monoubiquitination of PCNA to recruit translesion polymerases and repair damaged DNA.11-13 Polyubiquitination through Ub K11 or K48 generally targets protein for proteasomal degradation.14,15 K63-linked Ub chains can function in DNA repair pathways.11 Synthesis of linear polyubiquitin chains on the NFκB essential modulator (NEMO) by the LUBAC complex is important in the activation of the NFκB pathway.8-10,16 Structural studies reveal that linkage through different sites on ubiquitin generate distinct polyubiquitin structures, which can adopt compact or open conformations.17 This structural diversity allows proteins with specific Ub-binding domains (UBDs) to discriminate between alternate Ub structures17 and mediate distinct downstream events.
Despite the importance of protein-ubiquitin structural diversity in controlling specific fates of targeted proteins, the mechanisms that control generation of this diversity is not well-understood. While it is clear that E2s in combination with E3s are important for substrate selection and linkage specificity during polyubiquitination, how different sites are selected on proteins or on ubiquitin to generate chains of particular topology is not well understood. RING E3s bind substrate and position their lysine(s) toward the thioester bond of E2~Ub for efficient ubiquitination.18 In addition to the E3-mediated positioning of substrate lysine/s for optimal ubiquitination, emerging work shows that amino acids proximal to ubiquitinated lysine (s) are important determinants of their efficiency of ubiquitination.14,19,20 Studies with the E2, Ube2C and its cognate E3, the anaphase-promoting complex (APC) suggest that these amino acids constitute initiation motifs, which control the efficiency of substrate lysine ubiquitination to exert temporal control on their degradation during the mitotic cell cycle.21 Similar findings have been reported for the E2, Cdc34 and its cognate E3 ligase, the Skp, cullin and F-box (SCF) complex.19 In addition to RING E3-mediated selection of lysines on substrates, the targeting of specific lysines on Ub during polyubiquitination to generate chains of specific topology is generally controlled by structural aspects of E2s.14,22 Structural studies of the Mms2/Ubc13-Ub demonstrate that this complex assembles so that K63 of acceptor is positioned proximal to the Ubc13-Ub thioester bond during Ub chain formation.23 Similarly, studies with Ube2S, which generates K11-specific polyubiquitin chains, demonstrates that this E2 recognizes a surface around K11 of acceptor Ub.24
In the current study, we investigated mechanisms of lysine specificity by the ubiquitin-conjugating enzyme, Cdc34. In yeast, Cdc34 and its cognate E3 RING ligase Skp1-Cdc53/cullin-F box protein, Cdc4 (SCFCdc4) generates K48-linked polyubiquitin chains on the cyclin-dependent kinase (CDK) inhibitor Sic1,25 which targets this protein for proteasomal degradation. This is necessary for alleviating the inhibitory effect of this protein on cyclin-dependent kinases (CDKs) to promote G1-S phase cell cycle progression.25,26 We have previously shown that amino acids immediately adjacent to ubiquitinated lysines on Sic1 influence their rate of ubiquitination by Cdc34/SCFCdc4 to control the rate of Sic1 degradation and cell cycle progression.19 In the current study, to gain a detailed insight into the importance of sequence motifs in ubiquitination efficiency by Cdc34/SCFCdc4, the relative importance of different amino acids positioned −2, −1, +1 and +2 on the efficiency of ubiquitination of Sic1 lysine 50 (Sic1-K50) or lysines in Ub during polyubiquitination was evaluated. These studies showed that alternate amino acids differentially impacted on the ubiquitination of Sic1-K50. The −1 position plays a critical role, as mutation of this site generally decreased Sic1-K50 ubiquitination, while charged amino acids in the −2 position reduced ubiquitination. In contrast, changes to the +1 and +2 positions did not affect ubiquitination in most cases. Modeling studies suggest that charged residues in the −2 and −1 position alter the interactions between substrate and Cdc34 to misplace lysine 50 in the catalytic cleft of Cdc34. These studies indicate that amino acids proximal to substrate lysines play important roles in their selectivity and ubiquitination efficiency by Cdc34, suggesting that E2s utilize this mechanism to select substrates for optimal ubiquitination. We also evaluated the importance of amino acids surrounding lysine residues in Ub for their impact on ubiquitination efficiency and polyubiquitin chain extension. As reported previously, Cdc34 showed a strong preference for Ub K48 during polyubiquitination;27 however, K11 and K63 were also utilized less efficiently. Mutating the proximal sites surrounding K11 and K63 to mimic amino acids surrounding K48 did not improve their ubiquitination efficiency, indicating that other determinants play important roles. Modeling the structure of the acceptor Ub with its K48 positioned at the thioester bond of the Cdc34~Ub complex, together with structure-guided mutagenesis and in vitro ubiquitination assays, unveiled K6 and Q62 of acceptor Ub as important new determinants for Ub K48-mediated polyubiquitination by Cdc34.
Results
Charged amino acids −1 and −2 proximal to Sic1-K50 reduce its ubiquitination efficiency by Cdc34/SCFCdc4
We previously investigated mechanisms of lysine specificity during ubiquitination of the yeast CDK inhibitor, Sic1, by the E2, Cdc34 in association with its cognate E3, SCFCdc4. These studies demonstrated that Cdc34/SCFCdc4 ubiquitinates Sic1 with differential efficiency on different lysine residues in the N terminus. Concurrent mutation of amino acids in the −1 and +1 positions proximal to the ubiquitinated lysine critically affects ubiquitination efficiency.19 These studies suggest that sequence motifs surrounding the ubiquitinated lysine are important for regulating the efficiency of ubiquitination, as demonstrated for the E2, Ube2C, which ubiquitinates mitotic substrates.21 In addition, studies assessing the E2, Ubc9, demonstrates that most small ubiquitin-like modifier (SUMO)-modified proteins contain a consensus motif ØKX(D/E), where Ø is a large hydrophobic residue.28 X-ray crystallography studies of Ubc9 complexed to its substrate RanGAP1 suggest that these amino acids proximal to SUMOylated lysine make important contacts to residues in the catalytic core of Ubc9.29 We therefore performed a comprehensive analysis of the impact of different amino acids in the −2, −1, +1 and + 2 positions on the ubiquitination of Sic1-K50 to obtain information into the relative importance of the position and identity of particular amino acid in relation to Sic1-K50 ubiquitination efficiency. We mutated amino acids in these positions to residues representing the seven different classes of amino acids including alanine (neutral), cysteine (thiol), aspartate (acidic), phenylalanine (aromatic), asparagine (amide), arginine (basic) and serine (alcohol) (Fig. 1A). The rationale for using Sic1-K50 is that this site displays an intermediate level of ubiquitination, between the optimal Sic1-K53 site and the suboptimal Sic1-K32, -K36, -K84 and -K88 sites.19 Therefore, Sic1-K50 should be amenable to both increased and decreased ubiquitination and thus is suitable for evaluating any impact the surrounding amino acid mutations may cause.

Figure 1. Amino acids −2, −2, +1 and +2 positions relative to Sic1-K50 regulate its efficiency of ubiquitination. (A) Schematic of the sequence surrounding Sic1-K50 with amino acids in the −2, −1, +1 and +2 positions (top) and mutation of these amino acids to the different amino acids belonging to different functional classes (bottom). (B) Cdc34/SCFCdc4-mediated monoubiquitination of Sic1-K50 with Ub K0 and the indicated mutants with changes to the −2 (Sic1-K50 T48), −1 (Sic1-K50 T49), +1 (Sic1-K50 S51) or +2 (Sic1-K50 F52) positions. As a control, E1 was omitted from the reaction (lane 1). The level of monoubiquitination of the different mutants was normalized relative to the level of monoubiquitination of wild-type Sic1-K50, which was calculated as the ratio of monoubiquitinated Sic1 (Sic1-K50-Ub) relative to unmodified Sic1 substrate (Sic1-K50). The results are quantified as the mean and standard error of the mean from three independent experiments. (C) The six N-terminal lysines of Sic1 and their immediate sequence environment.
Mutation of Sic1 threonine 48 (T48) at the −2 position to the aforementioned alternate amino acids resulted in variable effects on Sic1-K50 ubiquitination. Namely, mutation to cysteine or serine did not impact Sic1-K50 ubiquitination (Fig. 1B). However, mutation to alanine, phenylalanine and asparagine increased Sic1-K50 ubiquitination (135–148%). Conversely, mutation to aspartate decreased activity to ~75%, while mutation to arginine resulted in the largest reduction in ubiquitination (~45%) (Fig. 1B). Therefore, mutation to alternate amino acid classes in this position differentially affects Sic1-K50 ubiquitination. Unlike the differential effects observed by mutation to different amino acids in the −2 position, mutation of threonine 49 in the −1 position resulted in a significant reduction in Sic-K50 ubiquitination with all the substitutions tested (Fig. 1B). This ranged from a minor reduction in ubiquitination to 77%, when threonine was mutated to alanine, to a major reduction to 28%, when this site was mutated to cysteine (Fig. 1B). Mutation to the other amino acids tested, including aspartate, asparagine, arginine and serine, reduced ubiquitination of Sic1-K50 to 46–60% (Fig. 1B). Therefore, the −1 position is critical in controlling Sic1-K50 ubiquitination. In contrast to the effects of mutation of the −1 and −2 positions, mutation of serine 51 in the +1 position did not affect Sic1 K50 ubiquitination in most cases. Mutation to alanine yielded the greatest change, with an increase in ubiquitination to 141% (Fig. 1B). All of the other mutations only had a minimal impact, with no mutation significantly reducing Sic1-K50 ubiquitination (Fig. 1B). Mutation of phenylalanine in the +2 position also resulted in modest effects on Sic1-K50 ubiquitination. Mutation to alanine reduced activity to 68% of wild-type activity. The other mutants tested, including cysteine, aspartate, tryptophan, asparagine and arginine resulted in Sic K50 ubiquitination ranging from 79–120% (Fig. 1B). Altogether, these studies show that altering the amino acids in the −2, −1, −1 and +2 positions can differentially and significantly impact on the efficiency of Sic1-K50 ubiquitination by Cdc34/SCFCdc4. The −1 and −2 positions are more important than the +1 and +2 positions, since mutations at these sites had a greater impact on Sic1-K50 ubiquitination. In particular, charged residues in the −1 and −2 positions, including aspartate and arginine, negatively impact on Sic1-K50 ubiquitination. Consistent with these findings, none of the six N-terminal lysines in Sic1, which are all ubiquitinated in S.cerevisiae,26 contain charged residues in the −1 and −2 position (Fig. 1C).
Computational model of the Cdc34~Ub complex bound to Sic148–52 substrate
To rationalize these results at a structural level, a ternary complex of Cdc34~Ub donor complex30 in association with the Sic148–52 was modeled by comparative modeling using as template the known structure of Ube2I and its substrate (pdb code: 2GRN)31 by Modeler v.9.11.32 Twenty different models were generated for each variant. The models differed mainly in their side-chain orientations at the interface of the complex. This allowed assessment of the reproducibility of the predicted effects induced by the mutations. Since mutation of the −2 and −1 positions, namely Sic148–52(T48R) and Sic148–52(T49D), yielded the greatest changes in Sic1-K50 ubiquitination (Fig. 1B), we focused on changes at these sites. The Sic148–52 was oriented within the catalytic site of the Cdc34~Ub donor complex, in agreement with the model of Sic1 bound to Cdc34/SCFCdc4,33 and the Sic1 S51 serine in +1 position aligned with the serine in +1 position observed in the 2GRN complex.
In agreement with the mechanism proposed for Ube2I, where N85, Y87 and D127 provide a suitable environment for alignment of the substrate lysine,31 the models suggest that the homologous Cdc34 residues, N87, Y89 and S139 are also important for Sic1-K50 alignment in the catalytic cleft. Mutagenesis of Cdc34 Y89 confirms the importance of this site for substrate lysine ubiquitination.19 The region surrounding the catalytic pocket of Cdc34 is rich in charged residues, arginines (colored in blue, Fig. 2A) and aspartate (colored in red, Fig. 2A). In particular Cdc34 R90, R93, D104 (which belongs to the acidic loop), D91 and arginine 74 in the C-terminal tail of ubiquitin. There are more than thirteen charged residues surrounding the Cdc34 catalytic core, and most are involved in well-defined intra- and intermolecular electrostatic networks in the native complex (Fig. 2B, left). In particular, two groups of charged residues located in positions that suggest the formation of salt-bridge interactions can be highlighted in the wild-type model. On one side of the catalytic cleft, Cdc34 R93 may be involved in a salt-bridge network with D91 and the lysine in the +3 position of the Sic1. On the other side, the two C-terminal Ub arginines are likely to form a network of electrostatic interactions with an aspartate of Ub itself and D104 of the Cdc34 acidic loop.

Figure 2. Model of the wild-type and mutant ternary complexes of Cdc34-Ub-Sic148–52. (A) A consensus model for the wild-type complex Cdc34-Ub-Sic148–52, as well as the distribution of the charged residues in the proximity of the Sic1 peptide. (B) The effects induced by the mutation T48R (middle panel), T49D (right panel) compared with the wild-type Sic148–52 (left panel). Ub-Rs denote ubiquitin arginines.
Mutation of Sic1 T48 to arginine likely induces repulsive electrostatic effects to misalign Sic1-K50 in the catalytic cleft. To assess this model of Sic1-K50 T48R Figure 2B (middle) was compared with the wild-type reference model (Fig. 2B, left). This modeling gives an estimate of locally induced effects and does not allow for large conformational rearrangements. The T48R mutation may induce electrostatic repulsion of Sic1 K50, causing a misplacement of its side chain in the catalytic cleft. This, in turn, in a cascade effect, may alter the interactions between Cdc34 R93 and D91 and the lysine in +3 position of Sic1. Since these are homology models and large conformational rearrangements are not allowed, an effect on perturbation of the arginines in the Ub tail cannot be ruled out. Modeling the Sic1-K50 T49D mutant (Fig. 2B, right) suggests that this mutation may impact on the Ub arginines by electrostatic interactions to alter the location of the Ub-tail with respect to Sic1 K50. This also induces misplacement of the Sic1-K50 side chain in the catalytic cleft. Ultimately, detailed information on the effect of mutations surrounding Sic1-K50 on their interaction with the Cdc34 catalytic site will require co-crystallization and structural determination of Sic1-K50 and its mutants with Cdc34.
Cdc34 generates Ub K48- in preference to K11- and K63-linked polyubiquitin chains
In addition to assessing the significance of the residues surrounding lysine/s in Sic1 substrate in regulating the efficiency of ubiquitination, the importance of amino acids proximal to Ub lysines in regulating polyubiquitin chain formation was investigated. Cdc34/SCFCdc4 is known to generate K48-linked polyubiquitin chains, leading to substrate proteasomal degradation.27 Previous studies showed that glycine 47 is critical for K48-linked polyubiquitination by Cdc34/SCFCdc4.19 To determine the role of residues surrounding Ub lysines in Ub lysine selection by Cdc34/SCFCdc4, we first investigated the relative specificity of Cdc34/SCFCdc4 toward different Ub lysines during polyubiquitination. For this, Sic1-K50 was used as substrate, which contains only one lysine at position 50, as described previously.19 This ensures that any higher Mr species observed following ubiquitination by Cdc34/SCFCdc4 are due to polyubiquitination, rather than ubiquitination on multiple lysine residues on Sic1. Wild-type Ub, or Ub where all lysine residues were mutated to arginine (Ub K0) or Ub mutants containing a single lysine were utilized. These studies showed that Cdc34/SCFCdc4 readily polyubiquitinated Sic1-K50 in the presence of wild-type Ub (Fig. 3, top panel). As expected, when Ub K0 was used, only monoubiquitinated Sic-K50 (Sic1-K50-Ub) was generated, as there were no lysines on Ub K0 for polyubiquitin chain extension (Fig. 3, top panel). Assays with Ub single lysine mutants showed that Cdc34/SCFCdc4 readily generated polyubiquitin chains in the presence of Ub K48, similar to that observed with wild-type Ub (Fig. 3, top panel). Conversely, assays in the presence of Ub mutants containing a single lysine at positions 6, 27, 29 or 33 generated monoubiquitinated Sic-K50 (Sic1-K50-Ub), which was indistinguishable from the pattern observed when Ub K0 was used (Fig. 3, top panel). Interestingly, when the Ub K11 and Ub K63 mutants were used, in addition to the generation of monoubiquitinated Sic1-K50 (Sic1-K50-Ub), diubiquitinated Sic1-K50 (Sic1-K50-Ub2) was also generated, although further polyubiquitin chain extension was impaired (Fig. 3, top panel). This data indicates that Cdc34/SCFCdc4 efficiently utilizes Ub K48 during polyubiquitination, but also that Ub K11 and K63 may be utilized, but at significantly lower efficiency.
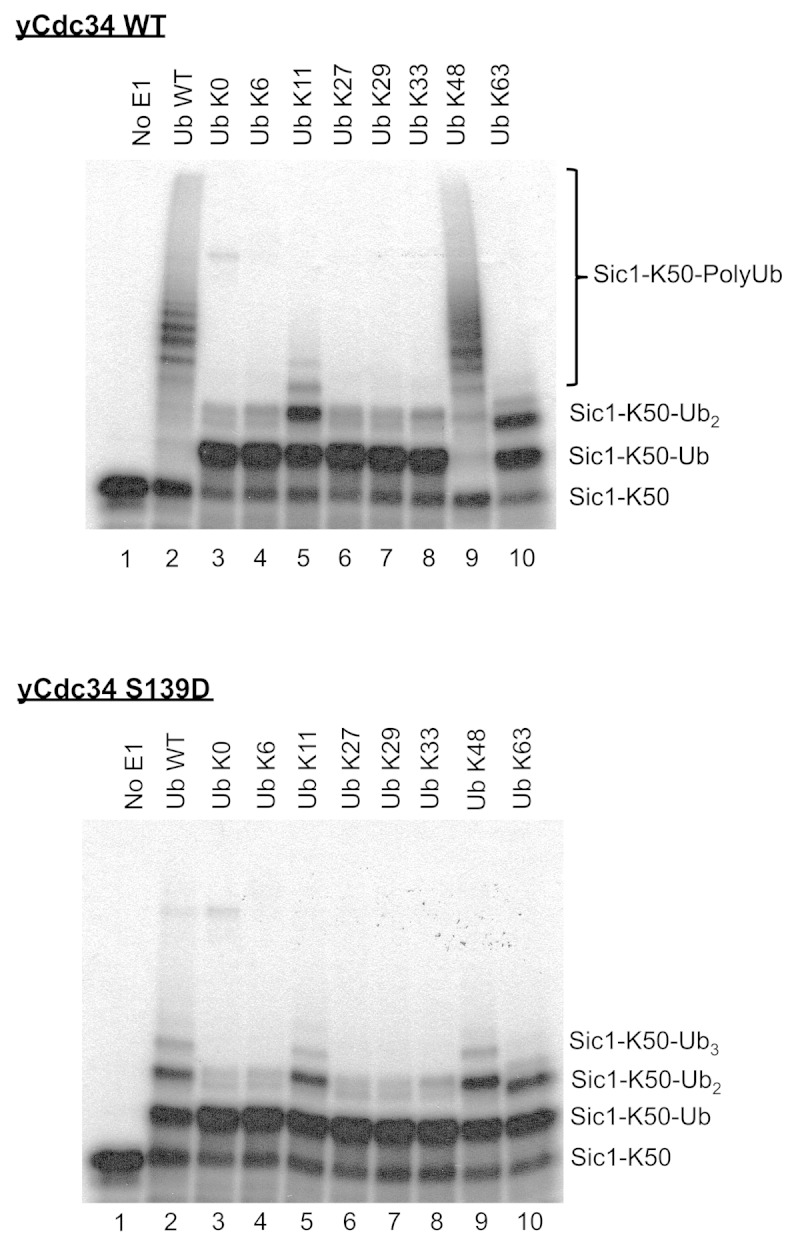
Figure 3. Cdc34 efficiently generates K48-linked polyubiquitin chains but also inefficiently utilizes Ub K11 and K63. Cdc34 (top) and Cdc34 S139D (bottom) were tested for their ability to generate polyubiquitin chains in the presence of SCFCdc4, with Sic1-K50 as substrate and wild-type Ub (Ub WT), Ub with no lysines (Ub K0) or Ub mutants with a single lysine in the designated position. Lane 1 is a control where E1 was omitted from the reaction. The position of Sic1-K50 substrate, monoubiquitinated Sic1-K50 (Sic1-K50-Ub), diubiquitinated Sic1-K50 (Sic1-K50-Ub2) and polyubiquitinated Sic1-K50 (Sic1 K50-PolyUb) is indicated on the right.
We have previously shown that recognition and utilization of Ub K48 by Cdc34 during polyubiquitination requires compatibility between amino acids surrounding Ub K48 and key amino acids in the catalytic core of Cdc34. In particular mutation of serine 139 of yeast Cdc34 to aspartate, which are the two alternate amino acids almost exclusively present at this position in different ubiquitin-conjugating enzymes, almost completely abolishes the ability of this Cdc34 mutant (Cdc34 S139D) to generate K48-linked polyubiquitin chains, but has no impact on its ability to monoubiquitinate different Sic1 lysines.19 However, polyubiquitination with the Cdc34 S139D mutant could be restored by mutation of residues proximal to Ub K48. Namely, the Ub Q49P and Ub L50S mutants restored polyubiquitination with the Cdc34 S139D mutant. Since our data shows that wild-type Cdc34 displays a strong preference for Ub K48 during polyubiquitination, but also can utilize Ub K11 and K63 inefficiently (Fig. 3, top panel), we investigated if the Cdc34 S139D mutant may display altered specificity toward different lysines of Ub. These data showed that unlike wild-type Cdc34, Cdc34 S139D was dramatically impaired in polyubiquitination with wild-type Ub. This was due to its inability to utilize Ub K48 efficiently, not unlike Ub K11 or Ub K63 (Fig. 3, bottom panel). Therefore, the Cdc34 S139D mutant inhibits its ability to utilize Ub K48, but does not change its preference toward other lysines in Ub.
Ub glycine 47 is critical for Cdc34/SCFCdc4-mediated polyubiquitination via Ub K48.19 Since Cdc34/SCFCdc4 efficiently utilizes Ub K48 during polyubiquitination but can inefficiently utilize Ub K11 and K63 (Fig. 3, top panel), we investigated if amino acids proximal to Ub K48 may be important and dominant determinants contributing to the specificity of Cdc34/SCFCdc4 for this lysine over Ub K11 and K63. We investigated if replacing the amino acids around Ub K11 and K63 with those present around Ub K48 could improve the ubiquitination of these lysines by Cdc34/CSF. Two Ub mutants were generated, where the −2, −1, +1 and +2 amino acids surrounding Ub K11 and K63 were mutated to those present around K48. Therefore, the wild-type sequence around K11 (TGK11TI) was changed to (AGK11QL), and the wild-type sequence around K63 (IQK63ES) was changed to (AGK63QL) (Fig. 4A). As expected, ubiquitination reactions using Cdc34/SCFCdc4 and Sic1-K50 as substrate, demonstrated that polyubiquitin chains were readily generated when wild-type Ub or Ub K48 was used, while Ub with only K11 and K63 were severely impaired in polyubiquitination (Fig. 4B, left panel). When the Ub K11 or K63 mutants containing the “K48-like” sequence context around their lysine (AGK11QL and AGK63QL) were utilized, polyubiquitination was also severely impaired and equivalent to that observed within their wild-type sequence context (Fig. 4B, left panel). When the Cdc34 S139D mutant was used in the same assays, there was no improvement in polyubiquitination with these Ub K11 and K63 mutants compared with their wild-type counterparts (Fig. 4B, right panel). Therefore, while amino acids proximal to K48, such as glycine 47, are critical for Cdc34/SCFCdc4-mediated Ub K48 polyubiquitination,19 other residues in addition to the −2, −1, +1 and +2 proximal to Ub lysines are important determinants of specificity during polyubiquitination.

Figure 4. Mutation of residues proximal to Ub lysines 11 and 63 to those present around Ub lysine 48 does not change Ub lysine specificity of Cdc34. (A) Schematic of the sequence surrounding Ub lysines 11 (Ub K11), 48 (Ub K48) and 63 (Ub K63) encompassing amino acids from the −10 +10 relative to lysine within their wild-type context is shown. Ub lysine 11 mutant (Ub K11 (K48-like) and Ub 63 mutant (Ub K63 (K48-like) were generated where their surrounding −2, −1, +1 and +2 amino acids (underlined) were changed to those present around Ub lysine 48. (B) Cdc34 (left) and Cdc34 S139D (right) were tested for their ability to generate polyubiquitin chains in the presence of SCFCdc4, with Sic1-K50 as substrate and wild-type Ub (Ub WT), Ub K11, Ub K48, Ub K63, Ub K11(K48-like) or Ub K63(K48-like). Lane 1 is a control where E1 was omitted from the reaction.
Structural model of the Cdc34~Ub donor complex with acceptor Ub
To gain a further insight into the basis of Ub K48 specificity during polyubiquitination by Cdc34, we modeled the overall binding interfaces and analyzed potentially important residues that interact between the Cdc34~Ub donor complex and K48 of the acceptor Ub. The crystal structures of human Cdc34 (PDB code: 2OB4)34 and ubiquitin (PDB code: 1UBQ),35 have been reported. The crystal structure of Cdc34 has the catalytic domain (resides 6–183) resolved, with a small loop region (residues 98–113) and the unstructured 53 amino acid C terminus tail missing. The C terminus plays a role in binding to the Cul1 subunit of the SCF complex.36,37 Previous NMR spectroscopy studies of the Cdc34~Ub donor complex show that a hydrophobic patch located on one side of Ub, comprising of residues L8, R42, L43, F45, G47, Q49, H68, L69, V70, L71, R72, L73, R74, G75 and G76, interact with the catalytic domain of Cdc34.38 Furthermore, in vitro ubiquitination assays using complementary charge swap mutants to map interacting residues of Ub and Cdc34, has provided more insight into the Cdc34~Ub donor complex.39 Based on these data, a refined model of the Cdc34~Ub complex was created with a thioester bond between G76 of Ub and the catalytic cysteine, C93 of Cdc34. This Cdc34~Ub complex was then computationally docked with the acceptor Ub (Fig. 5A). The model of the whole complex highlighted a number of protein-protein interactions that may be important in positioning the acceptor Ub K48 residue toward the Cdc34~Ub thioester bond. For example, three potential hydrogen bonds were identified between the acceptor Ub and Cdc34 (Fig. 5B). Importantly, this model predicted that, in addition to the important role of G47, residues such as K6 and Q62 in acceptor Ub, which are distal in the amino acid sequence but proximal in the 3-dimensional structure, may provide important contacts for orientating K48 of acceptor Ub toward the Cdc34~Ub thioester (Fig. 5B).
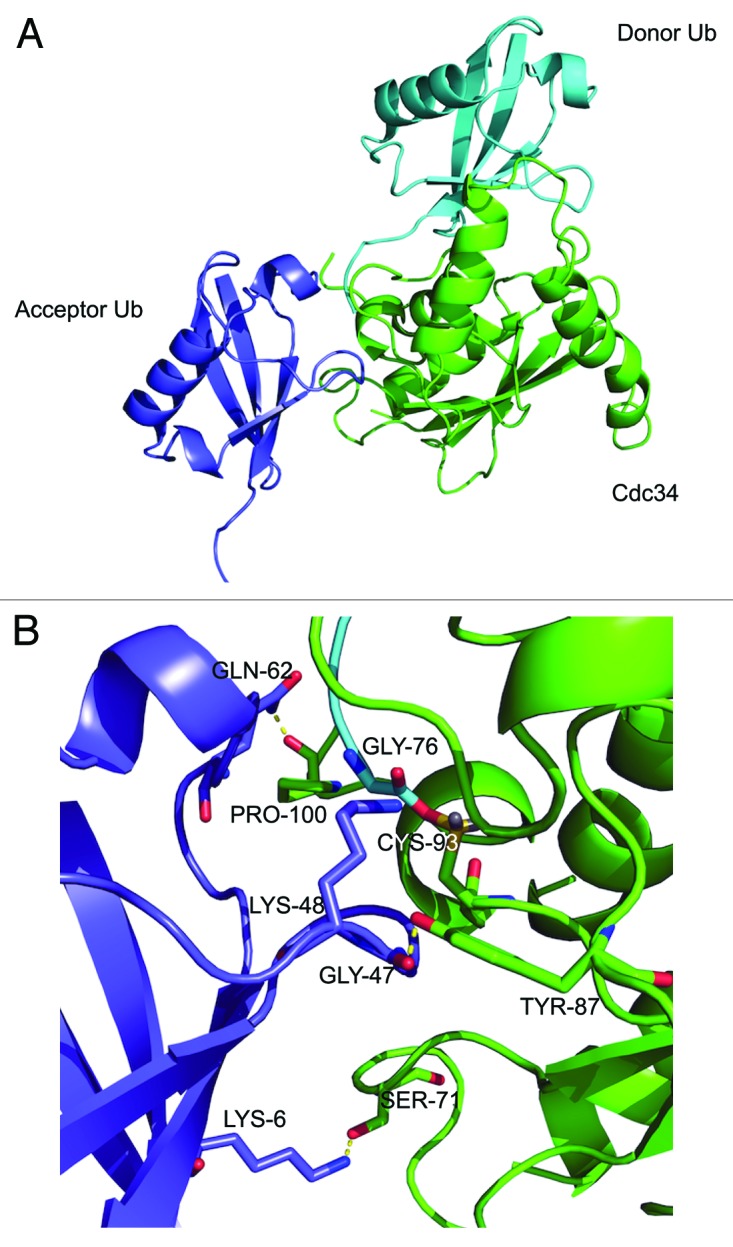
Figure 5. Model of the Cdc34~Ub donor complex in association with acceptor Ub. (A) Molecular model of Cdc34~Ub donor complex (Cdc34, green; donor Ub, cyan) in association with acceptor Ub (purple). Lysine 48 of the acceptor Ub is positioned toward the thioester bond of the Cdc34~Ub complex. (B) In the catalytic region of the Cdc34~donor Ub/acceptor Ub complex interface three potential hydrogen bonds are highlighted (yellow), together with the lysine 48 of acceptor Ub positioned toward the Cdc34~Ub thioester bond. Figure was constructed using pymol (http://pymol.org/).
Lysine 6 and glutamine 62 of acceptor Ub are important for K48-linked polyubiquitination by Cdc34
To assess the importance of these potential interactions, mutations in Ub were constructed and their activity assessed in the Cdc34/SCFCdc4-mediated polyubiquitination assays. The model in Figure 5 represents human Cdc34~Ub. The Ub pathway, Cdc34 and its E3 ligase SCF are conserved through evolution from yeast to humans. Cdc34 is essential for viability in S. cerevisiae, and human Cdc34 can functionally complement yeast deficient in Cdc34 function.40 Furthermore, human Cdc34 is active with the yeast SCFCdc4 ligase in polyubiquitination of Sic1 in vitro.41 Polyubiquitination assays were therefore performed using human Cdc34 and Ub and their mutant counterparts, together with yeast SCFCdc4 and Sic1-K50 as substrate.
Since Ub G47 is known to be important for K48-mediated polyubiquitination of Sic1 by Cdc34/SCFCdc4,19 we tested the impact of mutating the Ub K6 and Q62 sites. In polyubiquitination assays with Cdc34/SCFCdc4 and Sic1-K50, polyubiquitination was readily observed with wild-type Ub (Fig. 6A). Mutation of Ub K6 to aspartate, (Ub K6D) significantly reduced Sic1-K50 polyubiquitination (Fig. 6A). Conversely, conservative mutation of K6 to arginine (Ub K6R) did not impair polyubiquitination activity (Fig. 6A). Mutation of the Ub Q62 to asparagine (Ub Q62N) also significantly reduced Sic1-K50 polyubiquitination (Fig. 6A).

Figure 6. Mutation of acceptor Ub K6 and Q62 affects K48 polyubiquitination by Cdc34. (A) Effect of mutation of sites in Ub on its K48-mediated polyubiquitination. Polyubiquitination of Sic1-K50 with Cdc34/SCFCdc4 and wild-type Ub or the designated Ub mutants. Control lanes had no Ub added (Lane 1). Lanes 2–5: Ub wild-type, Ub K6D, Ub K6R and Ub Q62N, respectively. (B) The Ub K6 and Q62 mutants do not impair generation of the Cdc34~Ub thioester. Wild-type Ub or the designated Ub mutants were incubated with E1 and Cdc34 under ubiquitination conditions for 15 min. The reaction was stopped with sample buffer under non-reducing conditions (NR) or reducing (R) conditions by including β-mercaptoethanol in the sample buffer. Samples were then separated on SDS-PAGE, transferred to nitrocellulose and subjected to western blotting with an anti-penta His antibody to detect Cdc34 (Cdc34) or Cdc34~Ub thioester complex (Cdc34~Ub). (C) Acceptor Q62N and K6D Ub mutants impair discharge of Ub from the Cdc34~Ub thioester complex. Cdc34~Ub thioester complex was generated in the presence of E1, ATP and treated with NEM/EDTA to prevent subsequent Cdc34 recharging, as described in experimental procedures. Wild-type Ub or the designated Ub mutants were then added to degrade the Cdc34~Ub complex. At the indicated times, aliquots were removed and subjected to SDS-PAGE under non-reducing conditions and western blotting with an anti-penta His antibody to detect Cdc34 or the Cdc34~Ub thioester complex (left). The rate of degradation of the Cdc34~Ub thioester complex normalized relative to the level of Cdc34~Ub thioester complex at 0 min, which was calculated as the ratio of Cdc34~Ub thioester complex relative to Cdc34. The results are quantified as the mean and standard error of the mean from three independent experiments (right). (D) Model of Ub K6 and Q62 mutagenesis. (1) Wild-type Ub K6 (purple carbons) interacts (yellow dashed lines) with Cdc34 S71 (green carbons). Ub K6R mutation (magenta carbons) is still able to form this interaction; however, the distance to the shorter Ub K6D mutation (pink carbons) is too far. (2) Wild-type Ub Q62 (purple carbons) interacts (yellow dashed lines) with the backbone of Cdc34 P100 (green sticks). When mutated to the shorter Ub Q62N, the distance is beyond the acceptable length. Mutagenesis and figures were constructed in pymol (http://pymol.org/).
To ensure that this loss of activity was not due to alterations in the ability of these mutants to be charged with E1 and then transferred to the catalytic cysteine of Cdc34, we assessed thioester bond formation on Cdc34. These assays showed that there was no difference in the ability Cdc34 to be charged with these mutants compared with wild-type Ub (Fig. 6B).
Since polyubiquitination of Sic1-K50 involves attachment of Ub first to K50 of Sic1 and then polyubiquitin chain extension through Ub K48, it is possible that the alterations in Sic1-K50 polyubiquitination with the Ub mutants may be due to effects on one or both of these steps. As we are interested in assessing the effects of the Ub mutants on K48-mediated polyubiquitination, we performed assays to assess only the discharge of Ub from Cdc34~Ub donor to K48 of an acceptor Ub, as described previously.27 These assays involve generation of the Cdc34~Ub complex, which is then incubated with acceptor Ub to monitor the attack and disappearance of the Cdc34~Ub complex by SDS-PAGE/western blotting.27 This assay thus bypasses the potential confounding effects of attack of the Cdc34~Ub complex by Sic1-K50 and only measures the attack of this complex by acceptor Ub K48. Wild-type Ub can attack and degrade the Cdc34~Ub thioester complex, which is accelerated in the presence of SCF.27 These studies showed that the Cdc34~Ub thioester complex was degraded in the presence of wild-type Ub, with greater than 50% depleted after 20 min (Fig. 6C). The Ub K6R yielded similar results (Fig. 6C). The Ub K6D was slightly delayed, while the Ub Q62N was significantly delayed in its ability to degrade the Cdc34~Ub thioester. These data indicate that Ub Q62 and K6 of acceptor Ub are important during K48 attack of the Cdc34~Ub thioester, with the Q62 site playing a more prominent role.
The aforementioned mutations, Ub K6D, K6R and Q62N, all agree with the computational modeling, validating the model. For the K6D mutation, the modeling predicts a loss of a protein-protein polar interaction with Ser71 of Cdc34. However, the longer K6R mutant would still be able to form the polar interaction. Similarly, the Q62N mutant would result in a loss of a polar interaction with the backbone of Cdc34 P100 (Fig. 6D).
Discussion
Protein ubiquitination is a versatile post-translational modification that can decorate proteins with a vast array of different ubiquitin structures, generating protein~Ub structural diversity, which is important for targeting proteins to different fates.3 In the majority of cases, ubiquitin is attached to specific lysines on substrate or itself, thus understanding the mechanistic basis of lysine selectivity is critical toward understanding the basis of structural diversity during ubiquitination.
In terms of substrate lysine selectivity, RING E3s bind substrate to optimally position ubiquitinated lysine/s toward the thioester bond of the E2~Ub conjugate to catalyze ubiquitin transfer.18 In addition to E3-mediated positioning, emerging studies indicate that the sequence environment surrounding ubiquitinated lysine/s in substrates is critical for their ubiquitination efficiency. Work in our laboratory has demonstrated that amino acids −1 and +1 proximal to lysine residues are critical for their ubiquitination efficiency by the E2 Cdc34 in association with its cognate E3, SCF.19 Similarly, the E2 Ube2C in association with its cognate E3, the anaphase-promoting complex (APC/C) RING E3 recognizes a sequence motif near acceptor lysines, which is important for lysine selection.14 These motifs have been suggested to constitute “initiation motifs,” which regulate the efficiency of lysine ubiquitination by Ube2C to exert temporal control over their proteasomal degradation during cell cycle progression.21
In view of the emerging importance of sequence motifs in ubiquitination efficiency, a systematic evaluation of the relative importance of both the position and identity of different amino acids positioned −2, −1, +1 and +2 on the efficiency of ubiquitination of a substrate lysine was evaluated. These positions were chosen, since previous studies on the E2, Ubc9, which transfers the small ubiquitin-like modifier, SUMO, to lysines in substrates analogous to ubiquitin transfer on substrates by ubiquitin-conjugating enzymes, demonstrates that Ubc9 recognizes residues from the −1 to +2 region, with the consensus SUMOylation motif, KxE/D (is a bulky hydrophobic residue).29
We utilized the yeast E2 Cdc34 and its cognate E3 RING ligase (SCF) to ubiquitinate the physiological substrate Sic1, with one lysine at position 50. These studies showed both the relative position and identity of amino acids differentially impacted on the ubiquitination of Sic1-K50. Therefore, the −1 position plays a critical role, as mutation of this site generally decreased Sic1-K50 ubiquitination (Fig. 1). Mutation of T48 in the −2 position could either increase or decrease activity, depending on the identity of the amino acid (Fig. 1). It was notable that positively or negatively charged arginine or aspartate residues in the −2 and −1 positions significantly reduced Sic1-K50 ubiquitination (Fig. 1). Unlike the effects observed by mutation of the −1 and −2 positions, mutation of the +1 and +2 positions in most cases only modestly affected Sic1-K50 ubiquitination (Fig. 1). Therefore, these studies show that the −1 and −2 positions are important determinants of ubiquitination efficiency, with charged residues in these positions negatively impacting on Sic1-K50 ubiquitination. Previous studies have provided models of Sic1 bound to Cdc34/SCFCdc4,33 and of the Cdc34~Ub thioester complex.30 Our molecular modeling analysis provides a rationale for the importance of particular amino acids proximal to Sic1-K50. The native complex is rich in salt-bridge interactions, which are well-organized in several networks (Fig. 2). Therefore, the introduction of charged residues in positions proximal to the target lysine of Sic1 are likely to affect the interactions between the different components of the complex. In particular, the −2 and −1 positions seem to be critical. The replacement of the wild-type threonines with arginine and aspartate, respectively, is predicted to cause a misplacement of the target lysine and potentially the terminal residues in the Ub-tail in the Cdc34 catalytic cleft. Hence, modeling the Sic1-K50 T48R mutant suggests that introduction of arginine in this position induces repulsive electrostatic effects to misalign the Sic1-K50 side-chain in the catalytic cleft (Fig. 2B). Similarly, modeling the Sic1-K50 T49D mutant also suggests that aspartate in this position may lead to electrostatic interactions with arginines in the C terminus of donor Ub to alter the location of the Ub-tail with respect to Sic1 K50 and misplace the Sic1-K50 side-chain in the catalytic cleft (Fig. 2B). Consistent with these results, none of the six N-terminal lysines in Sic1, which are ubiquitinated by Cdc34/SCF contain charged residues in the −1 and −2 position (Fig. 1C). In addition to the effects of charged residues, the presence of uncharged residues in the −1 position was also detrimental to Sic1-K50 ubiquitination. For example, Sic1-K50T49N reduced ubiquitination to 54%, indicating that this site is particularly sensitive to alterations. This perturbation may be due to steric hindrance induced by the larger asparagine side chain compared with the threonine residue. This observation indicates that both the size and charge of side chains of amino acids in position 49 contribute to the efficiency of Sic1-K50 ubiquitination.
Our results are consistent with emerging studies demonstrating the importance of amino acids proximal to substrate lysines in regulating their efficiency of ubiquitination. Therefore, the E2 Ube2C recognizes sequence motifs on its substrates. Interestingly, unlike in our studies where charged residues in the −1 and −2 positions decreased ubiquitination of Sic1-K50, with Ube2C, the initiation motifs consist of charged residues, including arginine, lysine, aspartate and histidine.21 For example lysine 50 within the initiation motif sequence LSK50RKHR was a major ubiquitination site in geminin, and mutation of HR53/54 residues to alanine reduced ubiquitination of geminin,21 Ube2C and its cognate E3, the APC ubiquitinate mitotic substrates. Initiation motifs control the efficiency of substrate lysine ubiquitination and proteasomal degradation and thus temporally control their degradation during mitosis.21 Cdc34 in association with SCF is also a critical regulator of the G1-S phase cell cycle transition, mediated through polyubiquitination and degradation of critical cell cycle regulators, such as CDK inhibitors and cyclins.2,25,26 Although it is known that post-translational modification of substrates, such as phosphorylation is often an important trigger for targeting substrates for Cdc34/SCF-mediated polyubiquitination,2 our studies suggest that sequence motifs proximal to substrate lysines also play important roles in their ubiquitination efficiency. This may provide a mechanism for fine-tuning the selection of substrates for optimal ubiquitination and thus temporal control of their destruction during G1-S phase. This notion is supported by our previous studies, which show that mutation of residues proximal to Sic1 lysines 32 and 53 can regulate their ubiquitination efficiency to control their rate of ubiquitination and degradation to control the kinetics of G1-S phase cell cycle progression.19 Therefore, mutation of the −1 and +1 positions in Sic1 K53 to mimic those surrounding Sic1 K84, reduced ubiquitination of this site by 80% and reduced the rate of Sic1 degradation and G1-S phase cell cycle progression in S.cerevisiae.19 Whether sequence or initiation motifs generally contribute to controlling substrate ubiquitination efficiency by Cdc34/SCF for temporal control of their degradation during the cell cycle awaits further investigation.
In addition to assessing the importance of residues proximal to Sic1-K50, our studies analyzed the influence of residues in acceptor Ub, which are important for generating K48-linked polyubiquitin chains. As reported previously, Cdc34 showed a strong preference for Ub K48 during polyubiquitination;27 however, K11 and K63 were also utilized, albeit significantly less efficiently, and diubiquitinated Sic1-K50 substrate was mainly generated (Fig. 3). Although the determinants of Cdc34 and Ub, which direct this enzyme to generate K48-linked polyubiquitin chains, have not been fully elucidated, our work and previous studies have provided insights into this area. It is clear that residues proximal to Ub K48 are important for their ubiquitination efficiency. Therefore, glycine 47 is critical for Ub K48-mediated polyubiquitination by Cdc34.19,27 Although glycine 47 is critical, this site and the other residues −2, +1 and +2 proximal to Ub K48 are not sufficient or the dominant specificity determinants for directing this lysine toward the Cdc34~Ub thioester bond. This was exemplified by our studies mutating the −2, −1, +1 and +2 sites surrounding K11 and K63 to mimic those surrounding K48, which did not improve their ubiquitination efficiency by Cdc34 (Fig. 4B), indicating that other important interactions are important for K48 specificity. Although, the interaction interface between acceptor Ub and the Cdc34~Ub complex orienting K48 toward the Cdc34~Ub thioester bond has not been elucidated, previous studies demonstrate that the last seven amino acids in the C-terminal tail, isoleucine 44 located in a hydrophobic patch and threonine 12 of acceptor Ub are important for Ub K48-mediated polyubiquitination by Cdc34/SCFCdc4.27 Our modeling and mutagenesis studies highlighted the importance of Q62 and K6 of acceptor Ub for K48 attack the Cdc34~Ub thioester complex (Figs. 5 and 6), which interact with residues in the Cdc34 catalytic region (Fig. 5 and Table 1). Our model suggests that Cdc34 Y87 interacts with Ub G47 (Fig. 5B). Previous studies have shown that Y89 of yeast Cdc34, which is homologous to Y87 of human Cdc34, is important for Ub K48 attack of the Cdc34~Ub thioester during polyubiquitination.19 Furthermore, studies of the SUMO-conjugating enzyme Ubc9 in association with its substrate RanGAP1 suggest that this Y87 is important for aligning substrate lysine and suppressing the lysine pK to activate it as a nucleophile.31 In addition to Y89, Cdc34 S139 is critical for polyubiquitination via K48.19 Our current studies indicate that mutation of this site to aspartate does not alter the Ub lysine specificity of Cdc34. In addition to these key Cdc34 catalytic residues, an acidic loop region of Cdc34 near the catalytic site is critical for generating K48-linked polyubiquitin chains.27 This unstructured acidic loop region was not included in our model but indicates that the interface between acceptor Ub and Cdc34 includes further important interactions. In conclusion, our studies have provided important new insights into the interaction between acceptor Ub and Cdc34. The structural elucidation of the full repertoire of interactions between these proteins to generate K48-linked polyubiquitin chains awaits further studies.
Table 1. Putative interactions of amino acids between the Cdc34 catalytic region and acceptor Ub.
| Acceptor Ub | Cdc34 |
|---|---|
| LYS-6 | SER-71 |
| GLY-47 | TYR-87 |
| GLN-62 | PRO-100 |
Materials and Methods
Plasmid constructs
Yeast Cdc34 wild-type42 and a point mutant carrying Serine-139-Aspartate (yCdc34 S139D) substitution,19 were previously cloned into E. coli expression vector pET15b (Novagen). The open reading frame (ORF) of mutant SIC1 carrying lysine 50 only (Sic1-K50) was subcloned into the NdeI and XhoI sites of pET15b vector to produce recombinant His6-Sic1-K50 protein. Site-directed mutagenesis was performed according to the manufacturer’s instruction (Invitrogen) to introduce point mutations to the amino acids proximal to Sic1-K50 (Fig. 1A). Codon-optimized wild-type and mutants of human Cdc34 (hCdc34), human and yeast Ub were synthesized by GeneArt AG, and subcloned into the NdeI and XhoI sites of pET15b, to generate recombinant His6-tagged hCdc34, Ub proteins.
Expression and purification of recombinant proteins
Recombinant wild-type His6-yCdc34 and S139D mutant were expressed and purified as described previously.19 Recombinant His6-hCdc34 and His6-Sic1-K50 wild-type and mutant proteins were expressed in E. coli strain Rosetta BL21 (DE3) pLysS (Novagen) and grown in LB medium containing 100 g/ml ampicillin at 37°C to an OD600nm of ~0.8. Protein expression was induced with either 0.75 mM (His6-Sic1-K50) or 1 mM (His6-hCdc34) IPTG at 37°C for 3 h. Cells were then collected, lysed, and proteins were purified on Ni2+-NTA resin (Qiagen) according to the manufacturer’s instruction. Purified His6-hCdc34 proteins were dialyzed against pH 6.0-dialysis buffer [50 mM HEPES (pH 6.0), 150 mM NaCl, 5% (v/v) Glycerol, 1 mM DTT, 0.5 mM PMSF] to remove imidazole, while eluates containing His6-Sic1-K50 protein was passed through a PD-10 desalting column (GE Lifesciences) to remove imidazole. Similarly, His6-Ub wild type and mutants proteins were expressed in E. coli strain Rosetta BL21 (DE3) pLysS (Novagen), but grown in Turbo Prime Broth (AthenaES) containing 100 µ/ml ampicillin at 37°C to an OD600nm of ~0.8. Protein expression was induced with 1 mM IPTG at 15°C overnight. Following lysis, recombinant His6-Ub proteins were purified on a His-trap HP purification column/Acta P900 HPLC (GE lifesciences) and fractions containing the desired proteins were pooled and concentrated. All purified His6-Ub proteins were dialyzed against pH 8.0-dialysis buffer to remove excess imidazole.
In vitro SCFCdc4/Cdc34-mediated Sic1-K50 ubiquitination
Sic1-K50 and proximal mutants were phosphorylated with recombinant cyclin A/CDK2, in the presence of γ32P-ATP, as described previously.42-44 SCFCdc4/Cdc34-dependent Sic1-K50 ubiquitination assays were performed as described previously.19,42 Briefly, 100 nM UBE1 (E1, Boston Biochem), 1 μM wild-type yCdc34,:25 nM SCFCdc4, 40 μM wild type Ub or lysine-less Ub (Ub K0) (Boston Biochem) and 0.15 μM 32P-labeled Sic1-K50 were mixed with Ub-reaction buffer [50 mM HEPES (pH 8.0), 50 mM potassium acetate, 2.5 mM magnesium acetate, 1 mM dithiothreitol (DTT), 2 mM ATP] in a 30 μl total reaction volume. In vitro ubiquitination assays were performed at 26°C for 1 h and quenched by addition of SDS-laemmli buffer. Samples were boiled, and proteins were separated on SDS-PAGE gels. 32P-labeled Sic1-K50 was visualized by autoradiography and quantified by densitometry analysis.
In vitro Cdc34~Ub thioester formation assays
Two μM of Cdc34 was charged with 40 μM of wild type or mutant Ub, in the presence of 200 nM E1 in Ub-reaction buffer at 26°C for 15 min. The reaction was stopped by addition of non-reducing SDS-laemmli buffer. As control, samples were also resuspended in SDS-laemmli buffer containing 140 mM 2-β-mercaptoethanol. Samples were separated on SDS-PAGE and analyzed by immunoblotting using anti-His5 antibody (Qiagen).
In vitro Cdc34~Ub thioester discharge assays
Cdc34~Ub thioester was generated in the presence of Ub-charging buffer [50 mM HEPES (pH 8.0), 5 mM MgCl2, 2 mM ATP] as reported previously.27 The reaction was stopped by addition of 10 nM N-ethylmaleimide [NEM]/50 mM EDTA solution and left at room temperature for 15 min. Samples were then diluted in Ub-chase buffer [50 mM HEPES (pH 8.0), 100 mM NaCl, 5 mM MgCl2, 1 mM DTT] containing 80 μM of wild type Ub or Ub mutants and incubated at 26°C. Samples were removed at the indicated times and resuspended in non-reducing SDS-laemmli buffer. Following SDS-PAGE, immunoblotting was performed using anti-His5 antibody (Qiagen). The rate of discharge was quantified by densitometry analysis.
Computational model of Cdc34~Ub complex bound to Sic148–52 substrate or acceptor Ub
A ternary complex of Cdc34~Ub donor complex30 in association with the Sic148–52 was modeled by comparative modeling using as template the known experimental structure of Ube2I and its substrate (pdb entry 2GRN)31 by Modeler v.9.11.32 The Sic148–52 was oriented within the catalytic site of the Cdc34~Ub donor complex, in agreement with the model of Sic1 bound to Cdc34/SCFCdc4.33 The models generated were evaluated by Vadar,45 with no residues in disallowed region of the Ramachandran plot and low packing defects. For a more refined model of the Cdc34~Ub thioester complex in association with acceptor Ub, the crystal structures of human Cdc34 (PDB code: 2OB4)34 and ubiquitin (PDB code: 1UBQ)35 were downloaded from the Protein Data Bank (http://pdb.org/).46 A thioester bond between G76 of Ub and C93 of Cdc34 was created in Sybylx1.2 (http://tripos.com/), and the two proteins were manually orientated to allow for known Cdc34~Ub amino acid contacts.38,39 This complex (Cdc34~Ub donor) was minimized under Merck Molecular Force Field (MMFFs) for 10,000 iterations. The Cdc34~Ub donor model was used to rigidly dock the acceptor Ub using zdock (http://zdock.bu.edu), with the restriction that its K48 residue was close to the thioester bond of the Cdc34~Ub donor complex. The top 500 models were clustered according to the acceptor Ub’s general orientation on the Cdc34~Ub donor complex. A cluster of over 35 solutions correlated well with the biological data; namely, it allowed for the Lys48 of the acceptor Ub to be adjacent to the thioester bond of the Cdc34~Ub donor complex. From this cluster, the lowest energy solution was selected as a predictive model of the Cdc34~Ub donor Ub acceptor complex. This model was then visually analyzed in Pymol (http://pymol.org/) which suggested over 14 van der Waals interactions and three polar interactions between CDC34 and the acceptor Ub.
Acknowledgments
We thank Prof Mike Tyers, Samuel Lunenfeld Research Institute, Mount Sinai Hospital (Toronto, Ontario Canada) for Cdc4 baculovirus. This research was supported by grant 620205 from the National Health and Medical Research Council to B.S. and a National Health and Medical Research Council Research Fellowship to M.W.P. We acknowledge the support of the Victorian State Government Operational Infrastructure Support Program to St Vincent’s Institute.
Glossary
Abbreviations:
- Ub
ubiquitin
- E1
ubiquitin-activating enzyme
- E2
ubiquitin-conjugating enzyme
- E3
ubiquitin-ligase enzyme
- SCF
Skp, cullin, F-box
- Cdc
cell division cycle
- CDK
cyclin-dependent kinase
- K
lysine
- ATP
adenosine-5-triphosphate
- RING
really interesting new gene
- HECT
homologous to E6-AP C terminus
- LUBAC
linear ubiquitin chain assembly E3 ligase complex
- NEMO
NFκB essential modulator
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PCNA
proliferating cell nuclear antigen
- SUMO
small ubiquitin-like modifier
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24818
References
- 1.Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 2.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 3.Sadowski M, Suryadinata R, Tan AR, Roesley SN, Sarcevic B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life. 2012;64:136–42. doi: 10.1002/iub.589. [DOI] [PubMed] [Google Scholar]
- 4.Deffenbaugh AE, Scaglione KM, Zhang L, Moore JM, Buranda T, Sklar LA, et al. Release of ubiquitin-charged Cdc34-S - Ub from the RING domain is essential for ubiquitination of the SCF(Cdc4)-bound substrate Sic1. Cell. 2003;114:611–22. doi: 10.1016/S0092-8674(03)00641-X. [DOI] [PubMed] [Google Scholar]
- 5.Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. 2009;37:937–53. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 6.Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10:755–64. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25:4877–87. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeda F, Deribe YL, Skånland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NFκB activity and apoptosis. Nature. 2011;471:637–41. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, et al. SHARPIN is a component of the NFκB-activating linear ubiquitin chain assembly complex. Nature. 2011;471:633–6. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- 10.Iwai K, Tokunaga F. Linear polyubiquitination: a new regulator of NFkappaB activation. EMBO Rep. 2009;10:706–13. doi: 10.1038/embor.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458:461–7. doi: 10.1038/nature07963. [DOI] [PubMed] [Google Scholar]
- 12.Ulrich HD, Jentsch S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000;19:3388–97. doi: 10.1093/emboj/19.13.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–41. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 14.Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653–65. doi: 10.1016/j.cell.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of linear polyubiquitylation of NEMO in NFkappaB activation. Nat Cell Biol. 2009;11:123–32. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 17.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–29. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 18.Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–56. doi: 10.1016/S1097-2765(03)00234-X. [DOI] [PubMed] [Google Scholar]
- 19.Sadowski M, Suryadinata R, Lai X, Heierhorst J, Sarcevic B. Molecular basis for lysine specificity in the yeast ubiquitin-conjugating enzyme Cdc34. Mol Cell Biol. 2010;30:2316–29. doi: 10.1128/MCB.01094-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadowski M, Sarcevic B. Mechanisms of mono- and poly-ubiquitination: Ubiquitination specificity depends on compatibility between the E2 catalytic core and amino acid residues proximal to the lysine. Cell Div. 2010;5:19. doi: 10.1186/1747-1028-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williamson A, Banerjee S, Zhu X, Philipp I, Iavarone AT, Rape M. Regulation of ubiquitin chain initiation to control the timing of substrate degradation. Mol Cell. 2011;42:744–57. doi: 10.1016/j.molcel.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, et al. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007;282:17375–86. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 23.VanDemark AP, Hofmann RM, Tsui C, Pickart CM, Wolberger C. Molecular insights into polyubiquitin chain assembly: crystal structure of the Mms2/Ubc13 heterodimer. Cell. 2001;105:711–20. doi: 10.1016/S0092-8674(01)00387-7. [DOI] [PubMed] [Google Scholar]
- 24.Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, Rape M. The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell. 2011;144:769–81. doi: 10.1016/j.cell.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodge A, Mendenhall M. The cyclin-dependent kinase inhibitory domain of the yeast Sic1 protein is contained within the C-terminal 70 amino acids. Mol Gen Genet. 1999;262:55–64. doi: 10.1007/s004380051059. [DOI] [PubMed] [Google Scholar]
- 26.Petroski MD, Deshaies RJ. Context of multiubiquitin chain attachment influences the rate of Sic1 degradation. Mol Cell. 2003;11:1435–44. doi: 10.1016/S1097-2765(03)00221-1. [DOI] [PubMed] [Google Scholar]
- 27.Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123:1107–20. doi: 10.1016/j.cell.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 28.Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11:861–71. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108:345–56. doi: 10.1016/S0092-8674(02)00630-X. [DOI] [PubMed] [Google Scholar]
- 30.Papaleo E, Casiraghi N, Arrigoni A, Vanoni M, Coccetti P, De Gioia L. Loop 7 of E2 enzymes: an ancestral conserved functional motif involved in the E2-mediated steps of the ubiquitination cascade. PLoS ONE. 2012;7:e40786. doi: 10.1371/journal.pone.0040786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yunus AA, Lima CD. Lysine activation and functional analysis of E2-mediated conjugation in the SUMO pathway. Nat Struct Mol Biol. 2006;13:491–9. doi: 10.1038/nsmb1104. [DOI] [PubMed] [Google Scholar]
- 32.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–73. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mittag T, Marsh J, Grishaev A, Orlicky S, Lin H, Sicheri F, et al. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheng Y, Hong JH, Doherty R, Srikumar T, Shloush J, Avvakumov GV, et al. A human ubiquitin conjugating enzyme (E2)-HECT E3 ligase structure-function screen. Mol Cell Proteomics. 2012;11:329–41. doi: 10.1074/mcp.O111.013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vijay-Kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 A resolution. J Mol Biol. 1987;194:531–44. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- 36.Kleiger G, Hao B, Mohl DA, Deshaies RJ. The acidic tail of the Cdc34 ubiquitin-conjugating enzyme functions in both binding to and catalysis with ubiquitin ligase SCFCdc4. J Biol Chem. 2009;284:36012–23. doi: 10.1074/jbc.M109.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleiger G, Saha A, Lewis S, Kuhlman B, Deshaies RJ. Rapid E2-E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell. 2009;139:957–68. doi: 10.1016/j.cell.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spratt DE, Shaw GS. Association of the disordered C terminus of CDC34 with a catalytically bound ubiquitin. J Mol Biol. 2011;407:425–38. doi: 10.1016/j.jmb.2011.01.047. [DOI] [PubMed] [Google Scholar]
- 39.Saha A, Lewis S, Kleiger G, Kuhlman B, Deshaies RJ. Essential role for ubiquitin-ubiquitin-conjugating enzyme interaction in ubiquitin discharge from Cdc34 to substrate. Mol Cell. 2011;42:75–83. doi: 10.1016/j.molcel.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plon SE, Leppig KA, Do HN, Groudine M. Cloning of the human homolog of the CDC34 cell cycle gene by complementation in yeast. Proc Natl Acad Sci USA. 1993;90:10484–8. doi: 10.1073/pnas.90.22.10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ceccarelli DF, Tang X, Pelletier B, Orlicky S, Xie W, Plantevin V, et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145:1075–87. doi: 10.1016/j.cell.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 42.Sadowski M, Mawson A, Baker R, Sarcevic B. Cdc34 C-terminal tail phosphorylation regulates Skp1/cullin/F-box (SCF)-mediated ubiquitination and cell cycle progression. Biochem J. 2007;405:569–81. doi: 10.1042/BJ20061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarcevic B, Mawson A, Baker RT, Sutherland RL. Regulation of the ubiquitin-conjugating enzyme hHR6A by CDK-mediated phosphorylation. EMBO J. 2002;21:2009–18. doi: 10.1093/emboj/21.8.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suryadinata R, Sadowski M, Steel R, Sarcevic B. Cyclin-dependent Kinase-mediated Phosphorylation of RBP1 and pRb Promotes Their Dissociation to Mediate Release of the SAP30{middle dot}mSin3{middle dot}HDAC Transcriptional Repressor Complex. J Biol Chem. 2011;286:5108–18. doi: 10.1074/jbc.M110.198473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, et al. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003;31:3316–9. doi: 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]