

Abstract
Extracellular acidification, a mandatory feature of several malignancies, has been mainly correlated with metabolic reprogramming of tumor cells toward Warburg metabolism, as well as to the expression of carbonic anydrases or proton pumps by malignant tumor cells. We report herein that for aggressive prostate carcinoma, acknowledged to be reprogrammed toward an anabolic phenotype and to upload lactate to drive proliferation, extracellular acidification is mainly mediated by stromal cells engaged in a molecular cross-talk circuitry with cancer cells. Indeed, cancer-associated fibroblasts, upon their activation by cancer delivered soluble factors, rapidly express carbonic anhydrase IX (CA IX). While expression of CAIX in cancer cells has already been correlated with poor prognosis in various human tumors, the novelty of our findings is the upregulation of CAIX in stromal cells upon activation. The de novo expression of CA IX, which is not addicted to hypoxic conditions, is driven by redox-based stabilization of hypoxia-inducible factor-1. Extracellular acidification due to carbonic anhydrase IX is mandatory to elicit activation of stromal fibroblasts delivered metalloprotease-2 and -9, driving in cancer cells the epithelial-mesenchymal transition epigenetic program, a key event associated with increased motility, survival and stemness. Both genetic silencing and pharmacological inhibition of CA IX (with sulfonamide/sulfamides potent inhibitors) or metalloprotease-9 are sufficient to impede epithelial-mesenchymal transition and invasiveness of prostate cancer cells induced by contact with cancer-associated fibroblasts. We also confirmed in vivo the upstream hierarchical role of stromal CA IX to drive successful metastatic spread of prostate carcinoma cells. These data include stromal cells, as cancer-associated fibroblasts as ideal targets for carbonic anhydrase IX-directed anticancer therapies.
Keywords: cancer-associated fibroblasts, carbonic anhydrase IX, prostate cancer, epithelial-mesenchymal transition, acidity
Introduction
Carbonic anhydrases (CAs) are a family of zinc metalloenzymes that rapidly catalyze the hydration of carbon dioxide, producing bicarbonate and protons.1,2 At least thirteen human active CA isoenzymes are expressed in different tissues and/or subcellular compartments, thereby concurring in control of intracellular and extracellular pH (pHi/pHe), as well as regulation of metabolic pathways employing CO2/HCO3.3,4 CA IX is a transmembrane enzyme endowed with an extracellular membrane-bound catalytic domain that contributes to acidification of the outer microenvironment.2,5 The contribution of CA IX to acidification of the tumor environment has also been correlated with acquisition of metastatic phenotypes and chemoresistance to weakly basic anticancer drugs. Beside simple acid-base balancing, expression of CA IX was shown to be involved in several processes, such as cell adhesion and intercellular communication, and correlates with high incidences of metastasis and poor prognosis in various human tumors.6-8 Within tumors CA IX is mainly distributed in perinecrotic areas, likely due to its acknowledged regulation by hypoxia through hypoxia-inducible factor 1 α (HIF1-α).9,10 In keeping with its ability to sense hypoxia, CA IX-mediated extracellular acidification might be closely associated with breakdown of the extracellular matrix (ECM), growth factors and protease activation, although confirmatory data are still partial.
Metastasis, the hallmark of tumor malignancy and the most common cause of death for cancer patients, is the result of a complex series of steps, which starts with a loss of cell-cell adhesion, increased invasiveness into the surrounding tissues, crossing blood barriers and survival within bloodstream and colonization of a distant organ.11 The key event in such metastatic pathway is epithelial-mesenchymal transition (EMT), an epigenetic program leading cancer cells to engage a multifaceted escaping strategy to move from the hostile environment of primary tumor, granting survival and dissemination of epithelial cancers.12,13 Several components of the tumor microenvironment have been described to elicit or enhance the EMT of cancer cells, including intratumoral hypoxia and stroma cells as cancer-associated fibroblasts (CAFs).14 The latter are stromal cells behaving as a double-edged sword, as they physically and metabolically support cancer cells, but they are also reactive to cancer cell-secreted factors, thereby engaging a diabolic biunivocal relationship enhancing cancer cells aggressiveness. They mainly act mimicking a pro-inflammatory environment, to which cancer cells react engaging the EMT program.14-16
The aim of this work is to address the role of CA IX in EMT regulation in response to conditioning from tumor microenvironment effectors. We found that CA IX is dramatically upregulated in CAFs upon contact with cancer cells, thereby enhancing extracellular acidification, activation of MMP-2 and MMP-9-driven EMT and allowing spreading of spontaneous metastases in mice.
Results
CA IX is expressed in both PCa cells and CAFs
We have recently reported that stromal fibroblasts from aggressive prostate carcinoma cells undergo activation in response to factors secreted by cancer cells themselves, achieving a “reactive” state, allowing them to become CAFs. CAFs, in turn, secrete soluble factors, mainly identified as matrix metalloproteases (MMPs), able to elicit a pro-invasive behavior in prostate cancer (PCa) cells coincident with EMT.21 At the same time, we extracted human prostate fibroblasts (HPFs) from healthy individuals affected by prostate hyperplasia. We have already reported that in vitro the co-culture of HPFs with PCa cells, or HPFs treatment with PCa cells conditioned medium (CM), leads to HPFs activation toward the reactive state of CAFs.14,21 Here, we used HPFs and CAFs obtained by in vitro activation to analyze their role in extracellular acidification during the EMT process induced in PCa cells by CAFs contact. Immunoblot analysis on HPFs and CAFs revealed that only CAFs express CA IX, while HPFs do not express the enzyme (Fig. 1A). We also confirmed that CAFs isolated from patients bearing aggressive prostate carcinoma (for pathological parameters of individuals from which CAFs have been isolated, see ref. 22) express CA IX at similar levels with respect to in vitro activated CAFs (Fig. S1). Furthermore, PCa cells with different level of aggressiveness, i.e., LnCap, DU145 and PC3, show similar expression level of CA IX (data not shown). Nevertheless, the exposure of PCa cells to the CM from CAFs elicits an increase of expression of the enzyme (Fig. 1B).
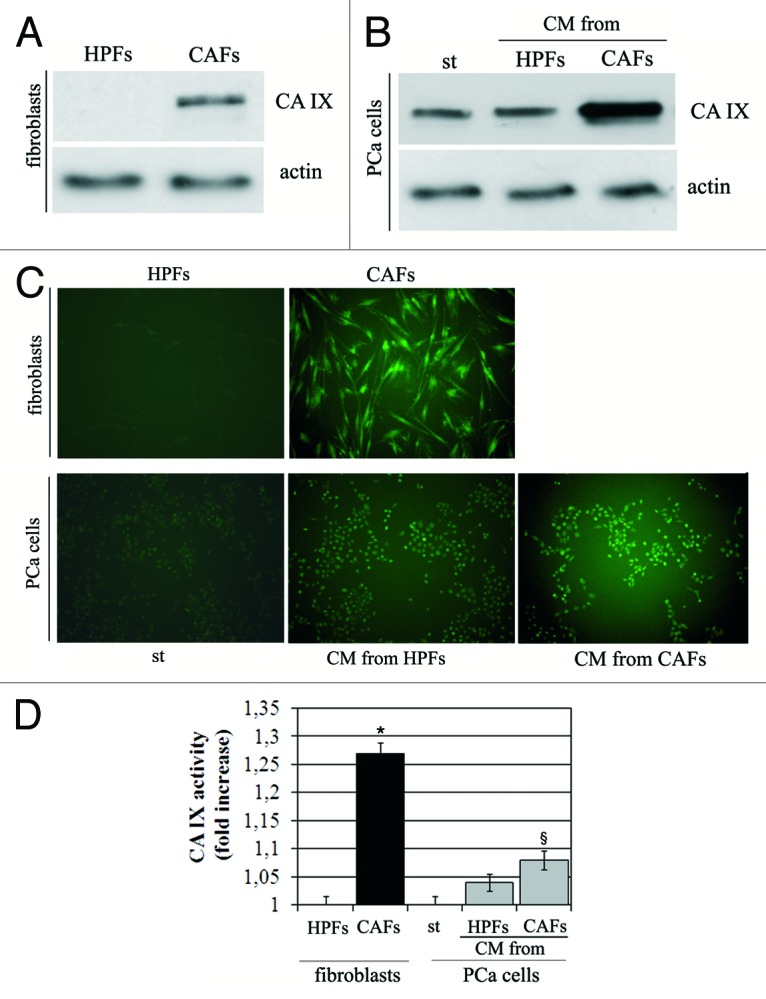
Figure 1. CAFs show high expression of activated CA IX. (A) Analysis of CA IX expression in HPFs and CAFs. Activation of fibroblasts was performed by treated HPFs with CM-PCa for 24 h. (B) Expression of CA IX in PCa cells. Prostate cells were treated with CM from HPFs or CAFs for 24 h. Actin immunoblot was used for normalization. (C) Analysis of CA IX activity by fluorescence microscopy and (D) by stop-flow analysis. *p < 0,001 vs. HPFs; §p < 0,01 vs. st.
The expression of CA IX in both CAFs or PCa cells upon exposure to their stromal cells is also correlated with increase in enzymatic activity of CA IX, as revealed by fluorescence analysis after the treatment of cells with the fluorescent probe FITC-labeled CAI#3, which targets the activated state of the extracellular catalytic domain of the enzyme (Fig. 1C), as well by stop-flow analysis (Fig. 1D). It should be mentioned that compound 3 is a potent CA IX inhibitor, with a KI of 24 nM against this isoform.18 It has also been proven that it binds the enzyme only in hypoxia and not normoxia.2 In both analyses, we observed that maximum activity is reached by CA IX expressed by CAFs.
HIF-1 activation mandatory for CA IX expression in both stromal CAFs and PCa cells
HIF-1 has already been reported as a master transcription factor involved in CA IX expression, although data on its involvement in the stromal counterpart are still at their infancy. We recently reported that, upon activation and contact with cancer cells, CAFs undergo a clear reprogramming toward Warburg metabolism, driven by a normoxic and redox-dependent stabilization of HIF-1.22 We therefore analyzed CA IX expression in CAFs and PCa cells in normoxia during HIF-1 inhibition following treatment with topotecan or N-acetyl cysteine (NAC), respectively, inhibiting the DNA transcriptional activity and the stability of the HIF1 transcription factor. Figure 2A and C shows that both treatments lead to the almost complete abolishment of CA IX expression in both stromal or cancer cells, respectively. In addition, we also silenced by RNA interfering HIF-1 in CAFs, confirming its mandatory role in CA IX expression control in prostate carcinoma microenvironment (Fig. 2B).

Figure 2. CA IX expression is HIF1-dependent both in CAFs and PCa cells. (A) Immunoblot analysis of ROS- and HIF1-dependance of CA IX in HPFs and CAFs. HPFs were cultured in serum-free medium or treated with CM-PCa for 24 h in the presence of NAC (20 mM) or topotecan (250 nM). (B) HIF1 silencing was performed in HPFs and after 48 h serum-free medium (for HPFs) or CM-PCa (for CAFs) were added to the cells for 24 h. (C) Immunoblot analysis of ROS- and HIF1-dependance of CA IX expression in PCa cells. NAC (20 mM) or topotecan (250 nM) were added to CM from HPFs or CM from CAFs and maintained throughout the experiment. In each immunoblot, actin was used for normalization.
CA IX expression leads to acidification of cancer-stromal environment
The contact between tumor cells and their stromal counterpart behaves as a propulsor factor to elicit a pro-invasive behavior of cancer cells, allowing them to lose cell-cell contacts, engage EMT and move away from primary tumor.14,21 To analyze the role of CA IX in cancer cell motility, we analyze the extracellular pH in normoxia in HPFs, CAFs and in PCa cells after the treatment with CM from HPFs or CAFs . We observed that the contact of CAFs with PCa cells gives rise to an increase in extracellular acidity, thereby suggesting that the reciprocal conditioning loop among stromal and cancer cells, involved in increasing motility, survival and stemness of cancer cells, also embraces acidification of tumor microenvironment (Fig. 3A). The use of two selective inhibitors of CA IX, namely compounds 1 and 2 (with an inhibition constant of 14 and 12 nM, respectively),19 leading to alcalinization of extracellular milieu, confirms that CA IX plays a mandatory role in such acidification (Fig. 3B). It should be mentioned that compound 1 is a membrane impermeant CAI due to its positively charged nature, whereas compound 2 is a membrane permeable compound.

Figure 3. CA IX induces the acidification of extracellular medium. (A) Measure of extracellular pH in HPFs, CAFs and in PCa cells after treatment with different CM. (B) Measure of extracellular pH in HPFs, CAFs and PCa cells after the treatment with CA IX inhibitors 1 (FC3-148 bis) and 2 (FC5–207A) for 48 h. *p < 0,001 vs. HPFs; #p < 0,01 vs. control; §p < 0,001 vs. control.
CA IX activity of CAFs enhances MMP-2 and MMP-9 secretion
We previously reported that the ability of CAFs to elicit a pro-invasive behavior of cancer cells is mainly due to a MMP-driven activation of a Rac1-dependent EMT program. Owing to the acknowledged sensitivity of MMPs to acidity, we investigated the involvement of an acidic environment due to CA IX expression in MMPs expression and activation. Our findings reveal that the ability of CAFs to secrete MMP-2 and MMP-9 is severely impaired during administration of the potent CA IX inhibitors 1 and 2 (Fig. 4A), as well as following elimination of CA IX expression by RNA interfering (Fig. 4B). Indeed, the activity of MMP-9, and to a smaller extent of MMP-2, is significantly decreased in CM collected from CA IX-silenced HPFs or CAFs, thereby confirming a key role played by CA IX in MMP-2 and MMP-9 secretion by CAFs (Fig. 4C).

Figure 4. The inhibition CA IX from CAFs decreases MMP-9. (A) Analysis of MMP-9 and MMP-2 in HPFs and CAFs after the treatment with the CA IX inhibitor 1 (FC3–148 bis) and 2 (FC5–207A). (B) CA IX silencing was performed in HPFs and CAFs, and then MMP-9 and MMP-2 were analyzed by zymography. Bar graphs show the level of MMP-2 and MMP-9 in HPFs and CAFs after the treatment with CA IX inhibitors. (C) Assay of MMP-2 and MMP-9 activity in CM from control- or CA IX-silenced CAFs. *p < 0,001 vs. control; #p < 0,01 vs. HPFs.
CA IX activity of CAFs is mandatory to drive activation of EMT in PCa cells
We observed that acidification of extracellular milieu, obtained omitting buffering of culture media for CAFs/PCa cells co-cultures, thereby allowing rapid acidification, greatly increased the ability of CAFs to drive the EMT program in PCA cells, as revealed by E-cadherin decrease and morphological analysis (Fig. 5A and B). The effects exerted by CAFs on cancer cells are intimately linked to the activation of an epigenetic program in cancer cells known as EMT and grant an invasive phenotype, survival to anoikis and achievement of stem-like traits.14,21 With the aim to dissect the role of CA IX-mediated acidification and MMPs secretion, we analyzed the sensitivity of CAFs-mediated EMT to genetic inhibition of CA IX. In keeping with morphological features (Fig. 5C) and E-cadherin expression (Fig. 5D), the modulation of CA IX expression is active on invasiveness of cancer cells (Fig. 5E). These data suggest that, within the tumor-stroma interplay, acidification due to CA IX expression plays a key role for activation of EMT, and backed on CAFs-expressed CA IX as the main source of the enzyme.

Figure 5. Modulation of CA IX expression affects EMT in PCa cells. (A) E-cadherin expression in HPFs, PCa cells and in PCa cells-CAFs co-colture cultured in DMEM buffered at pH 7 and in unbuffered medium (placed at 37°C without CO2) for 48 h. Cadherin expression was shown in bar graph. (B) Representative image of PCa cells treated as described in (A). (C) CA IX was silenced in HPFs and CAFs and CMs were harvested. PCa cells were then treated with serum-free medium (st) or different CMs for 72 h, and then cells were photographed. (D) Evaluation of E-cadherin expression in the same experimental setting described in (C). In each analysis actin immunoblot was used for normalization. a.u., arbitrary units. (E) Boyden assay of PCa cells after treatment for 18 h with different CMs obtained from HPFs or CAFs (control or silenced for CA IX). Bar graph represents the mean of invaded PCa cell (six fields for sample). *p < 0,001 vs. buffered medium; #p < 0,001 vs. control CAFs; §p < 0,001 vs. control CAFs.
MMP-9 from CAFs is a key player to drive activation of EMT in PCa cells
To confirm that MMP-2 and MMP-9 are mandatory factors in PCa cells invasiveness, we also used nanomolar competitive inhibitors of these two matrix proteases, namely GlcNAc-SLS-HA and Lac-SLS-HA. These inhibitors belong to an unprecedented family of atoxic, water-soluble arylsulfonamides characterized by a saccharidic residue (Glc, GlcNAc or Lactose) linked, through a spacer, to the sulfonamidic nitrogen.20 Except for MMP-1 and -7, all compounds showed low nanomolar Ki values for the MMPs tested (MMP-2, -8,-9,-12 and -13). This is not a surprise; as a matter of fact, given the active site topology of MMPs, most inhibitors share similar features, i.e., the ability to bind to the metal ion, to the hydrophobic pocket termed S1′, and to the substrate binding groove. Peculiarly, the inhibitors here tested present a hydrophilic chain that protrudes out of the protein toward the solvent region, allowing a complete water solubility without affecting the affinity for the enzymes. We observed that inhibition of MMP-2 and MMP9 is extremely active in impairing PCa cells invasiveness due to CAFs contact, preventing the elongation of the cells (Fig. 6A), the E-cadherin decrease (Fig. 6B) and, finally, PCa cell invasiveness (Fig. 6C).

Figure 6. The modulation of MMP-9 expression prevents EMT in PCa cells. (A) Representative images of PCa cells treated for 72 h with CM from HPFs and CAFs containing MMP-9 inhibitor GlcNAc-SLS-HA and Lac-SLS-HA. (B) Immunoblot of E-cadherin expression in the same experimental setting described in (A). (C) Invasion analysis of PCa cells in the presence of MMP-9 inhibitor GlcNAc-SLS-HA and Lac-SLS-HA. Bar graph represents the mean of invaded PCa cells (six fields for sample randomly chosen). (D) MMP-9 was silenced in HPFs and CAFs. Then, CM from HPFs or from CAFs (both control and MMP-9 silenced) were added to PCa cells for 72 h, and cells were photographed. (E) Immunoblot analysis of E-cadherin in PCa cells in the same experimental setting described in (D). Actin immunoblot is used for normalization. (a.u., arbitrary units). (F) Invasion assay of PCa cells treated with CM from control HPFs or CAFs or with CM from silenced-MMP-9 HPFs or CAFs. *p < 0,001 vs. control CAFs.
In addition, on the basis of the acknowledged hierarchy between MMP-2 and MMP-9, we also silenced MMP-9 and observed abolishment of EMT, as suggested by cell morphology (Fig. 6D; Fig. S2), stable maintenance of E-cadherin expression (Fig. 6E) and decreased invasiveness (Fig. 6F), thereby confirming the key role played by this protease in eliciting EMT and motility in PCa cells.
Furthermore, the addition of recombinant MMP-9 to CM collected from CA IX-silenced CAFs rescues the ability of PCa cells to undergo EMT, thereby confirming the hierarchy between CA IX and MMP-9 in EMT activation (Fig. S3).
Finally, we used a spontaneous metastasis assay in SCID mice to assess the role of CA IX expressed by stromal CAFs for tumor outgrowth and lung metastatic dissemination of cancer cells. We co-injected in the lateral flanks of mice PCa cells and CAFs (1:5 ratio). We already reported that in the absence of CAFs, PCa cells are unable to elicit tumors upon heterotopic injection, and that CAFs behave as synergistic bystanders eliciting survival and growth of cancer cells, allowing their escaping from primary tumors by EMT. We observed that CAFs silenced for CA IX elicit a delayed tumor onset and growth (Fig. 7A), and, more strikingly, that they are mostly unable to elicit metastatic dissemination of PCa cells to lungs (Fig. 7B), likely due to their inability to elicit EMT.

Figure 7. CA IX silencing in CAFs decreases tumor growth and lung micrometastasis formation. (A) Xenograft growth in SCID bg/bg mice of wild-type (control) or CA IX-silenced CAFs injected s.c. with PCa cells (CAFs:PCa cells ratio 1:5). (B) Bar graph shows the mean of lung micrometastasis. (C) Representative images of lung micrometastasis (arrows) in control and CA IX silenced treated mice. (D) Ca IX silencing by RNA interference in CAFs is shown. *p < 0,001 vs. control.
Discussion
CA IX has been found overexpressed by cancer cells from many tumor types, being a component of the pH regulatory system exploited by these cells to counteract the harmful effects of an highly glycolytic metabolism.1,23,24 Although the function of CA IX to maintain an intracellular pH favorable for tumor survival is widely acknowledged,1,25 its participation in the generation of an increasingly acidic extracellular environment and the consequential promotion of tumor cell invasiveness is still not completely understood.
So far, few indications suggest that CA IX contributes to cell adhesion and migration, vital processes for metastatic progression in human cancer. Expression of CA IX in normal epithelial cells leads to colocalization of CA IX with β-catenin and disruption of cell-cell adhesion.26 In keeping, in cervical carcinoma cells, the overexpression of exogenous human CA IX leads to weakened cytoskeletal remodeling, disassembled focal adhesion, cell-cell adhesion and increased cell motility, mainly acting through inactivation of Rho small GTPase.27 Although the authors do not dissect the molecular pathways leading CA IX-overexpressing cells to enhance their motility, they propose activation of EMT. Here we report a specific role of CA IX in the regulation of EMT of cancer cells, a key epigenetic program supporting the ability of tumor cells to evade the primary tumor microenvironment enhancing their motility, allowing their survival to a pro-oxidant and pro-inflammatory ambient, as well as favoring the expression of stem-like traits by cancer cells.12,28 In our model, EMT of cancer cells is the results of extracellular factors. We observed that prostate cancer cells actually undergo EMT in response to contact with their stromal fibroblasts, expressing CA IX as a result of their conversion to CAFs. Indeed, stromal CA IX leads to extracellular acidification, in turn enhancing CAFs MMP-2 and -9 activation and engaging EMT in cancer cells. We could argue that in the model proposed by Shin and colleagues,27 where CA IX is artificially overexpressed in cancer cells, Rho inactivation could be downstream with respect to extracellular acidification and activation of MMP-dependent EMT. In keeping with this idea, EMT drives cancer cells toward mesenchymal motility, a motility style depending on Rac small GTPase activation, driving cell polarization, lamellipodia formation, MMP activation and proteolysis of extracellular matrix.12,29,30 Indeed, Rho and Rac GTPases have been acknowledged to be reciprocally controlled through several mechanisms (30), and we have previously reported that PCa cells exposed to CAFs show a MMP-driven Rac activation and Rho inhibition, culminating in EMT and escaping from primary tumors.21,31
Although the upregulation of CA IX has already been reported for several cancer cells, mainly in response to intratumoral hypoxia,32,33 we now clearly indicate that the stromal fibroblasts infiltrating prostate carcinoma concur to extracellular acidification through de novo expression of CA IX in CAFs. CAFs are a relevant component of tumor microenvironment, being the most represented cells in tumor masses. They can either be fibroblasts resident in the target organ or recruited and differentiated by circulating mesenchymal stems cells of bone marrow origin.34,35 In any case, they respond to cancer-delivered factors able to enhance their reactivity toward a myo-fibroblast-like state called CAFs. In turn, CAFs are able to engage EMT in cancer cells, thereby affecting their motility, survival, stem-like traits expression and dissemination of metastatic colonies.14 A latere with respect to EMT, we recently involved CAFs in the metabolic reprogramming of prostate cancer cells. We described a circuitry in which PCa cells elicit a clear Warburg metabolism in CAFs which produce lactate uploaded by PCa cells showing a reverse Warburg phenotype through exploitation of lactate to fulfill anabolism and tumor growth.22 Hence, although CAFs can be view as acidifying cells through their dramatic production of lactate, extruded together with H+ using their overexpressed monocarboxylate transported-4, lactate and H+ are both rapidly removed by tumor microenvironment by cancer cells, expressing monocarboxylate transported-1 and uploading lactate for anabolic purposes.22 As acidification of tumor microenvironment is highly warranted for moving cells using mesenchymal motility style, due to MMP-sensitivity for low pH,36 the role of de novo expressed CA IX of CAFs appears mandatory. Indeed, membrane bound CA IX release H+ in the extracellular milieu, while the accompanying HCO3− is likely re-uploaded by the sodium bicarbonate co-transporter (NBCe1) associated with CA IX at the leading edges of moving cells.37 Extracellular acidity is mandatory to drive MMP-2 and MMP-9 secretion, rapidly leading to PCa cells EMT, enhancing their motility and metastatic dissemination in SCID mice. To our knowledge, this is the first report describing a specific role of the stromal component of tumors in the regulation of extracellular acidity, a feature commonly associated to tumor cells undergoing acidosis due to their Warburg metabolism. Relevantly, in tumors reprogrammed toward anabolism of lactate as prostate carcinoma,22 the stroma in place of tumor cells is forced to acidify the milieu to regulate MMP-driven EMT.
CA IX has been widely correlated with intratumoral hypoxia. Expression of CA IX by tumor cells is usually very low in normoxia, but its level is strongly increased during hypoxia due to a hypoxia-responsive element for HIF-1α located immediately upstream of the transcription start site of CA IX.24,32 Our data describe the expression of CA IX in stromal fibroblasts exposed to contact with prostate cancer cells, which elicits their activation toward a myofibroblast-like phenotype while they are maintained in normoxia. This finding is in keeping with the key role played by HIF-1 within tumor microenvironment, even in normoxic conditions.21,38,39 Indeed, stromal reactivity is under the strict control of HIF-1 transcriptional program. In breast and prostate carcinoma, both suffering of high stromal infiltration and undergoing constitutive oxidative stress, the activation of HIF-1 has been described to occur even in normoxia and to be dependent on redox-based stabilization of the transcription factor, allowed also without a strict decrease in oxygen concentration.21,38,39 The redox-based HIF-1 stabilization in CAFs suffering oxidative stress has been correlated with their metabolic reprogramming toward the Warburg phenotype22,40-42 or the ability to secrete stromal derived factor-1 and to attract endothelial precursors.22,31,39 We now involve the redox-based HIF-1 activation also in CA IX de novo expression in CAFs, mandatory for the acidification of extracellular environment and to elicit EMT through MMP-9 activation. All these events, although driven by HIF-1 transcription, do not strictly need hypoxia to occur, relying on other components of tumor microenvironment.
To date, at least in breast and renal carcinoma, CA IX-targeted therapeutic approaches based on monoclonal antibodies or small molecule inhibitors, have entered clinical trials and deserve promising expectations.43 In this light, our data have important pharmacological implications, as they include stromal fibroblasts as key regulators of extracellular acidification and adaptation strategies used by tumors, adding a further level of complexity to the system. CAFs, as they are non-transformed genetically stable cells, should be ideal pharmacological targets with respect to cancer cells,44,45 endowed with intrinsic adoptive features, especially if these CA IX-targeted agents will be administered in combination with conventional chemotherapy.
Materials and Methods
Materials
Unless specified, all reagents were obtained from Sigma. Antibodies anti-actin and E-cadherin, scramble siRNA-A (sc-37007), HIF1α-siRNA (sc-35561) and CA IX-siRNA (sc-29869), MMP-9-siRNA (sc-29400) were from Santa Cruz Biotechnolog. siRNA products from Santa Cruz generally consist of pools of three to five target-specific 19–25 nucleotide siRNAs designed to knockdown gene expression. Scramble shRNA plasmid (sc-108060) and CA IX shRNA plasmid (sc-29869-SH) from Santa Cruz was used for in vivo studies. Anti-CA IX antibodies were from Bioscience Slovakia. Anti-HIF-1α antibodies were from Becton Dickinson. Lipofectamine 2000 was from Invitrogen. The CA IX inhibitors 4-(2,4,6-trimethylpyridinium)-ethyl-benzenesulfonamide, perchlorate salt (compound 1) (FC3-148B bis), benzothiophene-3-ylmethylsulfamide (compound 2) (FC5-207A) and the fluorescent CAI (compound 3) were previously reported by one of our groups (19) (Fig. 8). Transwell system was from Costar. Diff-Quick solution for coloring boyden was from Medion Diagnostics AG.
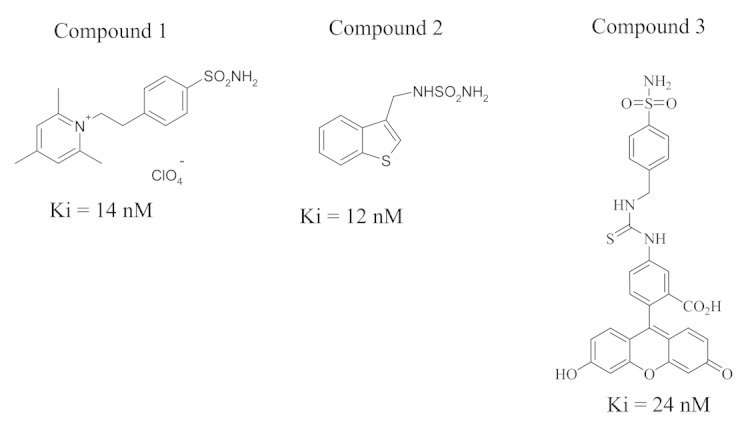
Figure 8. The chemical structure of the CA IX inhibitors used in the paper is shown.
Cell culture, silencing and treatment of the cells with CA IX and MMP-9 inhibitors
Human PCa cells (PC3) were from the European Collection of Cell Culture, were authenticated by PCR/STR (short tandem repeat) analysis (ECACC) and used within 6 mo of resuscitation of original cultures. Healthy human prostate fibroblasts (HPFs) and CAFs were isolated from surgical explantation of patients who signed informed consent in accordance with the Ethics Committee of Azienda Ospedaliera Universitaria Careggi and used until they reached ten passages. Tissues from patients affected by benign prostatic hyperplasia were used for obtaining HPFs.12 Conditioned media (CM) were obtained by 48-h serum-starved cells, clarified by centrifugation and used freshly. Co-culture was performed by plating HPFs and PCa cells in proportion 1:3. Silencing with siRNA or with CA IX shRNA plasmid was performed with Lipofectamine 2000 following manufacturer's instructions. At the end of each experiment, pH of the culture medium was immediately measured by pH meter. The Ca IX inhibitors 1 and 2 were added to cells at the beginning of the experiment at 100 uM final concentration. The MMP-9 inhibitors GlcNAc-SLS-HA and Lac-SLS-HA were added to CM (54 and 76 nM final concentration, respectively) and maintained throughout the experiment.
Fibroblasts and PCa cells activation
HPFs were grown to sub-confluence and treated for 24 h with CMPCa (obtained culturing PCa cells in serum-free medium for 48 h) to obtain in vitro activated CAFs that we already shown to behave similarly to in vivo extracted CAFs (from patients bearing aggressive carcinoma, see ref. 22). Fresh serum-free medium was added to HPFs for an additional 24 h before collection of CMHPF.
Western blot analysis
1 × 106 cells were lysed for 20 min on ice in 500 ul of complete radio-immunoprecipitation assay (RIPA) lysis buffer.17 Lysates were clarified by centrifugation, separated by SDS-PAGE and transferred onto PVDF. The immunoblots were incubated in 2% milk and probed with primary and secondary antibodies.
Analysis of CA IX activity
CA IX activity was analyzed either by confocal microscopy and fluorescence stop-flow. For confocal microscopy analysis, cells were cultured on glass coverslips and treated with appropriate medium depending on the experiments. After the treatment, cells were washed with PBS and the CA IX fluorescent probe FITC-labeled CAI#3 (1 mM, final) (compound 3)18 was added to living cells for 1 h. Cells were then washed extensively with PBS, mounted with glycerol plastine and observed with a fluorescence microscope (Leica TCS SP5).2 An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activity. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.4) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 sec. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionized water, and dilutions up to 0.001 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were pre-incubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3, as reported earlier, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in house as reported earlier.18,19
Metalloproteinase analysis
Zimography was performed using cultured media collected in our experimental conditions. Aliquots of cultured media were electrophoresed on 8% SDS-PAGE co-polymerized with 0.1% (w/v) type A gelatine. Gels were washed twice in 2.5% v/v Triton X-100 for 30 min and then incubated in 50 mM TRIS-HCl, pH 7.4, 200mM NaCl and 5 mM CaCl2 at 37°C for 24 h. After incubation, the gels were stained with 0.1% Coomassie brilliant blue in acetic acid, methanol and distilled water (1:2:3, respectively) for 60 min at room temperature. After destaining, the gels were immersed in distilled water and scanned immediately with Quantity-One Image Analysis software (Bio-Rad). Bands of gelatinase activity appeared as transparent areas against a blue background. Metalloproteinases activity was measured with AmpliteTM Universal Fluorimetric MMP Activity Assay Kit according to the manifacturer’s instructions.
Boyden invasion assay
Transwell system, equipped with 8-um pore polyvinylpirrolidone-free polycarbonate filters (6.5 mm diameter) were used. Cells were loaded into the upper compartment (1 × 105 PC3 cells in 200 uL of medium). The upper sides of the porous polycarbonate filters were coated with 50 ug/cm2 of reconstituted Matrigel and placed into 24-well culture dishes containing 500 ul of complete growth medium. After 18 h of incubation at 37°C, non-invading cells were removed mechanically using cotton swabs, and the micro porous membrane was stained with Diff-Quick solution. Chemotaxis was evaluated by counting the cells migrated to the lower surface of the filters (six randomly chosen fields).
Analysis of MMP-9 inhibitors by fluorimetric assays
The values of Ki for the compounds GlcNAc-SLS-HA and Lac-SLS-HA were determined by evaluating their ability to inhibit the hydrolysis of fluorescence-quenched peptide substrate Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 (Biomol, Inc). The analyses have been performed at 298 K in 50 mM HEPES buffer, containing 10 mM CaCl2, 0.05% Brij-35, at pH 7, using 1 nM of MMP catalytic domains and 1 uM of peptide. The experiments were performed with peptide concentration much lower than Ki. The fluorescence (excitation max 328 nm; emission max 393 nm) was measured for 3 min after the addition of the substrate using a Varian Eclipse fluorimeter. Fitting of rates as a function of inhibitor concentration provided the Ki values.20
Xenograft experiments
In vivo experiments were conducted in accordance with national guidelines and approved by the ethical committee of Animal Welfare Office of Italian Work Ministry and conform to the legal mandates and Italian guidelines for the care and maintenance of laboratory animals. Experiments were performed using 6–8-wk-old male severe combined immunodeficient (SCID)-bg/bg mice (Charles River Laboratories International). Animals (six per group) were monitored daily and tumor size was measured every 2–3 d by a caliper. Tumor volumes were determined by the length (L) and the width (W): V = (LW2)/2 (12).
Histology
Tissues were fixed in 4% (vol/vol) phosphate-buffered formalin and paraffin embedded. Consecutive sections with 5 μm thick were mounted on positively charged slides. Tissue sections were de-paraffinazed, re-hydrated and stained with hematoxylin-eosin or Azan-Mallory trichromic stainings. Metastases were counted using NIS ELEMENTS _ F-2.20 software combined with a microscope Nikon Eclipse 50i.
Statistical analysis
Data are presented as means ± SD from at least three independent experiments. Statistical analysis of the data was performed by Student’s t-test. p values of ≤ 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
Italian Association for Cancer Research (AIRC), The Tuscany Tumor Institute (ITT), the PORCREO Tuscany Project TUMAR and MIUR PRIN.
Glossary
Abbreviations:
- CA
carbonic anhydrase
- CAFs
cancer-associated fibroblasts
- CM
conditioned medium
- EMT
epithelial-mesenchymal transition
- HPF
human prostate fibroblasts
- PCa
prostate carcinoma
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24902
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24902
References
- 1.Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol. 2011;226:299–308. doi: 10.1002/jcp.22400. [DOI] [PubMed] [Google Scholar]
- 2.Švastová E, Hulíková A, Rafajová M, Zat’ovicová M, Gibadulinová A, Casini A, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004;577:439–45. doi: 10.1016/j.febslet.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 3.Thiry A, Dogné JM, Masereel B, Supuran CT. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci. 2006;27:566–73. doi: 10.1016/j.tips.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Zatovicova M, Sedlakova O, Svastova E, Ohradanova A, Ciampor F, Arribas J, et al. Ectodomain shedding of the hypoxia-induced carbonic anhydrase IX is a metalloprotease-dependent process regulated by TACE/ADAM17. Br J Cancer. 2005;93:1267–76. doi: 10.1038/sj.bjc.6602861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swietach P, Hulikova A, Vaughan-Jones RD, Harris AL. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene. 2010;29:6509–21. doi: 10.1038/onc.2010.455. [DOI] [PubMed] [Google Scholar]
- 6.Kim JY, Shin HJ, Kim TH, Cho KH, Shin KH, Kim BK, et al. Tumor-associated carbonic anhydrases are linked to metastases in primary cervical cancer. J Cancer Res Clin Oncol. 2006;132:302–8. doi: 10.1007/s00432-005-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee S, Shin HJ, Han IO, Hong EK, Park SY, Roh JW, et al. Tumor carbonic anhydrase 9 expression is associated with the presence of lymph node metastases in uterine cervical cancer. Cancer Sci. 2007;98:329–33. doi: 10.1111/j.1349-7006.2007.00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robertson N, Potter C, Harris AL. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res. 2004;64:6160–5. doi: 10.1158/0008-5472.CAN-03-2224. [DOI] [PubMed] [Google Scholar]
- 9.Chia SK, Wykoff CC, Watson PH, Han C, Leek RD, Pastorek J, et al. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J Clin Oncol. 2001;19:3660–8. doi: 10.1200/JCO.2001.19.16.3660. [DOI] [PubMed] [Google Scholar]
- 10.Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC, et al. Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res. 2001;61:7992–8. [PubMed] [Google Scholar]
- 11.Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell’s escape from oxidative stress. Cancer Metastasis Rev. 2010;29:351–78. doi: 10.1007/s10555-010-9225-4. [DOI] [PubMed] [Google Scholar]
- 12.Giannoni E, Parri M, Chiarugi P. EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxid Redox Signal. 2012;16:1248–63. doi: 10.1089/ars.2011.4280. [DOI] [PubMed] [Google Scholar]
- 13.Pani G, Giannoni E, Galeotti T, Chiarugi P. Redox-based escape mechanism from death: the cancer lesson. Antioxid Redox Signal. 2009;11:2791–806. doi: 10.1089/ars.2009.2739. [DOI] [PubMed] [Google Scholar]
- 14.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 15.Cat B, Stuhlmann D, Steinbrenner H, Alili L, Holtkötter O, Sies H, et al. Enhancement of tumor invasion depends on transdifferentiation of skin fibroblasts mediated by reactive oxygen species. J Cell Sci. 2006;119:2727–38. doi: 10.1242/jcs.03011. [DOI] [PubMed] [Google Scholar]
- 16.Chang J, Jiang Z, Zhang H, Zhu H, Zhou SF, Yu X. NADPH oxidase-dependent formation of reactive oxygen species contributes to angiotensin II-induced epithelial-mesenchymal transition in rat peritoneal mesothelial cells. Int J Mol Med. 2011;28:405–12. doi: 10.3892/ijmm.2011.683. [DOI] [PubMed] [Google Scholar]
- 17.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 18.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168–81. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 19.Alterio V, Hilvo M, Di Fiore A, Supuran CT, Pan P, Parkkila S, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA. 2009;106:16233–8. doi: 10.1073/pnas.0908301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calderone V, Fragai M, Luchinat C, Nativi C, Richichi B, Roelens S. A high-affinity carbohydrate-containing inhibitor of matrix metalloproteinases. ChemMedChem. 2006;1:598–601. doi: 10.1002/cmdc.200600020. [DOI] [PubMed] [Google Scholar]
- 21.Giannoni E, Bianchini F, Calorini L, Chiarugi P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid Redox Signal. 2011;14:2361–71. doi: 10.1089/ars.2010.3727. [DOI] [PubMed] [Google Scholar]
- 22.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130–40. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 23.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10:767–77. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 24.Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–83. [PubMed] [Google Scholar]
- 25.De Simone G, Supuran CT. Carbonic anhydrase IX: Biochemical and crystallographic characterization of a novel antitumor target. Biochim Biophys Acta. 2010;1804:404–9. doi: 10.1016/j.bbapap.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 26.Svastová E, Zilka N, Zat’ovicová M, Gibadulinová A, Ciampor F, Pastorek J, et al. Carbonic anhydrase IX reduces E-cadherin-mediated adhesion of MDCK cells via interaction with beta-catenin. Exp Cell Res. 2003;290:332–45. doi: 10.1016/S0014-4827(03)00351-3. [DOI] [PubMed] [Google Scholar]
- 27.Shin HJ, Rho SB, Jung DC, Han IO, Oh ES, Kim JY. Carbonic anhydrase IX (CA9) modulates tumor-associated cell migration and invasion. J Cell Sci. 2011;124:1077–87. doi: 10.1242/jcs.072207. [DOI] [PubMed] [Google Scholar]
- 28.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 30.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8:23. doi: 10.1186/1478-811X-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giannoni E, Taddei ML, Parri M, Bianchini F, Santosuosso M, Grifantini R, et al. EphA2-mediated mesenchymal-amoeboid transition induced by endothelial progenitor cells enhances metastatic spread due to cancer-associated fibroblasts. J Mol Med (Berl) 2013;91:103–15. doi: 10.1007/s00109-012-0941-9. [DOI] [PubMed] [Google Scholar]
- 32.Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011;71:3364–76. doi: 10.1158/0008-5472.CAN-10-4261. [DOI] [PubMed] [Google Scholar]
- 33.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168–81. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 34.Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012;31:195–208. doi: 10.1007/s10555-011-9340-x. [DOI] [PubMed] [Google Scholar]
- 35.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316:1324–31. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 36.Kato Y, Lambert CA, Colige AC, Mineur P, Noël A, Frankenne F, et al. Acidic extracellular pH induces matrix metalloproteinase-9 expression in mouse metastatic melanoma cells through the phospholipase D-mitogen-activated protein kinase signaling. J Biol Chem. 2005;280:10938–44. doi: 10.1074/jbc.M411313200. [DOI] [PubMed] [Google Scholar]
- 37.Svastova E, Witarski W, Csaderova L, Kosik I, Skvarkova L, Hulikova A, et al. Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J Biol Chem. 2012;287:3392–402. doi: 10.1074/jbc.M111.286062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiaschi T, Chiarugi P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int J Cell Biol. 2012;2012:762825. doi: 10.1155/2012/762825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med. 2010;2:211–30. doi: 10.1002/emmm.201000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiavarina B, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Tanowitz HB, Pestell RG, et al. Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle. 2012;11:3280–9. doi: 10.4161/cc.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, et al. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs “fertilizer”. Cell Cycle. 2011;10:2440–9. doi: 10.4161/cc.10.15.16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murri-Plesko MT, Hulikova A, Oosterwijk E, Scott AM, Zortea A, Harris AL, et al. Antibody inhibiting enzymatic activity of tumour-associated carbonic anhydrase isoform IX. Eur J Pharmacol. 2011;657:173–83. doi: 10.1016/j.ejphar.2011.01.063. [DOI] [PubMed] [Google Scholar]
- 44.Chometon G, Jendrossek V. Targeting the tumour stroma to increase efficacy of chemo- and radiotherapy. Clin Transl Oncol. 2009;11:75–81. doi: 10.1007/s12094-009-0317-y. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Liu J. Tumor stroma as targets for cancer therapy. Pharmacol Ther. 2013 doi: 10.1016/j.pharmthera.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.