

Abstract
Cyclin G2 (CycG2) and Cyclin G1 (CycG1), two members of the Cyclin G subfamily, share high amino acid homology in their Cyclin G boxes. Functionally, they play a common role as association partners of the B′γ subunit of protein phosphatase 2A (PP2A) and regulate PP2A function, and their expression is increased following DNA damage. However, whether or not CycG1 and CycG2 have distinct roles during the cellular DNA damage response has remained unclear. Here, we report that CycG2, but not CycG1, co-localized with promyelocytic leukemia (PML) and γH2AX, forming foci following ionizing radiation (IR), suggesting that CycG2 is recruited to sites of DNA repair and that CycG1 and CycG2 have distinct functions. PML failed to localize to nuclear foci when CycG2 was depleted, and vice versa. This suggests that PML and CycG2 mutually influence each other’s functions following IR. Furthermore, we generated CycG2-knockout (Ccng2−/−) mice to investigate the functions of CycG2. These mice were born healthy and developed normally. However, CycG2-deficient mouse embryonic fibroblasts displayed an abnormal response to IR. Dephosphorylation of γH2AX and checkpoint kinase 2 following IR was delayed in Ccng2−/− cells, suggesting that DNA damage repair may be perturbed in the absence of CycG2. Although knockdown of B′γ in wild-type cells also delayed dephosphorylation of γH2AX, knockdown of B′γ in Ccng2−/− cells prolonged this delay, suggesting that CycG2 cooperates with B′γ to dephosphorylate γH2AX. Taken together, we conclude that CycG2 is localized at DNA repair foci following DNA damage, and that CycG2 regulates the dephosphorylation of several factors necessary for DNA repair.
Keywords: Cyclin G2, DNA damage, PML, PP2A B'γ, γH2AX
Introduction
DNA is frequently damaged by environmental agents such as ionizing radiation (IR), UV radiation (UV) and chemicals and by intracellular factors such as replication stress. Gamma-irradiation induces double-strand breaks (DSBs). This is one of the most dangerous types of DNA damage, because both DNA strands are severed.1 DNA damage must be repaired to maintain the integrity of the genome and to prevent mutations being propagated. Defects in genome protection mechanisms lead to genome instability, which could potentially cause malignant tumors to form. DNA damage response (DDR) signaling is regulated by multiple proteins. The activation of these factors following DNA damage is controlled by changes in their cellular localization and by post-translational modifications, such as Ser/Thr phosphorylation.2 The MRN (Mre11, Rad50, Nbs1) complex accumulates at sites of DNA damage and recruits and activates the Ser/Thr kinase, ataxia telangiectasia mutated (ATM). Activated ATM phosphorylates H2AX, a variant of histone H2A, at Ser139.2,3 This phosphorylated form of H2AX is hereafter referred to as “γH2AX.” γH2AX localizes to a large region around the DSB and forms IR-induced foci.4 γH2AX recruits several DNA repair signaling factors and thereby amplifies the DNA repair signal. ATM subsequently phosphorylates checkpoint kinase 2 (CHK2) at Thr68.1,3 The phosphorylation of Thr68 is required for full activation of CHK2 kinase activity. Activated CHK2 transduces the DNA damage signal to downstream effectors such as p53, resulting in cell cycle arrest or cell death. After DNA repair is completed, γH2AX is dephosphorylated by protein phosphatase 2A (PP2A), PP4 and PP6, and is thereby inactivated.5-8
The promyelocytic leukemia (PML) nuclear body (PML-NB), also termed the PML oncogenic domain and nuclear domain 10, is a sub-nuclear compartment within mammalian cells. The PML-NB plays many roles in a variety of cellular processes, including transcriptional regulation, post-translational modification, DNA repair, apoptosis, senescence and viral infection.9 The PML-NB is a dynamic structure, and the size and number of PML-NBs varies during the cell cycle and following cellular stress.9,10 After DNA damage, the number of PML-NBs increases via a supermolecular fission mechanism.11 PML is the main component of PML-NB and acts as a scaffold for the other components. PML-deficient mice and cells are resistant to a variety of apoptotic stimuli such as IR.12 PML enhances p53 function by regulating post-translational modifications, such as CBP-dependent acetylation and CHK2-dependent phosphorylation.13,14 PML binds and activates CHK2 by mediating its autophosphorylation, thereby regulating the CHK2-p53 apoptotic pathway.15 PML recruits PP2A to the PML-NB, which prevents the nuclear localization and activation of phosphorylated AKT. Thus, PML activates FOXO3-dependent transcription of several pro-apoptotic and cell cycle-inhibitory genes.12,16 PML is phosphorylated by several kinases that are activated by DNA damage or stress, such as ATM, ATR and CHK2. This may regulate PML stability, PML-NB biogenesis and the association of PML with particular proteins.9
PP2A is a ubiquitously expressed Ser/Thr phosphatase that targets a wide range of substrates that have important roles in diverse cellular processes. PP2A is a heterotrimeric holoenzyme complex consisting of a catalytic subunit (PP2A C), a scaffolding subunit (PP2A A) and a regulatory subunit (PP2A B).17 The A and C subunits are each encoded by two genes (PPP2R1A and -B for PP2A A, PPP2CA and -B for PP2A C). By contrast, the B subunit is classified into four families: B (B55/PR55), B′ (B56/PR61), B′′ (PR48/PR72/PR130) and B′′′ (PR93/PR110), and each family consists of several isoforms that are generated by alternative splicing. The B subunit controls the substrate specificity, phosphatase activity and subcellular localization of PP2A.18 The mammalian B′ family includes five groups: B′α, -β, -γ, -δ and -ε. The B′γ (PPP2R5C) group consists of three splice isoforms called B′γ1, B′γ2 and B′γ3. B′γ3 is the longest isoform and is localized to the nucleus via its nuclear localization signal.19 PP2A containing B′γ is associated with p53-pS15, which is phosphorylated by ATM and dephosphorylates the Thr55 residue of p53, thereby promoting stabilization of p53 following DNA damage.8
Cyclin G2 (CycG2) is encoded by the CCNG2 gene and belongs to a family of cyclins that are homologous to Cyclin G1 (CycG1).20 CycG1 expression is directly correlated with the presence of active p53, and CycG2 expression is induced by p53-dependent and -independent mechanisms.21-23 Expression of CycG2 increases during mid-S to early G2 phase to regulate cell cycle progression.22 CycG2 expression is also induced during cell cycle arrest in response to hypoxia, endoplasmic reticulum stress and inhibitory growth signals.24-26 Moreover, expression of CycG1 and CycG2 is induced by DNA damage.27,28 Ectopic expression of CycG2 induces growth inhibition.28,29 Interestingly, CycG2 expression is downregulated in several cancers, including thyroid and oral cancers.30,31 Antitumor agents induce CycG2 expression in cancer cells and inhibit cell proliferation.32-34 CycG1 binds to PP2A through the B′γ subunit and recruits PP2A to its target proteins. Mdm2 is dephosphorylated by PP2A, and this leads to destabilization of p53.35 CycG2 also binds to PP2A through B′γ and induces G1/S arrest.36,37 CycG1 and CycG2 contribute to G2/M arrest during DDR, but they appear to perform different roles.27,28 However, little is known about the roles of CycG2, as a CycG2-knockout mouse has not been studied.
In this study, we established and analyzed CycG2-knockout (Ccng2−/−) mice and mouse embryonic fibroblasts (MEFs) isolated from these mice. The mice were born healthy and developed normally; however, Ccng2−/− MEFs displayed an abnormal response to IR. CycG2, but not CycG1, accumulated at DNA repair foci, and CycG2 co-localized with γH2AX and PML after DNA damage. These data suggest that CycG1 and CycG2 have distinct roles in DNA repair. Dephosphorylation of γH2AX and CHK2 after IR was delayed in Ccng2−/− MEFs, suggesting that CycG2 is required for DNA repair. Additionally, γH2AX dephosphorylation following IR was delayed when cells were depleted of B′γ. These results suggest that CycG2 and PP2A contribute to dephosphorylate, and thereby inactivate, DNA repair-related factors once DNA repair has been completed.
Results
CycG2, but not CycG1, is recruited to PML-NBs following IR
It remains unclear whether CycG1 and CycG2 play similar or distinct roles in the DDR. To examine whether the subcellular localizations of CycG1 and CycG2 are altered following DNA damage, we performed immunostaining of endogenous CycG1 and CycG2 in human osteosarcoma U2OS cells after IR. In non-treated (NT) cells, CycG2 was detected in the cytoplasm and nucleus in dispersed dots (Fig. 1A, NT panels). However, CycG2 was also detected in large and intense nuclear foci 2 h and 4 h after IR (Fig. 1A, left-most panels). Since these nuclear foci resembled PML-NBs, cells were co-labeled with an anti-PML antibody (Fig. 1A, middle panels). A proportion of the CycG2 foci co-localized with PML (Fig. 1A, right-most panels, andFig. 1B). This experiment was repeated using the TIG-1 cell line (normal human fibroblasts), and CycG2 was again detected in nuclear foci after IR (Fig. 1B). A representative magnified view of the nucleus of a U2OS cell 4 h after IR revealed that 54 and 63% of CycG2 foci and PML foci were co-localized, respectively (Fig. 1C). The degree of co-localization between CycG2 nuclear foci and PML nuclear foci in TIG-1 cells increased in a time-dependent manner following IR (Fig. 1D).

Figure 1. CycG2, but not CycG1, is recruited to PML-NBs following IR. U2OS (A) or TIG-1 (B) cells treated with IR (10 Gy) were fixed after 0.5, 2 or 4 h. NT cells were used as a negative control. Cells were immunostained with anti-CycG2 and anti-PML antibodies. (C) A representative magnified image of a U2OS nucleus 4 h after IR; CycG2 (green), PML (red) and merged image (yellow). The numbers below the images represent the number of CycG2 foci, PML foci and co-localized foci. A differential interference contrast (DIC) image is shown to illustrate the cell shape. (D) The graph shows the percentage of foci that were co-labeled with CycG2 and PML (purple) or with CycG1 and PML (red) vs. the total number of PML foci at different time points following IR in TIG-1 cells. More than four cells were observed in each case. The error bars denote the standard deviation (SD). (E and F) Cells were transfected with siRNA targeting CycG2 (E) or PML (F) and treated with IR (10 Gy). Cells were immunostained with anti-CycG2 and anti-PML antibodies. The turquoise and orange arrows in (E) indicate cells in which CycG2 was depleted. (G) A magnified image of cell at 4 h shown in (F). PML was not fully depleted in right cell, while PML was fully depleted in left cells. Bar = 10 μm. (H) Protein levels of PML in TIG-1 cells treated with GL2 or siCycG2. Western blot analysis was performed using anti-PML and anti-CycG2 antibodies. GAPDH was used as a loading control. (I) The mRNA level of PML in TIG-1 cells treated with GL2 or siCycG2.
By contrast, CycG1 was localized in the nuclei of NT cells, and this did not change after IR (Fig. S1A). It was previously reported that mitomycin C treatment induces the recruitment of CycG1 DNA replication/repair foci that contain anti-proliferating cell nuclear antigen (PCNA) in NT cells.38 Notably, CycG1 foci did not co-localize with PML foci (Fig. S1B). Moreover, unlike CycG1, CycG2 did not co-localize with PCNA (Fig. S2A). This suggests that CycG2 plays a role at PML-NBs in the response to IR, whereas CycG1 does not. CycG2 did not co-localize with PML-NBs following UV radiation (Fig. S2B). Consequently, we focused on the function of CycG2 in PML-NBs following IR.
PML is required for CycG2 to be localized to PML-NBs following IR
We next investigated whether CycG2 and PML function cooperatively in PML-NBs. We examined if the depletion of CycG2 interferes with the localization of PML to PML-NBs or vice versa. We performed siRNA-mediated depletion of CycG2 and studied the localization of PML before and 2 h after IR. PML was not detected following depletion of CycG2 (turquoise and orange arrows in Fig. 1E). This suggests that CycG2 is required for proper formation of PML-NBs by PML. Similarly, when PML was depleted and cells were observed at several time points after IR, the recruitment of CycG2 to PML-NBs decreased (Fig. 1F and G). CycG2 nuclear foci were only detected in cells in which PML was not fully depleted (right cell of Fig. 1G). Since the signal intensity of PML was attenuated by depletion of CycG2, we investigated the expression level of PML in cells treated with siCycG2 by western blotting (Fig. 1H). Interestingly, we found that the protein level of PML was decreased following depletion of CycG2. Furthermore, we analyzed the mRNA level by RT-PCR to examine whether CycG2 contributes to the regulation of the expression of PML mRNA (Fig. 1I). The results showed that the PML mRNA level in CycG2-depleted cells (siCycG2) was lower than that in the negative control cells (GL2). These results suggest that CycG2 regulates the PML gene transcription and PML-NB formation. Thus, CycG2 and PML might function together to form a positive feedback loop following IR.
CycG2 and γH2AX co-localize following IR
PML-NBs partially co-localize with γH2AX foci.9 Since we observed that CycG2 is recruited to PML-NBs, at least some CycG2 foci should co-localize with γH2AX foci. γH2AX foci represent sites of DNA repair following IR. Therefore it is possible that CycG2 accumulates at DNA repair foci in response to DNA damage.
To determine whether CycG2 foci co-localize with γH2AX foci before or after IR, we co-labeled U2OS cells with an anti-CycG2 antibody and an anti-γH2AX-pS139 antibody, which specifically detects H2AX-pS139 that accumulates at DNA repair foci. No co-localization was observed in NT cells, because very few γH2AX foci were present (NT in Fig. 2A). As expected, γH2AX foci rapidly appeared 0.5 h after IR, and this signal persisted for up to 2 h after IR, after which the signal decreased. Approximately 25% of γH2AX foci co-localized with CycG2 foci 0.5 h after IR (Fig. 2A and B). Almost all γH2AX foci co-localized with CycG2 foci 2 h after IR (Fig. 2A and B), when the intensity and number of CycG2 foci had increased (Fig. 2B). It should be noted that a proportion of CycG2 foci were not co-localized with γH2AX foci at this time point (Fig. 2A). The intensity and number of CycG2 foci decreased 4 h after IR, similar to γH2AX foci (Fig. 2A). These results suggest that at least a fraction of CycG2 accumulates at DNA repair foci after IR.

Figure 2. CycG2 co-localizes with γH2AX after IR. (A) Cells treated with IR were immunostained with anti-CycG2 and anti-γH2AX antibodies. DNA was stained with Hoechst 33258. DIC images are shown to illustrate the cell shape. Bar = 10 μm. (B) The graph shows the percentage of foci that were co-labeled with CycG2 and γH2AX vs. the total number of γH2AX foci during the IR time course. The error bars denote the SD.
Generation of CycG2-knockout mice
To understand the physiological role of CycG2 in more detail, we generated CycG2-knockout mice by performing homologous recombination to disrupt Ccng2, the CycG2 gene. To avoid confusion, we will refer to the mouse CycG2 gene as “Ccng2” and to the mouse CycG2 protein as “mCycG2” hereafter. A region from exon 2 to exon 4 in the genome was disrupted by insertion of a neomycin resistance cassette (PGK-neo), as illustrated in Figure 3A. Heterozygous embryonic stem (ES) cells (129S2) were screened using genomic Southern blot analysis, and three positive clones were obtained (Fig. 3B). These clones were independently injected into C57BL/6 blastocysts, which were then transferred to pseudopregnant females to generate chimeric mice.

Figure 3. CycG2 is required for dephosphorylation of γH2AX during DDR. Generation of Ccng2-knockout mice. (A) The targeting construct and mutant allele used to generate the knockout mice. Exons are indicated by open boxes. Coding sequences are indicated by filled boxes. (B) Southern blot analysis of ES cells following digestion of DNA with AseI. Probe A, which is indicated by the black bar in (A), detected 9.2 and 6.1 kb bands, representing the mutant and wild-type alleles, respectively. D3 is a negative control ES cell. (C) Genotype analysis of mice generated by mating Ccng2 heterozygotes. Genomic mouse DNA was analyzed by PCR using two sets of primers, one recognizing the wild-type gene (7637 Fw and 8684 Rv; 1,048 bp) and the other recognizing the gene with the neomycin insertion (7637F w and pMulti PGK-1; 668 bp). (D) RT-PCR analysis of mRNA isolated from MEFs. The equivalent amount of RNA samples loaded in each lane (24 or 4.8 ng) was used for Ccng2 and for analysis. GAPDH was used as a positive control; 12 or 2.4 ng of RNA samples was loaded in each lane. (E) IR-treated MEFs were incubated for up to 32 h and fixed at the indicated time points. Cells were stained with an anti-γH2AX antibody and Hoechst 33258. Bar = 10 μm. (F) The graph shows the percentage of cells that contained more than 10 γH2AX foci at various time points following IR. Three independent regions of the coverslip (50 cells per area) were counted to calculate the SD. The error bars denote the SD (G and H) Wild-type MEFs (G) or Ccng2−/− MEFs (H) were treated with IR for up to 48 h and harvested at the indicated times. The phosphorylation of H2AX was assessed by immunoblotting with anti-γH2AX and anti-H2AX antibodies. GAPDH was used as a loading control.
Heterozygous Ccng2 mutant mice were obtained by crossing chimeric mice with C57BL/6 mice, which were then screened by RT-PCR analysis. Homozygous Ccng2-knockout mice (Ccng2−/−) were obtained by mating heterozygous mice, and the genotypes of the offspring were analyzed by PCR using genomic DNA isolated from the tail (Fig. 3C). Ccng2−/− mice were healthy and fertile and had a normal lifespan. These mice were maintained until they were 12-mo-old, and they displayed no evidence of illness or spontaneous carcinogenesis, suggesting that the absence of Ccng2 does not increase the incidences of disease, including cancer (data not shown). MEFs were established from embryos at embryonic day 12.5–14.5, and expression of Ccng2 mRNA was analyzed by RT-PCR. Ccng2−/− MEFs did not express Ccng2 mRNA (Fig. 3D).
CycG2 regulates dephosphorylation of γH2AX following DNA damage
PP2A is required to dephosphorylate γH2AX, which promotes DNA repair.5 Since CycG2 binds the B′γ subunit of PP2A, CycG2 may regulate the function of PP2A.36,37 To examine whether CycG2 regulates dephosphorylation of γH2AX following DNA damage, we performed immunostaining of γH2AX in Ccng2−/− MEFs following IR. γH2AX foci appeared rapidly following IR in both wild-type (Ccng2+/+) and Ccng2−/− MEFs, and the signal intensity persisted for up to 8 h following IR (Fig. 3E). The percentage of cells containing γH2AX foci gradually decreased in wild-type cells, indicating γH2AX was gradually dephosphorylated (Fig. 3E and F). This occurred at a slower rate in Ccng2−/− MEFs, suggesting that dephosphorylation of γH2AX was significantly delayed in these cells. This result was confirmed by western blot analysis under the same experimental conditions. γH2AX expression persisted for longer in Ccng2−/− MEFs than in wild-type MEFs after IR (Fig. 3G and H).
Exogenous expression of mCycG2 rescues dephosphorylation of γH2AX in Ccng2−/− MEFs
We next examined whether the delay in dephosphorylation of γH2AX in Ccng2−/− MEFs is due to the absence of CycG2. A rescue experiment was performed using Ccng2−/− MEFs that stably expressed 6Myc-mCycG2. The expression level of exogenous mCycG2 was confirmed by western blot analysis with an anti-myc antibody, and two cell lines expressing different levels of 6Myc-mCycG2 were selected (Fig. 4A). Nuclear γH2AX foci were observed 2 h after IR in almost all cells in both these cell lines, whereas γH2AX was not detected in NT cells. Ccng2−/− MEFs with γH2AX foci were still observed 24 h after IR, whereas almost no γH2AX foci were detected in wild-type MEFs at this time point (Fig. 4B). When the same experiment was performed using Ccng2−/−, MEFs expressing 6Myc-mCycG2 (C1 and C2 cell lines), the percentage of cells with γH2AX foci 24 h after IR was reduced as compared with parent Ccng2−/− MEFs (Fig. 4B and C). This suggests that exogenous expression of mCycG2 in Ccng2−/− MEFs at least partly rescued the disappearance of γH2AX foci following IR. This result was confirmed by western blot analysis (Fig. 4D). The band intensity of γH2AX in Ccng2−/− MEFs expressing 6Myc-mCycG2 disappeared 16 h after IR (lanes 22–24), similar to the result observed in wild-type MEFs (lanes 6–8). The γH2AX signal in Ccng2−/− MEFs was still detected 16–32 h after IR (lanes 14–16). These results indicate that the delay in dephosphorylation of γH2AX in Ccng2−/− MEFs is due to the absence of CycG2.

Figure 4. Exogenous expression of mCycG2 in Ccng2−/− MEFs rescues the dephosphorylation of γH2AX. (A) Western blot analysis of exogenous mCycG2 expression in Ccng2−/− MEFs stably expressing 6Myc-mCycG2. (B) Representative images of wild-type MEFs, Ccng2−/− MEFs or Ccng2−/− MEFs expressing 6Myc-mCycG2 stained with an anti-γH2AX antibody at various time points following IR. DNA was stained with Hoechst 33258. Bar = 10 μm. (C) The graph indicates the percentage of cells containing γH2AX foci at various time points following IR. Data were calculated as described in the legend to Figure 3F. The error bars denote the SD. (D) Changes in H2AX phosphorylation at various time points after IR. Western blotting was performed using anti-γH2AX and anti-H2AX antibodies. GAPDH was used as a loading control.
B′γ is required for dephosphorylation of γH2AX
To examine whether B′γ contributes to the dephosphorylation of γH2AX in our experimental system, we depleted B′γ and stained for endogenous γH2AX. In control cells, γH2AX was first detected in the nucleus 0.5–1 h after IR. The signal peaked at 2 h after IR, after which it gradually disappeared (Fig. 5A, uppermost panels). When B′γ was depleted, γH2AX expression appeared and peaked at similar time points (Fig. 5A, third row panels); however, the decrease in γH2AX signal was perturbed. Whereas γH2AX expression began to decrease 4 h after IR in control cells, this was not observed following depletion of B′γ (Fig. 5A, third row panels). This observation was quantified by calculating the percentage of cells containing γH2AX foci at various time points following IR in cells treated with GL2 (a negative control) or B′γ siRNA. Depletion of B′γ delayed the decrease in the γH2AX signal following IR (Fig. 5A and B). These results indicate that B′γ is required for dephosphorylation of γH2AX. Furthermore, we examined whether CycG2 and B′γ function together in dephosphorylation of γH2AX. We performed immunostaining of γH2AX in wild-type MEFs and Ccng2−/− MEFs treated with GL2 or siB′γ following IR (Fig. 5C and D). If CycG2 and B′γ mutually regulate dephosphorylation of γH2AX in different pathways, depletion of B′γ in Ccng2−/− MEFs would be expected to prolong the delay of γH2AX dephosphorylation. γH2AX foci appeared rapidly following IR in each cell. In agreement with the result observed in U2OS, knockdown of B′γ in wild-type MEFs induced the delay of decrease in the γH2AX foci following IR as compared with GL2 (Fig. 5D, first and second bars from left). Moreover, in Ccng2−/− MEFs treated with GL2, the percentage of cells containing γH2AX foci was increased than that in wild-type MEFs treated with GL2 (first and third bars from left). Although knockdown of B′γ in wild-type cells also delayed dephosphorylation of γH2AX, knockdown of B′γ in Ccng2−/− cells prolonged this delay (second and fourth bars from left), suggesting that CycG2 and B′γ coordinately act in the same pathway to dephosphorylate γH2AX following IR.

Figure 5. CycG2 cooperates with B′γ in dephosphorylation of γH2AX. (A) U2OS cells treated with B′γ siRNA (siB′γ) or control siRNA (GL2) were exposed to IR and fixed at various time points. Representative images of cells stained with an anti-γH2AX antibody and Hoechst 33258 are shown. Bar = 10 μm. (B) The graph indicates the percentage of cells containing γH2AX foci at various time points following IR. Data were calculated as described in the legend to Figure 3F. (C) Wild-type MEFs and Ccng2−/− MEFs treated with siB′γ or GL2 were exposed to IR and incubated for up to 24 h (only the images for NT, 1 and 24 h are shown here). Representative images of cells stained with an anti-γH2AX antibody and Hoechst 33258 are shown. Bar = 10 μm. (D) The bar graph indicates the percentage of cells containing γH2AX foci at 24 h after IR. Data were calculated as described in the legend to Figure 3F. Values significantly different from GL2-treated or siB′γ-treated wild-type MEFs are indicated (*, **, ***, ****). *p = 0.0053, **p = 0.0017, ***p = 0.00056, ****p = 0.040 (Student’s t-test) Note: ns, non-significant. The error bars denote the SD.
CycG2 regulates the phosphorylation status of CHK2
The T68 residue of CHK2 is phosphorylated by ATM and is dephosphorylated by PP2A following DNA damage.39,40 We examined whether CycG2 is also involved in the dephosphorylation of CHK2-pT68 after DNA damage. We immunostained Ccng2−/− MEFs with an anti-CHK2-pT68 antibody and counted the number of cells with CHK2-pT68 foci at various time points after IR. CHK2-pT68 signal appeared 1 h after IR, and formation of foci peaked 4 h after IR in both wild-type and Ccng2−/− MEFs (Fig. 6A, fourth panels from top). CHK2-pT68 foci were not observed in wild-type MEFs 24 h after IR but were still detected in Ccng2−/− MEFs (Fig. 6A, bottom panels). This indicates that dephosphorylation of CHK2-pT68 was delayed in Ccng2−/− MEFs. Moreover, the percentage of cells containing CHK2-pT68 foci 24 h after IR was similar in wild-type MEFs and Ccng2−/− MEFs expressing 6Myc-mCycG2 (C2 cell line). This suggests that exogenous expression of mCycG2 restored the disappearance of CHK2-pT68 foci in Ccng2−/− MEFs (Fig. 6A, bottom fifth panel from left). This was confirmed by quantitative analysis of the data (Fig. 6B). These results suggest that CycG2 is required for dephosphorylation of the T68 residue of CHK2, which regulates CHK2 activity.
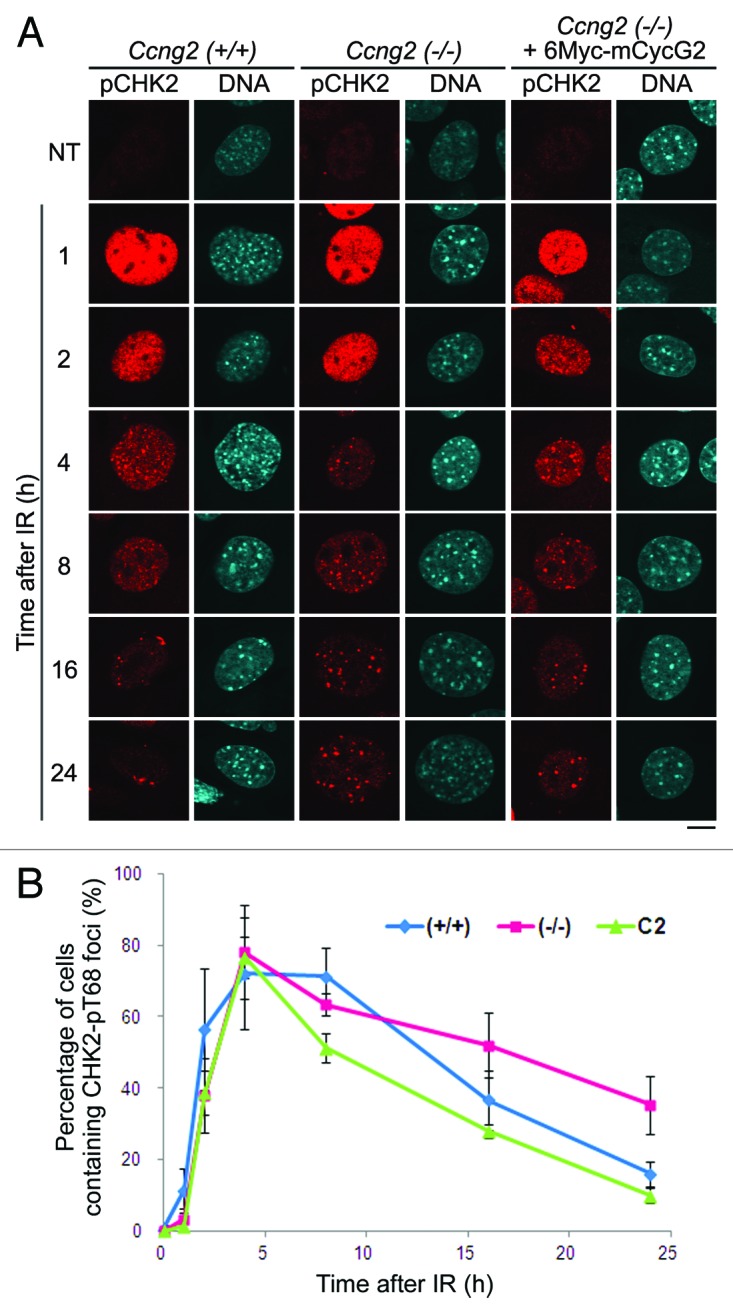
Figure 6. CycG2 is required for dephosphorylation of CHK2-pT68 after DNA damage. (A) Wild-type MEFs, Ccng2−/− MEFs or Ccng2−/− MEFs expressing 6Myc-mCycG2 MEF were treated with IR for up to 32 h and fixed at various time points. Representative images of cells stained with an anti-CHK2-pT68 antibody and Hoechst 33258. Bar = 10 μm. (B) The graph indicates the percentage of cells with CHK2-pT68 foci at various time points following IR. Data were calculated as described in the legend to Figure 3F. The error bars show the SD.
Discussion
CycG2 expression increases in response to various growth-inhibitory signals, including DNA damage. The level of CycG2 mRNA increased in U2OS and MEF cells following IR, and this was sustained for up to 24 h after IR (data not shown). Although some reports suggest that CycG2 contributes to the G1/S and G2/M checkpoints,28,37 the precise role of CycG2 in DDR is still unclear. The co-localization of CycG2 with PML and γH2AX following IR indicates that CycG2 localizes to DNA repair foci following DNA damage (Figs. 1 and 2). In addition, CycG2 failed to localize to nuclear foci when PML was depleted and vice versa (Fig. 1E and F). Moreover, depletion of CycG2 reduced the mRNA expression of PML (Fig. 1H and I). These results indicate that CycG2 and PML require each other to be localized to PML-NBs (Fig. 1E and F). Furthermore, CycG2 controls the dephosphorylation of the DNA repair-related factors γH2AX and CHK2-pT68 at DNA repair foci (Figs. 3 and 6).
CycG1 regulates the G2/M checkpoint, and CycG1 expression is increased following DDR.27 CycG1 associates with the MDM2-ARF complex, which disrupts the p53-MDM2 complex and activates p53 by preventing the degradation of p53.36 This is important for the accumulation of p53 in the initial period following IR. Degradation of p53 is promoted when CycG1 dissociates from ARF and binds B′γ.36 CycG1, unlike CycG2, did not co-localize with PML, indicating that CycG1 is not localized to DNA repair foci (Fig. S1). Moreover, dephosphorylation of γH2AX and CHK2-pT68 was delayed following IR in Ccng2−/− MEFs, which express CycG1 (Figs. 3 and 6). These results suggest that CycG1 cannot rescue the defects in DDR following IR in Ccng2−/− MEFs. CycG2 was not localized to PML-NBs after UV exposure, indicating that CycG2 specifically localizes to sites of double-strand breaks following IR (Fig. S2B). CycG1 localizes at replication foci after treatment with mitomycin C.38 CycG2 did not co-localize with PCNA after mitomycin C treatment in this study, indicating that CycG2 does not localize at replication foci (Fig. S2A). These results suggest that CycG1 and CycG2 have distinct roles; CycG2 functions in DDR following IR, whereas CycG1 functions at replication foci during the cell cycle.
γH2AX and CHK2 are dephosphorylated by PP2A, PP4 and PP6 during DDR.8 CycG2 associates with PP2A containing B′γ and regulates PP2A function,36,37 similar to CycG1.35 Here, we showed that CycG2 regulates dephosphorylation of γH2AX and CHK2 after DNA damage. Additionally, γH2AX foci (Fig. 5) and CHK2-pT68 foci (see ref. 41) persisted for longer after IR when B′γ was depleted. Notably, after IR, the delay in γH2AX dephosphorylation caused by B′γ depletion was prolonged in Ccng2−/− MEFs. These results suggest that CycG2 and B′γ functionally cooperate following IR. Furthermore, we previously reported that GAK, a CycG binding protein, phosphorylates B′γ at Thr104. This phosphorylation increases the affinity of B′γ for PP2A C, thereby regulating the localization of PP2A A and promoting PP2A phosphatase activity. The appearance of this phosphorylated form of B′γ at DNA repair foci coincides with dephosphorylation of γH2AX.41 These results suggest that CycG2 recruits PP2A containing B′γ to its targets at DNA repair foci, and that GAK activates PP2A by phosphorylating B′γ. Activation of PP2A promotes dephosphorylation of DNA repair-related factors such as γH2AX and CHK2 at the later stages of DDR.
Although the dephosphorylation of CHK2-pT68 was significantly delayed in Ccng2−/− MEFs, the delay in γH2AX dephosphorylation was more pronounced. This is likely, because CHK2-pT68 is also dephosphorylated by Wip1, as well as PP2A.42 Alternatively, CycG2 may have an indirect role in the dephosphorylation of CHK2-pT68. Ccng2−/− mice were born healthy and had a normal lifespan under normal laboratory conditions. Previous reports revealed that CycG2 plays a role in cell cycle arrest, and the data presented here suggest that CycG2 contributes to DNA repair. Negative regulation of DDR is important to prevent inappropriate checkpoint signaling and to allow cell cycle progression when DNA damage has been repaired. These results suggest that CycG2 has distinct functions in stressed and unstressed cells. CycG2 negatively regulates DDR following DNA repair.
Materials and Methods
Generation of CycG2-knockout mice
The targeting vector was designed to replace exons 2–4, including the initiation codon, with a PGK-neomycin gene (PGK-neo) as a positive selection marker. The 5′ short arm including exon 1 (XhoI fragment) and the 3′ long arm (AscI fragment), including exons 5–8, were cloned into the pMulti-ND plasmid. The targeting construct was electroporated into ES cells. Ccng2 mutant cells obtained by homologous recombination were identified by Southern blot analysis and PCR. Targeted ES cell clones were microinjected into C57BL/6 blastocysts, which were re-implanted into pseudopregnant females to generate chimeric mice. These mice were mated with C57BL/6 mice for germline transmission. Heterozygous offspring were intercrossed to generate homozygous embryos. All animal experiments were performed with the approval of the Animal Experiments Committee of Osaka University (permission number: BikenA-H19-36-0 and BikenA-H19-37-0).
Southern blot and PCR analysis
DNA extracted from embryos was digested with AseI and detected with probe A. The mutant and wild-type allele generated a 9.2 kb and a 6.1 kb band, respectively (see Fig. 3A). Genotyping PCR was performed using DNA extracted from tail snips with two sets of primers: one recognizing the wild-type gene (7637Fw, 5′-GGGCGCTGAGCCCCGCTGAGGAGC-3′ and 8684Rv, 5′-CAATCCGAAAAGCTGAACCCCTTC-3′; 1,048 bp) and the other recognizing the gene with the neomycin insertion (7637Fw and pMulti PGK-1, 5′-GGCTGGACGTAAACTCCTCTTCAG-3′; 668 bp). PCR was performed using Ex Taq polymerase (Takara) with the PCRx Enhancer System (Invitrogen) under the following conditions: a pre-heating step (94°C for 3 min), 40 reaction cycles (94°C for 30 sec, 58°C for 30 sec, 72°C for 1 min 40 sec) and a final elongation step (72°C for 6 min).
Generation of MEFs
Mouse embryos were removed from the uterus at embryonic day 12.5–14.5, and a section of the embryo was used for genotyping. Primary MEFs were obtained as described previously.43 The seeding of trypsinized MEFs onto a 60 mm dish was defined as passage 0, and the first re-plating onto a 100 mm dish was defined as passage 1. MEFs were cultured at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone, SV30014.03), 100 U/mL penicillin G, 100 mg/mL streptomycin sulfate (Nacalai Tesque) and 50 mM 2-mercaptoethanol (GIBCO).
RT-PCR
Total RNA was extracted from MEFs using an RNeasy kit (QIAGEN) and cDNA was synthesized from 3 μg of RNA using the High-Capacity cDNA Archive Kit (Applied Biosystems). PCR was performed with the following primer pairs: Ccng2: forward, 5′ -GTGTCCAGGATTGAGAAATGCC-3′, and reverse, 5′-GGCACAAGGCTAATACAGATGG-3′; PML: forward, 5′-ATGGAGCCTGCACCCGCCCGATCT-3′, and reverse, 5′-TGGCTTTCTTGGATACAGCTGCAT-3′, GAPDH: forward, 5′ -TCACCATCTTCCAGGAGCGAG-3′, and reverse, 5′-GCTGTAGCCGTATTCATTGTC-3′. PCR conditions were as follows: a pre-heating step (94°C for 2 min) followed by 30 reaction cycles [94°C for 30 sec, 55°C (PML; 58°C) for 30 sec, 72°C for 30 sec] and a final elongation step (72°C for 5 min). PCR products were subjected to agarose gel electrophoresis followed by ethidium bromide staining.
Antibodies
Polyclonal antibodies: anti-hCycG2 (MBL), anti-CycG1 (UBI), anti-H2A.X (ab11175, Abcam) and anti-CHK2-pT68 (#2661, Cell Signaling). Monoclonal antibodies: anti-PML (PM-G3), anti-γH2AX (JBW301), anti-GAPDH (Fitzgerald Industries International), anti-myc (PL14, MBL), anti-α-tubulin (B-5-1-2, Sigma) and PCNA (PC10).
Cells and transfection
U2OS and TIG-1 cells were maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin (Nacalai Tesque). Ccng2−/− MEFs stably expressing pCX4-bsr-6Myc-mCycG2 were maintained in MEF medium containing 5 μg/ml of blasticidin (InvivoGen). Plasmids were transfected using Lipofectamine Plus (Invitrogen) according to the manufacturer’s protocol. Cells were selected in the presence of blasticidin for 2 wk and the clones were isolated.
Immunostaining
Cells were cultured on coverslips and treated with 10 Gy of IR (Gammacell 40 Exactor), 60 J/m2 of UV irradiation (Stratalinker 2400, STRATAGENE, Agilent Technologies) or 10 μg/ml of mitomycin C. At the indicated times following treatment, cells were incubated for 10 min with 4% formaldehyde in PBS without calcium and magnesium [PBS(-)], 0.1% Triton X-100 in PBS(-) and 0.05% Tween-20 in PBS(-). Cells were blocked with 5% FBS in TBS-Tw (20 mM TRIS-HCl [pH 7.5], 150 mM NaCl, 0.05% Tween-20) for 1 h. Cells were incubated with the indicated antibodies for 3 h, followed by incubated with the appropriate secondary antibodies [AlexaFluor 488- and 594-conjugated anti-rabbit/mouse IgG (Molecular Probes)] for 90 min. Finally, the cells were stained with Hoechst 33258 (Sigma-Aldrich) to label the nuclei. All steps were performed at room temperature. Samples were imaged with a fluorescence BX51 microscope (Olympus) and a confocal laser scanning FV10i (Olympus) or LSM510 (Zeiss) microscope. Images were analyzed using Photoshop 7.0 software (Adobe).
Western blot analysis
Cells were lysed in N08 buffer [25 mM Tris-Cl (pH 7.6), 150 mM NaCl, 1% Nonidet P-40, 1% Na-deoxycholate, 1% sodium dodecyl sulfate] and samples were separated by SDS-PAGE and transferred onto a PVDF Immobilon-P membrane (Millipore). The membrane was blocked with 5% skimmed milk/TBS-Tw and probed with the relevant antibodies. Immunoreactive protein bands were visualized using Western Lightning Plus-ECL (PerkinElmer, Inc).
siRNA
siTRIO full set siRNA duplex cocktails were used to knockdown CycG2 and PML in human cells (THF27A-226 and THF27A-256, B-Bridge). To knockdown B′γ or GL2 the following chemically synthesized siRNA duplexes were used (Gene Design): GL2: 5′-CGUACGCG GAAUACUUCGAtt-3′ and 5′-UCGAAGUAUUCCGCGUACGtt-3′; B′γ: 5′-UACUGGCCAAAGACUCACAtt-3′, 5′-UGUGAGUCUUUGGCCAGUAtt-3′. siRNA duplexes were transfected using Oligofectamine (Invitrogen) according to the manufacturer’s protocol. Cells were exposed to IR 48 h after transfection.
Supplementary Material
Acknowledgment
We thank Dr Katsuyuki Tamai (MBL and CycLex Co., LTD) for providing anti-hCycG2 antibody and Mr Kosuke Torigata, Ms Kana Ooi and Ms Hisae Takema (DMG, RIMD, Osaka University) for technical assistance. We thank Mr Hiroaki Ito and Ms Akiko Kawai (Genome Info. Res. Center, Osaka University) for technical assistance in establishment of knockout mice. We also thank Drs Patrick Hughes (Bioedit, LTD) for critically reading the manuscript. This work was supported in part by Grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan to Y.N., N.Y. and H.N.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24878
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24878
References
- 1.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–9. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 3.Derheimer FA, Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010;584:3675–81. doi: 10.1016/j.febslet.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakamura AJ, Rao VA, Pommier Y, Bonner WM. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle. 2010;9:389–97. doi: 10.4161/cc.9.2.10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chowdhury D, Keogh M-C, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005;20:801–9. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Nakada S, Chen GI, Gingras A-C, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008;9:1019–26. doi: 10.1038/embor.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, et al. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46. doi: 10.1016/j.molcel.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng A, Maller JL. Serine/threonine phosphatases in the DNA damage response and cancer. Oncogene. 2010;29:5977–88. doi: 10.1038/onc.2010.371. [DOI] [PubMed] [Google Scholar]
- 9.Lallemand-Breitenbach V, de Thé H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2:a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–16. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 11.Dellaire G, Ching RW, Ahmed K, Jalali F, Tse KCK, Bristow RG, et al. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J Cell Biol. 2006;175:55–66. doi: 10.1083/jcb.200604009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinton P, Giorgi C, Pandolfi PP. The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ. 2011;18:1450–6. doi: 10.1038/cdd.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–10. doi: 10.1038/35021000. [DOI] [PubMed] [Google Scholar]
- 14.Louria-Hayon I, Grossman T, Sionov RV, Alsheich O, Pandolfi PP, Haupt Y. The promyelocytic leukemia protein protects p53 from Mdm2-mediated inhibition and degradation. J Biol Chem. 2003;278:33134–41. doi: 10.1074/jbc.M301264200. [DOI] [PubMed] [Google Scholar]
- 15.Yang S, Jeong J-H, Brown AL, Lee C-H, Pandolfi PP, Chung JH, et al. Promyelocytic leukemia activates Chk2 by mediating Chk2 autophosphorylation. J Biol Chem. 2006;281:26645–54. doi: 10.1074/jbc.M604391200. [DOI] [PubMed] [Google Scholar]
- 16.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–7. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, Claret FX. Phosphatases: the new brakes for cancer development? Enzyme Res. 2012;2012:659649. doi: 10.1155/2012/659649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalev P, Sablina AA. Protein phosphatase 2A as a potential target for anticancer therapy. Anticancer Agents Med Chem. 2011;11:38–46. doi: 10.2174/187152011794941172. [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Phiel C. Functions of B56-containing PP2As in major developmental and cancer signaling pathways. Life Sci. 2010;87:659–66. doi: 10.1016/j.lfs.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bates S, Rowan S, Vousden KH. Characterisation of human Cyclin G1 and G2: DNA damage inducible genes. Oncogene. 1996;13:1103–9. [PubMed] [Google Scholar]
- 21.Arachchige Don AS, Dallapiazza RF, Bennin DA, Brake T, Cowan CE, Horne MC. Cyclin G2 is a centrosome-associated nucleocytoplasmic shuttling protein that influences microtubule stability and induces a p53-dependent cell cycle arrest. Exp Cell Res. 2006;312:4181–204. doi: 10.1016/j.yexcr.2006.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martínez-Gac L, Marqués M, García Z, Campanero MR, Carrera AC. Control of Cyclin G2 mRNA expression by forkhead transcription factors: novel mechanism for cell cycle control by phosphoinositide 3-kinase and forkhead. Mol Cell Biol. 2004;24:2181–9. doi: 10.1128/MCB.24.5.2181-2189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 24.Ragel BT, Couldwell WT, Gillespie DL, Jensen RL. Identification of hypoxia-induced genes in a malignant glioma cell line (U-251) by cDNA microarray analysis. Neurosurg Rev. 2007;30:181–7, discussion 187. doi: 10.1007/s10143-007-0070-z. [DOI] [PubMed] [Google Scholar]
- 25.Thomas KC, Sabnis AS, Johansen ME, Lanza DL, Moos PJ, Yost GS, et al. Transient receptor potential vanilloid 1 agonists cause endoplasmic reticulum stress and cell death in human lung cells. J Pharmacol Exp Ther. 2007;321:830–8. doi: 10.1124/jpet.107.119412. [DOI] [PubMed] [Google Scholar]
- 26.Fu G, Peng C. Nodal enhances the activity of FoxO3a and its synergistic interaction with Smads to regulate Cyclin G2 transcription in ovarian cancer cells. Oncogene. 2011;30:3953–66. doi: 10.1038/onc.2011.127. [DOI] [PubMed] [Google Scholar]
- 27.Kimura SH, Ikawa M, Ito A, Okabe M, Nojima H. Cyclin G1 is involved in G2/M arrest in response to DNA damage and in growth control after damage recovery. Oncogene. 2001;20:3290–300. doi: 10.1038/sj.onc.1204270. [DOI] [PubMed] [Google Scholar]
- 28.Zimmermann M, Arachchige-Don AS, Donaldson MS, Dallapiazza RF, Cowan CE, Horne MC. Elevated Cyclin G2 expression intersects with DNA damage checkpoint signaling and is required for a potent G2/M checkpoint arrest response to doxorubicin. J Biol Chem. 2012;287:22838–53. doi: 10.1074/jbc.M112.376855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Cui ZS, Luo Y, Jiang L, Man XH, Zhang X. Effect of Cyclin G2 on proliferative ability of SGC-7901 cell. World J Gastroenterol. 2004;10:1357–60. doi: 10.3748/wjg.v10.i9.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito Y, Yoshida H, Uruno T, Nakano K, Takamura Y, Miya A, et al. Decreased expression of Cyclin G2 is significantly linked to the malignant transformation of papillary carcinoma of the thyroid. Anticancer Res. 2003;23(3B):2335–8. [PubMed] [Google Scholar]
- 31.Kim Y, Shintani S, Kohno Y, Zhang R, Wong DT. Cyclin G2 dysregulation in human oral cancer. Cancer Res. 2004;64:8980–6. doi: 10.1158/0008-5472.CAN-04-1926. [DOI] [PubMed] [Google Scholar]
- 32.Kasukabe T, Okabe-Kado J, Honma Y. Cotylenin A, a new differentiation inducer, and rapamycin cooperatively inhibit growth of cancer cells through induction of Cyclin G2. Cancer Sci. 2008;99:1693–8. doi: 10.1111/j.1349-7006.2008.00867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Padua MB, Hansen PJ. Changes in expression of cell-cycle-related genes in PC-3 prostate cancer cells caused by ovine uterine serpin. J Cell Biochem. 2009;107:1182–8. doi: 10.1002/jcb.22222. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Z, Liu Y, He H, Chen X, Chen J, Lu Y-C. Candidate genes influencing sensitivity and resistance of human glioblastoma to Semustine. Brain Res Bull. 2011;86:189–94. doi: 10.1016/j.brainresbull.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 35.Okamoto K, Li H, Jensen MR, Zhang T, Taya Y, Thorgeirsson SS, et al. Cyclin G recruits PP2A to dephosphorylate Mdm2. Mol Cell. 2002;9:761–71. doi: 10.1016/S1097-2765(02)00504-X. [DOI] [PubMed] [Google Scholar]
- 36.Kimura SH, Nojima H. Cyclin G1 associates with MDM2 and regulates accumulation and degradation of p53 protein. Genes Cells. 2002;7:869–80. doi: 10.1046/j.1365-2443.2002.00564.x. [DOI] [PubMed] [Google Scholar]
- 37.Bennin DA, Don AS, Brake T, McKenzie JL, Rosenbaum H, Ortiz L, et al. Cyclin G2 associates with protein phosphatase 2A catalytic and regulatory B′ subunits in active complexes and induces nuclear aberrations and a G1/S phase cell cycle arrest. J Biol Chem. 2002;277:27449–67. doi: 10.1074/jbc.M111693200. [DOI] [PubMed] [Google Scholar]
- 38.Reimer CL, Borras AM, Kurdistani SK, Garreau JR, Chung M, Aaronson SA, et al. Altered regulation of Cyclin G in human breast cancer and its specific localization at replication foci in response to DNA damage in p53+/+ cells. J Biol Chem. 1999;274:11022–9. doi: 10.1074/jbc.274.16.11022. [DOI] [PubMed] [Google Scholar]
- 39.Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–6. [PubMed] [Google Scholar]
- 40.Liang X, Reed E, Yu JJ. Protein phosphatase 2A interacts with Chk2 and regulates phosphorylation at Thr-68 after cisplatin treatment of human ovarian cancer cells. Int J Mol Med. 2006;17:703–8. [PubMed] [Google Scholar]
- 41.Naito Y, Shimizu H, Kasama T, Sato J, Tabara H, Okamoto A, et al. Cyclin G-associated kinase regulates protein phosphatase 2A by phosphorylation of its B′γ subunit. Cell Cycle. 2012;11:604–16. doi: 10.4161/cc.11.3.19114. [DOI] [PubMed] [Google Scholar]
- 42.Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–80. doi: 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 43.Yabuta N, Okada N, Ito A, Hosomi T, Nishihara S, Sasayama Y, et al. Lats2 is an essential mitotic regulator required for the coordination of cell division. J Biol Chem. 2007;282:19259–71. doi: 10.1074/jbc.M608562200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.