

Abstract
Fibroblast cultures from two individuals with biotin-responsive organicacidemia were found to have a pleiotropic deficiency of propionyl-CoA carboxylase, β-methylcrotonyl-CoA carboxylase, and pyruvate carboxylase activities after growth in biotin limited culture medium, conditions which do not affect the carboxylase activities of normal cells. All three enzyme activities were restored to normal levels after transferring the mutant strains to biotin-rich medium. Both patients excreted abnormal levels of an array of metabolic intermediates, including β-methylcrotonate, β-hydroxyisovalerate, β-hydroxypropionate, and lactate, which reflect metabolic blocks at all three carboxylase sites.
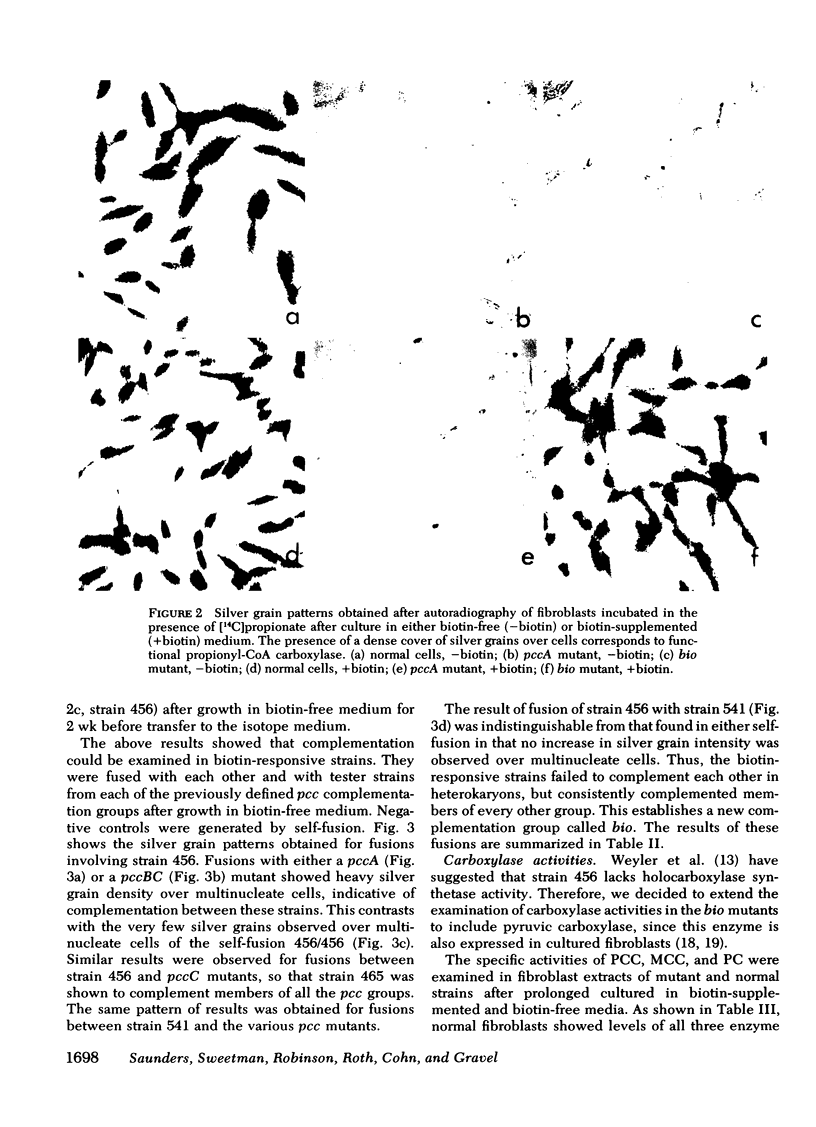
14 mutants deficient in only propionyl-CoA carboxylase activity from patients with propionicacidemia and the two biotin-responsive strains were examined for complementation with seven previously mapped pcc mutants. No new pcc complementation groups were identified. Nine of the mutants were mapped to group pccA. The remaining 12 mutants mapped to pccBC or its B or C subgroups, confirming the complex nature of this group. The biotin-responsive mutants failed to complement each other but did complement mutants from all the pcc groups. Thus biotin-responsive organicacidemia is defined by a new complementation group, bio. The results obtained in this study suggest that the bio mutants have a defect of either biotin transport or a common holocarboxylase synthetase required for the biotin activation of all three mitochondrial carboxylases.
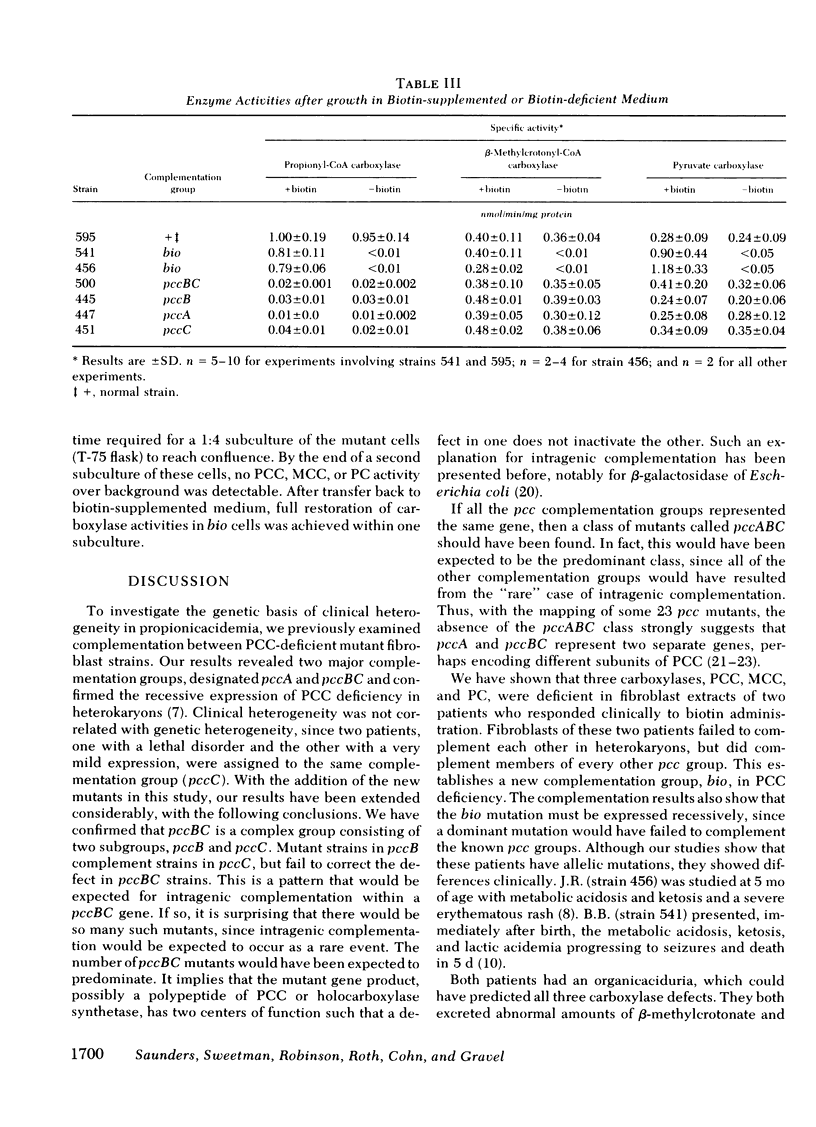
Full text
PDF





Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Achuta Murthy P. N., Mistry S. P., Kummerow F. A. Some aspects of biotin binding to protein catalyzed by biotin-deficient chicken liver preparations. Proc Soc Exp Biol Med. 1974 Feb;145(2):565–570. doi: 10.3181/00379727-145-37852. [DOI] [PubMed] [Google Scholar]
- Atkin B. M., Utter M. F., Weinberg M. B. Pyruvate carboxylase and phosphoenolpyruvate carboxykinase activity in leukocytes and fibroblasts from a patient with pyruvate carboxylase deficiency. Pediatr Res. 1979 Jan;13(1):38–43. doi: 10.1203/00006450-197901000-00009. [DOI] [PubMed] [Google Scholar]
- Bartlett K., Gompertz D. Combined carboxylase defect: biotin-responsiveness in cultured fibroblasts. Lancet. 1976 Oct 9;2(7989):804–804. doi: 10.1016/s0140-6736(76)90640-1. [DOI] [PubMed] [Google Scholar]
- Chalmers R. A., Bickle S., Watts R. W. A method for the determination of volatile organic acids in aqueous solutions and urine, and the results obtained in propionic acidaemia, beta-methylcrotonylglycinuria and methylmalonic aciduria. Clin Chim Acta. 1974 Mar;52(1):31–41. doi: 10.1016/0009-8981(74)90385-4. [DOI] [PubMed] [Google Scholar]
- Chalmers R. A., Lawson A. M., Watts R. W. Studies on the urinary acidic metabolites excreted by patients with beta-methylcrotonylglycinuria, propionic acidaemia and methylmalonic acidaemia, using gas-liquid chromatography and mass spectrometry. Clin Chim Acta. 1974 Mar;52(1):43–51. doi: 10.1016/0009-8981(74)90386-6. [DOI] [PubMed] [Google Scholar]
- GREEN N. M. AVIDIN. 1. THE USE OF (14-C)BIOTIN FOR KINETIC STUDIES AND FOR ASSAY. Biochem J. 1963 Dec;89:585–591. doi: 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompertz D., Bartlett K., Blair D., Stern C. M. Child with a defect in leucine metabolism associated with beta-hydroxyisovaleric aciduria and beta-methylcrotonylglycinuria. Arch Dis Child. 1973 Dec;48(12):975–977. doi: 10.1136/adc.48.12.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel R. A., Lam K. F., Scully K. J., Hsia Y. Genetic complementation of propionyl-CoA carboxylase deficiency in cultured human fibroblasts. Am J Hum Genet. 1977 Jul;29(4):378–388. [PMC free article] [PubMed] [Google Scholar]
- Guibaud P., Divry P., Marcon G., Cotton J. B., Collombel C., Larbre F. Une forme a révélation tardive de l'acidemie propionique. Arch Fr Pediatr. 1975 Mar;32(3):259–273. [PubMed] [Google Scholar]
- Hillman R. E., Keating J. P., Williams J. C. Biotin-responsive propionic acidemia presenting as the rumination syndrome. J Pediatr. 1978 Mar;92(3):439–441. doi: 10.1016/s0022-3476(78)80441-7. [DOI] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Lau E. P., Cochran B. C., Munson L., Fall R. R. Bovine kidney 3-methylcrotonyl-CoA and propionyl-CoA carboxylases: each enzyme contains nonidentical subunits. Proc Natl Acad Sci U S A. 1979 Jan;76(1):214–218. doi: 10.1073/pnas.76.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan R., Russell J. D., Arinze I. J. Pyruvate carboxylase and phosphoenopyruvate carboxykinase in cultured human fibroblasts. J Cell Physiol. 1977 Aug;92(2):285–292. doi: 10.1002/jcp.1040920217. [DOI] [PubMed] [Google Scholar]
- Roth K., Cohn R., Yandrasitz J., Preti G., Dodd P., Segal S. Beta-methylcrotonic aciduria associated with lactic acidosis. J Pediatr. 1976 Feb;88(2):229–235. doi: 10.1016/s0022-3476(76)80987-0. [DOI] [PubMed] [Google Scholar]
- Stanners C. P., Eliceiri G. L., Green H. Two types of ribosome in mouse-hamster hybrid cells. Nat New Biol. 1971 Mar 10;230(10):52–54. doi: 10.1038/newbio230052a0. [DOI] [PubMed] [Google Scholar]
- Sweetman L., Bates S. P., Hull D., Nyhan W. L. Propionyl-CoA carboxylase deficiency in a patient with biotin-responsive 3-methylcrotonylglycinuria. Pediatr Res. 1977 Nov;11(11):1144–1147. doi: 10.1203/00006450-197711000-00006. [DOI] [PubMed] [Google Scholar]
- UTTER M. F., KEECH D. B. PYRUVATE CARBOXYLASE. I. NATURE OF THE REACTION. J Biol Chem. 1963 Aug;238:2603–2608. [PubMed] [Google Scholar]
- Wadlington W. B., Kilroy A., Ando T., Sweetman L., Nyhan W. L. Hyperglycinemia and propionyl coA carboxylase deficiency and episodic severe illness without consistent ketosis. J Pediatr. 1975 May;86(5):707–712. doi: 10.1016/s0022-3476(75)80354-4. [DOI] [PubMed] [Google Scholar]
- Weyler W., Sweetman L., Maggio D. C., Nyhan W. L. Deficiency of propionyl-Co A carboxylase and methylcrotonyl-Co A carboxylase in a patient with methylcrotonylglycinuria. Clin Chim Acta. 1977 May 2;76(3):321–328. doi: 10.1016/0009-8981(77)90158-9. [DOI] [PubMed] [Google Scholar]
- Wolf B., Hsia Y. E., Rosenberg L. E. Biochemical differences between mutant propionyl-CoA carboxylases from two complementation groups. Am J Hum Genet. 1978 Sep;30(5):455–464. [PMC free article] [PubMed] [Google Scholar]
- Wolf B., Rosenberg L. E. Heterozygote expression in propionyl coenzyme A carboxylase deficiency. Differences between major complementation groups. J Clin Invest. 1978 Nov;62(5):931–936. doi: 10.1172/JCI109221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabin I., Villarejo M. R. Protein complementation. Annu Rev Biochem. 1975;44:295–313. doi: 10.1146/annurev.bi.44.070175.001455. [DOI] [PubMed] [Google Scholar]