

Ras-transformed cell lines require autophagy for mitochondrial function and engrafted tumor growth. Guo et al. now find that deleting autophagy-related-7 (atg7) caused K-rasG12D-driven tumors to accumulate dysfunctional mitochondria and induce proliferative arrest, reducing tumor burden. atg7 loss altered tumor fate from adenomas and carcinomas to oncocytomas. Lipid accumulation occurred in atg7-deficient tumors only when p53 was deleted. atg7 deficiency reduced fatty acid oxidation (FAO) and increased sensitivity to FAO inhibition, indicating that with p53 loss, Ras-driven tumors require autophagy for mitochondrial function and lipid catabolism.
Keywords: autophagy, K-ras, NSCLC, metabolism, mitochondria, oncocytoma, p53, fatty acid oxidation
Abstract
Macroautophagy (autophagy hereafter) degrades and recycles proteins and organelles to support metabolism and survival in starvation. Oncogenic Ras up-regulates autophagy, and Ras-transformed cell lines require autophagy for mitochondrial function, stress survival, and engrafted tumor growth. Here, the essential autophagy gene autophagy-related-7 (atg7) was deleted concurrently with K-rasG12D activation in mouse models for non-small-cell lung cancer (NSCLC). atg7-deficient tumors accumulated dysfunctional mitochondria and prematurely induced p53 and proliferative arrest, which reduced tumor burden that was partly relieved by p53 deletion. atg7 loss altered tumor fate from adenomas and carcinomas to oncocytomas—rare, predominantly benign tumors characterized by the accumulation of defective mitochondria. Surprisingly, lipid accumulation occurred in atg7-deficient tumors only when p53 was deleted. atg7- and p53-deficient tumor-derived cell lines (TDCLs) had compromised starvation survival and formed lipidic cysts instead of tumors, suggesting defective utilization of lipid stores. atg7 deficiency reduced fatty acid oxidation (FAO) and increased sensitivity to FAO inhibition, indicating that with p53 loss, Ras-driven tumors require autophagy for mitochondrial function and lipid catabolism. Thus, autophagy is required for carcinoma fate, and autophagy defects may be a molecular basis for the occurrence of oncocytomas. Moreover, cancers require autophagy for distinct roles in metabolism that are oncogene- and tumor suppressor gene-specific.
Autophagy captures, degrades, and recycles intracellular components in lysosomes to maintain protein and organelle quality and provide metabolic and synthetic substrates for cell growth and homeostasis (Kuma et al. 2004; Suzuki et al. 2011). Autophagy cargo includes aggregated proteins and organelles, particularly mitochondria, the accumulation of which in autophagy-deficient cells and tissues is toxic. Thus, autophagy is critical for homeostasis due to important roles in metabolism, organelle function, and proteotoxic waste removal.
Essential autophagy genes were first identified in yeast by virtue of their requirement for survival to nitrogen starvation (Tsukada and Ohsumi 1993; Suzuki et al. 2011). Starvation survival provided by autophagy-mediated cellular self-catabolism and recycling is a function conserved from yeast to mammals. Analogous to yeast, basal autophagy levels are low but are robustly induced by starvation in mice, and those deficient for the essential autophagy gene autophagy-related-5 (atg5) fail to survive the neonatal starvation period, and their tissues display signs of metabolic impairment (Kuma et al. 2004). Autophagy is clearly essential for normal cells and tissues by conferring metabolic stress tolerance during interruptions in nutrient availability.
Normal cells require autophagy to eliminate damaged mitochondria and other dysfunctional or superfluous organelles, which maintains the pool of functioning organelles, contributes substrates to metabolic pathways, and limits sources of toxic reactive oxygen species (ROS) such as damaged mitochondria. Autophagy also degrades aggregated proteins, particularly the autophagy cargo receptor and substrate p62/SQSTM1. The combination of p62 accumulation and aggregation is toxic, promotes oxidative stress, activates the DNA damage response, and causes tissue damage, cell death, and inflammation. Damage induced by p62 accumulation is particularly severe with autophagy ablation in liver and pancreas (Mathew et al. 2009; Komatsu et al. 2010; Li et al. 2013). Autophagy defects also cause neurodegeneration and muscle damage associated with defective protein and metabolic homeostasis, although the mechanisms responsible are specifically unknown (Itakura and Mizushima 2011).
Given the broad functional role of autophagy in protein and organelle quality control and metabolic homeostasis, it is not surprising that the role for autophagy in cancer appears context-dependent (White 2012). On the one hand, in settings in which autophagy defects promote chronic oxidative stress, tissue damage, and inflammation, the resulting genetic instability can promote cancer initiation in mouse models (Karantza-Wadsworth et al. 2007; Mathew et al. 2007; Takamura et al. 2011; White 2012), and in this context, autophagy can be tumor-suppressive. On the other hand, autophagy can promote tumor cell stress tolerance and survival, and in this context, autophagy can be tumor-promoting (White 2012). For example, tumor cells activate autophagy in hypoxic tumor regions where it is required for tumor cell survival, suggesting that autophagy inhibition may be an approach to sensitize tumor cells to metabolic stress (Degenhardt et al. 2006; Mathew et al. 2007; Rabinowitz and White 2010; Amaravadi et al. 2011; White 2012). Exploitation of autophagy modulation for either cancer prevention or therapy requires a specific understanding of the role of autophagy in the context of the microenvironment, therapeutic stress, and differing tissue physiology and upon the occurrence of distinct oncogenic events.
One oncogenic event that creates autophagy dependence is activation of Ras oncogenes. Autophagy is up-regulated in cancer cell lines with activating mutations in Ras and is required for growth, stress tolerance, and tumorigenesis (Lock et al. 2010; Guo et al. 2011; Yang et al. 2011). Autophagy impairment in these settings causes the accumulation of defective mitochondria, depletes key tricarboxylic acid cycle metabolites, suppresses mitochondrial respiration, and decreases energy charge. Autophagy may both maintain the pool of mitochondria and provide metabolic and biosynthetic substrates necessary for tumorigenesis (Rabinowitz and White 2010; Guo et al. 2011; Yang et al. 2011; White 2012). Ras-driven cancers are more autophagy-dependent than normal cells for survival to nutrient starvation, perhaps because Ras renders mitochondrial health particularly dependent on autophagy. Ras-driven cancers may thereby be susceptible to autophagy inhibition therapy (Kimmelman 2011; White 2012).
To examine the role of autophagy in Ras-driven cancers in a physiological setting, we used genetically engineered mouse models (GEMMs) of non-small-cell lung cancer (NSCLC). An oncogenic allele of Ras was spontaneously activated in the lung, and tumors arose and progressed in the context of a functional immune system. atg7 was either intact or deleted in tumor cells to assess its functional contribution to tumor initiation and progression from hyperplasia to adenomas to adenocarcinomas. Mice were also engineered to additionally delete the p53 tumor suppressor gene in tumor cells, which alters metabolism (Cheung and Vousden 2010; Goldstein and Rotter 2012) and accelerates progression to adenocarcinoma (Jackson et al. 2005), potentially influencing autophagy.
Here we report that atg7 deficiency in tumors arising in a GEMM for K-rasG12D-driven NSCLC accelerated induction of p53, suppressed proliferation, and markedly blunted tumor growth. Deficiency in atg7 in tumors caused tumor cells to accumulate defective mitochondria, and progression of tumors was diverted from adenomas and carcinomas to more benign oncocytomas, indicating that the functional status of autophagy dictates tumor fate. Despite the reduced tumor burden caused by atg7 deficiency, overall mouse survival was not increased because mice bearing atg7-null and deteriorating tumors died from desquamative interstitial pneumonia instead of cancer. While compound deletion of p53 partially relieved suppression of tumor cell proliferation with atg7 loss, tumors still incurred a proliferative block, accumulated defective mitochondria, and progressed to oncocytomas, which increased overall survival. Without p53, atg7 deficiency caused tumor cells to accumulate lipids, attributed to defective mitochondrial fatty acid oxidation (FAO). This suggests that mitochondrial function maintained by atg7 is critical for the metabolism and growth of Ras-driven NSCLC.
Results
Autophagy deficiency reduces K-ras-driven lung tumor burden
To test whether autophagy blockade suppressed tumorigenesis in spontaneously arising Ras-driven cancers in an immune competent setting, the LSL-K-rasG12D GEMM for human NSCLC (Jackson et al. 2001) was crossed to atg7flox/flox mice possessing a conditional deficiency in atg7 (Komatsu et al. 2005). Initiation of tumorigenesis by K-rasG12D/+ activation without and with atg7 deletion was achieved by intranasal delivery of adenovirus-Cre to K-rasG12D/+;atg7+/+ and K-rasG12D/+;atg7flox/flox mice. atg7 deletion and loss of Atg7 protein expression only in tumor cells and not in stroma throughout tumorigenesis was confirmed by immunohistochemistry (IHC) at 14 and 42 wk (Supplemental Figs. S1A, S8A), Western blot (Supplemental Fig. S1B), and PCR (data not shown), and tumors were positive for surfactant apoprotein-C (surfactant C protein [SP-C]) and negative for Clara cell antigen (Clara cell secretory protein promoter [CCSP]) (Supplemental Fig. S1C).
Autophagy was low in normal lungs (K-ras+/+;atg7+/+), indicated by a high level of microtubule-associated protein light chain 3-I (LC3-I) relative to the processed, autophagosome-associated LC3-II, whereas K-rasG12D-induced tumors had both increased LC3-II levels and punctation indicative of autophagosome formation (Fig. 1A,B), consistent with up-regulation of basal autophagy by Ras (Lock et al. 2010; Guo et al. 2011; Yang et al. 2011). LC3 punctation was less apparent in tumors at 6 wk but was widespread at 14 wk and increased thereafter, suggesting a time-dependent requirement for autophagy (Fig. 1A; Supplemental Figs. S2A, S8A). atg7 deficiency in tumors prevented LC3-I processing to LC3-II and caused accumulation of LC3-I and autophagy substrate p62 in large aggregates apparent at 6 wk that increased throughout tumorigenesis (Fig. 1A,B; Supplemental Figs. S2A,B, S8A). Abundant autophagosomes in K-rasG12D/+;atg7+/+ but not in K-rasG12D/+;atg7−/− tumors were confirmed by electron microscopy (Fig. 2B). The timing of autophagosome appearance in atg7+/+ tumors and autophagy substrates in atg7−/− tumors suggested that autophagy becomes critical at 6–14 wk and progressively thereafter.
Figure 1.

Atg7 deletion in K-ras-driven lung tumors blocks autophagy and reduces lung tumor size. (A) Representative IHC staining for LC3 and p62 in 14-wk-old tumors. Red arrows in K-rasG12D/+;atg7+/+ tissue point to autophagosomes, and black arrows in K-rasG12D/+;atg7−/− tissue point to LC3 or p62 aggregates. (B) Western blot of LC3-I-to-LC3-II processing and p62 accumulation in tumors at 18 wk after tumor initiation. Numbers identify individual mice. (C) Representative gross lung pathology (n = 5 mice for each time point in K-rasG12D/+ mice). (D) Graph of wet lung weight from C. The error bars represent the SEM; (*) P < 0.05 (t-test). (E) Representative histology (H&E) of lungs and tumor burden at the indicated times (n = 3 mice for each time point in K-rasG12D/+ mice). Full scans of all lobes are shown in Supplemental Figure S3. (F) Quantification of tumor burden from E. The error bar represents the SEM; (*) P < 0.05; (**) P < 0.01 (two-way ANOVA with Bonferroni post-test). (G) K-rasG12D/+;atg7+/+ and K-rasG12D/+;atg7−/− tumor-bearing mice (n = 3 for each time and genotype) were scanned by micro-CT at the indicated times to monitor lung tumor progression. K-ras+/+;atg7+/+ mice provided a normal lung control. The number in each panel represents the mouse identification number. The top panels show three-dimensional reconstruction of mouse lungs. Blue indicates lung airspace. The bottom panels show transverse section of the chest, with increased white areas indicating tumor. (H) Quantification of normal lung volume from G. Each spot represents an individual mouse. Normal lung volume decreased faster in mice with K-rasG12D/+;atg7+/+ tumors compared with those with atg7 deletion. (I) Quantification of Ki67-positive tumor cells. The error bar represents the SEM; (*) P < 0.05; (**) P < 0.01 (two-way ANOVA with Bonferroni post-test). Representative images of IHC are shown in Supplemental Figure S5C.
Figure 2.
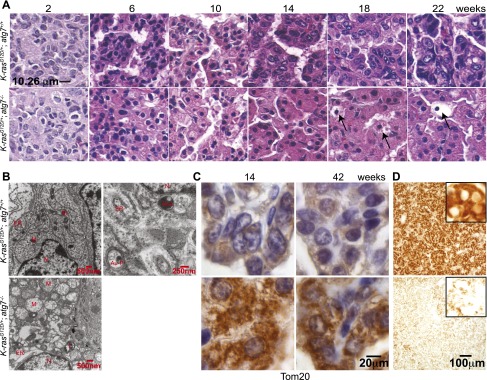
Atg7 deficiency converts adenomas/carcinomas to oncocytomas and causes accumulation of dysfunctional mitochondrial in K-ras-driven tumors. (A) Histology of lung tumors shows progression of K-rasG12D/+;atg7+/+ tumors to adenocarcinomas and of K-rasG12D/+;atg7−/− tumors to oncocytomas. Note the development over time of expansive cytoplasm in oncocytomas and necrotic cells (arrows) (n = 3 mice for each time point and genotype). (B) Representative electron microscope images of tumors at 18 wk. (N) Nuclear; (M) mitochondria; (Au-P) autophagosome; (Au-L) autolysosome; (L) lamellar body; (ER) endoplasmic reticulum. (C) Representative IHC of Tom20 shows accumulation of mitochondria in atg7-deficient tumors. (D) Cytochrome c oxidase activity in tumors at 25 wk after tumor induction.
Sporadic atg7 loss in normal lung (K-ras+/+) did not alter lung histology (Supplemental Fig. S3). Functional autophagy status did not alter K-rasG12D-induced tumorigenesis prior to 10 wk based on gross lung pathology, quantification of tumor numbers, wet lung weight, and histological tumor burden (Fig. 1C–F; Supplemental Figs. S2C, S3). The burden of atg7-deficient tumors from 14 to 18 wk was half that of wild type (Fig. 1E,F; Supplemental Fig. S3). Tumor burden assessed by microcomputed tomography (micro-CT) in individual mice followed over time also demonstrated reduced tumor growth and more lung airspace with atg7 deletion compared with wild type (Fig. 1G,H).
Autophagy deficiency converts adenomas and carcinomas to oncocytomas
Wild-type and atg7-deficient tumors were histologically similar at 2–6 wk, and both developed hyperplasia that progressed to adenoma (Fig. 2A). atg7 wild-type tumors progressed to adenocarcinomas, where natural acquisition of p53 mutations enables progression (Jackson et al. 2001). In contrast, beginning at 6–10 wk, all atg7-deficient adenomas progressed to low-grade, papillary, oncocytic neoplasms or oncocytomas composed of tumor cells with a swollen, grainy eosinophilic cytoplasm (oncocytes) (Fig. 2A). At later times (18–42 wk), many of the atg7-deficient tumors disintegrated, causing tumor atrophy (Fig. 2A; Supplemental Fig. S8A). Reduction of atg7-deficient tumor burden was coincident with reduced proliferation (Ki67) and decreased levels of phospho-MEK1/2 (P-MEK1/2) and P-ERK1/2 and p53 and p21 induction, compared with wild-type tumors (Fig. 1I; Supplemental Figs. S4A–C, S5A–C), suggesting that inhibition of tumor cell proliferation upon atg7 deletion is the main cause of reduced tumor burden. Senescence-associated β-galactosidase-positive cells were similarly observed in both atg7+/+ and atg7−/− tumors (Supplemental Fig. S6), indicating that senescence did not cause atg7−/− tumor proliferation defect or atrophy. Death of atg7-deficient tumor cells was morphologically necrotic without activation of caspase-3 (Fig. 2A, arrows; Supplemental Figs. S6B, S8A, arrows) and was coincident with DNA damage response activation (γ-H2AX-positive) (Supplemental Fig. S7). atg7-deficient tumors accumulated morphologically abnormal mitochondria (Fig. 2B; Supplemental Fig. S8C) and had increased staining for the mitochondrial marker Tom20 (Fig. 2C). Enzymatic activity for cytochrome c oxidase, an essential component of electron transport chain complex IV that is commonly defective in oncocytomas (Gasparre et al. 2011), was decreased in atg7-deficient compared with wild-type tumors (Fig. 2D). Thus, atg7 deficiency in lung tumors causes adenomas and carcinomas to progress to oncocytomas.
Inflammation, not tumor burden, causes death of mice with atg7-deficient tumors
Although atg7 deficiency in K-ras lung tumors reduced tumor burden by half, there was, surprisingly, no overall survival advantage (Fig. 3A). At 42 wk after tumor initiation, the lungs of mice with atg7+/+ tumors were enlarged with increased wet lung weight and tumor burden in comparison with lungs bearing atg7-deficient tumors (Fig. 3B–D). The sustained reduction in tumor burden at 42 wk caused by atg7 deficiency also correlated with further accumulation of LC3, p62, and abnormal mitochondria (Supplemental Fig. S8A,C). Since there was no evidence of metastasis, this suggested that tumor-specific atg7 deficiency caused death by means other than cancer.
Figure 3.

Inflammation is the major cause of death of mice bearing K-rasG12D/+;atg7−/− tumors. (A) Kaplan-Meier survival curve of mice with K-rasG12D/+ tumors that were atg7+/+, atg7+/−, and atg7−/− (P > 0.05, log-rank test). (B) Representative gross lung pathology at 42 wk after tumor initiation (n = 4 mice for K-rasG12D/+;atg7+/+; n = 5 mice for K-rasG12D/+;atg7−/−). (C) Quantification of wet lung weight from B. (*) P < 0.05 (t-test). (D) Quantification of tumor and crystalline macrophage burden in the lungs at 42 wk (n = 4 mice for K-rasG12D/+;atg7+/+; n = 5 mice for K-rasG12D/+;atg7−/−) (t-test). (E) Tumor and lung pathology at 42 wk. The top panels show representative lung lobes with K-rasG12D/+;atg7+/+ tumors or with inflammation in K-rasG12D/+;atg7−/− tumor-bearing lungs. Full scans of all lobes are shown in Supplemental Figure S3. The histology below shows typical adenocarcinoma (atg7+/+) or oncocytoma (atg7−/−) (arrow indicates necrotic cell). Below are representative IHC stainings for the macrophage marker CD68. The bottom panels show representative tumor with adjacent air sacs in mice with K-rasG12D/+;atg7+/+ tumors or inflammation in K-rasG12D/+;atg7−/− tumor-bearing lungs. The inset panels at right show representative histology and electron microscopy of crystalline macrophages in the lungs of mice with K-rasG12D/+;atg7−/− tumors. (F) The table shows the percentage of desquamative interstitial pneumonia in K-rasG12D/+;atg7+/+ and K-rasG12D/+;atg7−/− mice at the indicated times. (G) Western blot of YM1 in normal lung tissue and tumors. (H) Comparison analysis of the gene expression data showing pathways and molecular functions most significantly enriched in K-rasG12D/+ lung tumors compared with normal lung tissue (P = 0.05) (yellow line) by ingenuity pathway analysis. (I) Heat map diagram of differential gene expression in atg7+/+ and atg7−/− tumors compared with normal lung tissue. The bar represents log2-transformed fold change in signal intensities. (J) Quantification of cytokine expression levels in mouse lungs. Mouse cytokine array panels are shown in Supplemental Figure S9. (**) P < 0.01; (***) P < 0.001 (t-test).
Histological analysis of lungs at 42 wk (Fig. 3E; Supplemental Fig. S3) revealed large atg7+/+ tumors surrounded by airspace, whereas lungs bearing smaller atg7−/− tumors lacked airspace due to fulminant crystalline macrophage and lymphocyte infiltration, typical of murine pneumonia (Hoenerhoff et al. 2006), resembling desquamative interstitial pneumonia. Between 18 and 42 wk, crystalline intra-alveolar macrophage infiltration was observed in 63% of mice with atg7−/− tumors compared with 12% of mice with atg7+/+ tumors (Fig. 3F). At 42 wk, the lung area occupied by macrophages/tumor was 7%/13% in mice with atg7−/− tumors, compared with 0.6%/28% in mice with atg7+/+ tumors (Fig. 3D–F). Macrophages expressed Atg7, indicating that their accumulation was not due to autophagy defect (data not shown). Macrophages were CD68-positive and contained crystals of YM1 (Chi3L3) protein (Fig. 3E,G; Supplemental Fig. S8B), common features of murine infection-induced pneumonia or trauma-induced inflammation (Hoenerhoff et al. 2006). YM1 is a chitinatase-like protein secreted by activated macrophages during infection and traumatic injury, and its presence is associated with tissue damage, repair, and remodeling (Lee et al. 2011). YM1 was detected only in atg7−/− tumor tissue at 18 wk and thereafter, subsequent to manifestation of autophagy defects in tumor cells (Fig. 3G).
Functional enrichment analysis of the top 1000 most differentially expressed genes in tumor-bearing lung tissue compared with normal lung tissue at 18 wk revealed that molecular functions related to inflammatory responses were highly enriched by tumor-specific atg7 deletion (Fig. 3H). Half of the differentially expressed genes increased in atg7-deficient compared with wild-type tumor-bearing lungs were inflammatory response genes (Fig. 3I). Consistent with macrophage infiltration, cytokine array analysis of pooled lung tissue (Fig. 3J; Supplemental Fig. S9) revealed that macrophage colony-stimulating factor (M-CSF) and triggering receptor expressed on myeloid cell-1 (TREM-1) were increased by tumor-specific atg7−/− deletion. Thus, pneumonia rather than tumor burden caused mortality of mice bearing K-rasG12D/+;atg7−/− tumors.
Autophagy deficiency reduces lung tumor burden and extends life span in the context of p53 deletion
To test whether atg7 deficiency suppressed tumorigenesis in spontaneously arising oncogenic Ras and p53-deficient lung adenocarcinomas, the p53flox/flox;LSL-K-rasG12D/+ GEMM for human NSCLC (Johnson et al. 2001; Jackson et al. 2005) was crossed to atg7flox/flox mice possessing a conditional deficiency in atg7 (Komatsu et al. 2005). Intranasal delivery of adenovirus-Cre induced tumorigenesis by K-rasG12D activation and p53 deletion without and with atg7 deletion in p53flox/flox;K-rasG12D/+;atg7+/+ and p53flox/flox;K-rasG12D/+;atg7flox/flox mice. atg7 deletion and loss of Atg7 protein expression only in atg7−/− tumor cells was confirmed by IHC at 18 wk (Fig. 4A) and PCR (data not shown).
Figure 4.

atg7 deficiency suppresses p53−/−-K-rasG12D-driven NSCLC. (A) Representative IHC for Atg7, LC3, and p62 in tumors at 14 wk after tumor initiation. Red arrows are autophagosomes, and black arrows are LC3 or p62 aggregates. (B) Western blot of LC3-I-to-LC3-II processing and p62 accumulation in tumors at 14 wk after tumor initiation. (C) Representative lung images from tumor-bearing mice (n = 3 mice for each time point and genotype) by micro-CT scanning. A wild-type (p53+/+;K-ras+/+;atg7+/+) mouse provided a normal lung reference. The top panels show three-dimensional reconstruction of mouse lungs, with blue indicating airspace. The bottom panels show transverse section of the chest, with increased white areas indicating tumor. (D) Quantification of normal lung volume from micro-CT-scanned mice from C. Each spot represents an individual mouse. The error bar represents the SEM; (*) P < 0.05 (t-test). (E) Representative histology (H&E) of lungs and tumor burden at the indicated times (n = 3 mice for each time and genotype). Full scans of all lobes are shown in Supplemental Figure S10. (F) Quantification of tumor burden from E. The error bar represents the SEM; (*) P < 0.05; (**) P < 0.01 (two-way ANOVA with Bonferroni post-test). (G) Histology of lung tumors shows a representative adenocarcinoma in p53−/−;K-rasG12D/+;atg7+/+ tumors and oncocytoma in p53−/−;K-rasG12D/+;atg7−/− tumors. IHC of Tom20 shows mitochondrial accumulation in atg7-deficient tumors compared with wild type at 16.5 wk after tumor initiation (n = 3 mice for each genotype). The complete time course is shown in Supplemental Figure S11. (H) Representative IHC for Ki67 in tumors at 18 wk after tumor induction, with quantification below. (I) Kaplan-Meier survival curve of mice with p53−/−;K-rasG12D/+ tumors that were atg7+/+ or atg7−/− (P < 0.01, log-rank [Mantel-Cox] test).
Concurrent deletion of p53 in K-rasG12D-induced tumors increased LC3 puncta formation (Fig. 4A), suggesting that loss of p53 further increased autophagy compared with p53+/+ tumors. Autophagy was functionally blocked in tumors with atg7 deletion (p53−/−;K-rasG12D/+;atg7−/−), indicated by the accumulation of LC3-I relative to LC3-II compared with normal lungs (p53+/+;K-ras+/+;atg7+/+) and atg7 wild-type tumors (p53−/−;K-rasG12D/+;atg7+/+) (Fig. 4B). In addition to high LC3-II levels, p53−/−;K-rasG12D/+;atg7+/+-induced tumors had low levels of the autophagy substrate p62 (Fig. 4B), consistent with active autophagy. atg7-deficient tumors accumulated LC3-I and p62 in large aggregates similarly to tumors without p53 deletion (Fig. 4A,B). Thus, autophagy is active and important for protein quality control in NSCLC independent of p53 status.
We then assessed the impact of atg7 deletion on p53−/− and K-rasG12D-mediated tumorigenesis. In comparison with atg7 wild-type tumors, atg7 deletion reduced tumor burden measured by micro-CT (Fig. 4C,D). There was no difference in tumor number, but when the tumor area was quantified in lung cross-sections, atg7 deletion reduced tumor burden compared with atg7 wild-type tumors (Fig. 4E,F; Supplemental Fig. S10). Through 16.5 wk, atg7 wild-type tumors were healthy adenocarcinomas, whereas those with atg7 deleted progressed to oncocytomas characterized by an expansive cytoplasm with increased Tom20 staining, indicating mitochondrial accumulation (Fig. 4G; Supplemental Fig. S11). Although engineered p53 loss greatly accelerated K-rasG12D-mediated adenocarcinoma development, oncocytomas still developed with atg7 deletion (Fig. 4G) similarly to when p53 is initially intact (Fig. 2A). This also suggests that the accelerated and sustained induction of p53 in atg7-deficient tumors (Supplemental Fig. S5A) produces the proliferative block when p53 is intact.
To determine the cause of reduced tumor burden, proliferation (Ki67) and apoptosis (active caspase-3) were assessed. While there was no difference in apoptosis (data not shown), tumors with atg7 deletion had significantly fewer proliferating cells compared with those with atg7 (Fig. 4H). Thus, the primary reason that atg7 loss reduced tumor burden is decreased proliferation, and although p53 induction can provide an initial proliferative block, a second proliferative block is engaged by atg7 deficiency in tumors without p53.
In contrast to tumors with p53 initially intact where mouse survival was not impacted by atg7 deletion, in p53-deleted tumors, death was due to tumor burden even with atg7 loss. Consistent with slower tumor growth, deletion of atg7 increased overall mouse survival (P = 0.0035) (Fig. 4I). A possible reason for different outcomes is that, with p53 intact, atg7 loss causes extensive oncocyte death and inflammation and that this is prevented by p53 deletion and defective cell death. Nonetheless, irrespective of p53 status, atg7 deficiency impairs Ras-driven tumor growth.
Autophagy deficiency causes lipid accumulation in p53-deficient tumors
To confirm the accumulation of abnormal mitochondria characteristic of oncocytomas in atg7-deleted tumors, electron microscopy was performed. In addition to morphologically abnormal mitochondria, there was the striking accumulation of lipid droplets in atg7-deficient tumor cells (Fig. 5A). Oil red O (Fig. 5B,C) or Bodipy staining (Supplemental Fig. S12) for neutral lipids confirmed lipid droplet accumulation specifically in tumors with atg7 deletion. Lipid accumulation was dependent on p53 deficiency, since p53+/+;K-rasG12D tumors had scant lipid accumulation that was only slightly elevated by atg7 deficiency (Fig. 5C). Thus, independent of p53 status, atg7 deficiency in NSCLC causes accumulation of abnormal mitochondria and diverts progression of carcinomas to oncocytomas. In contrast, p53 deletion at the onset of tumorigenesis causes atg7-deficient tumors to specifically accumulate neutral lipids. This suggests that autophagy is essential for lipid homeostasis specifically with p53 deficiency in K-rasG12D-driven NSCLC. Since p53 deficiency increases glucose uptake, directs glucose to lipid storage, and suppresses FAO (Ide et al. 2009; Cheung and Vousden 2010; Goldstein and Rotter 2012), this may render tumors prone to lipid accumulation if lipid synthesis is increased or if FAO is inhibited further.
Figure 5.

atg7 deficiency promotes tumor lipid accumulation. (A) Representative electron microscope images of tumors showing lipid droplet accumulation with atg7 deletion at 18 wk after tumor initiation. (Nu) Nucleus; (M) mitochondria; (L) lamellar body; (G) glycogen; (LD) lipid droplet. (B) Representative low-magnification images of Oil red O staining of representative lung lobes (frozen sections) at 18 wk after tumor induction (n = 2 mice for each genotype). (C) Representative high-magnification images of Oil red O staining of tumor tissue at the indicated times (n = 2 mice for each genotype).
atg7-deficient tumor-derived cell lines (TDCLs) have defective lipid homeostasis and are sensitive to starvation
To address the mechanism by which autophagy mediates lipid homeostasis, atg7+/+ and atg7−/− TDCLs were generated from atg7 wild-type or atg7-deficient p53−/−;K-rasG12D tumors. atg7-deficient TDCLs displayed accumulation of LC3-I characteristic of autophagy defect (Fig. 6A) without altering growth in complete medium (Supplemental Fig. S13). In contrast to TDCLs with atg7, starvation (Hank's Balanced Salt Solution, HBSS) of atg7-deficient TDCLs prevented LC3-I-to-LC3-II conversion, caused p62 accumulation, activated apoptosis (Fig. 6B), and prevented clonogenic survival (Fig. 6C; Supplemental Fig. S14A), which is consistent with our previous findings in Ras-driven cancer cells with deletion of either atg5 or atg7 (Guo et al. 2011). In addition, atg7-deficient TDCLs were more sensitive to ischemia-induced cell death (Supplemental Fig. S14B). Thus, similar to engineered mouse and human cancer cell lines and yeast (Guo et al. 2011; Suzuki et al. 2011), TDCLs require atg7 to survive metabolic challenge.
Figure 6.
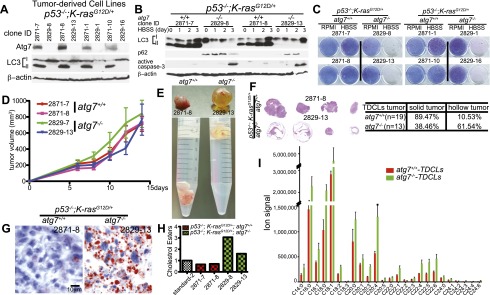
atg7-deficient TDCLs accumulate lipids and are sensitized to starvation-induced death. (A) Western blot for Atg7 and LC3 in TDCLs. (B) Western blot for LC3, p62, and active caspase-3 during starvation (HBSS) of TDCLs at the indicated times. (C) Clonogenic survival assay of TDCLs following 3 d of starvation (HBSS) and 4 d of recovery in normal medium (RPMI). (D) Growth of TDCL tumors in nude mice (n = 6 tumors for each TDCL). (E) Representative gross pathology of tumors derived from TDCLs from D. (F, left) Histology (H&E) of representative tumors from atg7 wild-type and atg7-deficient TDCL tumors showing hollow centers with atg7 deletion. (Right) Quantification of hollow tumor formation. (G) Oil red O staining of TDCL tumors (n = 4 tumors for each genotype). (H) Quantitation of cholesterol esters by thin-layer chromatography (TLC) (Supplemental Fig. S16A). (I) The graph shows the increased total pool size of FAs in atg7-deficient TDCLs compared with wild-type TDCLs under steady status. The error bar represents ±SD; P < 0.05 (t-test).
To determine whether the metabolic defect of atg7-deficient TDCLs manifested in tumorigenesis, growth of TDCLs in nude mice was assessed. Although the overall size of subcutaneous masses was not altered by atg7 deficiency (Fig. 6D), atg7 wild-type TDCLs formed solid tumors, whereas those deficient for atg7 formed hollow, fluid-filled cysts (Fig. 6E,F). Extensive lipid accumulation was apparent in atg7-deficient TDCL tumors by Oil red O staining (Fig. 6G) and the buoyancy of tissue in aqueous medium (Fig. 6E). As in the spontaneously arising tumors, those with atg7 were healthy, solid adenocarcinomas, and those without atg7 were largely cysts (Fig. 6F) composed of stroma with some lipid-filled tumor cells (Fig. 6G; Supplemental Fig. S15). As in the primary tumors, atg7-deficient TDCLs had high basal lipid levels, specifically cholesterol esters (Fig. 6H; Supplemental Fig. S16A,B), the levels of which were maintained in starvation (Supplemental Fig. S16C). The total pool sizes of saponified FAs as measured by mass spectrometry were significantly increased in atg7-deficient compared with atg7 wild-type TDCLs (Fig. 6I). Together, this suggests that atg7 is essential for p53−/−;K-rasG12D NSCLCs to tolerate metabolic stress and efficiently form tumors and that this may be due to a requirement for autophagy for lipid catabolism. When lipid catabolism is impaired, tumor cells may fail to survive interruptions in nutrient availability despite the presence of abundant lipid stores.
atg7 deficiency causes defective mitochondrial FAO
Mitochondria are predominantly responsible for FAO, and autophagy defects cause accumulation of defective mitochondria. To address whether lipid accumulation with atg7 deficiency was associated with defective mitochondria, mitochondrial mass (MM) and membrane potential (MMP) were assessed. atg7-deficient TDCLs had increased MM (Fig. 7A; Supplemental Fig. S17A), consistent with their increased Tom20 staining (Fig. 4G), but decreased MMP (Fig. 7A; Supplemental Fig. S17B), suggesting that loss of atg7 caused accumulation of defective mitochondria.
Figure 7.

Autophagy is required for FAO. (A) Total MM (top) and MMP (bottom) in TDCLs under normal conditions (atg7+/+: n = 10; atg7−/−: n = 9 independent clones). (**) P < 0.01; (***) P < 0.001 (t-test). (B) OCR of TDCLs (two independent clones for each genotype) under normal conditions without (left) or with (right) FCCP challenge. The error bar represents ±SD; P > 0.05 (t-test). (C) OCR of TDCLs (two independent clones from each genotype) under normal or starvation (HBSS for 4 h) following addition of FCCP (0.3 μM) to establish maximum respiratory capacity. The error bar represents ±SD; (*) P < 0.05 (t-test). (D) Clonogenic survival assay of TDCLs with glutamine addition (1 mM) under 3 d of starvation (HBSS) and 3 d of recovery in normal medium (RPMI). (E, top) OCR trace of TDCLs in response to palmitate (400 μM) showing relative percentage change upon palmitate addition. (Bottom) OCR of TDCLs from the top panel at 71 min. The error bar represents ±SD; (*) P < 0.05 (t-test). (F) The graph shows autophagy-dependent changes in intracellular metabolite levels (52 metabolites; log2-transformed ratios of ion signals) in two atg7-deficient compared with two atg7 wild-type TDCLs in normal conditions. (G) The graphs show representative increased amino acid levels in two atg7-deficient compared with two atg7 wild-type TDCLs at each indicated starvation (HBSS) time. Additional amino acids are shown in Supplemental Figure S19. (H) Clonogenic survival assay for TDCLs following starvation (HBSS for 15 h and 25 h) in the presence of the CPT1 inhibitor etomoxir (50 μm) with and without pyruvate (1 mM). The complete assay is shown in Supplemental Figure S20.
To address the mechanism by which autophagy prevented lipid accumulation and promoted tumorigenesis, we tested the hypothesis that autophagy is required to maintain functional mitochondria for FAO. The oxygen consumption rate (OCR) of atg7 wild-type and atg7-deficient TDCLs was measured. Although atg7 deficiency did not significantly alter the basal OCR under normal growth conditions (Fig. 7B), the reserve capacity (as measured by addition of the uncoupler FCCP) after brief (4 h) starvation was markedly reduced (Fig. 7C). A major consequence of the brief starvation is removal of glutamine, which is normally a major mitochondrial fuel supply. Apparently, autophagy deficiency augments the intrinsic propensity of Ras-driven tumors to depend heavily on glutamine to power oxidative metabolism. Indeed, glutamine supplementation rescued survival of the atg7-deficient TDCL in starvation (Fig. 7D).
To test the potential for autophagy-defective cells to engage in FAO, TDCLs were briefly starved and provided the FAO substrate palmitate, and OCR was measured. While there was little difference in OCR under normal growth conditions, atg7 deficiency significantly reduced palmitate-induced OCR, which is a measure of mitochondrial FAO (Fig. 7E). The inability of the autophagy-defective cells to oxidize free palmitate when provided in the medium rules out the primary defect being accessing lipid stores. Instead, these cells are fundamentally defective in FAO, and this defective mitochondrial function presumably explains the accumulation of lipids in atg7-deficient TDCLs and tumors.
To broadly assess the metabolic consequences to atg7 deficiency, we measured pool sizes of 159 intracellular metabolites in normal and nitrogen starvation conditions (i.e., removal of all media amino acids) in TDCLs (Supplemental Table S1). Prior to starvation, one-third of the metabolites showed autophagy-dependent changes that were statistically significant (false discovery rate [FDR] < 0.05) and consistent across both cell line pairs. Surprisingly, more metabolites were increased than decreased in the autophagy-defective cells, with a majority of the increased compounds being amino acids or related compounds (acetylated amino acids and amino acid catabolites) (Fig. 7F). This suggests an autophagy-induced defect in amino acid catabolism. Like the defect in lipid catabolism, the defective amino acid catabolism is presumably due to mitochondrial deficiency.
Following starvation, there were strong changes in metabolite levels that were largely consistent between the wild-type and autophagy-null cells (Supplemental Fig. S18), with the elevated levels of amino acids and related compounds in the autophagy-null cells persisting during starvation (Fig. 7G; Supplemental Fig. S19). In addition, glycolytic intermediates and NADH were higher in the autophagy-defective than wild-type cells after starvation (Supplemental Figs. S18, S19). The elevated levels of glycolytic intermediates may also result from defective mitochondria but through a different mechanism: When oxidative phosphorylation is decreased, glycolysis increases to maintain ATP levels, which are similar in autophagy wild-type and autophagy-defective cells.
To test whether atg7 function was required for transport of fatty acids (FAs) into mitochondria, TDCLs were assessed for sensitivity to etomoxir, which inhibits FAO by blocking CPT1. The survival of starved, atg7 wild-type TDCLs was somewhat decreased by etomoxir, and this decrease was rescued by the alternative mitochondrial substrate pyruvate. In contrast, atg7-deficient TDCLs were ablated by etomoxir even in the presence of pyruvate (Fig. 7H; Supplemental Fig. S20). Like FAs and unlike glutamine, entry of pyruvate into the TCA cycle is gated by antecedent oxidative steps in the mitochondrion to generate acetyl-CoA. These steps may be blocked in autophagy-defective cells. Thus, atg7 deficiency causes defective mitochondrial function, which in turn leads to greater sensitivity to further inhibition of FAO. This suggests combination therapy involving dual autophagy and FAO inhibition as a therapeutic strategy for p53-deficient cancers.
Discussion
The two models of spontaneous K-rasG12D-driven NSCLC analyzed here—one with and the other without p53—demonstrated that atg7 is critical for sustained tumor cell proliferation and progression of lung tumors to adenomas and carcinomas. Common to both models, atg7 deletion blocked tumor growth and caused tumors to accumulate defective mitochondria and progress to more benign oncocytomas instead of adenomas and carcinomas. These findings support the overall concept that autophagy facilitates the growth and metabolism of Ras-driven cancers (Lock et al. 2010; Guo et al. 2011; Yang et al. 2011). Consistent with previous work, evidence suggests that the mechanism behind the requirement for autophagy in tumorigenesis is that it is needed for proliferation and to maintain mitochondrial function and cellular metabolism necessary for survival to stress and starvation (Guo et al. 2011; Yang et al. 2011). What is new here is the revelation that autophagy-deficient tumors that accumulate defective mitochondria are oncocytomas and that the metabolic requirement for autophagy is influenced by the functional status of p53.
Oncocytomas are rare, predominantly benign neoplasms that arise in any organ but more often in kidney, salivary and parathyroid glands, and thyroid and are characterized by cells that accumulate respiration-defective mitochondria (Gasparre et al. 2011). Neither the origin of oncocytomas nor the functional significance of their mitochondrial impairment is known. It has been hypothesized that mitochondrial respiration defects trigger compensatory mitochondrial biogenesis that drives the accumulation of defective mitochondria. Our finding that atg7 deficiency diverts adenomas and carcinomas to oncocytomas instead suggests that loss of autophagy and mitochondrial quality control can produce oncocytomas and that mitochondrial function is required for efficient progression to carcinoma. Thus, autophagy dictates tumor cell fate, and this suggests a possible role for autophagy inhibitors in the treatment of NSCLC.
When p53 is intact during K-rasG12D tumor development, atg7 deletion causes premature p53 induction and a proliferation block that is the primary cause of reduced tumor burden. Alleviating this p53 barrier by deletion of p53 temporarily restored proliferation and growth of atg7-deficient tumors (Supplemental Figs. S3, S10). The mechanism by which p53 is induced in atg7-deficient tumors is not clear, but autophagy deficiency causes ROS production, a known activator of p53 and the DNA damage response (Supplemental Fig. S7; Karantza-Wadsworth et al. 2007; Mathew et al. 2007). Ultimately, however, atg7 deficiency even in the absence of p53 results in proliferative arrest that correlates with profound metabolic defects. Thus, autophagy loss can promote p53-dependent growth arrest that suppresses tumor growth, but ultimately, p53-independent growth arrest mechanisms likely tied to compromised metabolism are engaged.
Uniquely to the p53-intact setting, autophagy defects manifest early, corresponding with tumorigenesis impairment and oncocytoma development, which were followed by induction of inflammation. This suggests that tumor cell dysfunction and eventual tumor atrophy due to autophagy deficiency preceded a secondary inflammatory response. Deficiency in essential autophagy genes promotes inflammation in the liver (Komatsu et al. 2007, 2010; Mathew et al. 2009), gastrointestinal tract (Cadwell et al. 2008; Li et al. 2013), and tumors (Degenhardt et al. 2006) but not in lungs (Supplemental Fig. S3; Takamura et al. 2011). This indicates that inflammation is a response to the presence of autophagy-deficient tumor cells. Activation of K-rasG12D in the lung by CCSP-Cre increases the proportion of lung epithelial cells with activated K-ras, elevates tumor cell cytokine production, and induces early death due to immune infiltration (Ji et al. 2006) as described here in tumors with atg7 deletion. Autophagy may suppress a tumor cell-intrinsic inflammatory response by maintaining mitochondrial metabolism and preventing necrotic cell death induction (Degenhardt et al. 2006). In support of cell death being the trigger of pneumonia in atg7-deficient tumors, there was less cell death in the p53-deficient setting, where pneumonia did not occur.
Without p53, atg7 deletion in tumors still blocked tumor growth. Ultimately, a metabolic crisis and inability to tolerate stress caused by loss of autophagy may block proliferation even in the absence of p53. The p53-independent block to tumor growth caused by loss of either atg5 or atg7 was also seen in Ras-transformed cancer cell lines (Guo et al. 2011; Yang et al. 2011).
Uniquely to the p53-deficient setting, atg7 was required for lipid homeostasis. atg7-deficient TDCLs display lipid accumulation and defective mitochondrial respiration when provided a lipid substrate, acute sensitivity to inhibition of FAO, and exquisite sensitivity to metabolic stress. p53 loss alters metabolism, promoting lipid storage (Zhu and Prives 2009; Cheung and Vousden 2010; Chesney and Telang 2013). When p53 loss and autophagy deficiency co-occur at the onset of tumorigenesis, this combination of enhanced lipid storage and impaired degradation result in fat-filled tumors. Providing extracellular glutamine can rescue survival of atg7-deficient TDCLs, suggesting that in addition to preserving FAO, autophagy may supply glutamine from protein degradation to maintain metabolism. This is consistent with the requirement for autophagy to maintain the pool of functional mitochondria for FAO and possibly to also supply metabolic substrates to mitochondria, both of which may be important for maintaining tumor cell metabolism. More generally, autophagy inhibition tends to impair mitochondrial metabolism and thus tumor growth, but the metabolic outcome depends on the specific oncogene and tumor suppressor gene involved and the timing of their mutation.
Materials and methods
Mice
Animal studies followed protocols approved by the Institutional Animal Care and Use Committee. LSL-K-rasG12D mice (Jackson Laboratory), Tp53flox/flox mice (Jackson Laboratory), and atg7flox/flox mice (provided by Dr. M. Komatsu) were cross-bred to generate K-rasG12D/+;atg7flox/flox, K-rasG12D/+;atg7+/+, p53flox/flox;K-rasG12D/+;atg7flox/flox, or p53flox/flox;K-rasG12D/+;atg7+/+ mice. Mice were infected intranasally with recombinant, replication-deficient adenovirus expressing Cre recombinase (University of Iowa Adenoviral Core) at 4 × 107 plaque-forming units (pfu) per mouse at 6–8 wk of age.
Tumor/macrophage burden quantification
H&E-stained lung specimens were imaged under a 20× objective on a Trestle/Zeiss MedMicro imaging system. Matlab was used to build the segmentation protocol from a series of image processing operators. Tumor masks and whole-tissue masks were computed from low-resolution image maps, which were extracted from the whole slide images. The segmented masks were used to generate the area ratios for tumor and macrophage burden.
Gene expression analysis
Lung tumor samples from mice at 18 wk post-Cre and comparator normal lung tissue (K-ras+/+;atg7+/+) were snap-frozen for RNA extraction. Extracted RNAs were processed, labeled, and hybridized to the GeneChip Mouse Genome 430A 2.0 array (Affymetrix) and analyzed as described previously (Mathew et al. 2009).
Micro-CT and lung volume quantification
K-rasG12D/+;atg7+/+, K-rasG12D/+;atg7−/−, and K-ras+/+;Atg7+/+ mice (n = 3 mice for each genotype) at the indicated times were isofluorane-anesthetized. Respiratory-gated and low-resolution CT images of maximally ventilated mice were obtained using a Siemens Inveon PET/CT. Using INVEON Research Workplace software, reconstructed image data were processed through a Gaussian filter, tissue determined to be pulmonary was segmented, and its volume was calculated. Three-dimensional reconstruction pulmonary images and transverse sections of the chest were generated using Osirix software.
Electron microscopy
Lung tumors from K-rasG12D/+;atg7+/+ and K-rasG12D/+;atg7−/− mice were fixed in 0.1 M cacodylate buffer (pH 7.4) with 2.5% glutaraldehyde, 4% paraformaldehyde, and 8 M CaCl2. Samples were processed as described previously (Guo et al. 2011), and images were captured with an AMT XR41 digital camera at 80 kV on a JEOL 1200EX transmission electron microscope.
Histology and IHC
Five mice from each K-rasG12D/+-bearing mouse group (atg7+/+ or atg7−/−) were sacrificed at 2, 6, 10, 14, 18, 22, and 42 wk via cervical dislocation, and lungs were fixed in 10% buffered formalin solution (Formalde-Fresh, Fisher Scientific) for 4 h and then transferred to 70% ethanol for paraffin-embedded sections. For preparation of frozen sections, lungs were fixed in 10% buffered formalin solution for 4 h, then transferred to 15% sucrose for 2 h, and finally transferred to 30% sucrose overnight before processing.
For IHC, paraffin-embedded sections were stained with antibodies against Atg7 (Sigma, catalog no. A2856), LC3 (Nano Tools, catalog no. LC3-5F10), p62 (Enzo Life Sciences, catalog no. PW9860-0100), Tom20 (Santa Cruz Biotechnology, catalog no. sc11415), p53 (Leica, product code NCL-p53-CM5P), p21 (BD Biosciences, catalog no. 56431), Ki67 (Abcam, catalog no. ab-15580), γ-H2AX (Cell Signaling, catalog no. 2577), P-MEK1/2 (Cell Signaling, catalog no. 2338), phosphorylated p42/44 MAPK (P-ERK) (Cell Signaling, catalog no. 4376), SP-C (Seven Hills Bioreagents, catalog no. WRAB-SPC), CCSP (Seven Hills Bioreagents, catalog no. WRAB-CCSP), and CD68 (Abcam, catalog no. ab31630). For quantification of IHC for p21, p53, Ki67, and γ-H2AX, images (100×; one of six from each of three mice for each time point) representative of entire lung lobes were obtained. A minimum of 150 cells from each image were scored at each time point for each genotype. Statistical significance was calculated by two-way ANOVA with a Bonferroni post-test.
Cytochrome c oxidase staining
Cryostat lung tissue sections were incubated in 2.5 mL of reaction buffer (0.2 M sodium dibasic phosphate, 0.2 M sodium monobasic phosphate at pH 7.6), 20 μg of catalase (Sigma, C-10), 10 mg of cytochrome c (Sigma, C-2506), 750 mg of sucrose in 7.5 mL of deionized H2O, and 5 mg of 3, 3′diaminobenzidine tetrahydrochloride hydrate (DAB) (Sigma, D-5637); mounted; and photographed.
Western blotting
Tumor samples were ground in liquid nitrogen and lysed in cell destruction buffer (Ambion). The following antibodies were used for Western blots: p62 (Guo et al. 2011), LC3 (Novus Biologicals, catalog no. NB600-1384), P-MEK1/2 (Cell Signaling, catalog no. 2338), total MEK1/2 (Cell Signaling, catalog no. 4694), phosphorylated p42/44 MAPK (Cell Signaling, catalog no. 9101), total p42/44 MAPK (ERK) (Cell Signaling, catalog no. 9102), anti-mECF-L (YM1) (R&D Systems, AF2446), active caspase 3 (Cell Signaling, catalog no. 9661), and β-actin (Sigma, catalog no. A1978).
Senescence-associated β-galactosidase staining
A β-galactosidase staining kit from Cell Signaling (catalog no. 9860) was used to detect β-galactosidase activity. Briefly, lung frozen sections were fixed in 1× fixative solution for 15 min at room temperature, rinsed with PBS twice, and incubated with β-galactosidase staining solution overnight at 37°C. Images were captured by an Infinity1 camera under a 20× objective using a Zeiss/Primo Vert microscope.
Mouse lung cytokine array assay
A Proteome Profiler Array-Mouse Cytokine Array Panel A kit (R&D Systems, catalog no. ARY006) was used to assess mouse lung cytokine levels. Briefly, K-rasG12D/+;atg7+/+ and K-rasG12D/+;atg7−/− tumor-bearing lungs (n = 3 mice for each genotype) were sacrificed at 20 wk after adenovirus-Cre infection. Lungs without tumors from K-ras+/+;atg7+/+ or K-ras+/+;atg7−/− genotypes (n = 1 mouse for each genotype) were used as controls. All five lung lobes from each mouse were snap-frozen in liquid nitrogen, ground in liquid nitrogen, and lysed in PBS with protease inhibitors and 1% Triton X-100. Samples were then frozen at ≤70°C, thawed, and centrifuged at 10,000g for 5 min to remove cell debris to generate lung tissue lysates. Two-hundred micrograms of lung tissue lysate from each genotype was run on an array according to the provided protocol. K-rasG12D/+;atg7+/+ or K-rasG12D/+;atg7−/− lung tissue lysates were pooled equally from each of the three different sample sets to provide an average across different animals. For the data analysis, cytokine array images were scanned and quantified using ImageJ.
Cell culture and OCR analysis
TDCLs were generated from p53−/−;K-rasG12D/+ tumors with or without atg7 at 8 wk after tumor initiation. TDCLs were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1 mM sodium pyruvate, and 1 mM sodium bicarbonate at 38.5°C with 8.5% CO2. The TDCLs' OCR measurement was performed as described previously (Guo et al. 2011). For palmitate-induced OCR, a readout of FAO was measured in response to BSA-conjugated palmitate (400 μM) in Krebs-Henseleit Buffer (KHB) (110 mM NaCl, 4.7 mM KCl, 2 mM MgSO4, 1.2 mM Na2HPO4, 2.5 mM glucose at pH 7.4) supplemented with 0.5 mM carnitine.
Metabolomic analysis by liquid chromatography-mass spectrometry (LC-MS)
Soluble metabolites from atg7+/+ and atg7−/− TDCLs (two clones each analyzed in triplicate) in normal or HBSS conditions at the indicated times were extracted in 2 mL of −80°C 80:20 methanol:water. Insoluble pellets were re-extracted twice with 0.5 mL each of 80:20 methanol:water for 5 min on dry ice. The supernatants from the three rounds of extraction were combined, dried under N2, and reconstituted in LC-MS-grade water. The samples were analyzed by three separate LC-MS systems: (1) stand-alone orbitrap MS (Exactive, Thermo Scientific) operating in negative full-scan mode at 100,000 resolution coupled to C18 ultraperformance reversed-phase ion pair chromatography (Lu et al. 2010), (2) triple-quadrupole mass spectrometer (TSQ Quantum Discovery Max, Thermo Scientific) operating in negative multiple reaction-monitoring mode coupled to C18 high-performance reversed-phase ion pair chromatography, and (3) triple-quadrupole mass spectrometer (TSQ Quantum Ultra, Thermo Scientific) operating in positive multiple reaction-monitoring mode coupled to hydrophilic interaction liquid chromatography (HILIC). Metabolites were identified by accurate mass (<5 ppm deviation; Exactive) or characteristic fragmentation product (triple quads) in combination with retention time matched to validated standards using in-house software. Peak intensity data were further analyzed using Excel and Matlab.
Analysis of saponified FAs by LC-MS
Two atg7+/+ and atg7−/− TDCL clones were assessed in triplicate. Cells were cultured in 6-mm dishes in normal conditions and rinsed twice with 2 mL of 37°C PBS; 1 mL of cold (−20°C) 50:50 MeOH/0.1 M HCl solution with 0.1 M HCl was added to the cells, and the resulting liquid and cell debris were scraped into 1.5-mL microfuge tubes. Cold (−20°C) chloroform (0.5 mL) was added, the mixture was vortexed for 1 min and centrifuged at 18,000g for 5 min, and then the chloroform layer was transferred to a glass vial. One milliliter of 90% methanol and 0.3 M KOH were added to the glass vial and heated for 1 h at 80°C to saponify FAs. Next, 100 μL of formic acid was added, and the samples were extracted twice with 1 mL of hexane, dried under N2, and dissolved in chloroform/methanol/H2O (1/1/0.3) to a final concentration of 2 μL of cell volume per milliliter for LC-MS analysis. Separation was performed by reversed-phase ion-pairing chromatography on a C8 column coupled with negative-ion mode full-scan LC-MS at 1 Hz scan time and 100,000 resolving power (stand-alone orbitrap, Thermo Fischer Scientific). FAs were identified by accurate mass (<5 ppm deviation, Exactive) or characteristic fragmentation product (triple quads) in combination with retention time matched to validated standards using in-house software. Peak intensity data were further analyzed using Excel and Matlab.
Clonogenic survival assays
TDCLs were seeded in 12-well plates (0.07 × 106 per well) in normal growth medium (RPMI). To starve the cells, the following day, RPMI was removed and replaced with HBSS for 3 d. The cells were then returned to normal medium for 4 d, fixed with cold methanol for 10 min, and stained with Giemsa.
Tumor growth assays in nude mice
Tumor growth assays in nude mice were performed as described previously (Guo et al. 2011). Briefly, three mice per cell line and four independent TDCLs per genotype were used to assess tumor formation. Cells (107) were subcutaneously injected into nude mice, and tumor growth was monitored at the indicated times. Mice were sacrificed at day 14 after injection, and tumors were fixed for histology as described previously (Guo et al. 2011).
Oil red O and Bodipy staining
For Oil red O staining, fixed frozen tissue sections were air-dried and stained with prewarmed Oil red O solution (0.5 g of Oil Red O in 100 mL of propylene glycol) for 8–10 min in a 60°C oven, differentiated in 85% propylene glycol solution for 1 min, and rinsed with distilled water twice. Slides were then lightly stained with alum hematoxylin for 1 min, rinsed with distilled water, and mounted for microscopy.
For Bodipy staining, frozen tissue sections or TDCLs were fixed with 4% formaldehyde for 15 min, rinsed with PBS, and then stained with Bodipy (1 mg/mL) for 15 min. Nuclei were counterstained with DAPI, and the slides were rinsed with distilled water and mounted for microscopy.
MM and MMP
Live TDCLs were stained with 25 nM MitoTracker-Red CMXRos (Molecular Probes/Invitrogen) to determine MMP and 25 nM MitoTracker-Green FM (Molecular Probes/Invitrogen) to determine MM for 30 min under normal growth conditions, washed with growth medium twice, trypsinized, and analyzed by flow cytometry (BD Influx Cell Sorter, BD Biosciences). The mean of MM fluorescence is shown. Relative MMP was calculated by dividing the mean of MMP fluorescence with the mean of MM fluorescence.
Acknowledgments
We thank M. Komatsu for atg7flox/flox mice; N. Campbell for micro-CT analysis; W. Chen for histology quantification; R. Patel for electron microscopy; C. Krier for microarray analysis; R. Katz and A. Ghavami for genotyping; S. Khor and J. Park for cell culture; J. Fan, S. Zhang, S. Hackett, and A. Krishnan for help with metabolomics; and CINJ Tissue Analytical Services. J.J.K. is a Hope Funds for Cancer Research Fellow (HFCR-11-03-01). This work was supported by National Institutes of Health grants (R37 CA53370 and RO1 CA130893 to E.W., and RC1 CA147961 to E.W. and J.D.R), the Department of Defense (W81XWH-09-01-0394 to R.S.D. and E.W.), and the Val Skinner Foundation.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.219642.113.
References
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E 2011. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17: 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. 2008. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456: 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesney J, Telang S 2013. Regulation of glycolytic and mitochondrial metabolism by Ras. Curr Pharm Biotechnol 14: 251–260 [DOI] [PubMed] [Google Scholar]
- Cheung EC, Vousden KH 2010. The role of p53 in glucose metabolism. Curr Opin Cell Biol 22: 186–191 [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, et al. 2006. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10: 51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparre G, Romeo G, Rugolo M, Porcelli AM 2011. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim Biophys Acta 1807: 633–642 [DOI] [PubMed] [Google Scholar]
- Goldstein I, Rotter V 2012. Regulation of lipid metabolism by p53—fighting two villains with one sword. Trends Endocrinol Metab 23: 567–575 [DOI] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25: 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenerhoff MJ, Starost MF, Ward JM 2006. Eosinophilic crystalline pneumonia as a major cause of death in 129S4/SvJae mice. Vet Pathol 43: 682–688 [DOI] [PubMed] [Google Scholar]
- Ide T, Brown-Endres L, Chu K, Ongusaha PP, Ohtsuka T, El-Deiry WS, Aaronson SA, Lee SW 2009. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell 36: 379–392 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Itakura E, Mizushima N 2011. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 192: 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA 2001. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15: 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T 2005. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 65: 10280–10288 [DOI] [PubMed] [Google Scholar]
- Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, Tonon G, McNamara K, Marconcini LA, Hezel A, et al. 2006. K-ras activation generates an inflammatory response in lung tumors. Oncogene 25: 2105–2112 [DOI] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T 2001. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410: 1111–1116 [DOI] [PubMed] [Google Scholar]
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E 2007. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 21: 1621–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC 2011. The dynamic nature of autophagy in cancer. Genes Dev 25: 1999–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131: 1149–1163 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. 2010. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12: 213–223 [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N 2004. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036 [DOI] [PubMed] [Google Scholar]
- Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang MJ, He CH, Takyar S, Elias JA 2011. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol 73: 479–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wu X, Holzer RG, Lee JH, Todoric J, Park EJ, Ogata H, Gukovskaya AS, Gukovsky I, Pizzo DP, et al. 2013. Loss of acinar cell IKKα triggers spontaneous pancreatitis in mice. J Clin Invest 123: 2231–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J 2010. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 22: 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD 2010. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal Chem 82: 3212–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E 2007. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 21: 1367–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. 2009. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137: 1062–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E 2010. Autophagy and metabolism. Science 330: 1344–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki SW, Onodera J, Ohsumi Y 2011. Starvation induced cell death in autophagy-defective yeast mutants is caused by mitochondria dysfunction. PLoS ONE 6: e17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N 2011. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25: 795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M, Ohsumi Y 1993. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 333: 169–174 [DOI] [PubMed] [Google Scholar]
- White E 2012. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12: 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25: 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Prives C 2009. p53 and metabolism: The GAMT connection. Mol Cell 36: 351–352 [DOI] [PubMed] [Google Scholar]