

Abstract
Saturated free fatty acids (FFA) can activate inflammatory cascades including the toll-like receptor 4 (TLR4) pathway. TLR4 is expressed by hepatocytes and may help link FFA to altered hepatic gluconeogenesis in type 2 diabetes mellitus. This study examined the role of TLR4 in mediating palmitate effects on the expression of phosphoenolpyruvate carboxykinase (PCK1) and the catalytic subunit of glucose-6-phosphatase (G6PC), rate-determining gluconeogenic enzymes. Human hepatocellular carcinoma cells (HepG2 and HuH7) were incubated in media including 2% bovine serum albumin and 250 to 1000 μM palmitate for 24 h. Signaling mediated by TLR4 was blocked by a TLR4 decoy peptide or small interfering RNA knockdown of TLR4. Palmitate induced dose-dependent increases in PCK1 and G6PC mRNA abundance, which were prevented by the TLR4 decoy peptide. Palmitate doubled PCK1 promoter activity, and TLR4 knockdown ablated this response. Lipopolysaccharide and monophosphoryl lipid A also up-regulated G6PC and PCK1 transcript abundance in a TLR4-dependent manner. Addition of oleate attenuated palmitate-induced increases in G6PC and PCK1 mRNA abundance. Palmitate increased nuclear factor κ-light-chain-enhancer of activated B cells reporter gene activity, which was unaffected by TLR4 blockade, but increased mRNA abundance of hepatocyte-specific cyclic AMP response element binding protein, a transcriptional regulator of PCK1, in a TLR4-dependent manner. Finally, TLR4 activation by palmitate increased subsequent cellular uptake of palmitate, and inhibiting ceramide synthesis ablated palmitate effects on PCK1 mRNA abundance and promoter activity. These results suggest that TLR4 signaling could play a critical role in linking elevated saturated FFA to increased transcription of gluconeogenic genes.
Keywords: Gluconeogenesis, Palmitate, Toll-like receptor 4, Saturated fatty acid, Diabetes
1. Introduction
The pathology of type 2 diabetes mellitus (T2DM) is complex because it involves multiple organs, dietary factors and inflammatory pathways. Several reports indicate that excessive gluconeogenesis (GNG) contributes to hyperglycemia in diabetics [1–3], which is caused at least partially by hepatic insulin resistance [4]. Elevated free fatty acids (FFA), especially saturated FFA, promote insulin resistance and also increase basal GNG [5,6]. Coupled with the fact that obesity and high-fat diets increase plasma FFA concentrations, these findings suggest that FFA may play a causative role in diabetes [7]. In fact, epidemiological studies have identified high FFA levels [8] and more saturated FFA profiles [9] as independent predictors of diabetes risk.
In a critical report, Lee and colleagues demonstrated that saturated FFA can activate inflammatory pathways via toll-like receptor 4 (TLR4) [10], and subsequent findings have been consistent with the concept that saturated FFA are TLR4 agonists [11,12]. The recent finding that TLR4 is expressed by hepatocytes [13,14] represents a potential mechanism linking FFA to altered liver function. In fact, TLR4 has been identified as a critical component of systemic inflammatory responses to high-fat diets, and the activation of TLR4 signaling may contribute to lipid-induced insulin resistance [15,16]; however, the critical pathways linking chronically elevated FFA to excessive GNG remain controversial and elusive. The main objective of the present study was to examine whether TLR4 mediates saturated FFA effects on the expression of gluconeogenic genes in human hepatocellular carcinoma cells (HepG2 and HuH7). Our results show that the TLR4 pathway plays a critical role in linking palmitate to increased gluconeogenic gene transcription.
2. Materials and methods
2.1. Cell culture
HepG2 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA), and HuH7 cells were a gift from Dr. Kyeong-Ok Chang (Kansas State University). Hepatoma cells were maintained as a monolayer culture in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen Corp., Grand Island, NY USA) supplemented with 10% fetal bovine serum (FBS) (CELLect Gold; MP Biomedicals, Solon, OH, USA) and 1% penicillin–streptomycin (Invitrogen Corp.) until 70% confluent. For treatment, medium was changed to DMEM or M199 (Invitrogen Corp.; 5.6 M glucose in both) with 5% FBS, 1% antibiotics (including glutamine when M199 medium was used), 2% fatty-acid-free, low-endotoxin (guaranteed ≤0.1 ng/mg endotoxin) bovine serum albumin (BSA) (A-8806; Sigma-Aldrich, St. Louis, MO, USA) and treatment compounds as specified below.
2.2. Treatments
The TLR4 decoy peptide (sequence: RQIKIWFQNRRMKWKKLHYRDFIPGVAIAA) used in this study was designed to interrupt TLR4 signaling by interfering with BB-loop interactions on the intracellular domain of TLR4 [17]. The TLR4 decoy peptide was synthesized by the Biopolymer Core Facility of the University of Maryland (Baltimore, MD, USA). The purity of the peptide was N95% as determined by liquid chromatography–mass spectrometry, and the peptide was dissolved in dimethyl sulfoxide at an initial concentration of 10 mM. The relative cytotoxicity of TLR4 decoy peptide and palmitate was determined by metabolism of alamarBlue (Invitrogen Corp.) over 1 h. HepG2 cells were treated with different concentrations of TLR4 decoy peptide (1–100 μM) for the viability assay. For the remaining experiments, the TLR4 decoy peptide was added to appropriate wells at a concentration of 1 μM 30 min prior to addition of other treatments, including palmitate (0, 250, 500, 750 and 1000 μM), oleate and palmitate (2:1) mixtures (0, 375, 750 and 1125 μM), lipopolysaccharide (LPS; 10 and 100 ng/ml) and monophosphoryl lipid A(MLA; 0, 1, 10, and 100 μg/ml) and were incubated for 24 h. Fatty acids (N95% purity, Sigma-Aldrich) were weighed into distilled, deionized water at a final concentration of 100 mM and incubated at 37°C for 1 h. The solution was then inverted to disperse the lipid, and 1 N KOH (1% vol:vol) was added to dissolve the fatty acids. LPS (≥500,000 endotoxin U/mg from Salmonella enteritidis; product L2012) and MLA (from Escherichia coli F583, product L6638) were purchased from Sigma-Aldrich. For the fatty acid uptake experiment, cells were treated for 24 h ± palmitate and TLR4 decoy peptide as described above, washed and then incubated with 1-14C-palmitate (0.1 μCi/ml; Moravek Biochemicals, Brea, CA, USA) in the same media (free of any treatments) for 30 min to measure palmitate uptake. In the ceramide inhibition experiment, myriocin (product BML-SL226; Enzo Life Sciences, Farmingdale, NY, USA) was added at a concentration of 1 μM for 30 min prior to the addition of 1000 μM palmitate as in previous experiments.
2.3. Quantitative real-time polymerase chain reaction (PCR)
Total RNA and protein were extracted using a PARIS kit following the manufacturer’s instructions (Applied Biosystems, Carlsbad, CA, USA). Complementary DNA was synthesized using the High-Capacity cDNA RT Kit according to the manufacturer’s instructions (Applied Biosystems). Quantitative real-time PCR was performed in triplicate with 5% of the cDNA product in the presence of 200 nM gene-specific forward and reverse primers with real-time fluorescent detection (7500 Fast Real-Time PCR System, Applied Biosystems) using Fast SYBR Green Master Mix (Applied Biosystems). Primers were designed (http://www.ncbi.nlm.nih.gov/tools/primer-blast) using the GenBank sequences shown in Table 1. Relative mRNA abundance was quantified by the delta-Ct method with ribosomal protein subunit 9 (RPS9) used to normalize values. Efficiencies of PCR reactions were tested on each plate by dilution of a pooled sample, and efficiencies for the various target genes ranged from 83% to 97%.
Table 1.
Primers used for quantitative real-time PCR gene expression analysis
| Genea | Sequence of primer (5′ to 3′) | Accession numberb |
|---|---|---|
| PCK1 | Forward, ATCCCCAAAACAGGCCTCA | NM_002591.2 |
| Reverse, ATGACGTACATGGTGCGACCT | ||
| G6PC | Forward, CCCGTGAGACTGGACCAGGGAAA | NM_000151.2 |
| Reverse, AGACATTCAGCTGCACAGCCCAG | ||
| CD36 | Forward, ACCTGGCTCAAGCACAAACCAA | NM_001001548.2 |
| Reverse, TGGTCCTGGAGACTGTTGAGCA | ||
| TLR4 | Forward, AGCCCTGCGTGGAGGTGGTT | NM_138554.3 |
| Reverse, GAAGGGGAGGTTGTCGGGGA | ||
| GRP78 | Forward, TGGTTCGTGGCGCCTTGTGA | NM_005347.4 |
| Reverse, GCACAGGAGCACAGCGCAAT | ||
| CREBH | Forward, AGCTGGTGCTCACCGAGGAT | NM_032607.1 |
| Reverse, TGCTTTCTTGCGCCGACTGC | ||
| RPS9 | Forward, TGAAGCTGATCGGCGAGTATG | NM_001013.3 |
| Reverse, CCTTCTCATCAAGCGTCAGCA |
PCK1: phosphoenolpyruvate carboxykinase; G6PC: the catalytic subunit of glucose-6-phosphatase; CD36: fatty acid translocase; TLR4: toll-like receptor 4; GRP78: 78-kDa glucose-regulated protein; CREBH: hepatocyte-specific cyclic AMP response element binding protein; RPS9: ribosomal protein subunit.
From NCBI Entrez Nucleotide Database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucleotide).
2.4. siRNA and luciferase assays
HepG2 cells were transfected in serum- and antibiotic-free OPTI-MEM medium with 100 nM pooled TLR4 siRNA (sc-40260) or scrambled (Scr) siRNA (sc-37007) using siRNA Transfection Reagent (sc-29528) in siRNA Transfection Medium (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) according to the manufacturer’s recommendations. Cells treated with siRNA were incubated for 24 h prior to reporter plasmid transfection. TLR4 knockdown was confirmed by Western blot.
For luciferase assays, HepG2 cells were transfected with 100 ng reporter gene plasmids in OPTI-MEM reduced serum media (Invitrogen Corp.) using TransIT-LT1 transfection reagent (Mirus Bio LLC, Madison, WI, USA) and incubated for 16 h prior to treatment. The PCK1-Luc plasmid encoded the region 1.9 kb upstream to 0.9 kb downstream of the human PCK1 start site (S114233; SwitchGear Genomics, Menlo Park, CA, USA). Nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) activity was determined using the established pIgκB-Luc reporter construct [18], and a Renilla luciferase vector (pRL-SV40) was used as a control reporter plasmid (Promega Corp., Madison, WI, USA). Cells were treated for 24 h as described previously, and luciferase activity was measured using the luciferase assay system (Promega Corp.) according to the manufacturer’s instructions.
2.5. Western blotting
HepG2 cellular protein was recovered after lysis of cells using the PARIS kit (Applied Biosystems). Protease inhibitors were added to the lysis buffer (Protease inhibitor cocktail I; Calbiochem, Gibbstown, NJ, USA). The homogenate was centrifuged at 15,000g for 10 min at 4°C, and total protein concentration of the supernatant was measured [19]. Forty micrograms of total protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis on a 4%–12% Tris–HCl gel and dry-transferred onto nitrocellulose membranes (iBlot; Invitrogen Corp.). Membranes were blocked in Tris buffer (pH 7.4) with 5% dry milk powder for 2 h at room temperature and then incubated with a rabbit anti-TLR4 antibody (sc-10741 diluted 1:1000; Santa Cruz Biotechnology) for 1 h at room temperature. After washing, membranes were incubated for 1 h at room temperature with a secondary antibody (anti-rabbit IgG, sc-2301; Santa Cruz Biotechnology) diluted 10,000-fold in Tris buffer (pH 7.4). Immunodetection was performed by chemiluminescence (West-Dura; Thermo Scientific, Waltham, MA, USA), and band images were visualized using a photo documentation system (ChemiDoc-It Imaging System; UVP Inc., Upland, CA, USA).
2.6. Lipid analyses
Cells incubated with 1-14C-palmitate were washed and harvested for liquid scintillation counting (LS 6500, Beckman, Brea, CA, USA). Intracellular ceramide concentrations were determined in separate cell lysate samples by mass spectrometry. Data acquisition, analysis and lipid quantification in comparison to internal standards, using an automated electrospray ionization–tandem mass spectrometry approach, were carried out as previously described [20], except that an aliquot of 2 μl of extract in chloroform was used for analysis, and free fatty acids and acyl product ions were not analyzed. The molar results were normalized to the protein content of the sample to produce data in nmol/mg protein.
2.7. Statistics
Results were analyzed by analysis of variance, and pairwise differences were determined by two-tailed Student’s t tests. Transcript abundance data for 78-kDa glucose-regulated protein (GRP78) and hepatocyte-specific cyclic AMP response element binding protein (CREBH) were log-transformed for analysis to achieve normal residual distributions, and reported means are back-transformed.
3. Results
3.1. Model validation
Prior to beginning investigations into the effects of palmitate on transcriptional control of GNG, we first verified that this model system did not induce cytotoxicity. Incubating HepG2 and HuH7 cells with 1000 μM palmitate and 2% BSA for 24 h did not decrease cell viability as assessed by resazurin metabolism (data not shown; P>.24, n=6–8). A small, cell-permeable peptide that inhibits TLR4 dimerization and subsequent signaling was also used [17]. Because this peptide had not previously been used in HepG2 cells, the dose-dependent effects of the TLR4 decoy peptide on HepG2 cell viability were tested using concentrations ranging from 1 nM to 100 μM. No effects on viability (24 h) with concentrations up to 10 μM were observed, and the 100-μM dose significantly decreased viability (P<.01, data not shown). Our experiments were conducted using a dose of 1 μM, lower than the doses of 10–40 μM used to establish the effects of the TLR4 decoy peptide in murine macrophages [17].
3.2. Palmitate increases abundance of gluconeogenic transcripts in a TLR4-dependent manner
To assess the effects of palmitate on transcriptional regulation of GNG, mRNA abundance of two rate-determining enzymes in this pathway (PCK1 and G6PC) was measured. Exposing HepG2 cells to 500, 750 and 1000 μM palmitate for 24 h induced 8.5-, 27.6- and 33.3-fold increases in PCK1 and 4.7-, 12.5- and 21.1-fold increases in G6PC mRNA abundance, respectively (P<.05 for both PCK1 and G6PC at all concentrations above, Fig. 1A and B). In the presence of the TLR4 decoy peptide, each of these responses was ablated, suggesting the involvement of TLR4 in the effects of palmitate on abundance of these transcripts.
Fig. 1.

Palmitate, mixed fatty acids, MLA and LPS increase transcript abundance for gluconeogenic genes via TLR4 signaling in HepG2 cells. HepG2 cells were incubated in M199 medium with various concentrations of palmitate, mixed fatty acids, MLA or LPS for 24 h, with the TLR4 decoy peptide (TLR4d; 1 μM) added 30 min prior to addition of lipid treatments. All wells included 2% BSA. After 24 h, total RNA was isolated for quantitative reverse transcriptase (qRT)-PCR analysis of PCK1 and G6PC relative to the internal control gene RPS9. Values are means±S.E.M.; n=3 to 6. Significant differences (P<.05) are indicated as follows: *, differs from control (no treatment); #, differs between lipid and lipid with TLRd. (A, B) Palmitate-induced increases in G6PC and PCK1 mRNA abundance are blocked by TLR4 decoy peptide. (C, D) Mixtures of unsaturated and saturated fatty acids (2:1 oleate:palmitate) increase G6PC and PCK1 mRNA abundance; however, the mixed fatty acids had significantly less impact on these transcripts than palmitate alone.(E, F) MLA-induced increases in G6PC and PCK1 mRNA abundance are blocked by TLR4 decoy peptide. (G, H) LPS increases G6PC and PCK1 mRNA abundance, and these responses are blocked by TLR4 decoy peptide.
3.3. Oleate attenuates palmitate-induced increases of gluconeogenic transcript abundance in a TLR4-dependent manner
To determine whether responses to palmitate were specific to this saturated fatty acid or were a more general response to fatty acids, the effects of a 2:1 mixture of oleate and palmitate were assessed in HepG2 cells. This 2:1 ratio of unsaturated:saturated fatty acids approximates physiological ratios [9]. The mixed fatty acids increased PCK1 mRNA at 750- and 1125-μM concentrations and G6PC mRNA at 1125 μM compared with the control (P<.05, Fig. 1C and D), although only the G6PC response at 1125 μM was significantly different than the corresponding TLR4 decoy peptide treatment. More importantly, these responses were substantially attenuated compared with the effects of palmitate alone. The direct comparison of 750 μM palmitate vs. 750 μM of the mixed fatty acids showed a response more than 10-fold greater for palmitate (P<.05). These data indicate that mixtures of saturated and unsaturated fatty acids do not result in the same induction of gluconeogenic transcripts as palmitate alone.
3.4. MLA and LPS increase the abundance of gluconeogenic transcripts in a TLR4-dependent manner
Free fatty acids can alter cell signaling in many ways other than through TLR4. Therefore, TLR4 agonists LPS and MLA (a nontoxic derivative of LPS [21]) were tested for effects on gluconeogenic genes. HepG2 cells treated with 1, 10 and 100 μg/ml MLA for 24 h had 3.4-, 11.4- and 27.1-fold increases in PCK1 and 1.3-, 3.5- and 14.8-fold increases in G6PC mRNA abundance, respectively (Fig. 1E and F). LPS also dramatically increased both PCK1 and G6PC mRNA levels (Fig. 1G and H).These responses were also eliminated in the presence of the TLR4 decoy peptide.
3.5. Palmitate induces similar increases in gluconeogenic transcripts in HuH7 cells
To determine whether TLR4-mediated induction of gluconeogenic transcripts is an anomaly found only in HepG2 cells, we evaluated the dose–response to palmitate in HuH7 cells with and without TLR4 blockade. As in the HepG2 cells, palmitate dramatically increased both PCK1 and G6PC mRNA levels, with significant responses at doses of 250 μM and above for PCK1 and 750 μM and above for G6PC (Fig. 2). All responses were again prevented by the TLR4 decoy peptide.
Fig. 2.

Palmitate increases transcript abundance for gluconeogenic genes via TLR4 signaling in HuH7 cells. HuH7 cells were incubated in M199 medium with various concentrations of palmitate for 24 h, with the TLR4 decoy peptide (TLR4d; 1 μM) added 30 min prior to addition of palmitate. All wells included 2% BSA. After 24 h, total RNA was isolated for qRT-PCR analysis of PCK1 (A) and G6PC (B) relative to the internal control gene RPS9. Values are means±S.E.M.; n=4. Significant differences (P<.05) are indicated as follows: *, differs from control (no treatment); #, differs between lipid and lipid with TLRd.
3.6. Palmitate increases PCK1 promoter activity in a TLR4-dependent manner
To verify that the PCK1 response was mediated by effects on the transcriptional rate rather than mRNA stability only, the activity of the PCK1 promoter was analyzed by luciferase assay in HepG2 cells. Consistent with mRNA abundance results, palmitate induced a twofold increase in PCK1 promoter activity, and this response was blocked by either the TLR4 decoy peptide or siRNA knockdown of TLR4 (Fig. 3A and B).
Fig. 3.
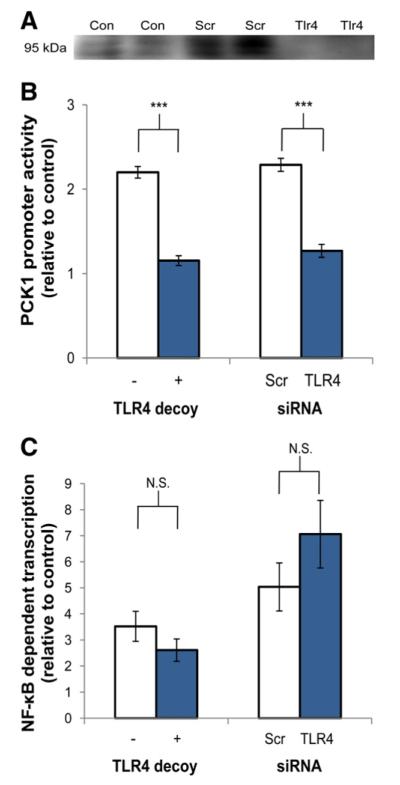
Palmitate-induced increases in PCK1 promoter activity are blocked by either TLR4 decoy peptide or TLR4 siRNA, but activation of NF-κB is TLR4 independent. For siRNA treatments, HepG2 cells were transfected with 100 nM pooled TLR4 siRNA or scrambled (Scr) siRNA and maintained for 24 h prior to additional treatments. (A) TLR4 knockdown was confirmed by Western blot. TLR4 protein was detected in untreated cells (Con) and cells treated with Scr siRNA, but not in cells treated with siRNA targeting TLR4. (B) Cells were transfected with 100 ng PCK1 or pRL-SV40 (control) luciferase reporter plasmids. After 16 h, TLR4 decoy peptide (1 μM) was added to appropriate wells 30 min prior to the addition of 1000 μM palmitate in DMEM with 2% BSA. After 24 h, promoter activity was quantified by luciferase assay. Palmitate alone significantly increased PCK1 promoter activity in both experiments (P<.001). (C) Cells were transfected with 100 ng pIgκB-Luc (NF-kB-binding promoter) or pRL-SV40 (control) luciferase reporter plasmids. After 16 h, TLR4 decoy peptide (1 μM) was added to appropriate wells 30 min prior to the addition of 1000 μM palmitate in DMEM with 2% BSA. After 24 h, promoter activity was quantified by luciferase assay. Palmitate alone significantly increased NF-kB activity in both experiments (P<0.001). Values are means±S.E.M. (n=3 to 6), representing the response to palmitate relative to controls (Scr alone in the siRNA experiment), which were normalized to 1. ***P<.001, N.S.=not significant.
3.7. Palmitate induces NF-κB-dependent transcription independent of TLR4
One key mediator of TLR4 signaling is NF-κB, and this transcription factor could mediate effects on gluconeogenic gene expression. Therefore, NF-κB activity was evaluated using the pIgκB-Luc reporter construct [18] in HepG2 cells. Palmitate significantly increased NF-κB reporter activity, but neither the TLR4 decoy peptide nor siRNA knockdown of TLR4 had a significant impact on this response to palmitate (Fig. 3C). Therefore, treatments that prevented the induction of PCK1 and G6PC gene expression by palmitate failed to alter NF-κB responses, suggesting that NF-κB activation alone does not explain the transcriptional responses.
3.8. Palmitate increases mRNA abundance of transcripts involved in lipid signaling and endoplasmic reticulum (ER) stress in a TLR4-dependent manner
In addition to activating inflammatory pathways [22], palmitate has been reported to initiate an ER stress response [23]. Therefore, the mRNA responses to palmitate and TLR4 blockade for ER stress mediators GRP78 and CREBH, as well as for TLR4 and the lipid scavenger fatty acid translocase (CD36), were also measured in HepG2 cells. Palmitate significantly increased mRNA abundance of TLR4, CD36, GRP78 and CREBH (Fig. 4). TLR4 blockade attenuated each of the significant responses to palmitate.
Fig. 4.

Palmitate increases mRNA abundance of transcripts involved in lipid signaling and ER stress in a TLR4-dependent manner. HepG2 cells were incubated in DMEM±1000 μM palmitate, with the TLR4 decoy peptide (1 μM) added 30 min prior to palmitate treatment. All wells included 2% BSA. After 24 h, total RNA was isolated for qRT-PCR analysis of (A) TLR4, (B) CD36, (C) GRP78 and (D) CREBH relative to the internal control gene RPS9. Values are means±S.E.M. (n=3 to 6).*P<.05, **P<.01, ***P<0.001, effect of palmitate compared with control; #P<0.05, †P<0.10, effect of palmitate+TLR4 decoy peptide compared with palmitate alone.
3.9. Activation of TLR4 by palmitate increases subsequent fatty acid uptake by HepG2 cells
Treatment effects on CD36 expression led us to assess fatty acid uptake after HepG2 cells were preincubated with palmitate for 24 h. Consistent with the observed effects on expression of the fatty acid transport protein, palmitate exposure increased subsequent uptake of 1-14C-palmitate, and the response was prevented by the TLR4 decoy peptide (Fig. 5).
Fig. 5.
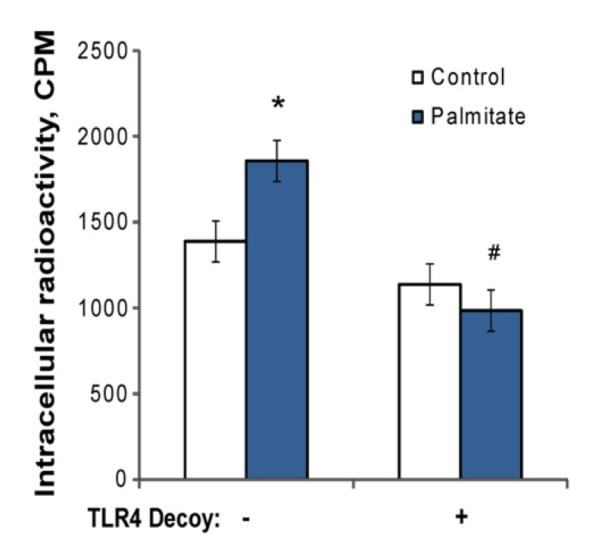
Palmitate exposure increases subsequent cellular uptake of palmitate in a TLR4-dependent manner. HepG2 cells were incubated in DMEM with 2% BSA±1000 μM palmitate, with the TLR4 decoy peptide (1 μM) added 30 min prior to palmitate treatment. After 24 h, cells were washed, and fresh media (DMEM with 2% BSA) including 0.1 μCi/ml 1-14C-palmitate were added to all wells for 30 min. Cells were washed and then harvested for detection of 14C. Values are means±S.E.M. (n=3).*P<0.05, effect of palmitate compared with control; #P<0.001, effect of palmitate +TLR4 decoy peptide compared with palmitate alone.
3.10. Ceramide signaling is involved in induction of gluconeogenic genes by palmitate
To determine if ceramides are also involved in palmitate-induced gluconeogenic gene expression, we used myriocin, an inhibitor of serine palmitoyltransferase, to block ceramide production. HepG2 cells treated with 1000 μM palmitate, palmitate with TLR4 decoy peptide and palmitate with myriocin had intracellular ceramide concentrations of 3.49, 2.01 and 0.28±0.24 nmol/mg protein, respectively. As shown in Fig. 6, increases in PCK1 mRNA abundance and PCK1 promoter activity induced by palmitate were ablated by myriocin. These results indicate that ceramides are involved in palmitate-induced gluconeogenic gene expression.
Fig. 6.
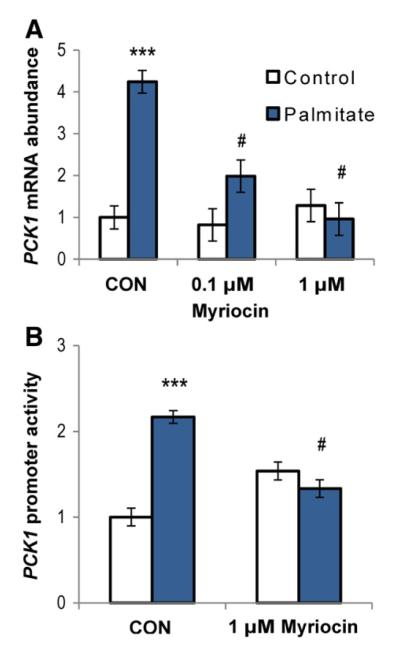
Ceramides are involved in induction of gluconeogenic genes by palmitate. (A) Palmitate-induced increases in G6PC mRNA abundance are suppressed in a dose-dependent manner by ceramide synthesis inhibition. HepG2 cells were incubated in DMEM with 2% BSA±1000 μM palmitate for 24 h, with the ceramide synthesis inhibitor myriocin added 30 min prior to addition of palmitate.(B) Palmitate-induced increases in PCK1 promoter activity are blocked by ceramide synthesis inhibition. HepG2 cells were transfected with 100 ng PCK1 or pRL-SV40 (control) luciferase reporter plasmids. After 16 h, myriocin (1 μM) was added to appropriate wells 30 min prior to the addition of 1000 μM palmitate in DMEM with 2% BSA. After 24 h, promoter activity was quantified by luciferase assay. Values are means±S.E.M. (n=3–6). ***P<0.001, effect of palmitate compared with control; #P<.001, effect of palmitate+myriocin compared with palmitate alone.
4. Discussion
Broad evidence indicates that elevated FFA can promote GNG, which leads to increased hepatic glucose output. High-fat diets or fasting-induced elevation of plasma FFA increases hepatic glucose production in rats [5,24]. Increased plasma FFA in fasting humans promote GNG, whereas this response is blocked by pharmacological suppression of FFA release [25]. Furthermore, FFA-induced increases in endogenous glucose production in humans are eliminated by the inhibition of GNG via ethanol infusion [6], indicating that FFA effects are mediated primarily by enhancement of GNG rather than glycogenolysis. In primary hepatocytes and HepG2 cells, GNG is increased in the presence of high FFA [26,27].
The mechanism by which FFA promote GNG remains elusive. A growing number of studies suggest that an important mechanism underlying this response is an increase in transcription of key gluconeogenic genes, particularly PCK1 and G6PC [26]. Increasing plasma FFA in rats increase hepatic G6PC mRNA abundance [28], and exposing primary hepatocytes to elevated FFA enhances mRNA abundance and the promoter activity of both PCK1 and G6PC [26,29], as well as PCK1 activity [30].We found that exposing HepG2 and HuH7 cells to various concentrations of palmitate for 24 h significantly increased PCK1 and G6PC mRNA abundance in a dose-dependent manner; PCK1 promoter activity in HepG2 cells was increased twofold after a 24-h incubation in the presence of 1000 μM palmitate. These results suggest that saturated FFA can directly promote the expression of genes encoding gluconeogenic enzymes. It should be noted that mRNA stability was not investigated in this study, and a reduction in mRNA degradation rate could also contribute to the increase in gluconeogenic transcript abundance.
The concentrations of FFA we used were within the physiological range of human plasma FFA; the reported mean fasting FFA concentration in healthy adults was 578 μM, with a range from 90 to 1473 μM [31]. Plasma contains a complex array of fatty acids, however, and the use of palmitate alone does not represent a physiological scenario. Nevertheless, the induction of gluconeogenic genes with higher concentrations of mixed FFA suggests that mechanisms identified with palmitate alone are relevant to more physiological scenarios.
The mechanisms linking saturated FFA to gluconeogenic gene expression are not fully understood. Numerous nuclear receptors, such as peroxisome proliferator-activated receptors (PPARs) and retinoid X receptor, bind FFA, but unsaturated FFA are generally far more potent activators of these factors [32], making it unlikely that saturated FFA responses are mediated solely by direct activation of transcription factors. Many studies have indicated that reactive oxygen species (ROS) or products of palmitate metabolism such as ceramides or diacylglycerides are critical mediators of transcriptional responses to saturated FFA. Each of these bioactive compounds is elevated in hepatocytes exposed to high concentrations of saturated FFA [7,23,33], and they may play a role in mediating or amplifying the effects of FFA on GNG gene expression, but no consensus model has emerged. In the present study, the increased PCK1 and G6PC mRNA abundance and elevated PCK1 promoter activity induced by palmitate were blocked by either the TLR4 decoy peptide or siRNA knockdown, suggesting that activation of TLR4 signaling plays a key role in stimulating the expression of gluconeogenic genes in response to palmitate. In fact, several studies have proposed that saturated FFA act as agonists for TLR4. Shi et al. [15] demonstrated that FFA are capable of activating TLR4 signaling and inflammatory responses in adipocytes and macrophages; deletion of TLR4 not only prevented lipid-induced insulin resistance in muscle and adipose tissue, but also prevented the increase in hepatic glucose output in response to lipid infusion. Likewise, Holland and colleagues [34] reported that TLR4-defective mice infused with lard oil retained the ability to down-regulate hepatic GNG in response to insulin, whereas wild-type mice infused with lard oil displayed severe hepatic insulin resistance. Perhaps more convincingly, recent findings suggest that saturated FFA-induced ROS promote TLR4 activation [11] and that TLR4 is required for palmitate-induced ceramide synthesis [35], which in turn is requisite for TLR4-induced insulin resistance [35]. Several lines of evidence indicate that ceramides impair glucose metabolism by inhibiting insulin-stimulated glucose uptake [36,37], and these effects were reversed by myriocin treatment. In the current study, we found that ceramide is involved in palmitate-induced gluconeogenic gene expression. Collectively, these data may suggest that ceramide is a key component of the networks that link saturated FFA-induced inflammatory pathways to both impaired insulin action and increased basal GNG, contributing the development of T2DM. In addition, we found that 1000 μM palmitate increased mRNA abundance of CD36, a class B scavenger receptor that facilitates the intracellular uptake and transport of FFA [38]. Coupled with the observation that palmitate preexposure increased the subsequent uptake of 14C-palmitate, we speculate that the up-regulation of CD36 may allow greater influx of lipid into the cell, potentially increasing intracellular products of fatty acid metabolism (e.g., ROS and ceramides) as well. The fact that a TLR4 blockade attenuated these responses indicates that palmitate induction of CD36 and FFA uptake may be mediated through the TLR4 pathway. Taken together, these data indicate that TLR4 might be a central molecular link among FFA, ceramides, inflammatory mediators and glucose metabolism. Strategies aimed at blocking hepatocyte TLR4 signaling may improve insulin sensitivity and GNG regulation.
The finding that mixed FFA decreased gluconeogenic gene expression compared with palmitate agrees with previous results demonstrating that saturated FFA activate TLR4 and unsaturated FFA inhibit saturated FFA-induced TLR4 activation and its downstream signaling pathways[39]. Up-regulation of NF-κB and cyclooxygenase-2 by saturated FFA and inhibition of this induction by unsaturated FFA are mediated through a common TLR4 pathway [10]. Although the underlying mechanism by which these FFA modulate the activation of TLR4 remains uncertain, acylation of saturated FA in LPS is absolutely required for its toxic effects and is critical for TLR4 receptor activation [40]. Thus, animals might have developed receptors to recognize the FA component of this highly conserved molecular structure and initiate immune responses [40]. More interestingly, recent evidence suggests that saturated and unsaturated FFA reciprocally modulate TLR4 activation by regulating the dimerization and recruitment of TLR4 into lipid rafts [11], which serve as organizing centers for the assembly of signaling molecules [41] and are essential for TLR4-mediated signal transduction [11]. Treatment of cells with unsaturated FFA may result in incorporation of these FFA into saturated FFA-enriched lipid rafts and inhibition of lipid raft formation, leading to displacement of signaling molecules from lipid rafts and attenuated TLR4 signaling [11]. Unsaturated FFA may thereby interfere with the ability of TLR4 to dimerize and signal effectively in the presence of palmitate. Oleate is also a natural ligand of PPAR-γ [42], which has recently been shown to suppress TLR4-mediated inflammation [43]; the interaction of this signal with TLR4-induced GNG gene expression would an interesting subject of future research.
Monophosphoryl lipid A, a derivative of the endotoxin LPS, lacks the toxic properties but retains the immunostimulatory function of its parent molecule [21,44]. Both MLA and LPS are ligands of TLR4, which signals through an elaborate downstream cascade initiated through two distinct adapter protein complexes, TRAM/TRIF (TRIF-related adaptor molecule/toll–interleukin-1 receptor domain-containing adaptor-inducing interferon-β) and MAL/MyD88 (MyD88-adapter-like/myeloid differentiation marker 88) [45]. In 2007, Mata-Haro et al. [21] first reported that MLA induced strong TRIF-associated but weak MyD88-associated responses through TLR4 when compared with those of LPS; this incomplete MyD88 signaling is proposed to account for reductions in inflammatory activities, whereas MLA is still able to trigger adaptive immunity via an almost intact TRIF-dependent pathway [21]. However, a later study by the same group demonstrated that MLA-induced signaling is not completely devoid of MyD88 involvement because MLA-induced p38 mitogen-activated protein kinase(p38 MAPK) activation depends on both MyD88 and TRIF pathways [46]. Our results showed that both MLA and LPS promoted the expression of gluconeogenic genes and these responses were TLR4 dependent. Although by which pathways MLA mediates these changes is not known, our data indicate that the toxic proinflammatory properties of ligands may not be required for the activation of TLR4-mediated gluconeogenic gene transcription. Recently, p38 MAPK was identified as a critical signaling component in FFA-induced transcription of key gluconeogenic genes and regulators, including PKC1, G6PC and peroxisome proliferator-activated-γ receptor coactivator α, an important stimulator of hepatic GNG [47] as well as PKC1 promoter activation [26]. Coupled with the fact that MLA is a potent stimulator of p38 MAPK [46], we speculate that MLA may promote gluconeogenic gene transcription through p38 MAPK-mediated signaling. To our knowledge, this is the first study to report the GNG-promoting potential of MLA. Further studies are needed to determine the specific role of p38 MAPK in MLA-induced gluconeogenic gene transcription and glucose production.
The potential mediators of the TLR4-dependent induction of gluconeogenic gene expression by palmitate are unclear. As mentioned above, TLR4 homodimers initiate two distinct adapter protein complexes, TRAM/TRIF and MAL/MyD88 [45]. These adaptor pairs couple with two distinct signaling pathways leading to the activation of interferon response factor 3 and NF-κB, respectively [45]. We found that palmitate significantly increased NF-κB reporter activity, but neither the TLR4 decoy peptide nor siRNA knockdown of TLR4 affected this response to palmitate. A previous study showed that the TLR4 decoy peptide at 40 μM prevented NF-κB activation by LPS [17]. In our model with a far lower dose of the TLR4 decoy peptide, TLR4 inhibition may have been inadequate to block the activation of NF-κB completely, or palmitate may have induced NF-κB activation via TLR4-independent pathways such as the TLR2 cascade [48]. Either way, the fact that TLR4 blockade completely eliminated the induction of PCK1 and G6PC by palmitate without preventing activation of NF-κB suggests that NF-κB activation alone does not explain the increase in gluconeogenic gene expression. In addition, the activation of two other downstream targets of TLR4, p38 MAPK and c-Jun NH2-terminal kinase, has been linked to high glucose production and hyperglycemia [49,50]. Overall, these data do not rule out the possibility that the response may be NF-κB related, but another transcription factor is almost certainly involved.
A recently identified regulator of gluconeogenic gene expression is CREBH. Hepatic CREBH expression is activated by elevated FFA in mice and by palmitate treatment in mouse primary hepatocytes [51]. Interestingly, CREBH also activates the PCK1 promoter; the active nuclear form of CREBH enhanced GNG by promoting both PCK1 and G6PC transcription. Conversely, acute depletion of CREBH leads to the reduction of blood glucose in mice [52]. Therefore, CREBH is an important physiological regulator of hepatic GNG. These findings are consistent with the idea that palmitate increases transcription and nuclear translocation of CREBH, which then acts in concert with other regulators of GNG to promote PCK1 and G6PC transcription. In addition, CREBH expression also can be activated by ER stress [53]. Recent studies have shown that ER stress may contribute to impaired regulation of glucose homeostasis and act as a potential link between obesity, impaired insulin sensitivity and the development of T2DM [23]. Many physiological and pathological processes that promote ER stress, including palmitate overloading [23], may induce metabolic responses through CREBH. Consistent with previous findings [51], we observed a 20-fold increase in CREBH mRNA in response to 1000 μM palmitate. Use of the TLR4 decoy peptide treatment significantly decreased this response. This novel observation suggests that palmitate induction of CREBH is mediated largely by TLR4 signaling, which agrees with previous reports that CREBH can be activated directly by LPS [54]. Although our data do not establish a causative role for CREBH in the increased GNG gene expression, the congruence between PCK1, G6PC and CREBH responses in our model is intriguing. Another key regulator of ER homeostasis is GRP78 due to its roles in protein folding and activation of ER stress sensors [55]. We found that palmitate significantly increased mRNA abundance of GRP78, as previously reported [22]. Similar to the CREBH response, mRNA abundance of GRP78 was not elevated by palmitate in the presence of the TLR4 decoy peptide (P=.08 vs. palmitate alone). Collectively, these responses indicate that the activation of TLR4 by palmitate can promote ER stress.
In conclusion, the present study demonstrates that TLR4 signaling plays a critical role in linking saturated FFA to increased transcription of gluconeogenic genes, and these effects are not explained by NF-κB responses. The mixtures of unsaturated and saturated fatty acids also induced gluconeogenic gene expression, although with far less potency than palmitate alone. Furthermore, both LPS and the nontoxic derivative of LPS, MLA, also promoted gluconeogenic gene transcription in a TLR4-dependent manner. Further studies are warranted to specify the signaling pathways downstream of TLR4 that mediate the effects of saturated FFA on GNG gene transcription, particularly the involvement of CREBH.
Acknowledgments
We thank Dr. Kyeong-Ok Chang at Kansas State University for kindly providing HuH7 cells.
Footnotes
Supported by the Kansas Agricultural Experiment Station; contribution no. 12-323-J. The project described was also supported by award number P20RR016475 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of National Center for Research Resources or the National Institutes of Health. Lipid analysis was performed at the Kansas Lipidomics Research Center, which was supported by NSF grants MCB 0455318 and 0920663 and DBI 0521587, and NSF EPSCoR grant EPS-0236913 with matching support from the State of Kansas through Kansas Technology Enterprise Corporation and Kansas State University.
References
- [1].Consoli A, Nurjhan N, Capani F, Gerich J. Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes. 1989;38:550–7. doi: 10.2337/diab.38.5.550. [DOI] [PubMed] [Google Scholar]
- [2].Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus.A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–7. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gastaldelli A, Baldi S, Pettiti M, Toschi E, Camastra S, Natali A, Ferrannini E, et al. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes. 2000;49:1367–73. doi: 10.2337/diabetes.49.8.1367. [DOI] [PubMed] [Google Scholar]
- [4].Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–6. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- [5].Lam TKT, Van de Werve G, Giacca A. Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab. 2003;284:E281–90. doi: 10.1152/ajpendo.00332.2002. [DOI] [PubMed] [Google Scholar]
- [6].Kehlenbrink S, Tonelli J, Koppaka S, Chandramouli V, Hawkins M, Kishore P. Inhibiting gluconeogenesis prevents fatty acid-induced increases in endogenous glucose production. Am J Physiol Endocrinol Metab. 2009;297:E165–73. doi: 10.1152/ajpendo.00001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am. 2008;37:635–46. doi: 10.1016/j.ecl.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pankow JS, Duncan BB, Schmidt MI, Ballantyne CM, Couper DJ, Hoogeveen RC, et al. Fasting plasma free fatty acids and risk of type 2 diabetes: the Atherosclerosis Risk in Communities Study. Diabetes Care. 2004;27:77–82. doi: 10.2337/diacare.27.1.77. [DOI] [PubMed] [Google Scholar]
- [9].Wang L, Folsom AR, Zheng Z-J, Pankow JS, Eckfeldt JH. Plasma fatty acid composition and incidence of diabetes in middle-aged adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Clin Nutr. 2003;78:91–8. doi: 10.1093/ajcn/78.1.91. [DOI] [PubMed] [Google Scholar]
- [10].Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through toll-like receptor 4. J Biol Chem. 2001;276:16683–9. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- [11].Wong SW, Kwon M-J, Choi AMK, Kim H-P, Nakahira K, Hwang DH. Fatty acids modulate toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009;284:27384–92. doi: 10.1074/jbc.M109.044065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Radin M, Sinha S, Bhatt B, Dedousis N, O’Doherty R. Inhibition or deletion of the lipopolysaccharide receptor toll-like receptor-4 confers partial protection against lipid-induced insulin resistance in rodent skeletal muscle. Diabetologia. 2008;51:336–46. doi: 10.1007/s00125-007-0861-3. [DOI] [PubMed] [Google Scholar]
- [13].Galloway E, Shin T, Huber N, Eismann T, Kuboki S, Schuster R, et al. Activation of hepatocytes by extracellular heat shock protein 72. Am J Physiol Cell Physiol. 2008;295:C514–20. doi: 10.1152/ajpcell.00032.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li L, Chen L, Hu L, Liu Y, Sun H-Y, Tang J, et al. Nuclear factor high-mobility group box1 mediating the activation of toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54:1620–30. doi: 10.1002/hep.24552. [DOI] [PubMed] [Google Scholar]
- [15].Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tsukumo DML, Carvalho-Filho MA, Carvalheira JBC, Prada PO, Hirabara SM, Schenka AA, et al. Loss-of-function mutation in toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–98. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- [17].Toshchakov VY, Fenton MJ, Vogel SN. Cutting edge: differential inhibition of TLR signaling pathways by cell-permeable peptides representing BB loops of TLRs. J Immunol. 2007;178:2655–60. doi: 10.4049/jimmunol.178.5.2655. [DOI] [PubMed] [Google Scholar]
- [18].Fujita T, Nolan GP, Liou H-C, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-κB p50 homodimers. Genes Dev. 1993;7:1354–63. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- [19].Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- [20].Sparkes BL, Slone EE, Roth M, Welti R, Fleming SD. Intestinal lipid alterations occur prior to antibody-induced prostaglandin E2 production in a mouse model of ischemia/reperfusion. Biochim Biophys Acta. 2010;1801:517–25. doi: 10.1016/j.bbalip.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. Thevaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–32. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- [22].Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185–94. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- [23].Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–81. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- [24].Dobbins RL, Szczepaniak LS, Myhill J, Tamura Y, Uchino H, Giacca A, et al. The composition of dietary fat directly influences glucose-stimulated insulin secretion in rats. Diabetes. 2002;51:1825–33. doi: 10.2337/diabetes.51.6.1825. [DOI] [PubMed] [Google Scholar]
- [25].Boden G, Chen X, Capulong E, Mozzoli M. Effects of free fatty acids on gluconeogenesis and autoregulation of glucose production in type 2 diabetes. Diabetes. 2001;50:810–6. doi: 10.2337/diabetes.50.4.810. [DOI] [PubMed] [Google Scholar]
- [26].Collins QF, Xiong Y, Lupo EG, Liu H-Y, Cao W. p38 mitogen-activated protein kinase mediates free fatty acid-induced gluconeogenesis in hepatocytes. J Biol Chem. 2006;281:24336–44. doi: 10.1074/jbc.M602177200. [DOI] [PubMed] [Google Scholar]
- [27].Gao D, Nong S, Huang X, Lu Y, Zhao H, Lin Y, et al. The effects of palmitate on hepatic insulin resistance are mediated by NADPH oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J Biol Chem. 2010;285:29965–73. doi: 10.1074/jbc.M110.128694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Massillon D, Barzilai N, Hawkins M, Prus-Wertheimer D, Rossetti L. Induction of hepatic glucose-6-phosphatase gene expression by lipid infusion. Diabetes. 1997;46:153–7. doi: 10.2337/diab.46.1.153. [DOI] [PubMed] [Google Scholar]
- [29].Xu C, Chakravarty K, Kong X, Tuy TT, Arinze IJ, Bone F, et al. Several transcription factors are recruited to the glucose-6-phosphatase gene promoter in response to palmitate in rat hepatocytes and H4IIE cells. J Nutr. 2007;137:554–9. doi: 10.1093/jn/137.3.554. [DOI] [PubMed] [Google Scholar]
- [30].Wan XD, Yang WB, Xia YZ, Wang JF, Lu T, Wang XM. Disruption of glucose homeostasis and induction of insulin resistance by elevated free fatty acids in human L02 hepatocytes. J Endocrinol Invest. 2009;32:454–9. doi: 10.1007/BF03346485. [DOI] [PubMed] [Google Scholar]
- [31].Baldeweg SE, Golay A, Natali A, Balkau B, Del Prato S, Coppack SW. Insulin resistance, lipid and fatty acid concentrations in 867 healthy Europeans. Eur J Clin Invest. 2000;30:45–52. doi: 10.1046/j.1365-2362.2000.00597.x. [DOI] [PubMed] [Google Scholar]
- [32].Jump DB, Botolin D, Wang Y, Xu J, Christian B, Demeure O. Fatty acid regulation ofhepatic gene transcription. J Nutr. 2005;135:2503–6. doi: 10.1093/jn/135.11.2503. [DOI] [PubMed] [Google Scholar]
- [33].Nakamura S, Takamura T, Matsuzawa-Nagata N, Takayama H, Misu H, Noda H, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284:14809–18. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Holland WL, Bikman BT, Wang L-P, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid–induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:1858–70. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bikman BT, Summers SA. Ceramides as modulators of cellularand whole-body metabolism. J Clin Invest. 2011;121:4222–30. doi: 10.1172/JCI57144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol. 1998;18:5457–64. doi: 10.1128/mcb.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–79. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- [38].Smith BK, Jain SS, Rimbaud S, Dam A, Quadrilatero J, Ventura-Clapier R, et al. FAT/CD36 is located on the outer mitochondrial membrane, upstream of long-chain acyl-CoA synthetase, and regulates palmitate oxidation. Biochem J. 2011;437:125–34. doi: 10.1042/BJ20101861. [DOI] [PubMed] [Google Scholar]
- [39].Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, et al. Reciprocal modulation of toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–51. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- [40].Lee JY, Zhao L, Hwang DH. Modulation of pattern recognition receptor-mediated inflammation and risk of chronic diseases by dietary fatty acids. Nutr Rev. 2010;68:38–61. doi: 10.1111/j.1753-4887.2009.00259.x. [DOI] [PubMed] [Google Scholar]
- [41].Simons K, Toomre D. Lipid rafts and membrane dynamics. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- [42].Ferré P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53:S43–50. doi: 10.2337/diabetes.53.2007.s43. [DOI] [PubMed] [Google Scholar]
- [43].Zhang LL, Gao CY, Fang CQ, Wang YJ, Gao D, Yao GE, et al. PPARγ attenuates intimal hyperplasia by inhibiting TLR4-mediated inflammation in vascular smooth muscle cells. Cardiovasc Res. 2011;92:484–93. doi: 10.1093/cvr/cvr238. [DOI] [PubMed] [Google Scholar]
- [44].Martin M, Michalek SM, Katz J. Role of innate immune factors in the adjuvant activity of monophosphoryl lipid A. Infect Immun. 2003;71:2498–507. doi: 10.1128/IAI.71.5.2498-2507.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Núñez Miguel R, Wong J, Westoll JF, Brooks HJ, O’Neill LA, Gay NJ, et al. A dimer of the toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signaling adaptor proteins. PLoS One. 2007;2:e788. doi: 10.1371/journal.pone.0000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cekic C, Casella CR, Eaves CA, Matsuzawa A, Ichijo H, Mitchell TC. Selective activation of the p38 MAPK pathway by synthetic monophosphoryl lipid A. J Biol Chem. 2009;284:31982–91. doi: 10.1074/jbc.M109.046383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2011;413:131–8. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- [48].Senn JJ. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem. 2006;281:26865–75. doi: 10.1074/jbc.M513304200. [DOI] [PubMed] [Google Scholar]
- [49].Cao W, Collins QF, Becker TC, Robidoux J, Lupo EG, Jr, Xiong Y, et al. p38 Mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J Biol Chem. 2005;280:42731–7. doi: 10.1074/jbc.M506223200. [DOI] [PubMed] [Google Scholar]
- [50].Nakatani Y, Kaneto H, Kawamori D, Hatazaki M, Miyatsuka T, Matsuoka TA, et al. Modulation of the JNK pathway in liver affects insulin resistance status. J Biol Chem. 2004;279:45803–9. doi: 10.1074/jbc.M406963200. [DOI] [PubMed] [Google Scholar]
- [51].Danno H, Ishii KA, Nakagawa Y, Mikami M, Yamamoto T, Yabe S, et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARα. Biochem Biophys Res Commun. 2010;391:1222–7. doi: 10.1016/j.bbrc.2009.12.046. [DOI] [PubMed] [Google Scholar]
- [52].Lee MW, Chanda D, Yang J, Oh H, Kim S, Yoon YS, et al. Regulation of hepatic gluconeogenesis by an ER bound transcription factor. CREBH. Cell Metab. 2010;11:331–9. doi: 10.1016/j.cmet.2010.02.016. [DOI] [PubMed] [Google Scholar]
- [53].Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–99. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- [54].Gentile CL, Wang D, Pfaffenbach KT. Fatty acids regulate CREBh via transcriptional mechanisms that are dependent on proteasome activity and insulin. Mol Cell Biochem. 2010;344:99–107. doi: 10.1007/s11010-010-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhou H, Zhang Y, Fu Y, Chan L, Lee AS. Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78) J Biol Chem. 2011;286:25687–96. doi: 10.1074/jbc.M110.212944. [DOI] [PMC free article] [PubMed] [Google Scholar]