

Abstract
Pulsatile GnRH release is essential to fertility and is modulated by gonadal steroids, most likely via steroid-sensitive afferents. Arcuate neurons coexpressing kisspeptin, neurokinin B (NKB), and dynorphin (KNDy neurons) are steroid-sensitive and have been postulated to both generate GnRH pulses and mediate steroid feedback on pulse frequency. KNDy neurons are proposed to interact with one another via NKB and dynorphin to activate and inhibit the KNDy network, respectively, and thus alter kisspeptin output to GnRH neurons. To test the roles of NKB and dynorphin on KNDy neurons and the steroid sensitivity of these actions, targeted extracellular recordings were made of Tac2(NKB)-GFP-identified neurons from castrate and intact male mice. Single-cell PCR confirmed most of these cells had a KNDy phenotype. The neurokinin 3 receptor (NK3R) agonist senktide increased action potential firing activity of KNDy neurons. Dynorphin reduced spontaneous KNDy neuron activity, but antagonism of κ-opioid receptors (KOR) failed to induce firing activity in quiescent KNDy neurons. Senktide-induced activation was greater in KNDy neurons from castrate mice, whereas dynorphin-induced suppression was greater in KNDy neurons from intact mice. Interactions of dynorphin with senktide-induced activity were more complex; dynorphin treatment after senktide had no consistent inhibitory effect, whereas pretreatment with dynorphin decreased senktide-induced activity only in KNDy neurons from intact but not castrate mice. These data suggest dynorphin-mediated inhibition of senktide-induced activity requires gonadal steroid feedback. Together, these observations support the hypotheses that activation of NK3R and KOR, respectively, excites and inhibits KNDy neurons and that gonadal steroids modulate these effects.
GnRH neurons form the final common pathway for the central regulation of fertility. GnRH is secreted in a pulsatile pattern (1–5) that is essential for pituitary synthesis and release of LH and FSH (5–9), which activate steroidogenesis and gametogenesis. Although gonadal steroids feed back to regulate GnRH pulse pattern (10), detection of steroid receptors other than estrogen receptor ß is rare in native GnRH neurons (11–14), suggesting feedback is largely mediated via steroid-sensitive afferents (15, 16).
One steroid-sensitive afferent population is the arcuate neurons coexpressing kisspeptin, neurokinin B (NKB, also known as Tac2), and dynorphin (KNDy neurons) (17–25). Several studies suggest these cells are important in reproductive neuroendocrine regulation; mutations in the kisspeptin receptor KISS1R, NKB, or the NKB receptor NK3R cause hypogonadotropic hypogonadism in humans (26–28), and knockout of the genes for kisspeptin (Kiss1) or KISS1R (Kiss1r) reduce or eliminate reproduction in mice (29, 30). Furthermore, κ-opioid receptor (KOR, the dynorphin receptor) antagonists increase LH release (31).
KNDy neurons have been hypothesized to regulate the pattern of GnRH release as a self-activating and self-inhibiting network (18, 32). In this model, kisspeptin provides excitatory KNDy network output to GnRH neurons, which express Kiss1r (33–39). NKB is postulated to stimulate KNDy neurons (26, 40, 41), generating kisspeptin release onto GnRH neurons to initiate a pulse of GnRH secretion (18, 42). NKB-induced activation of the KNDy network also putatively activates dynorphin release, and subsequent action of dynorphin is proposed to inhibit NKB-stimulated KNDy activation, stop kisspeptin output to GnRH neurons, and thus terminate the GnRH pulse (18, 31, 32, 43).
Functional studies of KNDy neurons are limited but have provided initial support for some elements of this model (31, 44, 45). Animal studies using the NK3R agonist senktide, however, have produced variable results, with both increases and decreases in LH depending on steroid milieu and species (32, 42, 44, 46–52). The present study examined how NKB and dynorphin interact to alter the activity of green fluorescent protein (GFP)-identified KNDy neurons in the arcuate nucleus from castrate and intact adult male mice directly at the central level.
Materials and Methods
Animals
Tac2-enhanced GFP BAC transgenic mice (015495-UCD/STOCK Tg [Tac2-EGFP]381Gsat) were obtained from the Mouse Mutant Regional Resource Center (http://www.mmrrc.org/). GnRH-GFP mice (54) were propagated in our colony. Mice were maintained under a 14-hour light,10-hour dark photoperiod with 2916 chow (Harlan, Indianapolis, Indiana) and water available ad libitum. Male mice aged 38 to 120 days were either left intact or were castrated under isoflurane anesthesia 3 to 7 days before study; bupivacaine was provided as an analgesic. All procedures were approved by the University Committee on the Use and Care of Animals at the University of Michigan.
Brain slice preparation
Reagents were purchased from Sigma Chemical Company (St. Louis, Missouri) unless noted. Solutions were bubbled with 95% O2/5% CO2 throughout experiments and for ≥15 minutes before use. Brain slices were prepared with modifications (55) as previously described (56). Brains were rapidly removed and placed in ice-cold high-sucrose saline solution containing (in mM) 250 sucrose, 3.5 KCl, 26 NaHCO3, 10 d-glucose, 1.25 Na2HPO4, 1.2 MgSO4, and 3.8 MgCl2. Coronal (300 μm) slices were cut with a Vibratome 3000 (Ted Pella, Inc, Redding, California). Slices were incubated 30 minutes at 30°C to 32°C in 50% high-sucrose saline and 50% artificial cerebrospinal fluid (ACSF) containing (in mM) 135 NaCl, 3.5 KCl, 26 NaHCO3, 10 d-glucose, 1.25 Na2HPO4, 1.2 MgSO4, and 2.5 CaCl2 (pH 7.4) and then transferred to 100% ACSF solution at room temperature for 0.5 to 7 hours before recording.
Cell harvest for single-cell PCR and cDNA synthesis
Patch pipettes (2–3 MΩ) were filled with 5–8 μL of a solution containing (in mM) 140 K-gluconate, 5 KCl, 10 HEPES, 5 EGTA, 4.0 Mg-ATP, 0.4 Na-GTP, and 1.0 CaCl2 (pH 7.3, 305 mOsm). The whole-cell configuration was obtained and cytoplasm aspirated into the pipette. Pipette contents were expelled into a microcentrifuge tube containing 2× reverse transcription buffer, and the volume was adjusted to 20 μL with molecular grade water (Invitrogen, Carlsbad, California). Similarly prepared pipettes (n = 5) were lowered into the slice, but positive pressure was maintained and no cell contents were intentionally harvested; these false harvests were processed as above and used to estimate background contamination (57). The components for the reverse transcription reaction were (in mM) 20 Tris (pH 8.4), 50 KCl, 5 MgCl2, 10 dithiothreitol, and 10 dNTPs plus 500 ng oligo dT (Invitrogen), 50 ng random hexamers (Roche Life Sciences, Indianapolis, Indiana), 200 U Superscript III (Invitrogen), and 40 U ribonuclease inhibitor (Roche). Mouse hypothalamic and liver RNA (10 ng diluted in pipette solution) and pipette solution stock were also reverse transcribed as positive and negative controls, respectively. Additionally, a standard curve of mouse hypothalamic RNA (5, 0.5, 0.05, and 0.005 ng/μL final concentration) was reverse transcribed. cDNA synthesis was performed as recommended. cDNA was stored at −80°C until PCR.
Single-cell PCR
Single-cell cDNA, controls, and the standard curve were preamplified using TaqMan PreAmp Master Mix (Invitrogen). TaqMan PrimeTime qPCR assays for mRNAs of Gapdh, Tac2, Kiss1, Pdyn, Oprk1, and Tac3r were purchased from Integrative DNA Technologies (Coralville, Iowa) (Table 1). All assay primers span an intron to minimize amplification of genomic DNA. PrimeTime qPCR assays were reconstituted to 20× (5μM probe, 10μM each primer) in Tris-EDTA. The components for the preamplification reaction were 5 μL cDNA, 0.05× final concentration of each primer-probe, 10 μL 2× preamplification buffer and water to a final volume of 20 μL. cDNA was preamplified for 18 cycles according to the manufacturer's recommended cycling conditions. The preamplified DNAs were then diluted 1:10 with Tris-EDTA; a second dilution to 1:50 was also created for more abundant transcripts (Gapdh and Tac2), both were stored at −20°C until used for quantitative PCR.
Table 1.
qPCR Assays for Single-Cell PCRa
| Gene | IDT Assay ID | Ref Seq No. | Exons | Amplicon, bp | Amplicon Location, bp |
|---|---|---|---|---|---|
| Kiss1 | Mm.PT.45.16269514 | NM_178260 | 1–2 | 105 | 66–170 |
| Pdyn | Mm.PT.45.9486062 | NM_018863 | 1–2 | 100 | 89–188 |
| Oprk1 | Mm.PT.42.8829407 | NM_001204371, v1 | 2–3 | 94 | 398–491 |
| NM_011011, v2 | 2a–3 | 94 | 368–461 | ||
| Tacr3 | Mm.PT.51.8434948 | NM_021382 | 4–5 | 114 | 1266–1379 |
| Gapdh | Mm.PT.39.1 | NM_008084 | 2–3 | 150 | 50–199 |
PrimeTime qPCR assays were from Integrated DNA Technologies, Coralville, Iowa (www.idtdna.com/pages/products/gene-expression/primetime-qpcr-assays-and-primers).
Quantitative PCR was performed using 2 to 5 μL of diluted preamplified DNA per reaction, in duplicate, for 40 to 50 cycles (TaqMan Gene Expression Master Mix; Invitrogen). PCR efficiencies were calculated from the slope of the standard curve. To confirm the linearity and parallelism of the preamplification step, 2 μL of non-preamplified standard curve cDNA was also amplified in singlicate. On average, the threshold cycle for preamplified standards was 10 cycles earlier than cDNA, but PCR efficiencies were nearly identical, indicating cDNAs were amplified linearly and without bias. Single cells were considered positive for a transcript if their threshold was a minimum of 4 cycles earlier than the preamplification blank. Amplicon size was confirmed by agarose gel electrophoresis.
Electrophysiological recordings
Targeted single-unit extracellular recordings were used, because this configuration has minimal impact on the cell's intrinsic properties, including response to fast synaptic transmission (58, 59). Recording pipettes (1.5–3 MΩ) were pulled from borosilicate glass (Schott no. 8250; World Precision Instruments, Sarasota, Florida) with a P-97 puller (Sutter Instrument, Novato, California). Pipettes were filled with HEPES-buffered solution containing (in mM) 150 NaCl, 10 HEPES, 10 glucose, 2.5 CaCl2, 1.3 MgCl2, and 3.5 KCl, and low-resistance (23 ± 3 MΩ) seals were formed between the pipette and neuron. Recordings were made in voltage clamp with a 0-mV pipette holding potential and signals filtered at 10 kHz using an EPC8 amplifier and PatchMaster software (version 2x42; HEKA Instruments Inc, Bellmore, New York).
Experimental designs
Slices were transferred to a recording chamber with constant perfusion of ACSF at 28°C to 33°C. All treatments were diluted in ACSF and administered by bath; vehicles had no effect on firing rate. After a ≥5-minute stabilization period, neuronal activity was recorded under control and treatment conditions as detailed below. At the end of each experiment, inactive cells were treated with high-potassium ACSF (20mM K+). Cells that exhibited action currents in response were verified to be alive, and all data, including quiescence, were used. For cells not responding to K+, data analysis was truncated at the last action current.
Experiment 1
To study effects of NKB on KNDy neurons and whether steroid milieu modifies effects, the NK3R agonist senktide (Phoenix Pharmaceuticals, Inc, Burlingame, California) (in ≤0.1% dimethylsulfoxide) was bath-applied to brain slices from castrate (1nM, 10nM, or 100nM) or intact adult male mice (10nM or 100nM) for 5 to 7 minutes. Senktide was used because it is more specific for NK3R than NKB (60); preliminary studies indicated senktide has similar effects to NKB in KNDy neurons (n = 3 cells from each castrate and intact male, not shown). Effects of 10nM senktide on GnRH neurons from castrate males were also determined and compared with untreated GnRH neurons.
Experiment 2
To examine whether activation of NK3R in KNDy neurons from castrate males requires fast synaptic transmission, ionotropic γ-aminobutyric acid (GABA) and glutamate receptors were blocked. Cells were first treated with 10nM senktide as above to verify response. In preliminary trials, repeated treatment of the same cell with 10nM senktide generated comparable responses under control conditions in KNDy neurons from both castrate and intact mice (n = 3 each, not shown). After washout (5–30 minutes to allow for return toward basal firing rate), a cocktail of blockers of GABAA (100μM picrotoxin in 0.2% ethanol), α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (40μM CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione in 0.2% dimethylsulfoxide), and N-methyl-D-aspartic acid (20μM APV, d(−)2-amino-5-phosphonovaleric acid) receptors was applied for 5 minutes before a second 5- to 7-minute treatment with 10nM senktide in the blocker cocktail.
Experiment 3
To study effects of dynorphin on KNDy neurons, 1μM dynorphin A (dynorphin; Tocris Bioscience, Ellisville, Missouri) was applied for 5 minutes to spontaneously active KNDy neurons from castrate and intact males. Spontaneously active neurons were used because we hypothesized dynorphin would inhibit KNDy neurons. Because extracellular recordings monitor firing activity, inhibition cannot be observed with this method in quiescent cells.
Experiment 4
To test the hypothesis that dynorphin reduces senktide-induced KNDy neuron activation in castrate males, senktide was applied for 5 to 7 minutes, followed by senktide alone, senktide and dynorphin (1μM), or senktide and the KOR agonist U69593 (1μM, in 0.01% ethanol) for an additional 5 to 7 minutes. To test whether endogenous KOR activation affected the firing pattern in the presence of prolonged senktide, cells were treated for 10 minutes with senktide and a KOR antagonist nor-binaltorphimine (norBNI, 10μM; Tocris Bioscience). To determine whether pretreatment with dynorphin blocked the ability of senktide to activate KNDy neurons, senktide was applied to confirm responsiveness (as in experiment 2), and then after the wash period, 1μM dynorphin was applied for 5 minutes, followed by 5 minutes of dynorphin plus 10nM senktide.
Experiment 5
To test the hypothesis that quiescent KNDy neurons are under suppression by endogenous dynorphin, 10μM norBNI was applied to quiescent KNDy neurons for 5 to 6 minutes. The ability of 10μM norBNI to block 1μM dynorphin-induced inhibition of firing was tested as a positive control.
Analyses
Targeted extracellular recordings detect action currents, which are the currents underlying action potentials. Thus, their frequency reflects action potential firing rate. Action currents were identified using custom software written in IgorPro (WaveMetrics, Inc, Lake Oswego, Oregon). Control action current frequency was averaged for the last 2 minutes before treatment. The first 3 minutes of treatment were not included in the analysis to allow time for solution exchange and drug penetration of the slice; the action current firing frequency was averaged during minutes 4 and 5 after initiating treatment. Cells with basal firing frequency ≤0.1 Hz were considered quiescent. Cells that remained quiescent during treatment were considered nonresponsive if they subsequently generated action currents in response to elevated K+. Spontaneously active cells with a change in firing frequency of >20% were considered responsive. The percentage of responsive cells is reported, but for statistical rigor, both responsive and nonresponsive cells were included in statistical analyses. No more than 3 cells from a given animal were included in the same experiment, and n indicates number of cells.
Data are reported as mean ± SEM. Nonparametric or parametric 2-tailed comparisons were used as appropriate for data distribution. Responses to single treatments were analyzed by Wilcoxon matched pairs test or paired t test. Data from cells exposed to multiple treatments were analyzed by 1-way Friedman test, followed by a Dunn's multiple-comparison test. Comparisons among groups were analyzed by Friedman 2-way repeated-measures ANOVA, Mann-Whitney U, or unpaired t test. All tests were 2-tailed, and significance was set at P < .05.
Results
GFP expression driven by the Tac2 promoter identifies KNDy neurons in the arcuate nucleus
Single-cell PCR was performed on Tac2-GFP-identified cells from the arcuate nucleus of castrate (23 cells) and intact (18 cells) males (Table 2). Single-cell cDNA was amplified for Gapdh to determine RNA quality; of 41 cells, 3 did not amplify for Gapdh and were eliminated from further analysis. No transcripts of interest were amplified from false harvests (n = 5). Tac2 mRNA was detected in 87% of all GFP-identified cells, indicating that GFP expression is highly correlated with endogenous Tac2 expression. The half-life of eGFP protein is ∼26 hours (61). It is thus likely that GFP-identified cells in intact males not expressing Tac2 mRNA when harvested had expressed this mRNA within the previous few days. All cells from castrate and 71% of cells from intact mice expressed Tac2, indicating steroid negative feedback suppresses Tac2 expression, consistent with earlier reports (32, 44, 62, 63). Consistent with this, 16 of 17 Tac2-GFP-identified cells from intact mice expressed either kisspeptin (Kiss1) or prodynorphin (Pdyn) mRNA, indicating a KNDy phenotype (32, 44). In contrast to a previous report (44), a substantial population of Tac2-GFP neurons that did not express Kiss1 was not observed (18% intact mice, 0% castrate mice). This discrepancy may be due to relative sensitivity of methods used or the mode of cell identification. The percentage of Pdyn mRNA-positive cells was 90% in castrate and 76% in intact mice. With regard to G protein-coupled receptors, 19% of cells from castrate and 41% of cells from intact mice expressed transcript for the dynorphin receptor KOR (Oprk1). Most (86%) cells from castrate mice expressed mRNA for the NKB receptor NK3R (Tac3r) vs 35% of cells from intact mice.
Table 2.
Single-Cell PCR Resultsa
| Transcript | Intact (n = 17) No. Cells Expressing Transcript, n (%) | Castrate (n = 21) No. Cells Expressing Transcript, n (%) |
|---|---|---|
| Tac2 | 12 (71) | 21 (100) |
| Kiss1 | 14 (82) | 21 (100) |
| Pdyn | 13 (76) | 19 (90) |
| Tac2 + Kiss1 | 11 (65) | 21 (100) |
| Tac2 + Pdyn | 10 (59) | 19 (90) |
| Pdyn + Kiss1 | 11 (65) | 19 (90) |
| Tac2 + Kiss1 + Pdyn | 10 (59) | 19 (90) |
| Kiss1 OR Pdyn | 16 (94) | 21 (100) |
| Oprk1 | 7 (41) | 4 (19) |
| Tac3r | 6 (35) | 18 (86) |
Results are shown as the number of cells expressing transcript (percentage of GFP-identified cells).
Spontaneous activity of KNDy neurons from intact and castrate males
Eighty percent of KNDy neurons from castrate mice (78 of 98) were quiescent during the control period (defined as a firing frequency of ≤0.1 Hz), whereas 57% (25 of 44) of KNDy neurons from intact mice were quiescent. The firing rate of spontaneously active KNDy neurons was not different between castrate (2.0 ± 0.3 Hz, n = 20 cells) and intact mice (1.9 ± 0.5 Hz, n = 19 cells).
The NK3R agonist senktide increases activity to a greater extent in KNDy neurons from castrate than those from intact males
To study the effects of activation of NK3R on KNDy neurons, the NK3R agonist senktide was bath-applied to brain slices from castrate adult males during extracellular recordings. Figure 1A shows a representative response to 10nM senktide in cells from castrate males. Senktide increased the frequency of action currents in a dose-dependent manner (1nM, P = .0625, n = 8; 10nM, P < .05, n = 11; 100nM, P < .05, n = 6; Figure 1C). The proportion of cells responding with >20% change in firing rate also increased with dose (3 of 8 at 1nM, 10 of 11 at 10nM, and 6 of 6 at 100nM).
Figure 1.
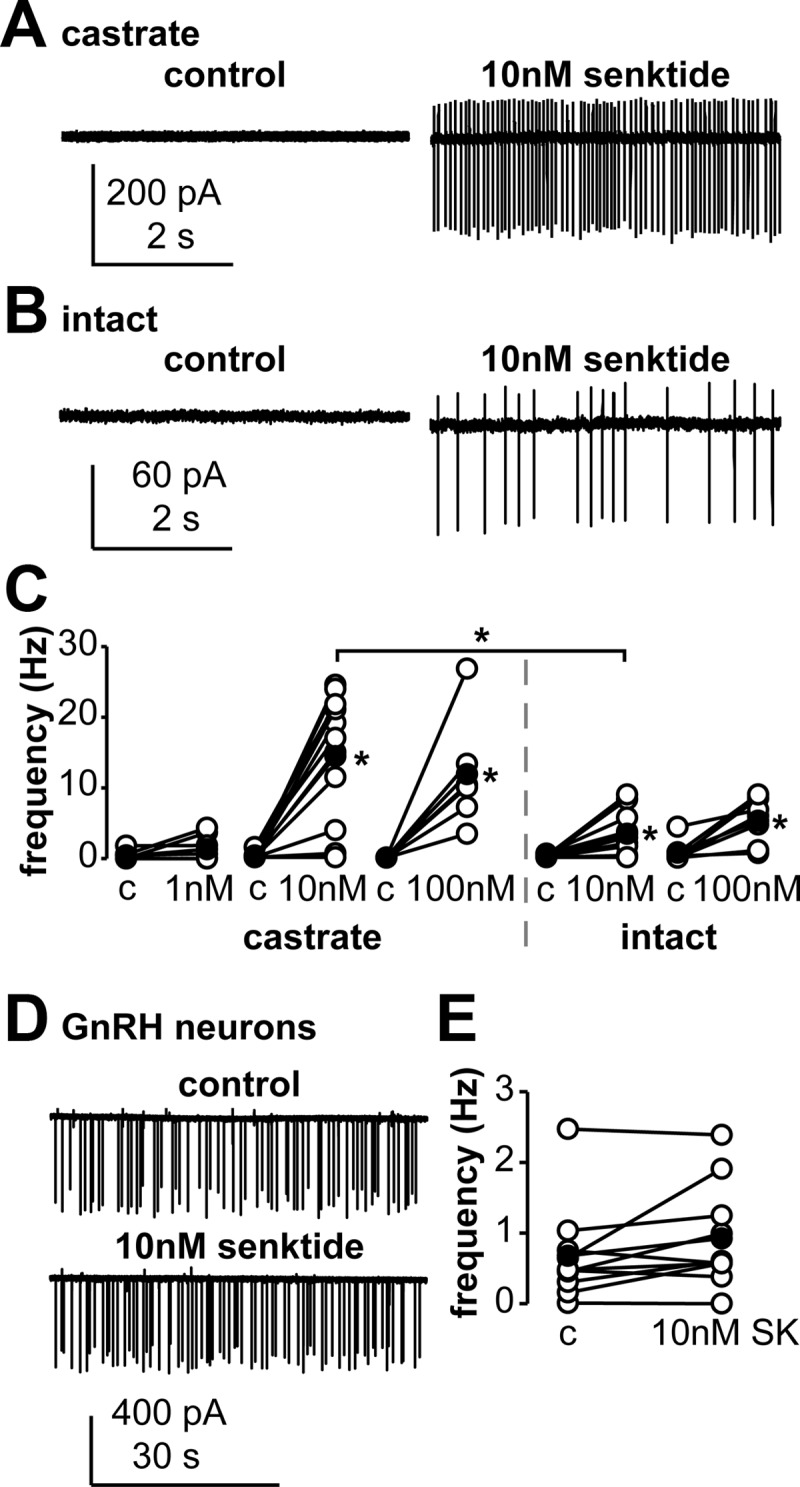
The NK3R agonist senktide increases the activity of KNDy neurons and does not change firing activity of GnRH neurons. Panels A and B, Representative extracellular recordings of a KNDy neuron from a castrate (panel A) or an intact (panel B, trace smoothed to reduce baseline drift) mouse during control (left) and 10nM senktide (right) conditions; variable current amplitudes reflect small changes in pipette position during the recording and do not provide information about changes in cell function. Panel C, Action current frequency of KNDy neurons during control (c) and senktide treatment at doses indicated in castrate (left) and intact (right) mice. Panel D, Representative extracellular recordings of a GnRH neuron from a castrate mouse during control (top) and 10nM senktide (bottom) conditions. Panel E, Action current frequency of GnRH neurons during control (c) and 10nM senktide (SK) treatment. Open circles show data from individual cells, filled circles show means, and lines connect data from the same cell. *, P < .05.
To test whether gonadal steroids affect the response to senktide in KNDy neurons, 10nM or 100nM senktide was bath-applied to brain slices from intact male mice; Figure 1B shows a representative response to 10nM senktide. Firing frequency increased in all KNDy neurons from intact mice in response to both 10nM (P < .05, n = 11, Figure 1C) and 100nM (P < .05, n = 7, Figure 1C) senktide. Senktide (10nM) caused a greater increase in KNDy neuron activity in cells from castrate mice versus those from intact mice (P < .05, Figure 1C), whereas the response to 100nM senktide was similar between cells from castrate and intact mice (Figure 1C). These data indicate that KNDy neurons from both castrate and intact mice are stimulated by activation of the NK3R but that neurons from castrate animals are more sensitive. For subsequent senktide experiments, 10nM was used because it was the lowest dose tested that consistently elicited a response, and this response was different between castrate and intact mice.
Senktide does not alter firing of GnRH neurons
To test whether senktide alters GnRH neuron activity, senktide was bath-applied to coronal preoptic area brain slices from castrate GnRH-GFP mice. Senktide (10nM) had no consistent effect on firing frequency of GnRH neurons (n = 10, Figure 1, D and E), with senktide treatment increasing (n = 5), decreasing (n = 2), or eliciting no change (n = 3) in firing frequency. Additionally, we compared senktide-treated cells with untreated GnRH neurons recorded for the same duration. Changes in firing frequency pattern in the absence of treatment (n = 5, not shown) were similar to those during senktide treatment. These data support the hypothesis that activation of NK3R within a coronal slice through the preoptic area does not consistently alter the activity of GnRH neurons at the level of the soma.
The senktide-induced response in KNDy neurons is independent of fast synaptic transmission
The activation of KNDy neurons by senktide could be direct and/or via NK3R-expressing afferents that remain in the brain slice. To begin to differentiate between these possibilities, we asked whether the senktide-induced firing activity in KNDy neurons required and/or was modulated by fast synaptic transmission. Cells were first verified to respond to senktide (10nM), washed with ACSF, and then treated a second time with senktide in the presence of blockers of ionotropic GABA and glutamate receptors (Figure 2). Blocking these receptors after the wash period did not alter firing rate of KNDy neurons (0.6 ± 0.4 Hz wash vs 0.9 ± 0.6 Hz blocked, n = 8), nor did it alter senktide-induced activity (8.5 ± 2.4 Hz unblocked vs 13 ± 3.0 Hz blocked, n = 8, Figure 2). These data suggest activation of NK3R excites KNDy neurons by a mechanism that does not require fast synaptic transmission.
Figure 2.

The senktide-induced activation of KNDy neurons is independent of fast synaptic transmission. Panel A, Representative extracellular recordings from a cell during control and treatment periods. Panel B, Action current frequency during various treatments. Open circles show data from individual cells, filled circles show means, lines connect data from the same cell, and dashed lines indicate a variable wash period with ASCF after the first senktide treatment. *, P < .05. Abbreviations: c, control; SK, 10nM senktide; block, blockers of ionotropic GABA and glutamate receptors (d(−)2-amino-5-phosphonovaleric acid, 6-cyano-7-nitroquinoxaline-2,3-dione, and picrotoxin).
Spontaneous activity of KNDy neurons is inhibited by dynorphin to a greater extent in cells from intact than castrate mice
To test the hypothesis that dynorphin decreases activity of KNDy neurons, we first examined spontaneously active (>0.1 Hz) KNDy neurons. Bath-applied dynorphin (1μM) decreased the firing frequency in 6 of 7 cells from castrate mice (n = 7, P < .05, Figure 3A) and in all cells examined from intact mice (n = 6, P < .05, Figure 3A). Dynorphin reduced the firing frequency of responsive cells from castrate mice by 50% and by 91% in cells from intact mice. The difference between the firing rate during dynorphin treatment vs the control firing frequency was greater in KNDy neurons from intact than those from castrate mice (P < .05, Figure 3B). The inhibitory effect of dynorphin was blocked by 10μM norBNI, a KOR antagonist, in all KNDy neurons from both castrate (4.5 ± 1.1 Hz norBNI vs 5.5 ± 1.2 Hz norBNI+dynorphin, n = 5) and intact (1.1 ± 0.6 Hz norBNI vs 1.5 ± 0.9 Hz norBNI+dynorphin, n = 3) mice. These data indicate endogenous activity of KNDy neurons is inhibited by dynorphin via KOR, and this inhibition is more pronounced in the presence of gonadal steroids.
Figure 3.
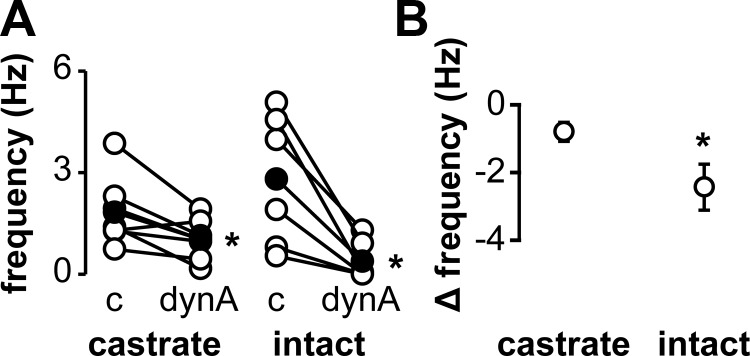
Dynorphin attenuates spontaneous activity in KNDy neurons. Panel A, Action current frequency during periods of control and treatment with 1 μM dynorphin in neurons from castrate (left) or intact (right) animals. Open circles show data from individual cells, filled circles show means, and lines connect data from the same cell. Panel B, Difference between action current frequency during treatment and control periods in cells from castrate or intact mice. *, P < .05. Abbreviations: c, control; dynA, 1μM dynorphin.
The KOR antagonist norBNI does not induce activity in quiescent KNDy neurons
Because most KNDy neurons from castrate mice were quiescent during the control period, and because dynorphin inhibited spontaneous activity in most KNDy neurons, we hypothesized that activity in quiescent cells was inhibited by endogenous dynorphin. To test this, quiescent KNDy neurons were treated with the KOR antagonist norBNI (10 μM). NorBNI did not induce activity (castrate: 0.0 ± 0.0 Hz control vs 0.0 ± 0.0 Hz treated, n = 5; intact: 0.0 ± 0.0 Hz control vs 0.0 ± 0.0 Hz treated, n = 5).
The pattern of firing activity in KNDy neurons induced by prolonged senktide treatment does not change with subsequent addition of KOR agonists in cells from castrate mice
We next tested the hypothesis that senktide-induced KNDy neuron activation is inhibited by dynorphin. KNDy neurons from castrate males were either treated with 10nM senktide for a full 10 minutes (n = 8) or were treated with senktide for 5 minutes followed by senktide plus dynorphin (1μM, n = 8) or senktide plus the KOR agonist U69593 (1μM, n = 9) for an additional 5 minutes. Neither dynorphin nor U69593 reduced the senktide-induced activation at the end of the second 5-minute treatment compared with activity at the end of the 10-minute senktide-only treatment (Figure 4).
Figure 4.
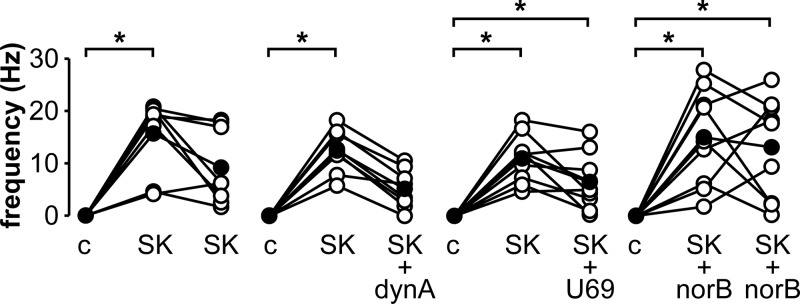
Activation of KOR does not reduce ongoing senktide-induced activity in KNDy neurons from castrate mice. Action current frequency during various treatments, which are detailed in text. Open circles show data from individual cells, filled circles show means; lines connect data from the same cell. *, P < .05. Abbreviations: c, control; dynA, 1μM dynorphin; norB, 10μM norBNI, a KOR antagonist; SK, 10nM senktide, an NK3R agonist; U69, 1μM U69593, a KOR agonist.
Examination of individual cells treated with dynorphin during the last 5 minutes of the 10-minute senktide treatment reveals that all cells exhibited >20% decrease in firing frequency after addition of dynorphin compared with senktide treatment alone (Figure 4). In contrast, only a subset of cells (4 of 8) treated with senktide alone for 10 minutes showed >20% decrease in firing frequency during similar time periods of recording (Figure 4). To test whether the decrease in firing frequency in this subset of cells is due to senktide-induced dynorphin release, norBNI (10μM) was included during a 10-minute senktide treatment (Figure 4). Similar to the observations with senktide alone, norBNI failed to maintain firing rate throughout the 10-minute senktide treatment, with 5 of 9 cells exhibiting >20% decrease in firing frequency.
Pretreatment with dynorphin blocks senktide-induced activation in KNDy neurons from intact but not castrate mice
To test whether treatment with dynorphin before, rather than during, senktide application is able to block senktide-induced activity, cells from both castrate and intact mice were first verified to respond to senktide (10nM), washed with ACSF, and then treated with dynorphin (1μM) followed by a second treatment with senktide in the presence of dynorphin (Figure 5). Senktide treatment increased firing frequency of KNDy neurons from castrate mice to the same extent regardless of whether or not dynorphin was present (0.0 ± 0.0 Hz control vs 18.1 ± 3.0 Hz senktide, P < .05; 2.1 ± 1.2 Hz dynorphin vs 13.4 ± 2.8 Hz senktide+dynorphin, P < .05, n = 7; Figure 5). In contrast, pretreatment with dynorphin decreased the senktide-induced firing frequency in KNDy neurons from intact mice (0.0 ± 0.0 Hz control vs 7.2 ± 2.9 Hz senktide, P < .05; 0.0 ± 0.0 Hz dynorphin vs 1.2 ± 0.8 Hz senktide+dynorphin, n = 6; Figure 5). Spontaneous activity was suppressed in KNDy neurons (n = 2 castrate, n = 2 intact, not shown) throughout a 10-minute treatment with dynorphin alone, confirming cells remained responsive to dynorphin throughout the duration tested. These data provide further support for enhancement of dynorphin-mediated inhibition by gonadal steroids.
Figure 5.
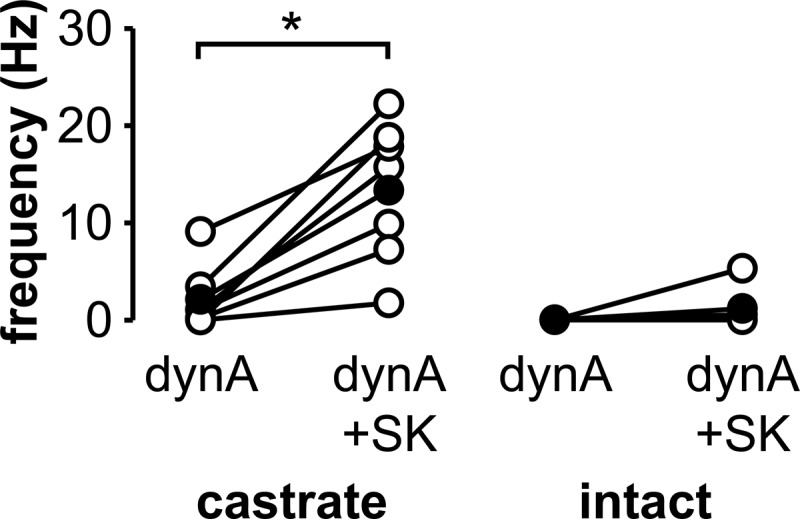
Pretreatment with dynorphin A decreases senktide-induced activity in KNDy neurons from intact but not castrate mice. Action current frequency in KNDy neurons from castrate (left) or intact (right) animals during treatments with 1μM dynorphin (dynA) and dynorphin plus 10nM senktide (SK); treatments shown follow a previous treatment to confirm responsiveness to senktide (not shown). Open circles show data from individual cells, filled circles show means, and lines connect data from the same cell. *, P < .05.
Discussion
Targeted extracellular recordings revealed that the NK3R agonist senktide activates KNDy neurons by a mechanism that is independent of fast synaptic transmission and is attenuated in cells from intact compared with castrate males. In contrast, dynorphin inhibited spontaneous and senktide-induced activity of KNDy neurons to a greater extent in cells from intact than castrate mice.
Based on genetics studies in infertile patients (26) and the coexpression of NKB and NK3R in arcuate neurons (40, 41), NKB has joined kisspeptin as a focus of studies of afferent control of GnRH release. Unlike the effect of kisspeptin, which consistently activates GnRH neurons, studies have yielded conflicting results concerning the role and site of NKB action. Peaks of multiunit activity (MUA) recorded near the arcuate nucleus in the medial basal hypothalamus are highly correlated with LH pulses, an indirect marker of GnRH release. Senktide increased the frequency of MUA peaks in multiple steroid milieus (48). In castrate and gonad-intact male nonhuman primates and in follicular-phase but not luteal-phase ewes, activation of NK3R caused robust single discharges of LH (42, 49). These data support a stimulatory role of NKB in several steroid conditions.
Rodent studies have had less consistent outcomes. Intracerebroventricular (icv) treatment with senktide decreased serum LH in ovariectomized (OVX) mice (32) and rats (47, 50) and in OVX rats replaced with estradiol in a model of negative feedback (46, 50, 51). Senktide (icv) also decreased the frequency of MUA peaks in OVX rats (50). In contrast, senktide (icv) increased serum LH in intact female rats and OVX female rats replaced with estradiol (morning vs afternoon not indicated) (47). In intact males, senktide (icv) was reported to have either no effect (52) or to increase serum LH (44). The lack of consistent changes in LH in response to senktide even within a species, sex, or steroid model may be due to differences in response at many levels including the KNDy neuron, the GnRH neuron, and/or the pituitary.
To avoid the complexities of these intermediate systems influencing the response to activation of NK3R, we examined the response to senktide directly at KNDy or GnRH neurons using electrophysiology to record activity at the soma of these cells in acute brain slices from the male mouse. Males were chosen for study because they are not subject to the variability of steroid feedback that occurs during the ovarian cycle in intact females. Furthermore, GnRH pulse generation is a feature of both sexes, and in males, this is unencumbered by coexistence of a surge mode of release. Although the steroids involved in feedback regulation of pulse frequency could differ between sexes, we postulate that many core mechanisms of pulse generation itself are shared between males and females. Senktide induced a dose-dependent increase in firing rate of KNDy neurons from castrate males but had no effect on GnRH neurons. These data are consistent with a recent electrophysiological study of kisspeptin-GFP-identified neurons in slices from castrate and intact adult male mice (44). The present data further identify a role for gonadal steroids in modulating the response to senktide because the activation induced in KNDy neurons from intact males was suppressed. This is contrary to the previous study, which found no evidence for steroid dependence of NK3R-mediated activation of KNDy neurons (44). This discrepancy may be attributable to recording configuration because the previous study used whole-cell recordings, which alter the intracellular milieu unlike extracellular recordings.
One possible mechanism for the reduced response to senktide observed in cells from intact males in the present study may be altered expression of NK3R in KNDy neurons. The transcript for NK3R (Tac3r) was expressed in most KNDy neurons from castrate animals but in less than a third of those from intact animals. Furthermore, a stimulatory effect of NK3R activation may be amplified by positive feedback from other KNDy neurons that synapse on the recorded cell (19, 24, 64).
In addition to possible network effects of NKB from other KNDy neurons, KNDy neurons receive other types of inputs, including those from non-KNDy cells that may be responsive to senktide. Some connections with such neurons could be maintained within the slice preparation, and thus, the effects observed may be due to action of senktide that is not directly on the neuron targeted for recording. One mode of signaling from these putative upstream neurons to KNDy neurons is fast synaptic transmission mediated by GABA and glutamate. KNDy neurons receive spontaneous GABAergic and glutamatergic transmission (65, 66), which may arise in part from KNDy neurons themselves because they express both the vesicular glutamate transporter 2 (vGLUT-2) (67, 68) and glutamic acid decarboxylase (GAD), an enzyme involved in GABA synthesis (68). The present observations show that NK3R-mediated stimulation of KNDy neurons is independent of fast synaptic transmission mediated by ionotropic glutamatergic and GABAergic signaling.
Gonadal steroids may have an effect on spontaneous activity in KNDy neurons. Eighty percent of cells from castrate mice vs 57% of those from intact mice were quiescent. The latter is similar to that observed in whole-cell recordings of arcuate kisspeptin neurons from intact males (66). Previous extracellular recordings of these neurons from intact males, however, reported only 10% were silent (69). This contrast may in part be due to a different definition of quiescence. Specifically, the present study considered cells to be quiescent if spontaneous firing frequency was <0.1 Hz, whereas the previous extracellular study considered cells to be silent only if the cell did not fire a single action potential throughout the recording duration, which would reduce the percentage of silent cells. Regardless, all datasets appear to challenge the hypothesis that gonadal steroids inhibit endogenous activity in KNDy neurons. It is important to point out that the present experiments were designed to examine the effects of neuromodulators on firing activity; recordings were thus of short duration. Changes in long-term firing pattern relevant to the frequency of GnRH release may well reveal effects of steroid feedback. It is nonetheless interesting to speculate that the regulation of mRNA expression and the regulation of firing activity by steroids in KNDy neurons may be different.
A potential cause for the quiescence observed in most KNDy neurons is endogenous dynorphin within the slice. Antagonists of KOR, the high-affinity receptor for dynorphin, support an inhibitory effect of dynorphin on LH release in mice, rats, and sheep (31, 32, 70), and KOR antagonists increase MUA in the medial basal hypothalamus in goats (48). In the present study in acutely prepared brain slices, the KOR antagonist norBNI did not initiate activity in KNDy neurons from either intact or castrate mice, suggesting that under these conditions, endogenous dynorphin is not responsible for quiescence.
Unlike the lack of effect of a KOR antagonist on quiescent KNDy neurons, the KOR agonist dynorphin inhibited spontaneously active KNDy neurons. Greater inhibition was observed in intact than castrate males, supporting the hypothesis that the inhibitory effect of dynorphin on KNDy neurons is greater in the presence of gonadal steroids. This may be due to higher responsiveness to dynorphin, because single-cell PCR revealed the percentage of KNDy neurons expressing KOR in intact mice was twice that in castrate mice in the present study. In contrast, in situ hybridization demonstrated no change in the percentage of cells expressing the transcript for KOR in castrate vs testosterone-replaced males (44). This discrepancy may result from a difference in feedback in intact vs testosterone-replaced mice or method of mRNA detection.
Dynorphin has been hypothesized to inhibit KNDy neurons after stimulation by NKB (18, 32, 43, 50, 51). Interestingly, in KNDy neurons from castrate males, neither pretreatment with dynorphin nor treatment with dynorphin subsequent to senktide treatment altered the average firing frequency pattern relative to that of senktide alone. In contrast to KNDy neurons from castrate mice, pretreatment with dynorphin attenuated senktide-induced activity in cells from intact males. These data further support stronger dynorphin-mediated inhibition in the presence of gonadal steroids. The inhibitory effect may be further enhanced by the reduced effectiveness of senktide in cells from intact mice. Because spontaneous activity in cells from both castrate and intact males was reduced by dynorphin, the lack of effect of dynorphin on senktide-induced activity in cells from castrate animals raises the question of what initiates spontaneous activity in KNDy neurons because dynorphin cannot counteract activation of NK3R. Other transmitters, such as glutamate or GABA, may play a role in this regard (67, 68).
When senktide treatment was extended to 10 minutes, some cells sustained or increased their firing frequency, whereas others decreased firing frequency. This raised the question of whether, in the latter subgroup, senktide stimulated release of endogenous dynorphin within the slice, thereby causing delayed inhibition of KNDy neuron activity during extended senktide treatment. In the presence of the KOR antagonist norBNI, however, a similar subpopulation of KNDy neurons decreased firing frequency after activation with senktide. This indicates activation of KOR by senktide-stimulated dynorphin release was not inhibiting the activity in this subpopulation. These observations suggest heterogeneity within the KNDy neuron population, in which some cells become desensitized to NK3R activation over time and others do not.
Disruption of the pulsatile pattern of GnRH release results in infertility in diseases including polycystic ovary syndrome and idiopathic hypogonadotropic hypogonadism (6, 53, 71); understanding mechanisms of control of GnRH release by steroid feedback is thus crucial to developing treatments. Our work begins to elucidate the role of KNDy neurons in this critical role. We provide evidence for regulation of KNDy neurons in males by activation of both NK3R and KOR in a gonadal steroid-dependent manner, with steroids reducing overall activity of KNDy neurons in response to these treatments. The electrophysiological recordings presented here used an extracellular configuration, which does not interfere with the intracellular environment but does not allow for exploration of intracellular mechanisms. Future work will test the mechanisms linking activation of KOR or NK3R to changes in activity.
Acknowledgments
We thank Elizabeth Wagenmaker and Dr Zhiguo Chu for excellent technical assistance, and Dr Richard DeFazio, Dr Carol Elias, Garrett Gaskins, Katarzyna Glanowska, and Elizabeth Wagenmaker for editorial comments.
This work was supported by National Institutes of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development R01HD34860 (to S.M.M.).
Disclosure Summary: K.A.R., L.L.B., and S.M.M. have nothing to disclose.
Footnotes
- ACSF
- artificial cerebrospinal fluid
- GABA
- γ-aminobutyric acid
- GFP
- green fluorescent protein
- icv
- intracerebroventricular
- KISS1R
- kisspeptin receptor
- KNDy
- arcuate neurons coexpressing NKB, dynorphin, and kisspeptin
- KOR
- κ-opioid receptor
- MUA
- multiunit activity
- NKB
- neurokinin B
- NK3R
- neurokinin 3 receptor
- norBNI
- nor-binaltorphimine
- OVX
- ovariectomized.
References
- 1. Carmel PW, Araki S, Ferin M. Pituitary stalk portal blood collection in rhesus monkeys: evidence for pulsatile release of gonadotropin-releasing hormone (GnRH). Endocrinology. 1976;99:243–248 [DOI] [PubMed] [Google Scholar]
- 2. Clarke IJ, Cummins JT. The temporal relationship between gonadotropin releasing hormone (GnRH) and luteinizing hormone (LH) secretion in ovariectomized ewes. Endocrinology. 1982;111:1737–1739 [DOI] [PubMed] [Google Scholar]
- 3. Levine JE, Pau KY, Ramirez VD, Jackson GL. Simultaneous measurement of luteinizing hormone-releasing hormone and luteinizing hormone release in unanesthetized, ovariectomized sheep. Endocrinology. 1982;111:1449–1455 [DOI] [PubMed] [Google Scholar]
- 4. Moenter SM, Brand RM, Midgley AR, Karsch FJ. Dynamics of gonadotropin-releasing hormone release during a pulse. Endocrinology. 1992;130:503–510 [DOI] [PubMed] [Google Scholar]
- 5. Levine JE, Duffy MT. Simultaneous measurement of luteinizing hormone (LH)-releasing hormone, LH, and follicle-stimulating hormone release in intact and short-term castrate rats. Endocrinology. 1988;122:2211–2221 [DOI] [PubMed] [Google Scholar]
- 6. Belchetz PE, Plant TM, Nakai Y, Keogh EJ, Knobil E. Hypophysial responses to continuous and intermittent delivery of hypothalamic gonadotropin-releasing hormone. Science. 1978;202:631–633 [DOI] [PubMed] [Google Scholar]
- 7. Haisenleder DJ, Katt JA, Ortolano GA, et al. Influence of gonadotropin-releasing hormone pulse amplitude, frequency, and treatment duration on the regulation of luteinizing hormone (LH) subunit messenger ribonucleic acids and LH secretion. Mol Endocrinol. 1988;2:338–343 [DOI] [PubMed] [Google Scholar]
- 8. Shupnik MA. Effects of gonadotropin-releasing hormone on rat gonadotropin gene transcription in vitro: requirement for pulsatile administration for luteinizing hormone-beta gene stimulation. Mol Endocrinol. 1990;4:1444–1450 [DOI] [PubMed] [Google Scholar]
- 9. Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Differential effects of gonadotropin-releasing hormone (GnRH) pulse frequency on gonadotropin subunit and GnRH receptor messenger ribonucleic acid levels in vitro. Endocrinology. 1997;138:1224–1231 [DOI] [PubMed] [Google Scholar]
- 10. Karsch FJ, Cummins JT, Thomas GB, Clarke IJ. Steroid feedback inhibition of pulsatile secretion of gonadotropin-releasing hormone in the ewe. Biol Reprod. 1987;36:1207–1218 [DOI] [PubMed] [Google Scholar]
- 11. Hrabovszky E, Steinhauser A, Barabás K, et al. Estrogen receptor-beta immunoreactivity in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology. 2001;142:3261–3264 [DOI] [PubMed] [Google Scholar]
- 12. Huang X, Harlan RE. Absence of androgen receptors in LHRH immunoreactive neurons. Brain Res. 1993;624:309–311 [DOI] [PubMed] [Google Scholar]
- 13. Lehman MN, Karsch FJ. Do gonadotropin-releasing hormone, tyrosine hydroxylase-, and beta-endorphin-immunoreactive neurons contain estrogen receptors? A double-label immunocytochemical study in the Suffolk ewe. Endocrinology. 1993;133:887–895 [DOI] [PubMed] [Google Scholar]
- 14. Herbison AE, Skinner DC, Robinson JE, King IS. Androgen receptor-immunoreactive cells in ram hypothalamus: distribution and co-localization patterns with gonadotropin-releasing hormone, somatostatin and tyrosine hydroxylase. Neuroendocrinology. 1996;63:120–131 [DOI] [PubMed] [Google Scholar]
- 15. Wintermantel TM, Campbell RE, Porteous R, et al. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52:271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mayer C, Acosta-Martinez M, Dubois SL, et al. Timing and completion of puberty in female mice depend on estrogen receptor α-signaling in kisspeptin neurons. Proc Natl Acad Sci U S A. 2010;107:22693–22698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oakley AE, Clifton DK, Steiner RA. Kisspeptin signaling in the brain. Endocr Rev. 2009;30:713–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lehman MN, Coolen LM, Goodman RL. Minireview: kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion. Endocrinology. 2010;151:3479–3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burke MC, Letts PA, Krajewski SJ, Rance NE. Coexpression of dynorphin and neurokinin B immunoreactivity in the rat hypothalamus: Morphologic evidence of interrelated function within the arcuate nucleus. J Comp Neurol. 2006;498:712–726 [DOI] [PubMed] [Google Scholar]
- 20. Goubillon ML, Forsdike RA, Robinson JE, Ciofi P, Caraty A, Herbison AE. Identification of neurokinin B-expressing neurons as an highly estrogen-receptive, sexually dimorphic cell group in the ovine arcuate nucleus. Endocrinology. 2000;141:4218–4225 [DOI] [PubMed] [Google Scholar]
- 21. Franceschini I, Lomet D, Cateau M, Delsol G, Tillet Y, Caraty A. Kisspeptin immunoreactive cells of the ovine preoptic area and arcuate nucleus co-express estrogen receptor α. Neurosci Lett. 2006;401:225–230 [DOI] [PubMed] [Google Scholar]
- 22. Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology. 2005;146:3686–3692 [DOI] [PubMed] [Google Scholar]
- 23. Smith JT, Dungan HM, Stoll EA, et al. Differential regulation of KiSS-1 mRNA expression by sex steroids in the brain of the male mouse. Endocrinology. 2005;146:2976–2984 [DOI] [PubMed] [Google Scholar]
- 24. Foradori CD, Coolen LM, Fitzgerald ME, Skinner DC, Goodman RL, Lehman MN. Colocalization of progesterone receptors in parvicellular dynorphin neurons of the ovine preoptic area and hypothalamus. Endocrinology. 2002;143:4366–4374 [DOI] [PubMed] [Google Scholar]
- 25. Ciofi P, Krause JE, Prins GS, Mazzuca M. Presence of nuclear androgen receptor-like immunoreactivity in neurokinin B-containing neurons of the hypothalamic arcuate nucleus of the adult male rat. Neurosci Lett. 1994;182:193–196 [DOI] [PubMed] [Google Scholar]
- 26. Topaloglu AK, Reimann F, Guclu M, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the Kiss1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–10976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627 [DOI] [PubMed] [Google Scholar]
- 29. d'Anglemont de Tassigny X, Fagg LA, Dixon JP, et al. Hypogonadotropic hypogonadism in mice lacking a functional Kiss1 gene. Proc Natl Acad Sci U S A. 2007;104:10714–10719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan YM, Broder-Fingert S, Paraschos S, et al. GnRH-deficient phenotypes in humans and mice with heterozygous variants in KISS1/Kiss1. J Clin Endocrinol Metab. 2011;96:E1771–E1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goodman RL, Coolen LM, Anderson GM, et al. Evidence that dynorphin plays a major role in mediating progesterone negative feedback on gonadotropin-releasing hormone neurons in sheep. Endocrinology. 2004;145:2959–2967 [DOI] [PubMed] [Google Scholar]
- 32. Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA. Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci. 2009;29:11859–11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han SK, Gottsch ML, Lee KJ, et al. Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci. 2005;25:11349–11356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang C, Roepke TA, Kelly MJ, Rønnekleiv OK. Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci. 2008;28:4423–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pielecka-Fortuna J, Chu Z, Moenter SM. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology. 2008;149:1979–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Messager S, Chatzidaki EE, Ma D, et al. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci U S A. 2005;102:1761–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gottsch ML, Cunningham MJ, Smith JT, et al. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology. 2004;145:4073–4077 [DOI] [PubMed] [Google Scholar]
- 38. Thompson EL, Patterson M, Murphy KG, et al. Central and peripheral administration of kisspeptin-10 stimulates the hypothalamic-pituitary-gonadal axis. J Neuroendocrinol. 2004;16:850–858 [DOI] [PubMed] [Google Scholar]
- 39. Smith JT, Li Q, Yap KS, et al. Kisspeptin is essential for the full preovulatory LH surge and stimulates GnRH release from the isolated ovine median eminence. Endocrinology. 2011;152:1001–1012 [DOI] [PubMed] [Google Scholar]
- 40. Amstalden M, Coolen LM, Hemmerle AM, et al. Neurokinin 3 receptor immunoreactivity in the septal region, preoptic area and hypothalamus of the female sheep: colocalisation in neurokinin B cells of the arcuate nucleus but not in gonadotrophin-releasing hormone neurones. J Neuroendocrinol. 2010;22:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krajewski SJ, Anderson MJ, Iles-Shih L, Chen KJ, Urbanski HF, Rance NE. Morphologic evidence that neurokinin B modulates gonadotropin-releasing hormone secretion via neurokinin 3 receptors in the rat median eminence. J Comp Neurol. 2005;489:372–386 [DOI] [PubMed] [Google Scholar]
- 42. Billings HJ, Connors JM, Altman SN, et al. Neurokinin B acts via the neurokinin-3 receptor in the retrochiasmatic area to stimulate luteinizing hormone secretion in sheep. Endocrinology. 2010;151:3836–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goodman RL, Parfitt DB, Evans NP, Dahl GE, Karsch FJ. Endogenous opioid peptides control the amplitude and shape of gonadotropin-releasing hormone pulses in the ewe. Endocrinology. 1995;136:2412–2420 [DOI] [PubMed] [Google Scholar]
- 44. Navarro VM, Gottsch ML, Wu M, et al. Regulation of NKB pathways and their roles in the control of Kiss1 neurons in the arcuate nucleus of the male mouse. Endocrinology. 2011;152:4265–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gottsch ML, Popa SM, Lawhorn JK, et al. Molecular properties of Kiss1 neurons in the arcuate nucleus of the mouse. Endocrinology. 2011;152:4298–42309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sandoval-Guzmán T, Rance NE. Central injection of senktide, an NK3 receptor agonist, or neuropeptide Y inhibits LH secretion and induces different patterns of Fos expression in the rat hypothalamus. Brain Res. 2004;1026:307–312 [DOI] [PubMed] [Google Scholar]
- 47. Navarro VM, Castellano JM, McConkey SM, et al. Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat. Am J Physiol Endocrinol Metab. 2011;300:E202–E210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wakabayashi Y, Nakada T, Murata K, et al. Neurokinin B and dynorphin A in kisspeptin neurons of the arcuate nucleus participate in generation of periodic oscillation of neural activity driving pulsatile gonadotropin-releasing hormone secretion in the goat. J Neurosci. 2010;30:3124–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramaswamy S, Seminara SB, Ali B, Ciofi P, Amin NA, Plant TM. Neurokinin B stimulates GnRH release in the male monkey (Macaca mulatta) and is colocalized with kisspeptin in the arcuate nucleus. Endocrinology. 2010;151:4494–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kinsey-Jones JS, Grachev P, Li XF, et al. The inhibitory effects of neurokinin B on GnRH pulse generator frequency in the female rat. Endocrinology. 2012;153:307–315 [DOI] [PubMed] [Google Scholar]
- 51. Grachev P, Li XF, Kinsey-Jones JS, et al. Suppression of the GnRH pulse generator by neurokinin B involves a κ-opioid receptor-dependent mechanism. Endocrinology. 2012;153:4894–4904 [DOI] [PubMed] [Google Scholar]
- 52. Corander MP, Challis BG, Thompson EL, et al. The effects of neurokinin B upon gonadotrophin release in male rodents. J Neuroendocrinol. 2010;22:181–187 [DOI] [PubMed] [Google Scholar]
- 53. Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update. 2006;12:351–361 [DOI] [PubMed] [Google Scholar]
- 54. Suter KJ, Song WJ, Sampson TL, et al. Genetic targeting of green fluorescent protein to gonadotropin-releasing hormone neurons: characterization of whole-cell electrophysiological properties and morphology. Endocrinology. 2000;141:412–419 [DOI] [PubMed] [Google Scholar]
- 55. Chu Z, Moenter SM. Endogenous activation of metabotropic glutamate receptors modulates GABAergic transmission to gonadotropin-releasing hormone neurons and alters their firing rate: a possible local feedback circuit. J Neurosci. 2005;25:5740–5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nunemaker CS, DeFazio RA, Moenter SM. Estradiol-sensitive afferents modulate long-term episodic firing patterns of GnRH neurons. Endocrinology. 2002;143:2284–2292 [DOI] [PubMed] [Google Scholar]
- 57. Xu C, Xu XZ, Nunemaker CS, Moenter SM. Dose-dependent switch in response of gonadotropin-releasing hormone (GnRH) neurons to GnRH mediated through the type I GnRH receptor. Endocrinology. 2004;145:728–735 [DOI] [PubMed] [Google Scholar]
- 58. Nunemaker CS, DeFazio RA, Moenter SM. A targeted extracellular approach for recording long-term firing patterns of excitable cells: a practical guide. Biol Proc Online. 2003;5:53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alcami P, Franconville R, Llano I, Marty A. Measuring the firing rate of high-resistance neurons with cell-attached recording. J Neurosci. 2012;32:3118–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wormser U, Laufer R, Hart Y, Chorev M, Gilon C, Selinger Z. Highly selective agonists for substance P receptor subtypes. EMBO J. 1986;5:2805–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Corish P, Tyler-Smith C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 1999;12:1035–1040 [DOI] [PubMed] [Google Scholar]
- 62. Danzer SC, McMullen NT, Rance NE. Dendritic growth of arcuate neuroendocrine neurons following orchidectomy in adult rats. J Comp Neurol. 1998;390:234–246 [PubMed] [Google Scholar]
- 63. Rance NE, Bruce TR. Neurokinin B gene expression is increased in the arcuate nucleus of ovariectomized rats. Neuroendocrinology. 1994;60:337–345 [DOI] [PubMed] [Google Scholar]
- 64. Krajewski SJ, Burke MC, Anderson MJ, McMullen NT, Rance NE. Forebrain projections of arcuate neurokinin B neurons demonstrated by anterograde tract-tracing and monosodium glutamate lesions in the rat. Neuroscience. 2010;166:680–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. DeFazio RA, Elias C, Moenter SM. Chloride homeostasis and response to GABA in kisspeptin neurons of the AVPV and arcuate nucleus. In: 2012 Neuroscience Meeting Planner New Orleans, LA: Society for Neuroscience; 2012 [Google Scholar]
- 66. Frazão R, Cravo RM, Donato J, Jr, et al. Shift in Kiss1 cell activity requires estrogen receptor α. J Neurosci. 2013;33:2807–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ciofi P, Leroy D, Tramu G. Sexual dimorphism in the organization of the rat hypothalamic infundibular area. Neuroscience. 2006;141:1731–1745 [DOI] [PubMed] [Google Scholar]
- 68. Cravo RM, Margatho LO, Osborne-Lawrence S, et al. Characterization of Kiss1 neurons using transgenic mouse models. Neuroscience. 2011;173:37–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. de Croft S, Piet R, Mayer C, Mai O, Boehm U, Herbison AE. Spontaneous kisspeptin neuron firing in the adult mouse reveals marked sex and brain region differences but no support for a direct role in negative feedback. Endocrinology. 2012;153:5384–5393 [DOI] [PubMed] [Google Scholar]
- 70. Gallo RV. κ-Opioid receptor involvement in the regulation of pulsatile luteinizing hormone release during early pregnancy in the rat. J Neuroendocrinol. 1990;2:685–691 [DOI] [PubMed] [Google Scholar]
- 71. Raivio T, Falardeau J, Dwyer A, et al. Reversal of Idiopathic Hypogonadotropic Hypogonadism. N Engl J Med. 2007;357:863–873 [DOI] [PubMed] [Google Scholar]