

Abstract
Objective
To examine the interaction of moderate and high dietary fat and ethanol with respect to formation of steatosis and regulation of the AMP-activated protein kinase (AMPK) pathway in a mouse model of chronic ethanol consumption.
Methods
Male C57BL/6J mice were pair-fed a modified Lieber-DeCarli diet composed of either moderate fat (30% fat-derived calories (MF)) or high fat (45% fat-derived calories (HF)) combined with increasing concentrations of ethanol (2–6%) for 6-weeks.
Results
Chronic ethanol consumption resulted in significant increases in plasma alanine aminotransferase in MF (1.84-fold) and HF mice (2.33-fold), yet liver triglycerides only increased significantly in the HF model (1.62-fold). Ethanol addition significantly increased plasma adiponectin under conditions of MF but not HF. In combination with MF, the addition of ethanol significantly decreased total and hepatic pThr172AMPKα and acetyl CoA Carboxylase (ACC). HF plus ethanol decreased pSer108AMPKβ yet a marked 1.5-fold increase in pThr172AMPKα occurred. No change was evident in pSer79ACC under conditions of ethanol and HF ingestion. In both models, nuclear levels of SREBP1c, ChREBP were decreased. Surprisingly, MF plus ethanol significantly elevated protein expression of MCAD, LCAD and VLCAD but did not significantly affect mRNA expression of other proteins involved in β-oxidation and fatty acid synthesis. HF plus ethanol significantly reduced mRNA expression of both SCD-1 and ELOVL5, but did not have an effect on MCAD or LCAD.
Conclusion
These data suggest that when co-ingested with ethanol, dietary fat differentially contributes to dysregulation of adiponectin-dependent activation of the AMPK pathway in the liver of mice.
Keywords: alcohol, dietary fat, AMPK, hepatic steatosis
1. Introduction
Chronic alcoholic liver disease (ALD) is a significant contributor to liver based morbidity in humans. Steatosis is an early consequence of chronic alcohol consumption in mammals and continued drinking of excessive amounts of alcohol can subsequently lead to severe liver injury in the form of steatohepatitis, hepatic fibrosis and cirrhosis [1]. While the molecular mechanisms involved in the formation of alcohol induced steatosis remain to be elucidated, current research implicates cellular processes involved in regulating fatty acid metabolism including β-oxidation, fatty acid synthesis and lipid transport. The liver kinase B1 (LKB1)/AMP-activated protein kinase (AMPK) regulatory pathway plays a critical role in fatty acid metabolism [2; 3]. Phosphorylation of AMPK by LKB1 increases the ability of AMPK to phosphorylate and inactivate a major rate-limiting enzyme in fatty acid synthesis, namely acetyl CoA carboxylase (ACC). ACC catalyzes the conversion of acetyl CoA to malonyl CoA [4] which is then utilized in the synthesis of saturated and unsaturated fatty acids. At high ratios of AMP/ATP, AMPK activity is increased and ACC activity is decreased leading to a decrease in cellular malonyl CoA. If AMP/ATP ratios are low, AMPK activity is decreased and excess malonyl CoA is used in the production of de novo fatty acid synthesis [5]. Fatty acid synthase (FASN) catalyzes the production of saturated fatty acids such as palmitate from malonyl CoA and palmitate is converted to stearate via fatty acid elongases such as ELOVL6 and desaturated via stearoyl CoA desaturase-1 (SCD-1) [6].
Previous reports of the effects of chronic ethanol on the regulation of AMPK and β-oxidation/fatty acid synthesis are conflicting. Early studies using rats indicated that chronic ethanol decreased the activity or expression of FASN and SCD-1 in the liver and ACC and FASN in adipose tissue [7; 8; 9]. In other studies, there was no difference in FASN or ACC activity suggesting that de novo fatty acid synthesis does not play a major role in ethanol induced fatty liver [10]. More recent studies have demonstrated equally opposing results. On one hand, in C57BL6 mice, chronic ethanol feeding decreased AMPK phosphorylation and activity and led to a corresponding increase in ACC activity, fatty acid synthesis and mRNA levels of SCD-1, ATP citrate lyase (ACLY) and FASN [11; 12; 13; 14]. Other studies have found either no change or an increase in AMPK activity promoting a decrease in fatty acid synthesis in mice [15; 16]. Saturated fat in combination with ethanol has been shown to have an effect on the AMPK pathway as well. In a comparison of the effects of ethanol and saturated or unsaturated high fat diets, 40% saturated fats were found to increase AMPK phosphorylation and ameliorated steatosis and liver damage in mice [17]. All of these studies used variable amounts of fat, ethanol concentration and length of ethanol feeding.
Recent reports have suggested that the daily recommended allowance for dietary fat in humans is approximately 25–35% of total caloric intake. In the present study, we examined the effect of unsaturated dietary fat and ethanol on the AMPK pathway and expression of enzymes involved in fatty acid synthesis was examined. Mice were fed an ethanol-containing diet with either a moderate fat (30%) or a high fat (45%) diet for 6 weeks. We report that the addition of increased dietary fat leads to an increase in liver damage in conjunction with steatosis and contributes to dysregulation of AMPK signaling. Furthermore, under both dietary conditions, an overall decrease in expression of enzymes involved in fatty acid synthesis occurs following chronic ethanol consumption.
2. Materials and methods
2.1 Animal Model and dietary information
C57BL/6J male mice (The Jackson Laboratory, Bar Harbor, ME), 6–8 weeks of age in groups of 12 were fed a modified Leiber-DeCarli diet (30% fat-derived calories (MF) or 45% fat-derived calories (HF) (Table 1)) (Bio-Serv, Frenchtown, NJ) consisting of isocaloric control and ethanol-treated animals [18]. Ethanol content was increased each week starting with 2% (v/v) ethanol, with the ethanol-derived calories increasing by 1% on a weekly basis until sacrifice; week 6 consisted of 6 % ethanol (v/v) or 31.8% ethanol-derived calories. In the control animals, calories derived from ethanol were replaced isocalorically by carbohydrates in the form of maltodextrin. Food consumption was monitored daily and body weights were measured once per week. Upon completion of the study, animals were anesthetized via intraperitoneal injection with sodium pentobarbital and euthanized by exsanguination. Blood was collected from the inferior vena cava and plasma separated via centrifugation at 4°C and assayed for alanine aminotransferase (ALT) activity (Sekisui Diagnostics, P.E.I., Canada). Excised livers were weighed and subcellular fractions obtained via differential centrifugation as previously described (6). All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the University of Colorado and were performed in accordance with published National Institutes of Health guidelines.
Table 1.
Diet composition of moderate fat and high fat diets ± ethanol
| Diet composition of different groups | MF | MF+E | HF | HF+E |
|---|---|---|---|---|
|
| ||||
| Ethanol (kcal/liter) | - | 355 | - | 355 |
| Protein (kcal/liter) | 150 | 150 | 150 | 150 |
| Fat (kcal/liter) | 302 | 302 | 450 | 450 |
| C18:2 Linoleic Acid (gram/liter) | 17.8 | 17.8 | 26.7 | 26.7 |
| C18:3 Linolenic Acid (gram/liter) | 0.4 | 0.4 | 0.6 | 0.6 |
| Saturated Fat (gram/liter) | 4.3 | 4.3 | 6.4 | 6.4 |
| monounsaturated fat (gram/liter) | 9.2 | 9.2 | 13.7 | 13.7 |
| polyunsaturated fat (gram/liter) | 18.2 | 18.2 | 27.2 | 27.2 |
2.2 Western Blotting
Proteins from either whole liver extracts or subcellular fractions were subjected to standard SDS-PAGE and transferred to PVDF (GE Healthcare, Picataway, NJ). Membranes were blocked for 60 minutes with a tris-buffered saline solution containing 1% Tween-20 (TBST) and 5% non-fat dry milk and probed overnight with primary antibodies directed against pSer428LKB1, pThr172AMPKα, pSer108AMPKβ, pSer79ACC, total LKB1, total AMPKα, total AMPKβ, total ACC (All from Cell Signaling, Danvers, MA), FASN (Pierce-Thermofisher, Rockford, IL), SCD-1 (Santa Cruz Biotechnology, Santa Cruz, CA), ELOVL5 (ABCAM, Cambridge, MA), ACLY (ABCAM), β-actin (Sigma Aldrich, St. Louis, MO). A horseradish peroxidase conjugated secondary (Jackson ImmunoResearch Inc. West Grove, PA) was then applied and membranes developed using ECL-Plus Reagent (GE Healthcare). Chemiluminescence was visualized using either film or a Storm 860 scanner from Molecular Dynamics (Sunnyvale, CA).
2.3 Triglycerides
Liver triglycerides were extracted from whole liver homogenates using chloroform: methanol (2:1) and measured using a kit from Sigma-Aldrich according to the manufacturer’s protocol. All values were normalized against tissue weight.
2.4 Histological analysis
Sections of freshly excised liver tissue were placed in 10% neutral buffered formalin (Sigma-Aldrich) for 16 hours, washed in 70% ethanol for 1 hr followed by incubation in 70% ethanol overnight. Samples were then processed, embedded in paraffin and mounted on slides by the UC Denver Histology Core. Hematoxylin and Eosin staining was performed by the UC Denver Histology Core. IHC was performed using a rabbit polyclonal antibody directed against CYP2E1 (Millipore, Billerica, MA) as previously described (7). Images of H&E stained or IHC stained liver sections were captured on an Olympus BX51 microscope equipped with a four megapixel Macrofire© digital camera (Optronics, Goleta, CA) using the PictureFrame© Application 2.3 (Optronics, Goleta, CA).
2.5 Serum Adiponectin
Serum isolated from each group was diluted 1:1000 and examined for adiponectin via ELISA according to the manufacturer’s instructions (Millipore, Billerica, MA). Absorbances for each sample were read at 450nm and 595nm using a microtiter plate reader (Molecular Devices, San Diego, CA) and
2.6 Statistical Analysis
Relative densitometry of Western blots was quantified using ImageJ (http://rsb.info.nih.gov/ij/). All data and statistical analysis was performed using Graph Pad Prism 4.02 for Windows (GraphPad Software, San Diego, CA). When comparing moderate fat control and ethanol samples with high fat control and ethanol samples a two-way analysis of variance followed by Bonferroni post hoc correction was utilized. All data are expressed as mean ± S.E.M. and p values <0.05 were considered significant.
3. Results
3.1 Effects of increased dietary fat and ethanol on liver weight and liver to body weight ratio
The data presented in Table 2 describes the effect of the four feeding regimens on body weight, overall liver weight and liver/body weight ratio. Comparing the MF and the MF+E groups, chronic ethanol resulted in a 1.91-fold decrease in total weight gain in the MF animals. Comparing the HF and HF+E animals, the increase in dietary fat in conjunction with ethanol led to a 3.17-fold decrease in total weight gain. Overall, HF+E animals gained less weight than their MF+E counterparts. In addition, the HF control animals consumed more and gained more than the MF control animals. Comparing the average daily energy intake, in the moderate fat and the high fat groups, the ethanol-fed animals consumed slightly higher total kcal compared to controls. There were no significant differences in ethanol consumption between groups (Table 2). Examining the overall change in liver to body weight ratio, only the HF+E exhibited a statistically significant −1.15-fold change in overall liver to body weight ratio compared to their respective controls.
Table 2.
Physical characteristics of MF+E and HF+E mice.
| Initial Weight | Final Weight | Weight Change | Liver Weight | Percent liver/Body weight | Caloric Intake | |
|---|---|---|---|---|---|---|
|
| ||||||
| 30% Fat Control | 22.32±0.576a | 30.03±1.40a | 7.72±0.957a | 1.15±0.060a | 3.85±0.137a | 16.74±0.174a |
| 30% Fat Ethanol | 23.32±0.350a | 27.37±1.161a | 4.05±1.031a,b | 1.15±0.031a | 4.24±0.172a | 17.38±0.268a |
| 45% Fat Control | 21.92±1.385a | 31.02±0.752a | 9.10±1.237a | 1.15±0.038a | 3.78±0.097a | 17.00±0.205a |
| 45% Fat Ethanol | 22.92±1.247a | 25.78±0.647a | 2.87±1.169a,b | 1.12±0.036a | 4.36±0.080b | 17.78±0.326b |
Data are means ± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (MF±E compared to HF±E groups). Means without a common superscript letter are significantly different (N=6 mice/group (p<0.05)).
3.2 Biochemical characterization of animals fed MF+E or HF+E diet
The effects of MF+E or HF+E on serum ethanol concentrations, ALT and liver triglycerides, were examined (Table 3). Both MF+E and HF+E animals had significantly higher ALT values than their respective controls. Compared to MF plus ethanol, the addition of HF plus ethanol led to a greater increase in ALT values (MF+E 1.84-fold, HF+E 2.33-fold. Using 2-way ANOVA, there was no significant interaction between the increased addition of dietary fat and ethanol. Compared to isocaloric controls, only the addition of a high fat diet plus ethanol led to a significant increase in liver triglycerides (HF+E 1.62-fold). Once again however, a significant interaction between increased dietary fat and ethanol was not evident. The effect of dietary fat on ethanol metabolism as measured by serum ethanol concentrations was assessed. From the data in Table 3, changes in dietary fat did not impact blood ethanol concentrations.
Table 3.
Biochemical analysis of serum and liver homogenates from MF+E and HF+E mice at the end of the 6 week feeding period. Serum ALT, liver triglycerides and blood ethanol content were determined as described in methods.
| ALT (U/ml) | Triglycerides (rnmol/L/mg tissue) | Blood ethanol mg/dl | |
|---|---|---|---|
|
| |||
| 30% Fat Control | 14.18±1.52a | 0.18±0.01a | |
| 30% Fat Ethanol | 26.13±3.28b | 0.20±0.01a | 254.4±30.7 |
| 45% Fat Control | 18.41±4.61a | 0.15±0.02a | |
| 45% Fat Ethanol | 42.99±4.52b | 0.24±0.03b | 245.4±33.8 |
Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (MF±E compared to HF±E groups). Means without a common superscript are significantly different (N=6 mice/group (p<0.05)).
3.3 Hematoxylin and Eosin staining show large cytoplasmic lipid droplets in both the MF+E and HF+E livers
Corresponding to an increase in liver triglycerides, hematoxylin and eosin staining of liver sections presented in Figure 1 indicate an increase in macrosteatosis in both MF+E and HF+E liver sections when compared to sections from their respective controls. Steatosis was predominantly located in zone 2 with minor accumulation of lipid droplets in zone 1.
Fig. 1.

Increased steatosis in zone 2 in MF+E and HF+E mice. Hematoxylin and eosin staining of tissue sections from MF+E and HF+E mice. (CV, central vein, PT, portal triad). Original Magnification 400X.
3.4 Induction of CYP2E1 in MF+E and HF+E livers
The ingestion of ethanol is well documented to induce hepatic Cytochrome 450 2E1 [19; 20]. To evaluate the extent of CYP2E1 induction, IHC was performed on tissue sections from MF+E and HF+E mice. The data presented in Figure 2A demonstrate substantial induction of CYP2E1 visible in=both the MF+E and HF+E mice. The MF+E animals exhibited slightly less intense staining than the HF+E animals. To validate these data, a Western blot was performed. As shown in Figure 2B and 2C, CYP2E1 expression is increased in the MF+E and HF+E groups. Subsequent quantification of CYP2E1 expression demonstrated a 2-fold increase in livers from MF+E and a 4-fold increase in the HF+E mice compared to pair-fed controls, which is consistent with the immunohistochemistry data. Two-way ANOVA indicated a significant interaction between dietary fat and ethanol in the induction of CYP2E1.
Fig. 2.


Chronic ethanol leads to increased CYP2E1 in MF+E and HF+E mice (A) Immunohistochemical analysis of CYP2E1 expression in MF, MF+E, HF and HF+E mice. Note positive staining in centrilobular region (zone 3) in both ethanol feeding models. (CV, central vein; PT, portal triad) Magnification 400X. (B) Western blotting analysis of CYP2E1 expression in MF, MF+E, HF and HF+E mice. (C) Quantification of the Western blots presented in Figure 2B (actin normalized). Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (MF±E compared to HF±E groups). Means without a common superscript letter are significantly different (N=6 mice/group (p<0.05)).
3.5 Dysregulation of AMPK signaling by dietary fat and ethanol in mice
The LKB1/AMPK pathway contributes to regulation of metabolic pathways in the liver. The effects of dietary fat on LKB1 and its downstream signaling intermediates were analyzed via total protein expression and phosphorylation of LKB1, AMPKα/β and ACC (Figure 3A and 3B). Compared to isocaloric controls, MF+E led to a decrease in total expression of LKB1, AMPKα and ACC. Increasing dietary fat content led to no significant change in expression of LKB1, AMPKα or ACC when compared to the respective HF control animals. The LKB1/AMPK pathway is regulated by phosphorylation. As shown in Figure 3C and 3D, pSer428LKB1 increased significantly in the MF+E mice and decreased significantly in HF+E mice. The immediate substrate of LKB1 is the heterotrimer AMPKα/β/γ [21]. In MF+E animals, levels of pThr172AMPKα significantly decreased. The addition of HF+E led to a significant increase in pThr172AMPKα but surprisingly, pSer108AMPKβ significantly decreased. AMPK phosphorylates Ser79ACC thereby inhibiting its activity and promoting β-oxidation. Phosphorylation of ACC significantly decreased in the MF+E but not the HF+E mice. Utilizing two-way ANOVA, a significant interaction was identified between dietary fat and ethanol for both pSer428LKB1 and pThr172AMPKα.
Fig. 3.

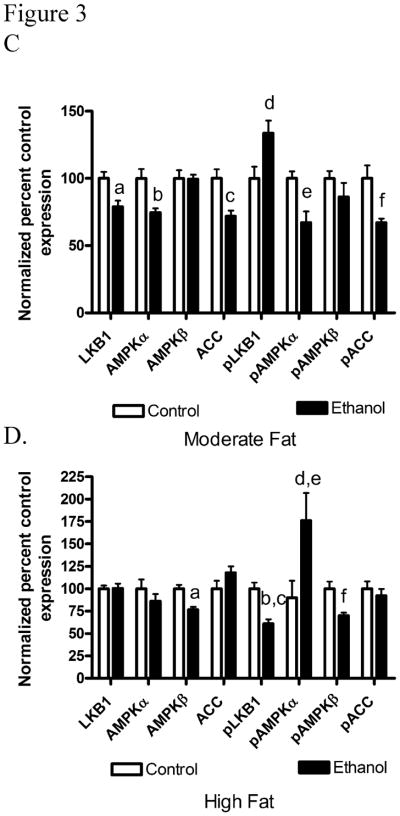

Chronic ethanol leads to changes in regulation of the AMPK pathway in MF+E and HF+E mice. Cytosolic fractions from both MF and HF control and ethanol groups were analyzed via standard immunoblotting and probed for total LKB1, AMPKα, AMPKβ, ACC expression and phosphorylation (pSer428 LKB1, pThr172 AMPKα, pSer108AMPKβ, pSer79 ACC) using rabbit polyclonal antibodies (A) MF+E, (B) HF+E, (C) Quantification of the Western blots (actin normalized). (D) Adiponectin levels in moderate fat and high fat ethanol models. Serum isolated from MF and HF control and ethanol groups was analyzed for adiponectin via ELISA. Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (MF±E compared to HF±E groups). Means without a common superscript letter are significantly different (N=6 mice/group (p<0.05)).
Serum adiponectin has been identified as an extracellular regulator of AMPK. Increased serum adiponectin correlates with increased AMPK phosphorylation and activation [22]. From Figure 3E, serum adiponectin was significantly increased in the MF+E animals. Ethanol did not have a significantly effect on adiponectin in the HF+E model. Using 2-way ANOVA, a significant interaction was present between increasing dietary fat but not with the addition of ethanol. Given that serum adiponectin was increased under MF+E conditions, relative mRNA expression was determined using a limited microarray analysis (Supplementary Table 1). From the array, there were no significant differences in hepatic mRNA expression of Acrp30, ADIPOR1, ADIPOR2 or PNPLA2 in either model.
3.6. Chronic ethanol-induced changes in mitochondrial acyl-CoA dehydrogenases in MF and HF mice
In other animal models, increases in AMPK phosphorylation have been implicated in increased expression of enzymes involved in mitochondrial β-oxidation. As described above, AMPK can promote β-oxidation by phosphorylating Ser79 of ACC and inhibiting its activity and phosphorylation of ACC significantly decreased in the MF+E but not the HF+E mice. In order to further characterize the effects of dietary fat in ALD we examined the effects of MF+E and HF+E on mitochondrial fatty acyl-CoA dehydrogenase (VLCAD, MCAD, LCAD, SCAD) expression by Western blotting. Compared to isocaloric controls, MF+E led to a significant increase in VLCAD, LCAD and MCAD. In the HF+E animals, only VLCAD was significantly elevated.
3.7 Chronic ethanol induced changes in nuclear ChREBP and SREBP1 in HF and MF mice
Nuclear localization of both SREBP1c and ChREBP are involved in regulation of de novo lipogenesis [6]. AMPK has been implicated in the regulation of both SREBP1 and ChREBP. Phosphorylation of ChREBP by AMPK prevents nuclear translocation [23]. Nuclear localization of SREBP1c and ChREBP were examined in both the MF and HF models. As shown in Figures 5A–C, compared to isocaloric controls, levels of nuclear SREBP1c trended down in MF+E and significantly decreased in HF+E mice. Nuclear localization of ChREBP significantly decreased in MF+E mice and trended down in HF+E mice. Corresponding to the decrease in ChREBP nuclear localization, cytosolic levels of ChREBP were increased in both models.
Fig. 5.

Downregulation of nuclear SREBP and ChREBP in MF+E and HF+E mice. Nuclear or cytoplasmic fractions from both MF and HF control and ethanol groups were analyzed via SDS PAGE, Western blotted and probed for nSREBP and nChREBP nuclear localization or cChREBP cytoplasmic localization using rabbit polyclonal antibodies (A) MF+E (B) HF+E. (C) Quantification of the Western blots (actin or lamin AC normalized). Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (MF±E compared to HF±E groups). Means without a common superscript letter are significantly different (N=6 mice/group (p<0.05)).
3.7 Chronic ethanol decreases expression of enzymes involved in de novo fatty acid synthesis in MF and HF mice
Decreased levels of nuclear SREBP and ChREBP have been demonstrated to result in decreased fatty acid synthesis. In addition, dephosphorylation of ACC promotes fatty acid synthesis. Overall protein expression of key enzymes in fatty acid synthesis ACLY, FASN, ELOVL5 and SCD-1 were examined by Western blotting. Consistent with the reduced nuclear localization of SREBP and ChREBP, quantification of the data in Figure 6A and 6B demonstrate that addition of ethanol reduced expression of ACLY and SCD-1 in both the moderate fat and the high fat groups (Figure 6C). Utilizing 2-way ANOVA, SCD-1 indicated a significant interaction with dietary fat and ethanol on expression.
Fig. 6.

Chronic ethanol leads to decreased expression of fatty acid synthetic enzymes in both MF+E and HF+E mice. Cytosolic or microsomal fractions from both MF and HF control and ethanol groups were analyzed via Western blotting and probed for ACLY, FASN, SCD-1 and ELOVL5 (A) MF+E (B) HF+E. (C) Quantification of the Western blots. Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (MF±E compared to HF±E groups). Means without a common superscript letter are significantly different (N=6 mice/group (p<0.05)). All blots were normalized to actin.
3.8 mRNA expression of proteins regulated by AMPK signaling
Activation or inactivation of AMPK can result in increased or decreased expression of proteins involved in both de novo lipogenesis, fatty acid transport and β-oxidation. To provide additional data with respect to AMPK signaling a limited microarray was performed using an arbitrary 2-fold cut-off in gene induction/repression [24]. As shown in Supplementary Table 1, examining de novo lipogenesis, in the MF+E fed animals there were no significant changes in mRNA expression in FAS, GPAT1, SCD-1, ME, ELOVL2,3,5 or 6. In the HF+E animal, chronic ethanol suppressed expression of SCD-1 (−3.51 fold), ELOVL5 (−2.00 fold) and ELOVL3 (−6.51 fold). All other proteins involved in de novo lipogenesis did not significantly differ from those observed in the controls. Examining β-oxidation, surprisingly, the MF+E animals did not exhibit significant differences in CPT-1α, ACOX1, MCAD, SCAD, LCAD, PPARα, or PGC1α mRNA expression. In the HF+E animals, only PPARα (−2.53 fold) exhibited a significant change in mRNA expression. Given that in both models there is increased lipid accumulation that may affect lipid transport expression of fatty acid transport genes was examined. From Supplementary Table 1, there were no significant differences in either model in mRNA expression of CD36 or MTTP.
4. Discussion
Depending on the amount of alcohol consumed, approximately 90% of chronic alcoholics will develop fatty liver or steatosis as an early marker of liver damage [25]. Of these, 10–35% will progress from simple steatosis to steatohepatitis and 5–15% will develop fibrosis. The biochemical mechanisms which lead to steatosis in ALD are not fully characterized but it is clear that multiple factors contribute to disease progression [26]. These include increased lipid peroxidation, oxidative stress and dysregulation of lipid metabolism [2; 27; 28; 29]. In this study, the effects of chronic ethanol feeding combined with either 30% or 45% dietary fat was examined. Consistent with previous reports, increasing the total amount of dietary fat significantly increased the amount of hepatic triglycerides and hepatocellular damage as determined by ALT [30]. Surprisingly, in the present study there is not a significant difference when the moderate fat groups are compared to the high fat groups.
In ALD, CYP2E1 is involved in the detoxification of ethanol [18; 31]. Previous reports have suggested an effect of dietary fat and CYP2E1 expression in rats [30; 32]. The addition of dietary medium-chain triglycerides instead of long chain triglycerides led to a decrease in overall liver damage and CYP2E1 expression [32]. In the present study, the relative composition of fatty acids remained the same (primarily long chain) and the total amount of long chain fatty acids was replaced with carbohydrates in the form of maltodextrin (MF+E). Further, MF+E exhibited less induction of CYP2E1, reduced triglyceride accumulation and hepatocellular damage when compared to HF+E. This is in agreement with previous studies that indicated that decreasing dietary fat along with increased carbohydrates resulted in reduced ALT, reduced triglyceride accumulation and reduced CYP2E1 induction [33]. During ALD, CYP2E1 induction occurs primarily in the perivenous region [31; 34]. In this study, tissue examination of the MF+E and HF+E mice revealed less intense staining of CYP2E1 in the MF+E animals when compared to HF+E animals. Concurrently, MF+E exhibited less reduction of CYP2E1 expression in the periportal region.
Cyclic AMP activated kinase is activated under conditions of low energy and its corresponding AMP/ATP ratio. Alternatively, increased serum adiponectin results in an increase in AMPK activation via adiponectin receptors 1 and 2 [22]. In the literature, reports concerning the effects of ethanol on activation of the AMPK pathway in mice vary depending on the amount of ethanol and the duration of feeding. In some studies, AMPK phosphorylation is increased whereas in others, AMPK phosphorylation is decreased [12; 14; 15; 35]. In one report, 40% saturated fat plus ethanol resulted in a 2-fold increase in AMPKα phosphorylation. Concurrently, 40% PUFA plus ethanol resulted in a slight decrease in AMPKα phosphorylation [17]. This further substantiates the role that dietary fat plays in regulation of AMPK by ethanol. In the present study, adiponectin levels are elevated in the MF+E model but this did not correlate with increased mRNA expression of ADIPOR1/2 or translate to an increase in pThr172AMPK and pSer79ACC. This has been demonstrated in another model that utilized hypertensive rats. In that model, increased hypertension led to elevated liver triglycerides and adiponectin but phosphorylation of AMPKα and ACC were decreased [36]. Feeding mice a high fat diet alone is known to reduce adiponectin and AMPK phosphorylation [37; 38]. In our model, HF+E did not result in an increase in adiponectin but there is an increase in AMPKα phosphorylation. Surprisingly this did not correspond to an increase in phosphorylation of AMPKβ and ACC. Clearly, in our model, the concomitant presence of high dietary fat with ethanol results in dysregulation of AMPK signaling. This effect has been previously identified in an overfeeding/obesity intragastric model, whereby AMPK phosphorylation was induced by ethanol yet ACC phosphorylation was decreased [15]. In that study, the authors did not examine AMPKβ phosphorylation. Phosphorylation of AMPKβ is required for full activation of the AMPK heterotrimer [39].
The effects of chronic ethanol administration on phosphorylation of LKB1 have not previously been examined. In this study, LKB1 phosphorylation remains decreased in both the MF+E and HF+E model whereas expression is only decreased in the MF+E model. One hypothesis is that phosphorylation of LKB1 at Ser428 is required for activation and subsequent AMPKα phosphorylation at Thr172 [40]. Other research has suggested that activation of LKB1 is independent of Ser428 phosphorylation [41]. Thus, based on our results, in the HF+E mice, the increase in AMPK phosphorylation may be independent of LKB1 phosphorylation.
In the liver, both ChREBP and SREBP regulate hepatic lipogenesis. Both transcription factors bind to promoters and positively affect transcription of lipogenic proteins such as ACC, FASN, SCD-1 and ELOVL6 [6; 42; 43; 44; 45; 46]. ChREBP is in part regulated by the ability of AMPKα to phosphorylate Ser568. Dephosphorylation results in increased nuclear localization of ChREBP [23; 47]. The decrease in AMPK downstream signaling may provide a mechanism for decreased ChREBP nuclear localization in MF+E and HF+E models.
In mouse models of nonalcoholic fatty liver disease, AMPKα is a negative regulator of SREBP1c. Increased AMPKα phosphorylation typically results in a decrease in nuclear SREBP1c. In both the intragastric model and the NAFLD human patient study, levels of SREBP1c were increased [3; 15]. This suggests an additional level of regulation between AMPKα/ACC and SREBP1c. Other studies however clearly indicate no change in SREBP following chronic ethanol in rats [48]. In the present study, levels of SREBP1c trend down in both MF+E and HF+E treated mice indicating dysregulation of signaling between AMPK and SREBP. Corresponding to the decrease in SREBP1c, nuclear localization of ChREBP is also decreased in HF+E and MF+E mice.
In the ALD literature, there are also opposing results with respect to activity and mRNA expression and protein expression of lipogenic proteins. Early studies demonstrated a decrease in SCD-1 and FASN enzymatic activities following chronic ethanol consumption in rats [7; 8]. More recently, Northern blotting and mRNA expression indicated ethanol-mediated increases in SREBP1c, Scd1, FASN, ACLY, ELOVL2, ELOVL3 and ACC expression [11; 13; 49]. These studies were all for 4 weeks and utilized either a 10% low fat diet (cocoa butter/safflower oil) in combination with ethanol or dietary fat content was not stated. The 10% low fat studies also found a substantial decrease in AMPKα phosphorylation. In the MF+E animals, AMPKα phosphorylation is reduced but not to the levels demonstrated by You et al.[12]. It may be that further decreasing dietary fat content may result in lower AMPKα activation. In summary, the discrepancy between previous low fat reports and our data may be due the length of ethanol administration, the quantity and source of dietary fat or a combination of both.
Examining the high fat model, another report using a 6-week chronic ethanol feeding regimen and the Lieber DeCarli diet suggests either no significant change (SREBP1c, FASN, ACC) or downregulation of mRNA expression (Scd1, ELOVL5) following chronic ethanol administration [50]. We also find that chronic ethanol feeding utilizing a similar feeding protocol, Scd-1 and ELOVL5 are suppressed. Thus increasing dietary fat content in combination with length of ethanol administration can lead to changes in expression of de novo lipogenesis (DNL) enzymes. The decrease in DNL expression in this study does not explain why there are increased levels of steatosis and triglycerides. Investigators have previously suggested that there are different pools of fatty acid CoA within hepatocytes and that ethanol can differentially regulate these pools [50]. Based on our data and others, it is possible that early steatosis formed during chronic ALD under high fat conditions is not due to de novo fatty acid synthesis or changes in expression of β-oxidation but due to alterations of other pathways [10; 50; 51]. There is data to support this hypothesis. Several studies have implicated defective VLDL secretion as a contributing factor in ethanol-induced steatosis [52]. In rats, chronic ethanol feeding resulted in decreased perivenous VLDL secretion [53]. The addition of betaine increased VLDL production and attenuated the development of alcoholic steatosis [54].
In summary, when compared to a moderate fat diet, the addition of a high fat diet plus ethanol leads to significant increases in liver damage, triglycerides and CYP2E1 expression. However, a high fat diet subtlety changes regulation of AMPK. As displayed in Figure 7, the increase in dietary fat may contribute to an increase in AMPKα phosphorylation but there is dysregulation of AMPK downstream signaling as shown by the lack of increased phosphorylation of ACC. This corresponds to no significant changes in mRNA of β-oxidation dependent genes. In the moderate fat animals, addition of ethanol resulted in increased serum adiponectin but phosphorylation of AMPKα did not increase. This suggests that regulation of adiponectin signaling may be defective under conditions of moderate fat plus ethanol. Both models display decreased nuclear localization of SREBP and ChREBP resulting in downregulation of fatty acid synthetic enzymes. Thus, depending on dietary fat content, ethanol may dysregulate hepatocyte signaling on different levels.
Fig. 7.

Summary of the effects of increased dietary fat plus ethanol on the AMPK pathway and expression of enzymes involved in fatty acid synthesis. In combination with ethanol, a moderate fat diet leads to increased LKB1 phosphorylation, decreased ACC phosphorylation, but no significant change in AMPK phosphorylation. When dietary fat is increased, chronic ethanol consumption decreases LKB1 phosphorylation, increases AMPK phosphorylation but has no significant effect on ACC phosphorylation. In both models, nuclear localization of SREBP1c and ChREBP are decreased corresponding to decreased expression of fatty acid synthetic enzymes. (dec-decreased, inc-increased, uc-unchanged).
Supplementary Material
Fig 4.

Effects of increasing dietary fat and ethanol on mitochondrial fatty acyl-CoA dehydrogenase expression. Whole cell lysates from both MF and HF control and ethanol groups were analyzed via standard immunoblotting and probed for VLCAD, LCAD, MCAD and SCAD expression using polyclonal antibodies. (A) MF+E, (B) HF+E, (C) Quantification of the Western blots (GAPDH normalized). Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (MF±E compared to HF±E groups). Means without a common superscript letter are significantly different (N=6 mice/group (p<0.05)).
Acknowledgments
Grants and funding: This research was funded by the following grants from the National Institutes of Health; 5F32AA018613-02 CTS, 5R37AA009300-16 DRP, 5R01DK074487-05 DRP
Abbreviations
- ACC
acetyl CoA carboxylase
- ALD
alcoholic liver disease
- ALT
alanine amino transferase
- AMPK
AMP-activated protein kinase
- ACLY
ATP citrate lyase
- ChREBP
carbohydrate response element binding protein
- CYP2E1
cytochrome p450 2E1
- ELOVL5
fatty acid elongase 5
- ELOVL6
fatty acid elongase 6
- FASN
fatty acid synthase
- LKB1
liver kinase B1
- LCAD
long chain acyl-CoA dehydrogenase
- MCAD
medium chain acyl-CoA dehydrogenase
- ME
malic enzyme
- PP2A
protein phosphatase 2A
- SCAD
short chain acyl-CoA dehydrogenase
- SREBP
sterol response element binding protein 1
- SCD-1
stearoyl CoA desaturase 1
- VLCAD
very long chain acyl CoA dehydrogenase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51:307–28. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 2.Sozio M, Crabb DW. Alcohol and lipid metabolism. Am J Physiol Endocrinol Metab. 2008;295:E10–6. doi: 10.1152/ajpendo.00011.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Enjoji M, Takayanagi R, Nakamuta M. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med. 2008;21:507–11. [PubMed] [Google Scholar]
- 4.Harris RA, Crabb DW. Inhibition of hepatic gluconeogenesis by dichloroacetate. Arch Biochem Biophys. 1978;189:364–71. doi: 10.1016/0003-9861(78)90223-0. [DOI] [PubMed] [Google Scholar]
- 5.Miller RA, Birnbaum MJ. An energetic tale of AMPK-independent effects of metformin. J Clin Invest. 2010;120:2267–70. doi: 10.1172/JCI43661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–38. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umeki S, Shiojiri H, Nozawa Y. Chronic ethanol administration decreases fatty acyl-CoA desaturase activities in rat liver microsomes. FEBS Lett. 1984;169:274–8. doi: 10.1016/0014-5793(84)80332-4. [DOI] [PubMed] [Google Scholar]
- 8.Rao GA, Lew G, Larkin EC. Alcohol ingestion and levels of hepatic fatty acid synthetase and stearoyl-CoA desaturase activities in rats. Lipids. 1984;19:151–3. doi: 10.1007/BF02534507. [DOI] [PubMed] [Google Scholar]
- 9.Wilson JS, Korsten MA, Donnelly LP, Colley PW, Somer JB, Pirola RC. Chronic ethanol administration depresses fatty acid synthesis in rat adipose tissue. Biochem J. 1988;251:547–51. doi: 10.1042/bj2510547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tijburg LB, Maquedano A, Bijleveld C, Guzman M, Geelen MJ. Effects of ethanol feeding on hepatic lipid synthesis. Arch Biochem Biophys. 1988;267:568–79. doi: 10.1016/0003-9861(88)90064-1. [DOI] [PubMed] [Google Scholar]
- 11.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277:29342–7. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 12.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 13.Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–42. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Villafranca J, Guillen A, Castro J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie. 2008;90:460–6. doi: 10.1016/j.biochi.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 15.Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, Ji C, Tsukamoto H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55:673–82. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergheim I, Guo L, Davis MA, Lambert JC, Beier JI, Duveau I, Luyendyk JP, Roth RA, Arteel GE. Metformin prevents alcohol-induced liver injury in the mouse: Critical role of plasminogen activator inhibitor-1. Gastroenterology. 2006;130:2099–112. doi: 10.1053/j.gastro.2006.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.You M, Considine RV, Leone TC, Kelly DP, Crabb DW. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–77. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lieber CS, DeCarli LM. Animal models of chronic ethanol toxicity. Methods Enzymol. 1994;233:585–94. doi: 10.1016/s0076-6879(94)33061-1. [DOI] [PubMed] [Google Scholar]
- 19.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–38. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Y, Zhuge J, Wang X, Bai J, Cederbaum AI. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology. 2008;47:1483–94. doi: 10.1002/hep.22222. [DOI] [PubMed] [Google Scholar]
- 21.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283:27628–35. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heiker JT, Kosel D, Beck-Sickinger AG. Molecular mechanisms of signal transduction via adiponectin and adiponectin receptors. Biol Chem. 2010;391:1005–18. doi: 10.1515/BC.2010.104. [DOI] [PubMed] [Google Scholar]
- 23.Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem. 2002;277:3829–35. doi: 10.1074/jbc.M107895200. [DOI] [PubMed] [Google Scholar]
- 24.Basseri S, Lhotak S, Fullerton MD, Palanivel R, Jiang H, Lynn EG, Ford RJ, Maclean KN, Steinberg GR, Austin RC. Loss of TDAG51 Results in Mature-Onset Obesity, Hepatic Steatosis, and Insulin Resistance by Regulating Lipogenesis. Diabetes. 2012 doi: 10.2337/db12-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010;105:14–32. doi: 10.1038/ajg.2009.593. quiz 33. [DOI] [PubMed] [Google Scholar]
- 26.Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–8. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 27.Baraona E, Lieber CS. Effects of ethanol on lipid metabolism. J Lipid Res. 1979;20:289–315. [PubMed] [Google Scholar]
- 28.Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 29.Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. In Vitro and in Silico Characterization of Peroxiredoxin 6 Modified by 4-Hydroxynonenal and 4-Oxononenal. Chem Res Toxicol. 2008 doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- 30.Lieber CS, DeCarli LM. Quantitative relationship between amount of dietary fat and severity of alcoholic fatty liver. Am J Clin Nutr. 1970;23:474–8. doi: 10.1093/ajcn/23.4.474. [DOI] [PubMed] [Google Scholar]
- 31.Cohen PA, Mak KM, Rosman AS, Kessova I, Mishin VM, Koivisto T, Lieber CS. Immunohistochemical determination of hepatic cytochrome P-4502E1 in formalin-fixed, paraffin-embedded sections. Alcohol Clin Exp Res. 1997;21:1057–62. [PubMed] [Google Scholar]
- 32.Lieber CS, Cao Q, DeCarli LM, Leo MA, Mak KM, Ponomarenko A, Ren C, Wang X. Role of medium-chain triglycerides in the alcohol-mediated cytochrome P450 2E1 induction of mitochondria. Alcohol Clin Exp Res. 2007;31:1660–8. doi: 10.1111/j.1530-0277.2007.00475.x. [DOI] [PubMed] [Google Scholar]
- 33.Korourian S, Hakkak R, Ronis MJ, Shelnutt SR, Waldron J, Ingelman-Sundberg M, Badger TM. Diet and risk of ethanol-induced hepatotoxicity: carbohydrate-fat relationships in rats. Toxicol Sci. 1999;47:110–7. doi: 10.1093/toxsci/47.1.110. [DOI] [PubMed] [Google Scholar]
- 34.Orlicky DJ, Roede JR, Bales E, Greenwood C, Greenberg A, Petersen D, McManaman JL. Chronic ethanol consumption in mice alters hepatocyte lipid droplet properties. Alcohol Clin Exp Res. 2011;35:1020–33. doi: 10.1111/j.1530-0277.2011.01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen Z, Liang X, Rogers CQ, Rideout D, You M. Involvement of Adiponectin-SIRT1-AMPK Signaling in the Protective Action of Rosiglitazone against Alcoholic Fatty Liver in Mice. Am J Physiol Gastrointest Liver Physiol. 2009 doi: 10.1152/ajpgi.00456.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez A, Catalan V, Becerril S, Gil MJ, Mugueta C, Gomez-Ambrosi J, Fruhbeck G. Impaired adiponectin-AMPK signalling in insulin-sensitive tissues of hypertensive rats. Life Sci. 2008;83:540–9. doi: 10.1016/j.lfs.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 37.Duval C, Thissen U, Keshtkar S, Accart B, Stienstra R, Boekschoten MV, Roskams T, Kersten S, Muller M. Adipose tissue dysfunction signals progression of hepatic steatosis towards nonalcoholic steatohepatitis in C57BL/6 mice. Diabetes. 2010;59:3181–91. doi: 10.2337/db10-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frederico MJ, Vitto MF, Cesconetto PA, Engelmann J, De Souza DR, Luz G, Pinho RA, Ropelle ER, Cintra DE, De Souza CT. Short-term inhibition of SREBP-1c expression reverses diet-induced non-alcoholic fatty liver disease in mice. Scand J Gastroenterol. 2011;46:1381–8. doi: 10.3109/00365521.2011.613945. [DOI] [PubMed] [Google Scholar]
- 39.Warden SM, Richardson C, O’Donnell J, Jr, Stapleton D, Kemp BE, Witters LA. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J. 2001;354:275–83. doi: 10.1042/0264-6021:3540275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952–62. doi: 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeqiraj E, Filippi BM, Deak M, Alessi DR, van Aalten DM. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 2009;326:1707–11. doi: 10.1126/science.1178377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dentin R, Pegorier JP, Benhamed F, Foufelle F, Ferre P, Fauveau V, Magnuson MA, Girard J, Postic C. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279:20314–26. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- 43.Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Harada K, Gotoda T, Ishibashi S, Yamada N. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem. 1999;274:35832–9. doi: 10.1074/jbc.274.50.35832. [DOI] [PubMed] [Google Scholar]
- 44.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101:7281–6. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–32. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moon YA, Shah NA, Mohapatra S, Warrington JA, Horton JD. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J Biol Chem. 2001;276:45358–66. doi: 10.1074/jbc.M108413200. [DOI] [PubMed] [Google Scholar]
- 47.Kabashima T, Kawaguchi T, Wadzinski BE, Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci U S A. 2003;100:5107–12. doi: 10.1073/pnas.0730817100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ronis MJ, Hennings L, Stewart B, Basnakian AG, Apostolov EO, Albano E, Badger TM, Petersen DR. Effects of long-term ethanol administration in a rat total enteral nutrition model of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G109–19. doi: 10.1152/ajpgi.00145.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yin HQ, Je YT, Kim M, Kim JH, Kong G, Kang KS, Kim HL, Yoon BI, Lee MO, Lee BH. Analysis of hepatic gene expression during fatty liver change due to chronic ethanol administration in mice. Toxicol Appl Pharmacol. 2009;235:312–20. doi: 10.1016/j.taap.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 50.Clugston RD, Jiang H, Lee MX, Piantedosi R, Yuen JJ, Ramakrishnan R, Lewis MJ, Gottesman ME, Huang LS, Goldberg IJ, Berk PD, Blaner WS. Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: a targeted lipidomic and gene expression study. J Lipid Res. 2011;52:2021–31. doi: 10.1194/jlr.M017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smathers RL, Fritz KS, Galligan JJ, Shearn CT, Reigan P, Marks MJ, Petersen DR. Characterization of 4-HNE Modified L-FABP Reveals Alterations in Structural and Functional Dynamics. PLoS One. 2012;7:e38459. doi: 10.1371/journal.pone.0038459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venkatesan S, Ward RJ, Peters TJ. Effect of chronic ethanol feeding on the hepatic secretion of very-low-density lipoproteins. Biochim Biophys Acta. 1988;960:61–6. doi: 10.1016/0005-2760(88)90009-4. [DOI] [PubMed] [Google Scholar]
- 53.Guzman M, Castro J. Zonal heterogeneity of the effects of chronic ethanol feeding on hepatic fatty acid metabolism. Hepatology. 1990;12:1098–105. doi: 10.1002/hep.1840120504. [DOI] [PubMed] [Google Scholar]
- 54.Kharbanda KK, Todero SL, Ward BW, Cannella JJ, 3rd, Tuma DJ. Betaine administration corrects ethanol-induced defective VLDL secretion. Mol Cell Biochem. 2009;327:75–8. doi: 10.1007/s11010-009-0044-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.