

Abstract
We posit a bottom-up sleep regulatory paradigm in which state changes are initiated within small networks as a consequence of local cell activity. Bottom-up regulatory mechanisms are prevalent throughout nature, occurring in vastly different systems and levels of organization. Synchronization of state without top-down regulation is a fundamental property of large collections of small semi-autonomous entities. We posit that such synchronization mechanisms are sufficient and necessary for whole organism sleep onset. Within brain we posit that small networks of highly interconnected neurons and glia, e.g. cortical columns, are semi-autonomous units oscillating between sleep-like and wake-like states. We review evidence showing that cells, small networks, and regional areas of brain share sleep-like properties with whole animal sleep. A testable hypothesis focused on how sleep is initiated within local networks is presented. We posit that the release of cell activity-dependent molecules, such as ATP and nitric oxide, into the extracellular space initiates state changes within the local networks where they are produced. We review mechanisms of ATP induction of sleep regulatory substances (SRS) and their actions on receptor trafficking. Finally, we provide an example of how such local metabolic and state changes provide mechanistic explanations for clinical conditions such as insomnia.
Keywords: ATP, cytokine, receptor, cerebral blood flow, brain imaging
Introduction
The minimum amount of nervous tissue necessary and sufficient for sleep to occur has yet to be identified. Regardless, despite millions of cases of lesioned brains, not a single case of a surviving individual or animal completely lacking sleep has been reported. This strongly suggests that sleep is a fundamental property of nervous tissue and that sleep is self-organizing. These observations led to theories of sleep initiation and regulation that require universal mechanisms widely applicable to damaged nervous systems or viable nervous tissue components whether in vivo or in vitro (Krueger and Obal, 1993; Kavanau, 1994; Tononi and Cirelli, 2003). The prevailing theories of sleep regulation are top-down regulatory mechanisms; they posit that sleep regulatory circuits impose sleep on the brain. Top-down regulatory systems are characteristic of engineered systems, for example the air traffic control system, but are fragile if disturbed. However, the top-down theories persist despite the robust evidence that lesion of any one of the sleep regulatory circuits fails to eliminate sleep, although after such lesions sleep is often abnormal in timing, behavior and electrophysiologically. Such sleep center theories have inadequately addressed many known sleep phenomena including sleep-dependent optimum performance, recovery of sleep after lesions, sleep inertia and clinical conditions associated with state dissociations (Mahowald et al., 2011). Further, the top-down theories have failed to provide insight into how brain activity is translated into sleep regulatory signals or how the waking brain transitions into sleep states. Logically, if one posits top-down sleep regulation, for instance by a sleep regulatory center, it leads to an infinite regress. Thus, if a sleep regulatory center is posited to impose sleep on the entire brain, then one has to ask “What is telling the center to impose sleep?” If an answer is provided, then one must ask; “Who is telling the teller?” etc. Bottom-up mechanisms avoid this logic pitfall.
A new sleep bottom-up regulatory paradigm is developing. Bottom-up mechanisms are fundamentally different from top-down regulation, lacking a central command. Within biology, bird flocking is an example of a dynamic bottom-up mechanism regulating state changes (direction of flock flight). The individual birds are semi-autonomous entities loosely connected via sight. No individual bird is dictating flock direction. Instead, the responses of an individual bird to another bird’s change in flight direction leads to the entire flock rapidly changing direction. Bottom-up mechanisms are prevalent in nature occurring in vastly different systems and levels of organization. Semi-autonomous units spontaneously synchronize without central control (Strogatz, 2004). We apply those ideas to sleep by positing that small highly interconnected networks of neurons and glia, such as cortical columns, are fundamental semi-autonomous units, loosely connected via electrical and chemical signaling, oscillating between states and synchronizing with each other. As such sleep is viewed as a distributed process in the brain being initiated within local small circuits as a consequence of prior activity and the associated changes in metabolism and cerebral blood flow within those circuits (Krueger et al., 2008; Rector et al., 2009). In these views, state oscillations occur within any viable neuronal/glia network and organism sleep emerges as local circuits synchronize state. The known sleep regulatory circuits may orchestrate the spatial and temporal coordination of state in multiple smaller networks. This organization in turn contributes to niche-appropriate predictive and preemptive behavior. Herein we address the issue of state changes occurring at different levels of tissue organization and weave a testable theory of sleep initiation and regulation.
Sleep-like phenotypes at different levels of tissue organization
Do neurons or glia sleep? This question raises definitional problems. It is not a simple matter of an inactive neuron being asleep and an active neuron being awake. There are many brain phenomena where inactivity provides or fine-tunes information, e.g., inhibitory surround areas within sensory receptive fields. Further, the separation of glia and neuron involvement in sleep regulation is difficult because astrocytes are intimately involved in sleep (Halassa et al., 2009). Other cell types in the brain may also be involved, e.g. capillary endothelial cells or arteriolar smooth muscle cells controlling cerebral blood flow. Regardless, thalamic and cortical neurons display a burst-pause action potential firing pattern during NREMS. Bursts of action potentials lasting about 500 milliseconds are followed by roughly 500 milliseconds of silence. These burst-pause patterns unfold as an animal is going to sleep and when synchronized with each other lead to a slow 1 Hz component of the electroencephalogram (EEG). High amplitude 0.5-4 Hz EEG waves characterize slow wave sleep (SWS). Thus one could use a burst-pause firing pattern as a definition for a neuron going to sleep as long as the caveats mentioned are attached to the definition, e.g. the unknown role of glia. Further, burst-pause activity can occur when an animal is awake (Pigarev et al., 1997), although the synchronized pattern of populations of neurons exhibiting the burst-pause pattern is absent during waking (Vyazovskiy et al., 2011a).
State oscillations in mixed neuronal/glia cultures
If we define a network state as a stereotypic output in response to a stable input, then a new state occurs when the same input leads to a different output. For instance, waking stimuli that induce explicit conscious perceptions often do not during sleep. Extending this logic to small networks, including cultured neurons and glia, it is predicted that: a) Characteristics of state oscillations, frequency, amplitude and synchronization, are activity-dependent. In neuronal/glial cultures action potentials are most often used to characterize state but other measures such as field potentials, gene expression, and neurotransmitter/modulator release are also used. b) State oscillations are regulated by activity-dependent genes. c) Unlike whole animals, cultures lack dynamic changes in blood flow and afferent input to drive activity; thus the nature of exogenously supplied input will dictate gene expression and state oscillation characteristics. d) If small networks are connected either electrically or chemically, the respective state oscillations will synchronize and coalesce into states expressing emergent properties. Predictions a-c have already been tested as outlined below; the experimental investigation of prediction d will likely provide major insights into emergent properties of brain tissue including sleep.
Mature neuronal/glial tissue cultures form active networks that display periodic bursts of action potentials. Although the inter-burst intervals in culture are longer than the inter-burst intervals that characterize thalamic and cortical neurons during sleep, the firing patterns suggest a sleep-like default state (Corner, 2008; Hinard et al., 2012). Slow frequency (0.05–0.2 Hz) electrical stimulation of such cultures entrains the spontaneous network bursts and recruits more active channels to participate in network-wide bursts, suggesting a sleep-like state (Chiappalone et al., 2007; Wagenaar, et al., 2005). In contrast, higher frequency stimulation (4–10 Hz) induces more random firing with fewer bursts than occur during baseline spontaneous conditions (Wagenaar, et al., 2005). Tafti and colleagues (Hinard et al., 2012) showed that the default sleep-like state could be transformed to a tonic firing pattern similar to that observed in wakefulness by stimulating the cultures with a cocktail of excitatory neurotransmitters. In addition, the chemically-stimulated cultures exhibited a transcriptome similar to the transcriptome of cortical cells harvested from sleep-deprived mice. Studies of neuronal culture state oscillations are not expected to provide a full understanding of sleep as a whole animal global behavior state unfolding in time and space. However, they are just in their infancy and already they indicate that the fundamental electro-chemical and genetic mechanisms responsible for network state oscillations and the consequences of state oscillations can be studied under controlled conditions in vitro. These results also lead to the issue of whether small networks in vivo demonstrate state oscillations, and if so, whether there is any connection of those oscillations to sleep and performance.
Cortical column sleep
A seminal paper by Pigarev et al. (1997) connects cortical neuron firing pattern to local cortical network state. The investigators measured firing patterns of individual occipital cortex/visual cortex neurons of monkeys as they were falling asleep yet still performing a visual task. During the wake-sleep transition, some of the cells started to fail to fire in response to stimulation whereas other cells did not. Importantly, the topographic location of those neurons within their visual receptive field predicted when the neuron would stop responding. Neurons in the peripheral part of the receptive stop responding before those in the central part of the receptive field. This strongly suggest that the change in local state is a network phenomenon because the order of recruitment is topographic dependent. Pigarev concluded that sleep was a property of local networks.
Cortical columns are anatomically defined, experimentally accessible, examples of small neuronal/glial networks. The rat somatosensory cortex is organized into columns with afferent nerve fibers from facial whiskers mapping onto specific columns and individual columns primarily receiving input from a specific whisker. Repeated single whisker stimulation and analysis of individual traces demonstrates evoked response potential (ERP) patterns that exhibit stereotypical temporal patterns. Electrical activity of single columns in response to evoked afferent input can be isolated then correlated with whole animal sleep (Rector et al., 2005). When the animal is awake, the ERP amplitude is smaller than when the rat is in NREMS if low sensory stimulus intensities are used (Phillips et al., 2012). When the animal is asleep, roughly 80% of the ERPs are sleep-like in that they have a high-amplitude. Conversely, when the animal is awake, most of the ERPs exhibit low amplitudes suggesting they are wake-like. Further, the longer a cortical column is in the wake-like state, i.e. has a low ERP magnitude, the higher the probability over the next few minutes it would enter the sleep-like state. This property is similar to a whole animal sleep in that the longer wakefulness persists, the higher the future probability of sleep. Further, since cortical neurons are more active during waking (Vyazovskiy et al., 2009), these results suggest that the probability of sleep occurring is activity-dependent. Most importantly, Rector and colleagues (Walker et al., 2005; Topchiy et al., 2009) showed that the cortical column state made a difference in the animal’s performance. He trained rats to lick in response to the stimulation of a single whisker, and then measured the ERP to characterize the state of that whisker’s column. When the ERP was of low amplitude, suggesting the column was in the wake-like state, the animal made the correct response (Figure 1). In contrast, if the ERP was of high amplitude, suggesting a sleep-like state, errors of omission or co-mission occurred. Further, the local application of tumor necrosis factor alpha (TNF), a sleep regulatory substance, to cortical columns promotes the sleep-like state determined using ERPs, (Churchill et al., 2008). We conclude from these experiments that; a) there is a behavioral manifestation of cortical column state that parallels whole animal state-dependency of behavior; b) sleep does not occur uniformly even within functionally coherent brain regions; and c) the oscillations of columns between states is dependent upon prior cell activity within the column.
Figure 1.

Response of a rat trained to lick a sucrose solution after stimulation of a specific facial whisker. Animals were trained to lick for a sweetened water reward when a particular whisker was stimulated (Stim Reward), but not to lick when a different whisker was stimulated (Sham No Reward). Training occurred over a 3 to 6 month period. Before training, a 64 channel electrode array was also implanted over the surface of the somatosensory cortex, and individual whisker cortical columns (barrels) were mapped and associated with the particular whiskers being stimulated. Once animals achieved greater than 80% correct responses, the electrical evoked responses from each electrode corresponding to the Stim and Sham whisker were sorted into those trials that were correct (Stim Reward with Licks or Sham No Reward no Licks) and those that were incorrect (Stim Reward with no Licks or Sham No Reward with Licks), and averaged across trials. Evoked responses were over 200 uV larger before incorrect responses, suggesting that the cortical column for each whisker was in its sleep-like state during the trial that resulted in an incorrect response.
Our model posits that a local network oscillate between states whenever there is an activity-dependent build-up of sleep regulatory substances. If the animal/human happens to be doing a task at that moment, and since the input/output relationship of the network is altered, the result can be an error in data processing resulting in performance degradation. Thus one could easily envision a requirement of multiple networks being engaged in a behavioral task; if one or more of the local networks oscillated into the sleep-like state the collective output of multiple networks would be changed – perhaps leading to an error. Further, it is parsomonious to posit that all sub-cortical networks, including known sleep regulatory networks, also are subject to activity-dependent molecular and genetic mechanisms as are cortical columns. A logical extension of this hypothesis is that both NREMS and REMS are a consequence of these mechanisms but mechanistically involve different levels of the central nervous system (Krueger and Obal 1993).
Sleep in larger parts of the brain
In animals, surgically-prepared cortical islands, lacking thalamic input but with intact blood flow, exhibit oscillations between EEG-slow waves and wake-like states, independent of behavioral sleep-wake state (Kristiansen and Courtois, 1949). The sleep regulatory substances (SRSs), interleukin-1 beta (IL1), TNF, growth hormone releasing hormone, and brain derived neurotrophic factor (BDNF), act locally within the cortex to alter EEG δ power, a sleep phenotype. After unilateral application of these substances to the cortical surface, EEG δ power during NREMS on the ipsilateral side, but not on the contralateral side, is enhanced, suggesting a more intense sleep on the side receiving these SRSs (Faraguna et al., 2008; Yasuda et al., 2005, 2007; Yoshida et al., 2004). Ipsilateral EEG δ power is not altered by these substances during waking or REMS. These SRS-induced unilateral EEG δ power changes correlate with enhanced cFos and IL1 immuno-reactivity in the corresponding cortical areas and reticular thalamus (Hallett et al., 2010). These results are consistent with and implicate the involvement of known biochemical sleep regulatory pathways (Krueger, 2008) and known cortico-thalamic sleep regulatory circuitry (Steriade, 2003) in these uni-hemispheric sleep phenotypes. Further, coupled with what is known about use-dependent production of IL1, TNF, and BDNF, these data strongly support the idea that sleep is targeted to active circuits and is initiated at a local network level.
Local to global
The observation of state oscillations at various levels of tissue organization suggests that sleep and some sleep phenotypes emerge from the integration and synchronization of smaller local network states and that sleep is dependent upon prior cell activity. Several sleep regulatory circuits, such as the hypothalamic ventrolateral preoptic area, are well characterized and reviewed extensively elsewhere (Saper et al., 2005; Szymusiak and McGinty, 2008; Jones, 2003; Datta and Maclean, 2007). Less well known are the observations that any coupled oscillators will spontaneously self-organize and synchronize oscillations. Oscillation frequency and amplitude are dependent upon system characteristics such as the strength of coupling between the semi-autonomous units. Synchrony of such networks occurs spontaneously throughout nature even in the absence of top-down regulatory control (Strogatz, 1997; Strogatz, 2004). As described above, cortical columns are semi-autonomous networks that oscillate between states (Rector et al., 2005) and are coupled electrically and biochemically to each other. Thus, we develop the idea that sleep results from the spontaneous local network state synchronization (Krueger et al., 2008) and this hypothesis has been modeled mathematically (Roy et al., 2008). Whole organism sleep is viewed as an emergent property of synchronized states of smaller local networks. The idea that sleep-like states are a fundamental property of local networks and are dependent upon the activity of such networks developed from our knowledge of the biochemical mechanisms of sleep (Krueger and Obal 1993). Such a view is not dissimilar to the modern view of the emergence of whole animal circadian rhythms from multiple semi-autonomous cells, each expressing the genes that collectively make the molecular clock (Lowrey and Takahashi, 2011). We now turn to sleep biochemical mechanisms before further integrating the newer concepts of brain organization of sleep into sleep-related synaptic connectivity.
Indexing brain activity to track prior activity and conversion to a sleep regulatory signal
A basic tenet of our local use-dependent sleep hypothesis is that CNS cell activity predicts sleep need (Krueger and Obal, 1993). This section addresses what is known of mechanisms and gene expressions involved in the initiation of state oscillations. Figure 2 outlines a cellular molecular mechanism that is posited to lead to state oscillations within small networks and whole animals1. ATP concentration in synaptic vesicles is 10-50 times higher than in the cytosol. ATP is released from GABAergic, cholinergic, noradrenergic and glutamatergic neurons and glia as a consequence of cell activity into the surrounding extracellular space (Burnstock, 2006, 2007). ATP is thus considered a neuro- and gliotransmitter. Some of the extracellular ATP is converted to adenosine via the ectonucleotidases CD39 and CD73. Extracellular adenosine from this source could contribute to sleep although extracellular adenosine, like extracellular ATP, is rapidly metabolized and removed. Nevertheless, extracellular adenosine is a key signal for induction of cerebral blood vessel dilatation and consequent increases in blood flow. Further, extracellular adenosine increases K+ permeability to hyperpolarize cells via purine type I receptors. This cellular hyperpolarization, called the down-state, leads to the action potentials pauses that characterize thalamic and cortical cells in sleeping animals mentioned earlier. This is a low energy demand condition that, coupled with the adenosine-enhanced blood flow, provides an opportunity for energy restoration.
Figure 2.
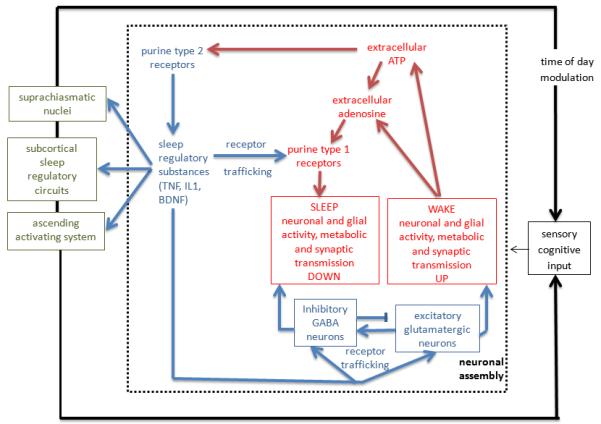
A proposed mechanism for the indexing of prior brain activity and its translation into a sleep regulatory mechanism. The steps within the broken line box are envisioned to occur within small networks such as cortical columns (see text). Those steps result in a state shift within the local network and in the production of molecules that interact with known sleep regulatory circuits (far left small boxes) that in turn coordinate state of larger areas of brain for niche-adaptation purposes. For instance, local application of IL1 to the cortex induces local cortical delta waves (Yasuda et al., 2005) and fos expression in the ventrolateral preoptic hypothalamic area (Yasuda et al., 2007), a well know sleep regulatory circuit. Neuronal activity is greater during waking than sleep (Vyazovskiy et al., 2009). Cell activity is associated with release of ATP into the extracellular (ex) space. ATPex either rapidly is catabolized to adenosine via ectonucleotidases or it binds to purine type 2 receptors. Ex-Adenosine binds to purine type 1 receptors and thereby rapidly and transiently hyperpolarizes neurons. In contrast, activation of the purine type 2 receptor P2X7 releases cytokines and neurotrophins from glia or neurons (see text); these molecules affect receptor trafficking including purine type 1, GABA, and glutamate receptors (Imeri and Opp, 2009; Reyes-Vazquez et al., 2012; Krueger et al., 2008). Cytokines also interact with known sleep regulatory circuits (far left) and thereby affect sleep (Nistico et al., 1992; Santello et al., 2011). For instance, IL1 inhibits serotonergic neurons and enhances GABAergic signaling in the dorsal raphe nucleus (Brambilla et al., 2007, 2010; Manfridi et al., 2003). IL1 also modulates locus coeruleus (DeSarro et al 1997), preoptic, basal forebrain and cholinergic laterodorsal tegmental neurons to affect sleep (Alam et al., 2004) and the suprachiasmatic nucleus to affect clock genes (Cavadini et al., 2007). → indicates stimulation; indicates inhibition.
Conversion of extracellular ATP to adenosine is rapid and does not appear to be self-sustaining. As such it could be involved in rapid state transitions but is unlikely to be involved in long-term sleep regulation. A slower process is also initiated by the transient changes in extracellular ATP. These changes are detected by membrane purine type 2 receptors and these receptors, e.g. the P2X7 receptor, in turn, cause the release of activity-dependent sleep regulatory substances such as TNF, IL1 and BDNF (Bianco et al., 2005; Ferrari et al., 2006; Hide et al., 2000; Solle et al., 2001; Suzuki et al., 2004; Verderio et al., 2006). ATP agonists enhance NREMS while ATP antagonists inhibit sleep in mice (Krueger et al., 2010). Further, mice lacking the P2X7 receptor exhibit reduced duration of NREMS and EEG δ wave power during NREMS compared to control mice after sleep deprivation. ATP-modulated release of IL1, TNF and BDNF provides for the translation of the transient ATP signal into local longer-lived (minutes to hours) regulatory signals capable of indexing prior cell activity. IL1, TNF and BDNF are pleiotropic having several activities via their respective receptors and signaling cascades, including promotion of sleep and acting on synaptic efficacy. For instance, they activate nuclear factor kappa B, (NFkB). Expression of purine type 1 receptors (Khoa et al., 2001; St Hilaire et al., 2009) and the glutamate AMPA receptor-gluR1 mRNAs (Furakawa and Mattson, 1998; O’Mahony et al., 2006; Yu et al., 2002) are affected by TNF and/or NFkB activation. Activation of NFkB in mammals promotes NREMS (Kubota et al 2000; Chen et al., 1999) and in fruit flies relish, the homolog gene for mammalian NFkB, is involved in sleep (Kuo et al., 2010; Williams et al 2007). The respective receptors and their subsequent cell membrane expression would change sensitivity of the postsynaptic neurons. Such receptor population change mechanisms take place over longer periods and they are proto-typical of synaptic scaling because the expression of postsynaptic receptors is modulated by presynaptic neuron activity as initiated by the release of ATP. Thus, postsynaptic neuron sensitivity is scaled to the prior use of the presynaptic synapse. TNF and BDNF are firmly linked to synaptic scaling (Beattie et al., 2002; Stellwagon and Malenka, 2006; Turrigiano 2008). These ATP/adenosine/cytokine events occur locally within active networks and the collective electro-chemical changes result in a shift in input-output relationships within the networks, i.e., a state shift.
The evidence implicating cytokines in sleep regulation was developed independently of the ATP literature; by way of example the TNF-sleep literature is briefly summarized (Krueger, 2008). If TNF is injected almost all of the consequences associated with prolonged sleep loss are induced. These include sensitivity to pain stimuli; cognitive, memory, and performance impairments; and symptoms of depression, sleepiness, fatigue, prolonged sleep. TNF injection also leads to symptoms associated with chronic sleep loss-associated pathologies such as metabolic syndrome, chronic inflammation, and cardiovascular disease. Further, a clinically available blocker of TNF (Enbrel) alleviates many of the sleep loss-associated symptoms, e.g. fatigue and sleepiness (Franklin, 1999; Vgontzas et al., 2004). In animals, TNF injections promote sleep, and inhibition of TNF using antibodies or soluble receptors reduces sleep. Mice lacking TNF receptors have reduced NREMS and REMS. In humans and animals, sleep deprivation up regulates TNF levels in blood or brain. Local application of TNF to cortical columns drives the columns into a sleep-like state (Churchill et al., 2008), and application of TNF to one side of the cortex enhances EEG SWA on the ipsilateral side but not on the contralateral side during sleep but not during wakefulness (Yoshida et al., 2004). In contrast, inhibition of spontaneous cortical TNF expression using a TNF small interfering (si)RNA has the opposite effects (Taishi et al., 2007). TNF immune-reactivity in neurons is enhanced after neuronal activity. An independent literature has characterized the involvement of TNF in synaptic scaling involving receptor trafficking as mentioned.
Sleep regulation and receptor trafficking: functional implications
By mobilizing neurotrophins, cytokines and synaptic receptors, which are the center-stage molecular substrates for neural plasticity, the sleep regulatory mechanism tap into the common paths that mediate experience-dependent plasticity, such as those that occur during learning and memory. Indeed, sleep-induced receptor trafficking has been directly compared to synaptic long-term potentiation or depression (LTP/D), which are best known as cellular substrates of learning and memory (Bredt and Nicoll, 2003). For example, in the rodent cerebral cortex and hippocampus sleep is accompanied by a general down regulation of synaptic AMPA receptors (AMPARs) as well as LTD-like molecular signatures including decreased phosphorylation of the AMPAR subunit GluR1 and CaMKII (Vyazovskiy et al., 2008; Hagewoud et al., 2010). Further, AMPAR responsiveness to unitary vesicular release of glutamate is decreased, suggesting decreased transmission efficacy (Liu et al., 2010). Consequently, after sleep, there is an enhanced capacity to induce long-term potentiation of AMPAR transmission in the cortex (Vyazovskiy, et al., 2008) and after acute or prolonged sleep loss LTP is inhibited (Davis et al., 2003; Vyazovskiy et al., 2011b). In an extension of those findings, removal of AMPARs during sleep in layer V pyramidal neurons occurs in the somatosensory cortex; this involves specifically the removal of Ca2+-permeable AMPARs (Lante et al., 2011). In the hippocampus, sleep deprivation results in reduced surface expression of NMDA receptors (NMDARs) in CA1 pyramidal neurons, which leads to impaired induction of NMDAR-dependent AMPAR LTP, thus affecting AMPAR trafficking by regulating synaptic NMDARs (McDermott et al., 2006, 2003; Kopp et al., 2006). Interestingly, when sleep-regulated plasticity genes were examined independent of circadian time, a number of the key players, including phosphorylated CREB, BDNF and Arc, were regulated by the ebb and flow of noradrenergic innervations (NE) during the sleep-wake cycle (Cirelli and Tononi, 2000). An important link was then provided in the context of emotion-charged learning; NE facilitates synaptic delivery of AMPARs and LTP induction by driving GluR1 phosphorylation (Hu et al., 2007). Thus, NE may represent one of the global regulators in setting the general tone of AMPAR down-regulation during sleep.
The above observations lead to the impression that sleep is generally accompanied by an LTD-like process in the cortex and hippocampus, which facilitates LTP induction during wakefulness and thus supports the hypothesis that sleep restores neural plasticity mechanisms and benefits subsequent learning and memory (Tononi and Cirelli, 2003; Krueger and Obal, 1993). However, this generalization may be premature. Within the cortex, there is good evidence for a LTP-like processes occurring during sleep that mediates memory consolidation rather than erasure. In the cat primary visual cortex, LTP-like changes in protein phosphorylation occur during sleep following prior visual experience, including GluR1 of AMPAR, ERK1/2 187, and CaMKIIα/β. These changes are accompanied by synaptic strengthening that consolidates the waking visual experience (Aton et al., 2009). In layer V/VI pyramidal cells of the medial prefrontal cortex, an enhanced miniature excitatory post synaptic current has been recorded after sleep, again suggesting strengthened AMPA transmission during sleep (Winters et al., 2011). Evidence along this line not only exemplifies the molecular and cellular substrates in support of the long-standing view that memory consolidation can occur during sleep (Walker and Stickgold, 2004; Marshall et al., 2006; Aton et al,, 2009; Mander et al., 2011), but also underscores the importance of specifying brain regions and local neural ensembles in defining the mechanisms or functional consequences of sleep.
The seemingly opposing results showing both strengthening and weakening of AMPAR transmission during sleep suggest a degree of complexity governing the sleep regulatory effects on synaptic transmission across brain regions. A key variable affecting the magnitude and direction of AMPAR transmission may be local network activity. For example, cortical pyramidal neurons fire action potentials either as semi-random individual spikes or as bursts of distinct frequencies. Both the timing of single action potentials firing and the frequency of action potential bursts within the range of sleep-related EEG oscillations can determine the direction (LTP or LTD) and extent of plasticity (Birtoli and Ulrich, 2004; Czarnecki et al., 2007). Nonetheless, sleep slow oscillations during NREM sleep are posited to be responsible both for LTD-like synaptic depression (Tononi and Cirelli, 2003; Czarnecki et al., 2007), and for LTP-like potentiation in the rodent somatosensory cortex (Chauvette et al., 2012), suggesting yet another layer of unidentified local regulatory mechanisms. If regulation of sleep is dependent on prior wake activity, and if both wake activity and sleep regulation has a local component, there is good reason to predict that sleep – from the molecular regulatory machinery to the functional consequence – inevitably reflects local needs.
Long-term synaptic reorganization achieved during sleep is best characterized in the cerebral cortex and the hippocampus, though it may go far beyond. In studying the nucleus accumbens (NAc) in rodents, a critical forebrain region at the ventral striatum that mediates reward processing, we found that the GABA transmission onto NAc principle neurons was enhanced relative to glutamate transmission following acute sleep deprivation (unpublished data). This may have direct implications to the altered reward-seeking behaviors that result from sleep deprivation (Steiner and Ellman, 1972; Aalto and Kiianmaa, 1986; Everson et al., 1989; Puhl et al., 2009; Hanlon et al., 2010), as GABA transmission in the NAc is shown to be critical for the regulation of both food reward (Hanlon, et al., 2004) and intracranial self-stimulation (Cheer et al., 2005). In humans, although sleep is well-recognized for its involvement in regulating emotional and motivational behaviors (Walker, 2010; Gujar et al., 2011a; Gujar et al., 2011b; Mingkel et al., 2011; Hanlon and Van Cauter, 2011; Hasler et al., 2012; Crum et al., 2004; Drummond et al., 1998; Hasler et al., 2012; Venkatraman et al., 2011), the underlying cellular and molecular mechanisms remain poorly understood. Knowledge about sleep-induced synaptic reorganization in brain reward circuitry will not only provide mechanistic insights for reward behavioral research, but also benefit sleep research by offering a common ground for a mechanistic comparison of sleep across brain regions. Thus, although the molecular details may differ significantly across different brain regions, sleep-induced synaptic (and network) remodeling has direct and diverse functional implications for the subsequent wake period. How those changes are affected by or may contribute to local as well as global sleep regulation are only beginning to be understood.
Sleep mechanisms are inseparable from brain plasticity mechanisms
Within the brain the connectivity process is regulated in part by gene expression; changes in gene expression affect the functional properties of the neuronal/glial networks. For instance, changes in receptor populations within a network will change that network’s responsiveness to appropriate stimuli. Many of these connectivity genes are activity-dependent in that their expression is directly altered by cell activity, e.g. action potentials. Further, activity-dependent brain expression of many of those genes, e.g. IL1, TNF, and neurotrophins such as BDNF, is also characterized as being involved in neuronal connectivity including synaptic scaling and as sleep regulatory substances (Krueger et al., 2008; Kushikata et al., 1999; Turrigiano, 2008). Because these sleep-regulatory genes are activity-dependent they provide a mechanism to target active networks/cells/synapses. Such mechanisms are thus bottom-up function-dependent regulatory events targeted to the networks and synapses used. Use-dependent connectivity changes are established epigenetic brain plasticity mechanisms. They also provide positive feedback leading to enhanced strength of connectivity thus to greater re-use of the active synapses/circuits whether they are excitatory or inhibitory. With time and greater use and re-use such a mechanism would be potentially destabilizing, leading, for example, to rigid non-plastic networks (Kavanau et al., 1994; Turrigiano, 2008). These observations suggest the presence of stabilizing mechanisms to preserve the brain’s epigenetic use-dependent plasticity (Krueger and Obal, 1993). We propose that these mechanisms are activity-dependent, involve the same molecules as regulate sleep, occur within the used local networks, provide a means for the brain to track prior activity, target network state changes to the active areas, and lead to emergent sleep within larger network collectives.
Human brain imaging studies; implications for regional sleep and application to a clinical condition
Human functional neuroimaging studies of NREMS and REMS have examined regional cerebral blood flow (CBF) using H 152O positron emission tomography (PET), and relative regional glucose metabolism (RRGM) using [18F] fluorodeoxyglucose (FDG) PET. NREMS, and slow wave sleep (SWS) in particular, are characterized by widespread reductions in CBF and glucose metabolic rate. The reductions are particularly marked in subcortical structures including thalamus, brainstem (including pontine and mesencephalic tegmentum), basal ganglia, and basal forebrain; and in cortical regions including orbital, medial, and dorsolateral prefrontal cortex, medial parietal cortex (precuneus and posterior cingulate), mesiotemporal cortex, and anterior cingulate (Maquet, 2000; Dang-Vu et al., 2010). Within SWS, phasic EEG features such as sleep spindles and slow waves are associated with more specific decreases in regional CBF: Within the thalamus for sleep spindles, and within medial frontal and parietal cortex for slow waves (Hofle et al., 1997; Dang-Vu et al., 2005). Subcortical changes during SWS likely reflect deactivation of arousal and sleep modulatory structures, as well as disfacilitation of the thalamus. On the other hand, widespread reductions in cortical blood flow and metabolism during SWS correspond to structures and circuits involved in waking functions such as cognitive control (frontal), emotion regulation (anterior cingulate, medial and orbitofrontal, mesial temporal), and self-awareness (precuneus, posterior cingulate, medial frontal).
Human functional neuroimaging studies of REM sleep show a very different pattern. Significant increases in CBF and RRGM (relative to SWS and wake) occur in subcortical structures including the pontine tegmentum and thalamus, in limbic structures (amygdala, hippocampus, and anterior cingulate cortex) and in temporal-occipital cortex. In contrast, persistent deactivation occurs in cortical structures including dorsolateral prefrontal, and medial and lateral parietal cortices, including the precuneus/posterior cingulate (Maquet, 2000; Dang-Vu et al., 2010). This pattern of activation and deactivation could be interpreted as indicating a state of arousal (brainstem, limbic system) accompanied by a relative lack of cognitive control (prefrontal cortex) or self-awareness (precuneus/posterior cingulate). This pattern could also be interpreted as being consistent with typical dream characteristics such as prominent visual and auditory perception, emotionality, lack of insight and temporal organization, and poor recall (Maquet et al., 1996).
In summary, NREMS and REMS in humans show dramatically different patterns of regional brain activation and deactivation. While both states are characterized by reduced activity in fronto-parietal cortex, NREMS is additionally characterized by reduced subcortical and limbic system activity, and REMS is characterized by increased activity in these structures.
The concept that regions of brain semi-independently express state as evidenced by imaging studies may also be clinically applicable. FDG PET studies suggest that individuals with insomnia have smaller wake-NREMS reductions of metabolism in arousal centers (brainstem, thalamus, amygdala, basal forebrain-hypothalamus, cingulate) compared to good sleepers (Nofzinger et al., 2004). Thus, higher RRGM occurs in these structures during NREMS compared to controls. RRGM in these structures during NREMS correlates with self-reported wakefulness after sleep onset (Nofzinger et al., 2006). More recent analyses also indicate that individuals with insomnia have higher RRGM in the precuneus, posterior cingulate and medial frontal cortex (Figure 3) (Buysse et al., 2011). These are central structures in the brain’s “default mode network” (DMN). The DMN is most active during passive resting wakefulness, and deactivates during tasks requiring focused attention (Raichle,2010; Fox and Raichle, 2007; Buckner et al., 2008; Broyd et al., 2009; Boly et al., 2008; Cavanna and Trimble, 2006). DMN activity is increased during unfocused, exploratory monitoring of the environment, internal mentation drawing on personal memory, and self-referential thinking (Buckner et al., 2008; Cavanna and Trimble, 2006). Increased DMN activity during NREMS is consistent with symptoms of enhanced awareness and “mind-wandering” among individuals with insomnia, even during EEG-defined sleep, and with observations that mind-wandering is associated with both enhanced DMN activity (Mason et al., 2007) and unhappiness (Killingsworth and Gilbert, 2010). Individuals with insomnia typically report more severe subjective symptoms (e.g., longer sleep latency and greater wakefulness after sleep onset) than is measured with concurrent polysomnography, a phenomenon sometimes known as sleep state misperception (Rosa and Bonnet, 2000; Krystal et al., 2002; Edinger and Krystal, 2003). Persistent DMN activity during NREMS may be a functional correlate of this phenomenon. Conversely, reduced activity of the DMN during NREMS in good sleepers may be consistent with a lack of self-awareness that leads this group to underestimate sleep latency and wakefulness after sleep onset (Manconi et al., 2010).
Figure 3.
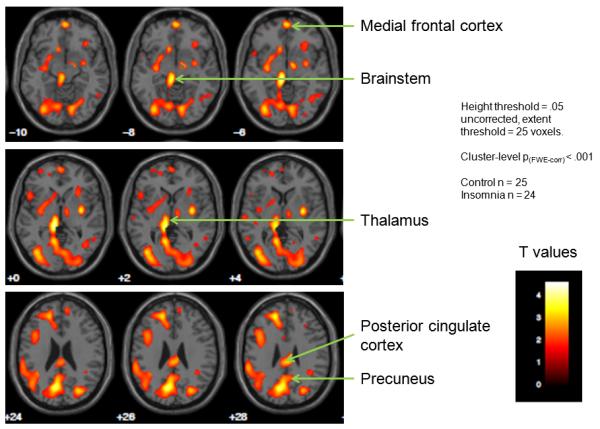
Subtraction images of [18F]-fluorodeoxyglucose positron emission tomography (FDG PET) scans conducted in participants with chronic insomnia and good sleepers. Participants in each group had FDG PET scans during morning wakefulness and the first non-rapid eye movement sleep (NREMS) period. Statistical Parametric Mapping version 8 was used to examine the interaction of sleep-wake state with participant group. The images above indicate areas in which the wake-NREMS differences were smaller for participants with chronic insomnia versus controls. Relatively smaller wake-sleep changes were observed for insomnia participants in regions related to introspective, self-focused mental activity (precuneus, posterior parietal cortex), emotion regulation (medial frontal cortex) and sleep-wake regulation (thalamus, brainstem). The color scale corresponds to t values for individual voxels in the SPM comparison. Height extent = threshold value for considering voxels significantly different. Extent threshold = number of contiguous voxels required to consider a cluster of voxels significantly different.
The hypothesis that sleep manifestations vary among brain regions is also relevant to a number of other common clinical sleep disorders and phenomena (Mahowald and Schenck, 2005). Patients with parasomnias arising from NREMS (e.g., sleepwalking, sleep terrors, confusional arousals) exhibit synchronized SWA characteristic of NREMS concurrent with wake-like behaviors such as walking, talking, and eating (Schenck et al., 1998). Case studies demonstrate waking EEG patterns in motor and cingulate cortices concurrent with sleep-like delta EEG activity in association cortex (Terzhagi et al., 2009), a pattern similar to that observed in a single case study with SPECT imaging (Bassetti et al., 2000). An even more dramatic example occurs in REM sleep behavior disorder (RBD), in which patients have vivid, often violent dreams associated with motor behaviors ranging from simple to complex; injury to the patient or bed partner is common (Schenck et al., 1993; Boeve, 2009). The behavior is attributed to loss of the usual atonia during REM sleep, mediated by several brainstem regions including the peri-locus coeruleus alpha. RBD often occurs in conjunction with dementia with Lewy bodies or other synucleinopathies, neurodegenerative disorders which lead to widely-distributed cortical and subcortical lesions (Boeve et al., 2001; Ferrini-Strambi et al., 2005). The human disorder of RBD was predicted by lesion studies in cats, in which the extent of behavioral disinhibition was proportional to the size and location of the brainstem lesion (Hendricks et al., 1982). Narcolepsy, a neurological disorder characterized by neuron loss in the orexinergic cells of the lateral hypothalamus, offers another example of how clinical symptoms may arise from the simultaneous occurrence of multiple sleep-wake states. In addition to sleep episodes intruding into wakefulness, patients with narcolepsy experience cataplexy (episodes of muscle atonia during wakefulness), sleep-related hallucinations, and sleep paralysis (muscle atonia during sleep-wake transitions). Each of these last three symptoms represent dissociated sleep-wake states characterized by simultaneous features of wakefulness (alertness, awareness of the environment) and REM sleep (atonia, dream-like imagery). Finally, some normal sleep-related phenomena can be seen as manifestations of local variations in sleep-wake states. The process of awakening from sleep is a gradual and regionally-variable process (Balkin et al., 2002). Regional blood flow first increases in brainstem, thalamus, and other centrencephalic structures, whereas medial and dorsolateral cortex shows increased blood flow over the first 20 minutes of awakening. This may relate to the phenomenon of sleep inertia, or sleep drunkenness, characterized by difficulty achieving the sleep-wake transition. Lucid dreaming, in which the individual is aware that he is dreaming in real time, could also represent a manifestation of local sleep. During lucid dreaming compared to REM sleep in general, EEG gamma activity and EEG coherence are higher in frontal and frontolateral regions (Voss et al., 2009).
Conclusion and implications for sleep function
After prolonged activation of glia/neurons within local networks, ATP, IL1, TNF, BDNF, and possibly additional SRS release would change the network’s responsiveness to inputs and cause a state shift (Figure 2). If the original input-output relationships of the active network were environmentally relevant and adaptive, then the new output associated with the new state, because they are qualitatively and quantitatively different from the original output, could be maladaptive if the animal remained responsive to environmental input. Further the state changes of multiple local networks would likely synchronize spontaneously. Thus, after the shift in input–output relationships, the outputs would lack the integrated network-activity patterns needed to induce environmentally adaptive cognitive or motor outputs. The classic sleep–wake regulatory circuits may coordinate the activity of multiple small networks, thereby ensuring the absence of whole-organism behavior at such times.
Local sleep mechanisms are inseparable from synaptic efficacy/connectivity regulatory functions of sleep. These local activity-dependent mechanisms also cause network output that characterizes the altered consciousness that pervades sleep. The environmentally disconnected outputs occurring during sleep, which are an alternative network pattern of synaptic use, could serve to preserve adaptive plasticity within the networks. Further, the mechanisms outlined herein would insure an alternative pattern of neuronal activation occurring periodically as a consequence of cell activity. As a consequence of the synaptic efficacy and connectivity use rules and synaptic mechanisms, such as scaling, the ability of the brain to retain its capacity to change is maintained. This may be the critical primordial function of sleep. If so, then sleep function cannot be separated from sleep mechanisms.
Acknowledgements
This work was supported in part by grants from the National Institutes of Health (NS025378 and HD036520 to JMK, MH60263 to DMR, MH024657 and AG020677 to DJB and DA029565 to YH) and by a grant from the W.M. Keck Foundation.
Footnotes
For ease of conception we limit our model to glial and neuronal release of ATP because ATP induces sleep regulatory substances and is catabolized to adenosine, a vasodilator that induces sleep. Other molecules and processes that are likely involved (Krueger et al., 2008) include NO (Kapas et al., 1994), cytokine production (Willenborg et al., 2007) and cerebral blood flow.
References
- Aalto J, Kiianmaa K. REM-sleep deprivation-induced increase in ethanol intake: role of brain monoaminergic neurons. Alcohol. 1986;3:377–381. doi: 10.1016/0741-8329(86)90057-1. [DOI] [PubMed] [Google Scholar]
- Alam MN, McGinty D, Bashir T, Kumar S, Imeri L, Opp MR, Szymusiak R. Interleukin-1 beta modulates state-dependent discharge activity of preoptic area and basal forebrain neurons: Role in sleep regulation. Eur. J. Neurosci. 2004;20:207–216. doi: 10.1111/j.1460-9568.2004.03469.x. [DOI] [PubMed] [Google Scholar]
- Aton SJ, Seibt J, Dumoulin M, Jha SK, Steinmetz N, Coleman T, Naidoo N, Frank MG. Mechanisms of sleep-dependent consolidation of cortical plasticity. Neuron. 2009;61:454–466. doi: 10.1016/j.neuron.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkin TJ, Braun AR, Wesensten NJ, Jeffries K, Varga M, Baldwin P, et al. The process of awakening: a PET study of regional brain activity patterns mediating the re-establishment of alertness and consciousness. Brain. 2002;125:2308–2319. doi: 10.1093/brain/awf228. [DOI] [PubMed] [Google Scholar]
- Bassetti C, Vella S, Donati F, Wielepp P, Weder B. SPECT during sleepwalking. Lancet. 2000;356:484–485. doi: 10.1016/S0140-6736(00)02561-7. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNF alpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bianco F, Pravettoni E, Colombo A, Schenk U, Moller T, Matteoli M, Verderio C. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol. 2005;174:7268–7277. doi: 10.4049/jimmunol.174.11.7268. [DOI] [PubMed] [Google Scholar]
- Birtoli B, Ulrich D. Firing mode-dependent synaptic plasticity in rat neocortical pyramidal neurons. J. Neurosci. 2004;24:4935–4940. doi: 10.1523/JNEUROSCI.0795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF. REM sleep behavior disorder: Updated review of the core features, the REM sleep behavior disorder-neurodegenerative disease association, evolving concepts, controversies, and future directions. Ann.N.Y.Acad.Sci. 2010;1184:15–54. doi: 10.1111/j.1749-6632.2009.05115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Parisi JE, Dickson DW, Ferman TJ, Benarroch EE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61:40–45. doi: 10.1212/01.wnl.0000073619.94467.b0. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007;130:2770–2788. doi: 10.1093/brain/awm056. [DOI] [PubMed] [Google Scholar]
- Boly M, Phillips C, Tshibanda L, Vanhuadenhuyse A, Schabus M, Dang-Vu TT, Moonen G, Hustinx R, Maquet P, Laureys S. Intrinsic brain activity in altered states of consciousness: how conscious is the default mode of brain function? Ann. N. Y. Acad. Sci. 2008;1129:119–129. doi: 10.1196/annals.1417.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla D, Barajon I, Bianchi S, Opp MR, Imeri L. Interleukin-1 inhibits putative cholinergic neurons in vitro and REM sleep when microinjected into the rat laterodorsal tegmental nucleus. Sleep. 2010;33:919–929. doi: 10.1093/sleep/33.7.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla D, Franciosi S, Opp MR, Imeri L. Interleukin-1 inhibits firing of serotonergic neurons in the dorsal raphe nucleus and enhances GAGAergic inhibitory post-synaptic potentials Eur. J. Neurosci. 2007;26:1862–1869. doi: 10.1111/j.1460-9568.2007.05796.x. [DOI] [PubMed] [Google Scholar]
- Broyd SJ, Demanuele C, Debener S, Helps SK, James CJ, Sonuga-Barke EJ. Default-mode brain dysfunction in mental disorders: a systematic review. Neurosci. Biobehav. Rev. 2009;33:279–296. doi: 10.1016/j.neubiorev.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann. N. Y. Acad. Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic signaling – An overview. Novartis Found. Symp. 2006;276:26–48. [PubMed] [Google Scholar]
- Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007;87:659–797. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- Buysse DJ, Germain A, Hall M, Monk TH, Nofzinger EA. A Neurobiological Model of Insomnia. Drug Discov. Today Dis. Models. 2011;8:129–137. doi: 10.1016/j.ddmod.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavadini G, Petrzilka S, Kohler P, Jud C, Tobler I, Birchler T, Fontana A. TNF-α suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl. Acad. Sci. 2007;104:12843–12848. doi: 10.1073/pnas.0701466104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanna AE, Trimble MR. The precuneus: a review of its functional anatomy and behavioural correlates. Brain. 2006;129:564–583. doi: 10.1093/brain/awl004. [DOI] [PubMed] [Google Scholar]
- Chauvette S, Seigneur J, Timofeev I. Sleep oscillations in the thalamocortical system induce long-term neuronal plasticity. Neuron. 2012;75:1105–1113. doi: 10.1016/j.neuron.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheer JF, Heien ML, Garris PA, Carelli RM, Wightman RM. Simultaneous dopamine and single-unit recordings reveal accumbens GABAergic responses: implications for intracranial self-stimulation. Proc Natl Acad Sci USA. 2005;102:19150–19155. doi: 10.1073/pnas.0509607102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Gardi J, Kushikata T, Fang J, Krueger JM. Nuclear factor kappa B-like activity increases in murine cerebral cortex after sleep deprivation. Am. J. Physiol. 1999;45:R1812–R1818. doi: 10.1152/ajpregu.1999.276.6.R1812. [DOI] [PubMed] [Google Scholar]
- Chiappalone M, Vato A, Berdondini L, Koudelka-Hep M, Martinoia S. Network dynamics and synchronous activity in cultured cortical neurons. International J. Neural Systems. 2007;17:87–103. doi: 10.1142/S0129065707000968. [DOI] [PubMed] [Google Scholar]
- Churchill L, Rector DM, Yasuda K, Fix C, Rojas MJ, Yasuda T, Krueger JM. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neurosci. 2008;156:71–80. doi: 10.1016/j.neuroscience.2008.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Tononi G. Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J. Neurosci. 2000;20:9187–9194. doi: 10.1523/JNEUROSCI.20-24-09187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corner MA. Spontaneous neuronal burst discharges as dependent and independent variables in the maturation of cerebral cortex tissue cultured in vitro: a review of activity-dependent studies in live ‘model’ systems for the development of intrinsically generated bioelectric slow-wave sleep patterns. Brain Res. Rev. 2008;59:221–244. doi: 10.1016/j.brainresrev.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Crum RM, Storr CL, Chan YF, Ford DE. Sleep disturbance and risk for alcohol-related problems. Am. J. Psychiatry. 2004;161:1197–1203. doi: 10.1176/appi.ajp.161.7.1197. [DOI] [PubMed] [Google Scholar]
- Czarnecki A, Birtoli B, Ulrich D. Cellular mechanisms of burst firing-mediated long-term depression in rat neocortical pyramidal cells. J. Physiol. 2007;578:471–479. doi: 10.1113/jphysiol.2006.123588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang-Vu TT, Schabus M, Desseilles M, Sterpenich V, Bonjean M, Maquet P. Functional neuroimaging insights into the physiology of human sleep. Sleep. 2010;33:1589–1603. doi: 10.1093/sleep/33.12.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang-Vu TT, Desseilles M, Laureys S, Degueldre C, Perrin F, Phillips C, Maquet P, Peigneux P. Cerebral correlates of delta waves during non-REM sleep revisited. Neuroimage. 2005;28:14–21. doi: 10.1016/j.neuroimage.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Datta S, Maclean RR. Neurobiological mechanisms for the regulation of mammalian sleep-wake behavior: Reinterpretation of historical evidence and inclusion of contemporary cellular and molecular evidence. Neurosci. Biobehav. Rev. 2007;31:775–824. doi: 10.1016/j.neubiorev.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CJ, Harding JW, Wright JW. REM sleep deprivation-induced deficits in the latency-to-peak induction and maintenance of long-term potentiation within the CA1 region of the hippocampus. Brain Res. 2003;973:293–297. doi: 10.1016/s0006-8993(03)02508-3. [DOI] [PubMed] [Google Scholar]
- De Sarro G, Gareri P, Sinopoli VA, David E, Rotiroti D. Comparative, behavioural and electrocortical effects of tumor necrosis factor-alpha and interleukin-1 microinjected into the locus coeruleus of rat. Life Sci. 1997;60:555–564. doi: 10.1016/s0024-3205(96)00692-3. [DOI] [PubMed] [Google Scholar]
- Drummond SP, Gillin JC, Smith TL, DeModena A. The sleep of abstinent pure primary alcoholic patients: natural course and relationship to relapse. Alcohol Clin. Exp. Res. 1998;22:1796–1802. [PubMed] [Google Scholar]
- Edinger JD, Krystal AD. Subtyping primary insomnia: Is sleep state misperception a distinct clinical entity. Sleep Med. Rev. 2003;7:203–214. doi: 10.1053/smrv.2002.0253. [DOI] [PubMed] [Google Scholar]
- Everson CA, Bergmann BM, Rechtschaffen A. Sleep deprivation in the rat: III. Total sleep deprivation. Sleep. 1989;12:13–21. doi: 10.1093/sleep/12.1.13. [DOI] [PubMed] [Google Scholar]
- Faraguna U, Vyazovskiy VV, Nelson AB, Tononi G, Cirelli C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J. Neurosci. 2008;28:4088–4095. doi: 10.1523/JNEUROSCI.5510-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat. Rev. Neurosci. 2007;8:700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- Frank MG, Issa NP, Stryker MP. Sleep enhances plasticity in the developing visual cortex. Neuron. 2001;30:275–287. doi: 10.1016/s0896-6273(01)00279-3. [DOI] [PubMed] [Google Scholar]
- Franklin CM. Clinical experience with soluble TNF p75 receptor in rheumatoid arthritis. Semin. Arthritis Rheum. 1999;29:171–181. doi: 10.1016/s0049-0172(99)80028-6. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Mattson MP. The transcription factor NFkB mediates increases in calcium currents and decreases in NMDA- and AMPA/kainate-induced currents induced by tumor necrosis factor alpha in hippocampal neurons. J. Neurochem. 1998;70:1876–1886. doi: 10.1046/j.1471-4159.1998.70051876.x. [DOI] [PubMed] [Google Scholar]
- Gujar N, McDonald SA, Nishida M, Walker MP. A role for REM sleep in recalibrating the sensitivity of the human brain to specific emotions. Cereb. Cortex. 2011a;21:115–123. doi: 10.1093/cercor/bhq064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar N, Yoo SS, Hu P, Walker MP. Sleep deprivation amplifies reactivity of brain reward networks, biasing the appraisal of positive emotional experiences. J. Neurosci. 2011b;31:4466–4474. doi: 10.1523/JNEUROSCI.3220-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagewoud R, Havekes R, Novati A, Keijser JN, Van der Zee EA, Meerlo P. Sleep deprivation impairs spatial working memory and reduces hippocampal AMPA receptor phosphorylation. J Sleep Res. 2010;19:280–288. doi: 10.1111/j.1365-2869.2009.00799.x. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett H, Churchill L, Taishi P, De A, Krueger JM. Whisker stimulation increases expression of nerve growth factor- and interleukin-1beta-immunoreactivity in the rat somatosensory cortex. Brain Res. 2010;1333:48–56. doi: 10.1016/j.brainres.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon EC, Baldo BA, Sadeghian K, Kelley AE. Increases in food intake or food-seeking behavior induced by GABAergic, opioid, or dopaminergic stimulation of the nucleus accumbens: is it hunger? Psychopharmacology (Berl) 2004;172:241–247. doi: 10.1007/s00213-003-1654-0. [DOI] [PubMed] [Google Scholar]
- Hanlon EC, Benca RM, Baldo BA, Kelley AE. REM sleep deprivation produces a motivational deficit for food reward that is reversed by intra-accumbens amphetamine in rats. Brain Res. Bull. 2010;83:245–254. doi: 10.1016/j.brainresbull.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon EC, Van Cauter E. Quantification of sleep behavior and of its impact on the cross-talk between the brain and peripheral metabolism. Proc. Natl. Acad. Sci. 2011;108(Suppl 3):15609–15616. doi: 10.1073/pnas.1101338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler BP, Smith LJ, Cousins JC, Bootzin RR. Circadian rhythms, sleep, and substance abuse. Sleep Med. Rev. 2012;16:67–81. doi: 10.1016/j.smrv.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks JC, Morrison AR, Mann GL. Different behaviors during paradoxical sleep without atonia depend on pontine lesion site. Brain Res. 1982;239:81–105. doi: 10.1016/0006-8993(82)90835-6. [DOI] [PubMed] [Google Scholar]
- Hide I, Tanaka M, Inoué A, Nakajima K, Kohsaka S, Inoué K, Nakata Y. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J. Neurochem. 2000;75:965–972. doi: 10.1046/j.1471-4159.2000.0750965.x. [DOI] [PubMed] [Google Scholar]
- Hinard V, Mikhail C, Pradervand S, Curie T, Houtkooper RH, Auwerx J, Franken P, Tafti M. Key electrophysiological, molecular, and metabolic signatures of sleep and wakefulness revealed in primary cortical cultures. J. Neurosci. 2012;32:12506–12517. doi: 10.1523/JNEUROSCI.2306-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofle N, Paus T, Reutens D, Fiset P, Gotman J, Evans AC, Jones BE. Regional cerebral blood flow changes as a function of delta and spindle activity during slow wave sleep in humans. J, Neurosci. 1997;17:4800–4808. doi: 10.1523/JNEUROSCI.17-12-04800.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat. Rev .Neurosci. 2009;10:199–210. doi: 10.1038/nrn2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B. Arousal systems. Front. Biosci. 2003;8:s438–s451. doi: 10.2741/1074. [DOI] [PubMed] [Google Scholar]
- Kapas L, Fang J, Krueger JM. Inhibition of nitric oxide synthesis inhibits rat sleep. Brain Res. 1994;664:189–196. doi: 10.1016/0006-8993(94)91969-0. [DOI] [PubMed] [Google Scholar]
- Kavanau JL. Sleep and dynamic stabilization of neural circuitry: A review and synthesis. Behav. Brain Res. 1994;63:111–126. doi: 10.1016/0166-4328(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Khoa ND, Montesinos MC, Reiss AB, Delano D, Awadallah N, Cronstein BN. Inflammatory cytokines regulate function and expression of adenosine A(2A) receptors in human monocytic THP-1 cells. J. Immunol. 2001;167:4026–4032. doi: 10.4049/jimmunol.167.7.4026. [DOI] [PubMed] [Google Scholar]
- Killingsworth MA, Gilbert DT. A wandering mind is an unhappy mind. Science. 2010;330:932. doi: 10.1126/science.1192439. [DOI] [PubMed] [Google Scholar]
- Kopp C, Longordo F, Nicholson JR, Luthi A. Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J. Neurosci. 2006;26:12456–12465. doi: 10.1523/JNEUROSCI.2702-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen K, Courtois G. Rhythmic electrical activity from isolated cerebral cortex. Electroencephalogr. Clin. Neurophysiol. 1949;1:265–272. [PubMed] [Google Scholar]
- Krueger JM, Obál F., Jr. A neuronal group theory of sleep function. J. Sleep Res. 1993;2:63–69. doi: 10.1111/j.1365-2869.1993.tb00064.x. [DOI] [PubMed] [Google Scholar]
- Krueger JM, Rector DM, Roy S, Van Dongen HPA, Belenky G, Panksepp J. Sleep as a fundamental property of neuronal assemblies. Nat. Rev. Neurosci. 2008;9:910–919. doi: 10.1038/nrn2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM. The role of cytokines in sleep regulation. Curr. Pharm. Des. 2008;14:3408–3416. doi: 10.2174/138161208786549281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Taishi P, De A, Davis CJ, Winters BD, Clinton J, Szentirmai E, Zielinski MR. ATP and the purine type 2 X7 receptor affect sleep. J. Appl. Physiol. 2010;109:1318–1327. doi: 10.1152/japplphysiol.00586.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal AD, Edinger JD, Wohlgemuth WK, Marsh GR. NREM sleep EEG frequency spectral correlates of sleep complaints in primary insomnia subtypes. Sleep. 2002;25:630–640. [PubMed] [Google Scholar]
- Kubota T, Kushikata T, Fang J, Krueger JM. A nuclear factor kappa β (NFkB) inhibitor peptide inhibits spontaneous and interleukin-1β-induced sleep. Am. J. Physiol. 2000;279:R404–R413. doi: 10.1152/ajpregu.2000.279.2.R404. [DOI] [PubMed] [Google Scholar]
- Kushikata T, Fang J, Krueger JM. Brain-derived neurotrophic factor enhances spontaneous sleep in rats and rabbits. Am. J. Physiol. 1999;276:R1334–1338. doi: 10.1152/ajpregu.1999.276.5.R1334. [DOI] [PubMed] [Google Scholar]
- Kuo TH, Pike DH, Beizaeipour Z, Williams JA. Sleep triggered by an immune response in Drosophila is regulated by the circadian clock and requires the NFkappB relish. BMC Neurosci. 2010;11:17. doi: 10.1186/1471-2202-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lante F, Toledo-Salas JC, Ondrejcak T, Rowan MJ, Ulrich D. Removal of synaptic Ca(2)+-permeable AMPA receptors during sleep. J. Neurosci. 2011;31:3953–3961. doi: 10.1523/JNEUROSCI.3210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZW, Faraguna U, Cirelli C, Tononi G, Gao XB. Direct evidence for wake-related increases and sleep-related decreases in synaptic strength in rodent cortex. J. Neurosci. 2010;30:8671–8675. doi: 10.1523/JNEUROSCI.1409-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Takahashi JS. Genetics of circadian rhythms in mammalian model organisms. Adv. Genet. 2011;74:175–230. doi: 10.1016/B978-0-12-387690-4.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald MW, Bornemann MAC, Schenck CH. State dissociation, human behavior and consciousness. Current Topics in Medicinal Chemistry. 2011;11:2392–2402. doi: 10.2174/156802611797470277. [DOI] [PubMed] [Google Scholar]
- Mahowald MW, Schenck CH. Insights from studying human sleep disorders. Nature. 2005;437:1279–1285. doi: 10.1038/nature04287. [DOI] [PubMed] [Google Scholar]
- Manconi M, Ferri R, Sagrada C, Punjabi NM, Tettamanzi E, Zucconi M, Oldani A, Castronovo V, Ferini-Strambi L. Measuring the error in sleep estimation in normal subjects and in patients with insomnia. J. Sleep Res. 2010;19:478–486. doi: 10.1111/j.1365-2869.2009.00801.x. [DOI] [PubMed] [Google Scholar]
- Mander BA, Santhanam S, Saletin JM, Walker MP. Wake deterioration and sleep restoration of human learning. Current Biol. 2011;21:R183–R184. doi: 10.1016/j.cub.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfridi A, Brambilla D, Bianchi S, Mariotti M, Opp MR, Imeri L. Interleukin-1 beta enhances non-rapid eye movement sleep when microinjected into the dorsal raphe nucleus and inhibits serotonergic neurons in vitro. Eur. J. Neurosci. 2003;18:1041–1049. doi: 10.1046/j.1460-9568.2003.02836.x. [DOI] [PubMed] [Google Scholar]
- Maquet P. Functional neuroimaging of normal human sleep by positron emission tomography. J. Sleep Res. 2000;9:207–231. doi: 10.1046/j.1365-2869.2000.00214.x. [DOI] [PubMed] [Google Scholar]
- Maquet P, Peters J, Aerts J, Delfiore G, Degueldre C, Luxen A, Franck G. Functional neuroanatomy of human rapid-eye-movement sleep and dreaming. Nature. 1996;383:163–166. doi: 10.1038/383163a0. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadottir H, Molle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–613. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- Mason MF, Norton MI, Van Horn JD, Wegner DM, Grafton ST, Macrae CN. Wandering minds: the default network and stimulus-independent thought. Science. 2007;315:393–395. doi: 10.1126/science.1131295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott CM, LaHoste GJ, Chen C, Musto A, Bazan NG, Magee JC. Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J. Neurosci. 2003;23:9687–9695. doi: 10.1523/JNEUROSCI.23-29-09687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott CM, Hardy MN, Bazan NG, Magee JC. Sleep deprivation-induced alterations in excitatory synaptic transmission in the CA1 region of the rat hippocampus. J. Physiol. 2006;570:553–565. doi: 10.1113/jphysiol.2005.093781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkel J, Htaik O, Banks S, Dinges D. Emotional expressiveness in sleep-deprived healthy adults. Behavioral Sleep Med. 2011;9:5–14. doi: 10.1080/15402002.2011.533987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistico G, De Sarro G, Rotiroti D. Behavioral and electrocortical spectrum power changes of interleukins and tumor necrosis factor after microinjection into different areas of the brain. In: Smirne S, Francesch M, Ferini-Stambi L, editors. Sleep, hormones, and immunological system. Mason; Milan (Italy): 1992. pp. 11–22. [Google Scholar]
- Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, Kupfer DJ. Functional neuroimaging evidence for hyperarousal in insomnia. Am. J. of Psych. 2004;161:2126–2131. doi: 10.1176/appi.ajp.161.11.2126. [DOI] [PubMed] [Google Scholar]
- Nofzinger EA, Nissen C, Germain A, Moul D, Hall M, Prince JC, Miewald JM, Buysse DJ. Regional cerebral metabolic correlates of WASO during sleep in insomnia. J. of Clin. Sleep Med. 2006;2:316–322. [PubMed] [Google Scholar]
- O’Mahony A, Raber J, Montano M, Foehr E, Han V, Lu SM, Kwon H, LeFevour A, Chakraborty-Sett S, Greene WC. NF-kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol. Cell Biol. 2006;26:7283–7298. doi: 10.1128/MCB.00510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DJ, Schei JL, Meighan PC, Rector DM. State-dependent changes in cortical gain control as measured by auditory evoked responses to varying intensity stimuli. Sleep. 2012;34:1527–1537. doi: 10.5665/sleep.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigarev IN, Nothdurft HC, Kastner S. Evidence for asynchronous development of sleep in cortical areas. NeuroReport. 1997;28:2557–2560. doi: 10.1097/00001756-199707280-00027. [DOI] [PubMed] [Google Scholar]
- Puhl MD, Fang J, Grigson PS. Acute sleep deprivation increases the rate and efficiency of cocaine self-administration, but not the perceived value of cocaine reward in rats. Pharm. Biochem. Beh. 2009;94:262–270. doi: 10.1016/j.pbb.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME. Two views of brain function. Trends Cogn. Sci. 2010;14:180–190. doi: 10.1016/j.tics.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Rector DM, Schei JL, Van Dongen HPA, Belenky G, Krueger JM. Physiological markers of localized sleep. Eur. J. Neurosci. 2009;29:1771–1778. doi: 10.1111/j.1460-9568.2009.06717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector DM, Topchiy IA, Carter KM, Rojas MJ. Local functional state differences between rat cortical columns. Brain Res. 2005;1047:45–55. doi: 10.1016/j.brainres.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Reyes-Vazquez C, Prieto-Gomez B, Dafny N. Interferon modulates central nervous system function. Brain Res. 2012;1442:76–89. doi: 10.1016/j.brainres.2011.09.061. [DOI] [PubMed] [Google Scholar]
- Rosa RR, Bonnet MH. Reported chronic insomnia is independent of poor sleep as measured by electroencephalography. Psychosom Med. 2000;62:474–482. doi: 10.1097/00006842-200007000-00004. [DOI] [PubMed] [Google Scholar]
- Roy S, Krueger JM, Rector DM, Wan Y. Network models for activity-dependent sleep regulation. J. Theor. Biol. 2008;253:462–468. doi: 10.1016/j.jtbi.2008.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Bezzi P, Volterra A. TNF alpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron. 2011;69:988–1001. doi: 10.1016/j.neuron.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;473:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Hurwitz TD, Mahowald MW. REM sleep behaviour disorder: an update on a series of 96 patients and a review of the world literature. Journal of Sleep Research. 1993;2:224–231. doi: 10.1111/j.1365-2869.1993.tb00093.x. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Pareja JA, Patterson AL, Mahowald MW. Analysis of polysomnographic events surrounding 252 slow-wave sleep arousals in thirty-eight adults with injurious sleepwalking and sleep terrors. J.Clin.Neurophysiol. 1998;15:159–166. doi: 10.1097/00004691-199803000-00010. [DOI] [PubMed] [Google Scholar]
- Solle M, Labsi J, Perragaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA. Altered cytokine production in mice lacking P2X(7) receptors. J. Biol. Chem. 2001;276:125–32. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Steriade M. The corticothalamic system in sleep. Front. Biosci. 2003;8:d878–d899. doi: 10.2741/1043. [DOI] [PubMed] [Google Scholar]
- St Hilaire C, Carroll SH, Chen H, Ravid K. Mechanisms of induction of adenosine receptor genes and its functional significance. J. Cell Physiol. 2009;218:35–44. doi: 10.1002/jcp.21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strogatz SH. Spontaneous synchronization in nature; Proc. of 1997 IEEE Internatl. Freq. Control Symposium; Orlando, FL. 1997. [Google Scholar]
- Strogatz S. Sync The Emerging Science of Spontaneous Order. Hyperion; New York: 2004. [Google Scholar]
- Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J. Neurosci. 2004;24:1–7. doi: 10.1523/JNEUROSCI.3792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Ann. N. Y. Acad. Sci. 2008;1129:275–286. doi: 10.1196/annals.1417.027. [DOI] [PubMed] [Google Scholar]
- Taishi P, Churchill L, Wang M, Kay D, Davis CJ, Guan X, De A, Yasuda T, Liao F, Krueger JM. TNFα siRNA reduces brain TNF and EEG delta wave activity in rats. Brain Res. 2007;1156:125–132. doi: 10.1016/j.brainres.2007.04.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzaghi M, Sartori I, Tassi L, Didato G, Rustioni V, LoRusso G, et al. Evidence of dissociated arousal states during NREM parasomnia from an intracerebral neurophysiological study. Sleep. 2009;32:409–412. doi: 10.1093/sleep/32.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep and synaptic homeostasis: a hypothesis. Brain Res. Bull. 2003;62:143–150. doi: 10.1016/j.brainresbull.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Topchiy IA, Wood RM, Peterson B, Navas JA, Rojas MJ, Rector DM. Conditioned lick behavior and evoked responses using whisker twitches in head restrained rats. Behav. Brain Res. 2009;197:16–23. doi: 10.1016/j.bbr.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-turning neuron: Synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman V, Huettel SA, Chuah LY, Payne JW, Chee MW. Sleep deprivation biases the neural mechanisms underlying economic preferences. J. Neurosci. 2011;31:3712–3718. doi: 10.1523/JNEUROSCI.4407-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Bianco F, Blanchard MP, Bergami M, Canossa M, Scarfone E, Matteoli M. Cross talk between vestibular neurons and Schwann cells mediates BDNF release and neuronal regeneration. Brain Cell Biol. 2006;35:187–201. doi: 10.1007/s11068-007-9011-6. [DOI] [PubMed] [Google Scholar]
- Vgontzas AN, Zoumakis E, Lin HM, Bixler EO, Trakada G, Chrousos GP. Marked decrease in sleepiness in patients with sleep apnea by etanercept, a tumor necrosis factor-α antagonist. J. Clin. Endocrinol. Metab. 2004;89:4409–4413. doi: 10.1210/jc.2003-031929. [DOI] [PubMed] [Google Scholar]
- Voss U, Holzmann R, Tuin I, Hobson JA. Lucid dreaming: a state of consciousness with features of both waking and non-lucid dreaming. Sleep. 2009;32:1191–1200. doi: 10.1093/sleep/32.9.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nature Neurosci. 2008;11:200–208. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Lazimy YM, Faraguna U, Esser SK, Williams JC, Cirelli C, Tononi G. Cortical firing and sleep homeostasis. Neuron. 2009;63:865–878. doi: 10.1016/j.neuron.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature. 2011a;472:443–447. doi: 10.1038/nature10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Tononi G. Electrophysiological correlates of sleep homeostasis in freely bhaving rats. Prog. Brain Res. 2011b;193:17–38. doi: 10.1016/B978-0-444-53839-0.00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Madhavan R, Pine J, Potter SM. Controlling bursting in cortical cultures with closed-loop multi-electrode stimulation. J. Neurosci. 2005;25:680–688. doi: 10.1523/JNEUROSCI.4209-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AJ, Topchiy I, Kouptsov K, Rector DM. ERP differences during conditioned lick responses in the rat. Sleep. 2005;28:A15. [Google Scholar]
- Walker MP. Sleep, memory and emotion. Prog. Brain Res. 2010;185:49–68. doi: 10.1016/B978-0-444-53702-7.00004-X. [DOI] [PubMed] [Google Scholar]
- Walker MP, Stickgold R. Sleep-dependent learning and memory consolidation. Neuron. 2004;44:121–133. doi: 10.1016/j.neuron.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Willenborg DO, Staykova M, Fordham S, O’Brien N, Linares D. The contributin of nitric oxide and interferon gamma to the regulation of the neuro-inflammation in experimental autoimmune encephalomyelitis. J. Neuroimmunology. 2007;191:16–25. doi: 10.1016/j.jneuroim.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Williams JA, Sathyanarayanan S, Hendricks JC, Sehgal A. Interactions between sleep and the immune response in Drosophila: a role for the NFkappaB relish. Sleep. 2007;30:389–400. doi: 10.1093/sleep/30.4.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BD, Huang YH, Dong Y, Krueger JM. Sleep loss alters synaptic and intrinsic neuronal properties in mouse prefrontal cortex. Brain Res. 2011;1420:1–7. doi: 10.1016/j.brainres.2011.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME. Regulation of AMPA receptor trafficking in the nucleus accumbens by dopamine and cocaine. Neurotoxicity Res. 2010;18:393–409. doi: 10.1007/s12640-010-9176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Churchill L, Yasuda T, Blindheim K, Falter M, Krueger JM. Unilateral cortical application of interleukin-1β (IL1β) induces asymmetry in Fos- and IL1β-immunoreactivity: Implications for sleep regulation. Brain Res. 2007;1131:44–59. doi: 10.1016/j.brainres.2006.11.051. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Yoshida H, Garcia-Garcia F, Kay D, Krueger JM. Interleukin-1β has a role in cerebral cortical state dependent electroencephalographic slow-wave activity. Sleep. 2005;28:177–184. doi: 10.1093/sleep/28.2.177. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Peterfi Z, Garcia-Garcia F, Kirkpatrick R, Yasuda T, Krueger JM. State-specific asymmetries in EEG slow wave activity induced by local application of TNFalpha. Brain Res. 2004;1009:129–136. doi: 10.1016/j.brainres.2004.02.055. [DOI] [PubMed] [Google Scholar]
- Yu Z, Cheng G, Wen X, Wu GD, Lee WT, Pleasure D. Tumor necrosis factor alpha increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase- and NF-kappaB-dependent mechanism. Neurobiol. Dis. 2002;11:199–213. doi: 10.1006/nbdi.2002.0530. [DOI] [PubMed] [Google Scholar]