

MicroRNAs and histone modifiers are key epigenetic regulators of gene expression, yet little is known about their roles in mammalian neural morphogenesis. Here, Zhou and colleagues identify microRNA-138 (miR-138) as a suppressor of mammalian axon regeneration. They show that the histone deacetylase SIRT1 negatively regulates miR-138 transcription in response to peripheral nerve injury in adult sensory neurons. This study thus uncovers a mutual negative feedback loop between the epigenetic regulators miR-138 and SIRT1 to control axon regeneration.
Keywords: microRNA, miR-138, SIRT1, histone deacetylase, axon regeneration, axon growth
Abstract
Regulated gene expression determines the intrinsic ability of neurons to extend axons, and loss of such ability is the major reason for the failed axon regeneration in the mature mammalian CNS. MicroRNAs and histone modifications are key epigenetic regulators of gene expression, but their roles in mammalian axon regeneration are not well explored. Here we report microRNA-138 (miR-138) as a novel suppressor of axon regeneration and show that SIRT1, the NAD-dependent histone deacetylase, is the functional target of miR-138. Importantly, we provide the first evidence that miR-138 and SIRT1 regulate mammalian axon regeneration in vivo. Moreover, we found that SIRT1 also acts as a transcriptional repressor to suppress the expression of miR-138 in adult sensory neurons in response to peripheral nerve injury. Therefore, miR-138 and SIRT1 form a mutual negative feedback regulatory loop, which provides a novel mechanism for controlling intrinsic axon regeneration ability.
Axon growth is achieved through coordinated gene expression in the neuronal soma, the transport of synthesized molecules along the axon, and the actual assembly of the axon by the cytoskeletal and membrane machinery at the distal axon. Regulation of gene expression during axon growth not only provides the raw materials for axon assembly, but also controls the intrinsic axon growth ability, which is greatly diminished in mature neurons of the mammalian CNS (Liu et al. 2010b). Thus, modulation of gene expression that governs the intrinsic axon growth ability has been a key approach for promoting axon regeneration after CNS injuries. However, our understanding of the molecular mechanisms by which gene expression is controlled during axon growth is very limited.
Epigenetic regulation independent of changes in DNA sequences is emerging as a key cellular mechanism to control gene expression, among which microRNAs and histone modifications are two major epigenetic mechanisms. To date, we know very little about the roles of epigenetic regulations in axon growth and regeneration. In the nervous system, microRNAs are known to play important roles in neural precursors to control neurogenesis (Fineberg et al. 2009; Liu and Zhao 2009; Li and Jin 2010) and in mature neurons to control synaptic function (Vo et al. 2010; Siegel et al. 2011). In contrast, the roles of microRNAs in the regulation of neuronal morphogenesis, including axon growth and regeneration, are much less studied. Two recent studies have reported the involvement of microRNAs in controlling axon growth from embryonic neurons in vitro (Dajas-Bailador et al. 2012; Franke et al. 2012). In mature animals, one study found that sensory axon regeneration in vivo was impaired in animals lacking the Dicer protein, which is crucial for microRNA processing (Wu et al. 2012), suggesting that microRNAs are potential novel regulators of axon regeneration. Indeed, several genetic profiling studies (Strickland et al. 2011; Zhang et al. 2011; Zhou et al. 2011) have shown that the expression levels of many microRNAs are changed in adult mouse sensory neurons after the peripheral nerve injury, which leads to enhanced intrinsic axon growth capacity and robust axon regeneration. However, to date, no study has ever reported the roles of microRNAs in the regulation of mammalian axon regeneration in vivo. Similarly, we know very little about the roles of histone modification in axon regeneration. To our knowledge, to date, only two recent studies have shown the involvement of histone acetyltransferase p300 in the regulation of axon regeneration (Gaub et al. 2010, 2011).
Here, we report that microRNA-138 (miR-138), a highly expressed microRNA in the nervous system (Obernosterer et al. 2006), functions to regulate axon growth during development and regeneration by acting as a molecular repressor. We further identify the NAD-dependent histone deacetylase (HDAC) SIRT1 as a downstream molecular target of miR-138. More importantly, we provide the first in vivo evidence that miR-138 and SIRT1 function to suppress and promote mammalian axon regeneration, respectively. Interestingly, we found that SIRT1 also acts as a transcriptional repressor to directly suppress the expression of miR-138 in response to peripheral nerve injury. Collectively, we demonstrate that mammalian peripheral nerve injury leads to robust axon regeneration by inducing the formation of a mutual negative feedback loop between two epigenetic factors: miR-138 and the HDAC SIRT1.
Results
miR-138 is developmentally regulated during cortical development and controls axon growth of embryonic cortical neurons
We first investigated whether miR-138 regulated axon growth using cultured mouse embryonic cortical neurons, which are a well-established model system for studying axon growth. Using mature microRNA-specific quantitative real-time PCR (qRT-PCR), we found that the expression level of endogenous miR-138 gradually increased in the cortical tissues during development, reaching the highest level in adult animals (Fig. 1A). As cortical neurons lose their intrinsic ability to support axon growth after maturation (Liu et al. 2012a), this result suggests that miR-138 might be a negative regulator of axon growth. To test this idea, we transfected embryonic day 15 (E15) cortical neurons with either the miR-138 mimics, which are double-stranded oligonucleotides designed to mimic the function of endogenous mature microRNA, or the miR-138 inhibitor, which is RNA oligonucleotides with a novel secondary structure (hairpin) designed to inhibit the biogenesis of endogenous microRNA (Dharmacon miRIDIAN microRNA reagents). Expression of the miR-138 mimics drastically increased the level of miR-138 in neurons, whereas expression of the miR-138 inhibitor markedly reduced the level of endogenous miR-138 (Fig. 1B). Functionally, overexpression of the miR-138 mimics significantly impaired axon growth, while expression of the inhibitor promoted axon growth (Fig. 1C,D). Similar results (Supplemental Fig. S1) were obtained with a DNA plasmid encoding miR-138 or a miR-138 sponge construct that expresses competitive inhibitors of miR-138 (Ebert et al. 2007). Together, these results indicate that miR-138 negatively regulates axon growth, likely through suppressing the intrinsic axon growth ability.
Figure 1.

miR-138 is developmentally regulated during cortical development and controls axon growth of embryonic cortical neurons. (A) Relative miR-138 expression levels in mouse cortical tissues during development from E15 to adult. n = 3. (B) miR-138 expression levels after transfection of the miR-138 mimics (left panel) or the miR-138 inhibitors (anti-138; right panel) in E15 cortical neurons. n = 4; (**) P < 0.01; (***) P < 0.001. (C) E15 mouse cortical neurons were transfected with EGFP (control), miR-138 mimics plus EGFP, and miR-138 inhibitor plus EGFP (anti-138) as indicated. The cells were fixed at 4 d in vitro (DIV4), and the axon lengths were measured. Note that overexpression of the miR-138 mimics inhibited axon growth, whereas overexpression of the miR-138 inhibitor promoted axon growth. n = 3; (**) P < 0.01. (D) Representative images of DIV4 cortical neurons transfected with EGFP, miR-138 mimics/EGFP, or anti-miR-138 inhibitors/EGFP. Bar, 100 μm.
Down-regulation of miR-138 after axotomy is necessary for regenerative axon growth of adult sensory neurons
We next studied the role of miR-138 in the regulation of axon regeneration using adult sensory neurons from the dorsal root ganglion (DRG), which regenerate robustly after peripheral nerve injury by reactivating their intrinsic axon regeneration capacity (Zhou et al. 2006). Similarly, we first examined the expression level of endogenous miR-138 in adult DRG neurons during peripheral axotomy-induced axon regeneration using qRT-PCR. The result showed that miR-138 was significantly down-regulated in DRG neurons 1 wk after sciatic nerve injury (Fig. 2A), consistent with its role in suppressing axon growth. Dissociation (in vitro axotomy) and in vitro culture of adult DRG neurons have also been shown to mimic in vivo axotomy to increase regeneration capacity (Smith and Skene 1997; Saijilafu and Zhou 2012). Consistent with this, the expression level of miR-138 was also drastically down-regulated in dissociated DRG neurons after 3 d in culture (Fig. 2B). These results are in line with a recent genetic profiling study in which miR-138 was among a group of microRNAs that are down-regulated in DRGs after peripheral nerve injury (Strickland et al. 2011). To determine the functional role of miR-138 in axon regeneration, we used a recently developed cell-replating model (Saijilafu and Zhou 2012). Specifically, dissociated DRG neurons were electroporated with the miR-138 mimics together with EGFP to label transfected cells and cultured for 3 d. The cells were then resuspended and replated for axon growth analysis 20–24 h later. The result showed that miR-138 overexpression markedly suppressed regenerative axon growth from adult DRG neurons (Fig. 2C,D), similar to that of embryonic cortical neurons (see Fig. 1C). Because the level of endogenous miR-138 had already been decreased in adult DRG neurons after 3 d in culture (see Fig. 2B), further down-regulation of miR-138 function with the microRNA inhibitor did not promote additional axon growth of adult DRG neurons (Supplemental Fig. S2). These findings suggest that miR-138 functions to suppress axon regeneration.
Figure 2.

Down-regulation of miR-138 after axotomy is necessary for regenerative axon growth of adult sensory neurons. (A) miR-138 expression level in adult DRGs was significantly decreased 7 d after sciatic nerve injury. The miR-138 level was quantified using qRT-PCR and normalized to the expression of the U6B small nuclear RNA gene (RNU6B). n = 4; (*) P < 0.05. (B) miR-138 expression level in dissociated adult DRG neurons was significantly reduced after 3 d in culture. n = 4; (**) P < 0.01. (C,D) Adult DRG neurons were transfected with EGFP (control) or miR-138 mimics plus EGFP. After 3 d in culture, the neurons were resuspended, replated, and cultured overnight for axon growth analysis. Quantification of axon length is shown in C (n = 4; [*] P < 0.05), and representative images of replated neurons transfected with EGFP and miR-138 mimics are shown in D. Bar,100 μm.
Down-regulation of miR-138 is required for peripheral axotomy-induced sensory axon regeneration in vivo
To extend the in vitro findings to an in vivo model of axon regeneration, we investigated the role of miR-138 in the regulation of peripheral axon regeneration of adult sensory neurons. Because peripheral nerve injury down-regulates endogenous miR-138 levels in adult DRG neurons (see Fig. 2A), we tested whether up-regulation of miR-138 was able to prevent peripheral axotomy-induced axon regeneration. By using our recently developed in vivo electroporation technique (Fig. 3A), which allows acute regulation of gene expression in DRG neurons of adult mice (Hur et al. 2011a; Saijilafu et al. 2011), we overexpressed the miR-138 mimics and EGFP in adult mouse DRG neurons. Two days later, the mice were subjected to a sciatic nerve crush procedure, and axon regeneration was assessed 3 d later. The result showed that overexpression of the miR-138 mimics markedly elevated the level of miR-138 in adult DRGs (Fig. 3B). Functionally, sensory axons of miR-138/EGFP-overexpressing neurons displayed significantly impaired axon regeneration in vivo compared with those of control neurons (Fig. 3C–E), demonstrating that axotomy-induced miR-138 down-regulation is necessary for in vivo axon regeneration.
Figure 3.

Down-regulation of miR-138 is required for peripheral axotomy-induced sensory axon regeneration in vivo. (A) Schematics of the protocol for in vivo electroporation and sciatic nerve regeneration experiments. The miR-138 mimics were electroporated together with EGFP into adult mouse DRGs (L4/5) in vivo. Two days later, the mice were subjected to a sciatic nerve crush procedure, and axon regeneration was assessed 3 d later. (B) qRT-PCR data indicating miR-138 level in adult DRGs in vivo 3 d after electroporation of the miR-138 mimics. n = 4; (*) P < 0.05. (C) Average lengths of regenerating sciatic nerve axons. n = 7 mice for the control group; n = 16 mice for the miR-138 group; (***) P < 0.001. (D) Cumulative distribution of the lengths of all individual axons measured. n = 249 for control; n = 378 for miR-138. (E) Representative images of EGFP-labeled regenerating axons in the whole-mount sciatic nerves. The crush sites were marked by the epineural suture (red arrows). Bar, 1 mm.
SIRT1 is a downstream target of miR-138 in adult sensory neurons in vitro and in vivo
To investigate the molecular mechanism by which miR-138 regulates axon regeneration, we searched for potential mRNA targets of miR-138 by cross-referencing several widely used programs (TargetScan and miRanda). Among several candidates, we selected SIRT1 as a potential target of miR-138 because SIRT1 has been shown to control axon growth and degeneration (Araki et al. 2004; Guo et al. 2011) and to be highly expressed in mouse DRGs (Sakamoto et al. 2004). To validate that SIRT1 expression is regulated by miR-138, we made a luciferase reporter construct by inserting the full-length mouse SIRT1 3′ untranslated region (UTR) containing the predicted miR-138 target site and flanking sequences into the 3′ of a Renilla luciferase (R-luc) reporter gene (Fig. 4A). Either the miR-138 mimics or its inhibitor was coexpressed with the SIRT1 3′ UTR in a mouse CNS catecholaminergic cell line, Cath. a differentiated (CAD) cells, which allowed high-efficiency transfection of the SIRT1 luciferase reporter plasmid. We found that overexpression of the miR-138 mimics repressed the expression of R-luc, whereas expression of the miR-138 inhibitor enhanced R-Luc expression (Fig. 4A). In contrast, when a mutant SIRT1 R-luc reporter that contains a mutated miR-138-binding site was used, neither miR-138 mimics nor its inhibitor was able to affect the R-luc expression (Fig. 4B). These results demonstrate that miR-138 specifically represses SIRT1 expression through the predicted target site in the SIRT1 3′ UTR. We then tested whether miR-138 regulated the endogenous SIRT1 in adult DRG neurons. First, the miR-138 mimics were electroporated into dissociated adult DRG neurons, and the SIRT1 expression level was examined by Western blot analysis after 3 d in culture. The result showed that miR-138 overexpression markedly reduced the protein level of SIRT1 in cultured adult DRG neurons (Fig. 4C). Next, we electroporated the miR-138 mimics directly into adult mouse DRGs in vivo, and the mice were subjected to a sciatic nerve crush procedure in the meantime. Three days later, the transfected DRGs were collected to detect SIRT1 expression. We found that overexpression of the miR-138 mimics, which antagonized peripheral axotomy-induced down-regulation of endogenous miR-138, markedly reduced the protein level of endogenous SIRT1 (Fig. 4D), indicating that miR-138 targets SIRT1 in adult DRG neurons in vivo. Taken together, these data indicate that SIRT1 is a physiological target of miR-138 in adult DRG neurons during axon regeneration.
Figure 4.

SIRT1 is a downstream target of miR-138 in adult sensory neurons during axon regeneration. (A) R-luc activity assay in CAD cells coexpressing the luciferase reporter containing the full-length SIRT1 3′ UTR and the miR-138 mimics or inhibitor. Note that expression of miR-138 mimics inhibited, while expression of the miR-138 inhibitor enhanced, the luciferase activity. n = 3; (**) P < 0.01. (B) Mutation of the miR-138 targeting site in the SIRT1 3′ UTR abolished the regulation of luciferase activity by miR-138 mimics or its inhibitor. n = 3. (C) Overexpression of the miR-138 mimics in cultured adult DRG neurons led to decreased endogenous SIRT1 protein level. (D) Electroporation of the miR-138 mimics into adult DRGs in vivo led to decreased endogenous SIRT1 protein level.
SIRT1 controls sensory axon regeneration in vitro and in vivo
To determine the role of SIRT1 in the regulation of sensory axon regeneration, we first detected the localization of SIRT1 in adult DRG neurons. Immunostaining results showed that SIRT1 was mainly localized in the nuclei of adult DRG neurons (Supplemental Fig. S3), suggesting that it might be involved in the regulation of gene expression during axon regeneration. We then examined the expression of SIRT1 in adult DRG neurons during peripheral axotomy-induced axon regeneration. We found that both the mRNA and the protein levels of SIRT1 were markedly increased in adult DRGs 7 d after the peripheral axotomy (Fig. 5A,B). To determine whether SIRT1 regulated axon growth from adult DRG neurons, we treated the cultured adult DRG neurons with EX527, a specific inhibitor of SIRT1 deacetylase activity (Peck et al. 2010). We found that application of EX527 significantly blocked regenerative axon growth from adult DRG neurons (Supplemental Fig. S4A). To confirm the pharmacological data, we knocked down endogenous SIRT1 with a group of four different siRNAs that are designed to minimize the off-target effects (ON-TARGETplus, Thermo Scientific Dharmacon). We found that acute depletion of SIRT1 also resulted in impaired regenerative axon growth from adult DRG neurons (Supplemental Fig. S4B). We also used a mutant SIRT1 lacking the deacetylase activity (H363Y) (Brunet et al. 2004; Gao et al. 2010), which acted in a dominant-negative manner to inhibit the activity of the endogenous SIRT1. The results showed that expression of this catalytically inactive mutant of SIRT1 significantly blocked axon growth from adult DRG neurons (Supplemental Fig. S4C) to an extent similar to those of EX527 and SIRT1 siRNAs. Moreover, expression of the SIRT1 mutant and application of EX527 at the same time did not result in additional inhibitory effects on axon growth (Supplemental Fig. S4D), confirming the specificity of EX527 and the SIRT1 mutant on endogenous SIRT1 activity. Consistent with down-regulation of endogenous miR-138 in cultured adult DRG neurons, endogenous SIRT1 was up-regulated correspondingly (data not shown). As a result, overexpression of SIRT1 did not further promote axon growth of adult DRG neurons (Supplemental Fig. S5A). However, when neurons were cultured on a low concentration of laminin, which provided a less favorable condition for axon growth, overexpression of SIRT1 resulted in a significant increase in axon growth (Supplemental Fig. S5B), suggesting that SIRT1 has the ability to promote axon growth. In support of this, previous studies have shown that overexpression of SIRT1 can drastically promote axon growth of embryonic cortical neurons (Guo et al. 2011; Li et al. 2013), which do not up-regulate SIRT1 automatically in culture.
Figure 5.

SIRT1 regulates sensory axon regeneration in vivo. (A,B) Both SIRT1 mRNA (n = 9; [**] P < 0.01) and protein levels were increased in adult DRGs 1 wk after sciatic nerve lesion compared with the naïve uninjured DRGs. (C) In vivo electroporation of SIRT1 siRNA oligos (siSIRT1) markedly knocked down SIRT1 protein level in DRGs after 3 d. (D) Average lengths of regenerating sciatic nerve axons. n = 7 mice for the control group; n = 15 mice for the SIRT1 siRNA group: (***) P < 0.001. (E) Cumulative distribution of the lengths of all individual axons measured. n = 249 for control; n = 545 for SIRT1 siRNA. (F) Representative images of EGFP-labeled regenerating axons in the whole-mount sciatic nerves. The crush sites were marked by the epineural suture (red lines). Bar, 1 mm.
To determine whether SIRT1 controlled axon regeneration in vivo, we directly electroporated SIRT1 siRNAs and EGFP into adult DRGs in vivo. The sciatic nerve was crushed 2 d later, and sensory axon regeneration was assessed 3 d thereafter (see Fig. 3A). The results showed that SIRT1 siRNAs markedly knocked down the endogenous SIRT1 in vivo (Fig. 5C). Functionally, down-regulation of SIRT1 significantly impaired axon regeneration in vivo compared with those of control neurons (Fig. 5D–F), demonstrating that axotomy-induced SIRT1 up-regulation is necessary for in vivo axon regeneration.
SIRT1 represses miR-138 expression in adult DRG neurons during regeneration
In addition to being a target of microRNAs, SIRT1 can also acts as a transcription repressor to control gene expression, including microRNA expression (Yamakuchi 2012). For instance, SIRT1 has been shown to regulate miR-134 expression in neural progenitors by direct binding to the genomic DNA regions upstream of the pre-miR-134 sequence (Gao et al. 2010). We therefore tested whether SIRT1 was able to regulate miR-138 expression in adult DRG neurons. We found that the expression of endogenous miR-138 is drastically up-regulated in cultured adult DRGs when SIRT1 was knocked down (Fig. 6A). We also examined whether overexpression of SIRT1 could repress miR-138 expression using a neuronal cell line, CAD cells. Indeed, we found that overexpression of SIRT1 led to significantly reduced expression of miR-138 (Supplemental Fig. S6). Interestingly, overexpression of the catalytically inactive mutant of SIRT1 (H363Y) resulted in elevated miR-138 levels, mimicking that of SIRT1 knockdown, indicating that SIRT1 represses miR-138 expression through its deacetylase activity. To determine whether SIRT1 repressed miR-138 expression in vivo during axon regeneration, we knocked down SIRT1 in vivo by electroporation and at the same time performed the sciatic nerve crush. We examined the expression of miR-138 3 d later and found that knocking down of SIRT1 markedly elevated the level of miR-138 (Fig. 6B). Together, these results suggest that axotomy-induced up-regulation of SIRT1 is necessary for miR-138 down-regulation in response to axotomy.
Figure 6.
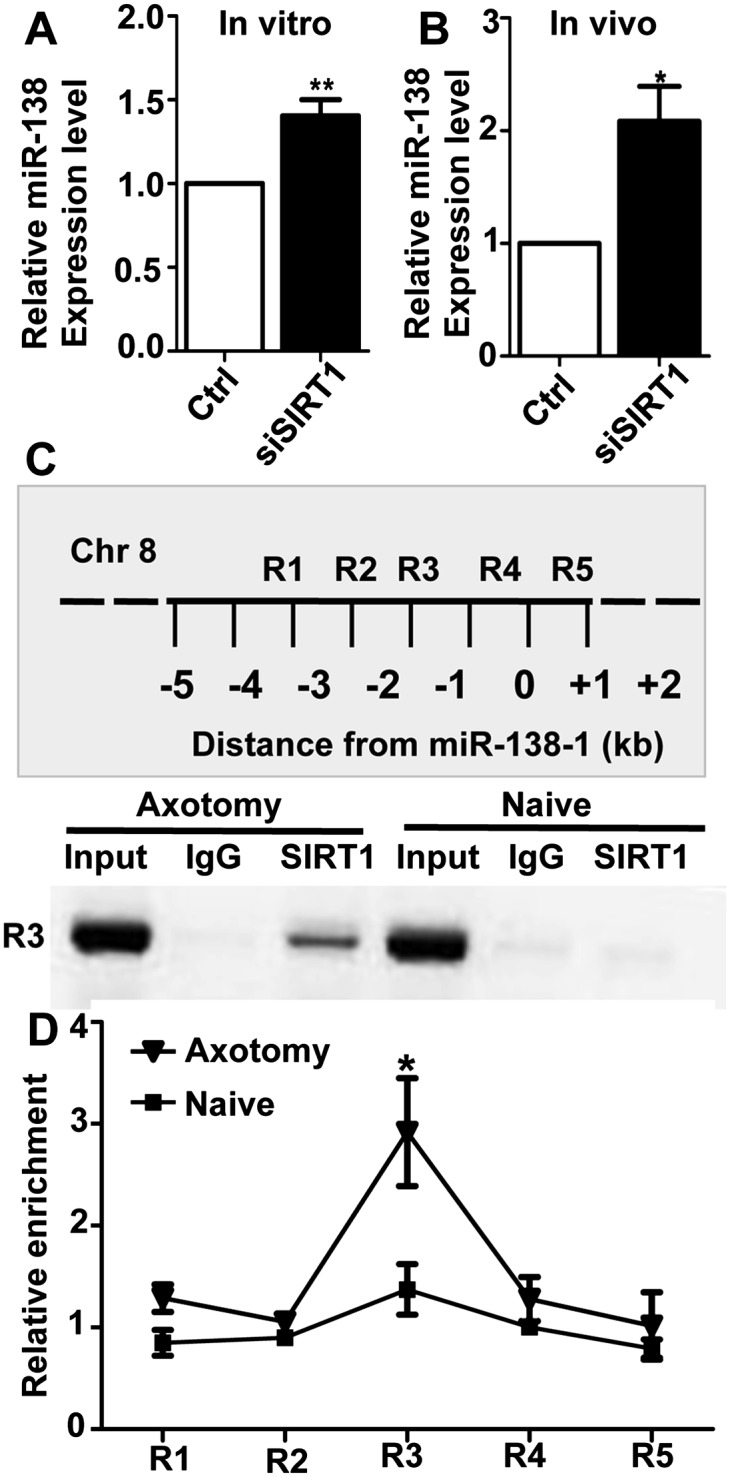
SIRT1 represses miR-138 transcription in adult DRG neurons. (A) Knocking down SIRT1 with siRNA (siSIRT1) in cultured adult DRG neurons led to increased expression of miR-138 in vitro. n = 7; (**) P < 0.01. (B) Knocking down SIRT1 in adult DRGs in vivo led to increased expression of miR-138 in vivo. n = 4; (*) P < 0.05. (C, top) Schematic drawing of the 4-kb genomic regions (R1–R5) proximal to the miR-138-1 gene on chromosome 8 that were assayed in the ChIP experiment. (Bottom) In the SIRT1 ChIP experiment, the genomic region R3 was amplified from injured adult DRG tissues, but not the naive tissues, to bind to SIRT1. (D) RT-PCR data quantifying the enrichment of the binding between SIRT1 and the R3 genomic region of the miR-138-1 locus in adult DRG tissues. n = 4; (*) P < 0.05. Note that SIRT1 only binds to the R3 region in response to the peripheral axotomy.
To determine whether SIRT1 regulates miR-138 expression by directly interacting with genomic regions proximal to miR-138, we performed chromatin immunoprecipitation (ChIP) using an anti-SIRT1 antibody in naïve (uninjured) or peripheral axotomized DRGs. We then analyzed the interaction between SIRT1 and five genomic regions (R1 to R5) spanning −4 kb upstream of to +1 kb downstream from the position of the pre-miR-138 sequence (Fig. 6C). The result showed that SIRT1 interacted with the R3 region (from −2 kb to −1 kb upstream) specifically in peripheral axotomized DRGs but not naïve DRGs (Fig. 6C,D). As a control, immunoprecipitation with IgG did not detect interactions with any regulatory regions surrounding pre-miR-138. The qRT-PCR data showed that the binding of SIRT1 with the R3 region was increased significantly in peripheral axotomized DRGs compared with that in naïve DRGs (Fig. 6D). Collectively, these data support the idea that SIRT1 represses miR-138 expression in adult DRG neurons by directly binding to the regulatory sequence upstream of pre-miR-138 in response to peripheral nerve injury.
SIRT1 is both the input and output signals of the miR-138/SIRT1 regulatory loop during axon regeneration
We showed that both miR-138 and SIRT1 change their expression in response to peripheral axotomy and functionally form a mutual negative feedback loop to regulate axon regeneration. To further determine how miR-138 and SIRT1 interact during peripheral axotomy-induced axon regeneration, we first examined the time courses of miR-138 and SIRT1 expression in response to peripheral axotomy. The results showed that SIRT1 was significantly up-regulated 12 h after axotomy (Fig. 7A), when the miR-138 level remained largely unchanged (Fig. 7B). Both SIRT1 up-regulation and miR-138 down-regulation peaked at 3 d after axotomy and then started to gradually subside. This time course result suggests that axotomy-induced SIRT1 up-regulation precedes the down-regulation of miR-138. Second, we overexpressed the miR-138 mimics and at the same time treated the cells with the SIRT1 inhibitor EX527. The results showed that inhibition of SIRT1 did not have an additive effect on miR-138-induced inhibition of regenerative axon growth (Fig. 7C), confirming that SIRT1 and miR-138 function in the same pathway to regulate axon regeneration. Third, to determine whether SIRT1 mediates the effect of miR-138 to control axon regeneration, a SIRT1 construct lacking the endogenous 3′ UTR (SIRT1-Δ3′ UTR) was used (Brunet et al. 2004). Expression of SIRT1-Δ3′ UTR alone did not affect regenerative axon growth from adult DRG neurons (see Supplemental Fig. S5). However, when SIRT1-Δ3′ UTR was coexpressed with the miR-138 mimics, it completely reverted the axon growth inhibition induced by miR-138 overexpression (Fig. 7D), indicating that SIRT1 acts downstream from miR-138 to regulate axon regeneration. Conversely, coexpression of the miR-138 inhibitor and SIRT1 siRNA still resulted in impaired regenerative axon growth, although expression of the miR-138 inhibitor partially rescued regenerative axon growth inhibited by knocking down of SIRT1 (Fig. 7E). Taken together, these results suggest that, functionally, SIRT1 is the main input and output molecule of the regulatory loop that controls axon regeneration, whereas miR-138 functions to modulate the SIRT1 level through a mutual negative feedback loop (Supplemental Fig. S7), which ensures more efficient SIRT1 up-regulation in response to peripheral axotomy.
Figure 7.

SIRT1 is both the input and output signals of the miR-138/SIRT1 regulatory loop to control axon regeneration. (A) qRT-PCR data indicating endogenous SIRT1 mRNA levels in adult DRGs after peripheral nerve injury. Note that SIRT1 expression was significantly up-regulated at 0.5 d after peripheral axotomy compared with the uninjured naïve DRGs. n = 8 for each time point; (*) P < 0.05; (**) P < 0.01. (B) qRT-PCR data indicating endogenous miR-138 levels in adult DRGs after peripheral nerve injury. Note that miR-138 expression was significantly down-regulated at 1 d after peripheral axotomy compared with the uninjured naïve DRGs. n = 8 for each time point; (*) P < 0.05; (**) P < 0.01. (C) Overexpression of miR-138 and inhibition of SIRT1 activity with the inhibitor EX527 had no additive inhibitory effects on regenerative axon growth from adult DRG neurons. n = 3; (**) P < 0.01. (D) Expression of SIRT1 lacking the 3′ UTR, which cannot be targeted by miR-138, was able to fully rescue axon growth of adult DRG inhibited by miR-138 expression. n = 3; (*) P < 0.05. (E) Inhibition of miR-138 with the inhibitor (anti-138) only partially rescued regenerative axon growth inhibited by knockdown of SIRT1 expression. n = 7; (**) P < 0.01.
Discussion
Axon growth is regulated by coordinated gene expression in the soma and local axon assembly at the distal axon (Liu et al. 2012a). In the mammalian nervous system, peripheral axotomy is known to induce a transcription-dependent genetic switch that underlies the subsequent peripheral axon regeneration. To date, the molecular mechanism underlying the switch remains elusive. Epigenetic modification is emerging as a major mechanism in the regulation of gene expression during many biological processes. However, its role in the regulation of axon growth and regeneration has rarely been studied. In this study, we revealed that axon regeneration is regulated by two epigenetic factors—SIRT1 and miR-138—forming a mutual negative signaling loop (Supplemental Fig. S7).
Several microRNAs are highly enriched in the brain tissue, such as miR-9, miR-124, and miR-138 (Obernosterer et al. 2006), indicating that microRNAs play important roles in controlling neuronal function. To date, most studies of microRNAs in the nervous system focus on their roles in regulation of neurogenesis in progenitors or synaptic function in mature synapses. Very few studies have examined the roles of microRNAs in post-mitotic neurons to control neuronal morphogenesis. Here we show for the first time that miR-138 negatively regulates axon growth from developing cortical neurons and, more importantly, in vivo axon regeneration from adult sensory neurons, likely through controlling gene expression in the neuronal soma. Interestingly, a recent study has shown that miR-9 also negatively regulates axon extension of embryonic cortical neurons (Dajas-Bailador et al. 2012) by targeting the cytoskeletal protein MAP1b locally in the axon. Together, these results show clearly that microRNAs provide a novel regulatory mechanism of axon growth and regeneration.
Each microRNA usually has multiple target genes (Lewis et al. 2005), and the same gene can be targeted by multiple microRNAs, depending on the specific cellular context in which the microRNA is expressed. A previous study has identified miR-138 as a negative regulator of the dendritic spine size by targeting the depalmitoylation enzyme acyl protein thioesterase 1 (APT1) (Siegel et al. 2009). In nonneuronal cells, miR-138 is able to target cyclin D1, a regulator of CDK kinases (Liu et al. 2012b); EZH2, a histone methyltransferase (Kisliouk et al. 2011); and p53, a cell cycle regulator (Ye et al. 2012). In human primary keratinocytes, miR-138 has been shown to target SIRT1 to control cell senescence (Rivetti di Val Cervo et al. 2012). In this study, we provide clear in vitro and in vivo evidence that SIRT1 is a functional target of miR-138 in adult DRG neurons to control axon regeneration. First, the expression levels of miR-138 and SIRT1 in adult DRGs showed reciprocal changes in vivo upon peripheral nerve injury. Second, overexpression of miR-138 in adult DRG neurons suppressed the endogenous SIRT1 level in vitro and in vivo. Third, overexpression of SIRT1 without the 3′ UTR fully rescued axon growth inhibited by miR-138 expression. Several previous studies have shown that miR-34a can target SIRT1 in different tissues (Yamakuchi 2012). However, the expression of miR-34a does not seem to be altered in adult sensory neurons upon peripheral axotomy (Strickland et al. 2011; Zhang et al. 2011; Zhou et al. 2011), suggesting that it is unlikely to target SIRT1, which is up-regulated drastically upon axotomy.
Here we also provide the first and strong evidence that SIRT1 functions to support axon regeneration in vitro and in vivo. Specifically, we showed that blocking SIRT1 activity with a pharmacological inhibitor or a dominant negative mutant or knocking down SIRT1 with siRNA inhibited regenerative axon growth of adult sensory neurons in vitro. These results are consistent with previous studies in which SIRT1 has been shown to support neurite outgrowth in PC12 cells (Sugino et al. 2010) and developing cortical neurons (Guo et al. 2011). More importantly, by knocking down SIRT1 in vivo, we provide the first in vivo evidence that SIRT1 functions to regulate axon regeneration.
How do miR-138 and SIRT1 control axotomy-induced axon regeneration of adult sensory neurons? Recent studies have suggested that one of the major functions of microRNAs is to act as a reinforcer to ensure transcription-dependent transition between two biological states by forming regulatory loops with their targets (Ebert and Sharp 2012). Indeed, here we found that SIRT1 not only was the target of miR-138, but also functioned to repress miR-138 transcription, therefore forming a mutual negative feedback loop. The expression time courses of SIRT1 and miR-138 in response to peripheral axotomy indicate that peripheral axotomy induces SIRT1 up-regulation first, which then represses the transcription of miR-138, suggesting that SIRT1 is the input signal of the regulatory loop. Interestingly, the epistasis analysis results place SIRT1 functionally downstream from miR-138 to regulate axon regeneration, suggesting SIRT1 as the major output signal as well. Therefore, the function of miR-138 is to reinforce the SIRT1 up-regulation through the mutual negative feedback loop (Supplemental Fig. S7). Based on these results, we think that in uninjured naïve adult sensory neurons, the high level miR-138 suppresses the expression of SIRT1, resulting in low intrinsic axon regeneration ability. Peripheral axotomy induces up-regulation of SIRT1, which represses the transcription of miR-138, therefore initiating the regulatory loop to further increase the SIRT1 level and boost the intrinsic axon regeneration ability (Supplemental Fig. S7). Although how SIRT1 up-regulation leads to increased regeneration ability is unknown, it is presumably through a genetic-based mechanism, similar to the role of SIRT1 in protecting axotomy-induced axonal degeneration (Araki et al. 2004). Future studies to identify the downstream effector genes of SIRT1 will be of great interest to elucidate how SIRT1 regulates the peripheral axotomy-induced genetic program supporting peripheral axon regeneration.
Because endogenous miR-138 and SIRT1 are autonomously down-regulated and up-regulated in dissociated and cultured adult DRG neurons, respectively, we were unable to further promote axon growth of adult DRG neurons by inhibiting miR-138 or overexpressing SIRT1. However, we showed that in embryonic cortical neurons, where the expressions of endogenous miR-138 and SIRT1 do not change, inhibiting miR-138 led to increased axon growth. Similarly, two previous studies have also shown that overexpression of SIRT1 in cortical neurons was sufficient to promote axon growth (Guo et al. 2011; Li et al. 2013). Thus, manipulation of miR-138 and SIRT1 expression is an effective approach to promote axon growth. Importantly, we showed that the miR-138 expression level in the cortical tissue increased significantly from development to adult, similar to those of negative regulators of intrinsic axon growth ability, such as Pten (Park et al. 2008) and KLF4 (Moore et al. 2009). In contrast, the expression of SIRT1 is known to be high in embryonic brains and DRGs (Sakamoto et al. 2004). Together, it suggests that modulating miR-138 and/or its target SIRT1 would be a novel approach to boost the intrinsic axon regeneration ability of mature mammalian CNS neurons.
Materials and methods
Primary neuron culture and in vitro transfection
All experiments involving animals were performed in accordance with the animal protocol approved by the Institutional Animal Care and Use Committee of Johns Hopkins University. Dissection and culture of mouse embryonic cortical and adult DRG neurons were performed as described previously (Hur et al. 2011a,b). In brief, DRGs were dissected from 8- to 12-wk-old adult CF-1 mice and digested with collagenase A (1 mg/mL) for 1.5 h, followed by trypsin-EDTA for 20 min at 37°C. The DRGs were then washed three times with MEM and dissociated with the culture medium (MEM supplemented with 5% fetal bovine serum [FBS] and antimitotic agents [20 μM 5-fluoro-2-deoxyuridine, 20 μM uridine]). For experiments involving RNA extraction, dissociated DRGs were plated on plastic culture dishes coated with poly-D-lysine (100 μg/mL) and laminin (10 μg/mL). For axon growth experiments, the dissociated neurons were first plated on plastic culture dishes coated with poly-D-lysine and laminin. Three days later, the neurons were resuspended and replated onto glass coverslips coated with poly-D-lysine and laminin (Saijilafu and Zhou 2012). Cortical neurons were prepared from E15 mouse embryos. The dissected cortical tissue was digested with trypsin-EDTA for 10 min at 37°C. The tissue was then washed three times with MEM plus 10% FBS and dissociated with the culture medium (neurobasal medium supplemented with B27, antibiotic agents penicillin/streptomycin, and GlutaMAX). The dissociated cortical neurons were plated on glass coverslips coated with poly-D-lysine (100 μg/mL).
DNA constructs or RNA oligos were transfected into dissociated neurons via electroporation using the nucleofector from Lonza as previously described (Hur et al. 2011a,b). Briefly, dissociated neurons were centrifuged to remove the supernatant and resuspended in 80–100 μL of specified Amaxa electroporation buffer with plasmid DNA (10–20 μg and 2–3 μg for DRG and cortical neurons, respectively) or RNA oligos (siRNA and microRNA mimics and inhibitor, 4 μL at 50 μM). Suspended cells were then transferred to a 2.0-mm cuvette and electroporated with the Amaxa Nucleofector apparatus. After electroporation, cells were immediately mixed with the desired volume of prewarmed culture medium and transferred to the culture dish. After neurons fully attached to the substrates (2–4 h), the medium was changed to remove the remnant transfection buffer.
Immunostaining, fluorescence microscopy, and image analysis
Neurons were fixed with 4% PFA for 20 min at room temperature. Fixed neurons were washed with PBS and blocked in blocking solution (2% BSA, 0.1% Triton X-100, 0.1% sodium azide in PBS). Primary and secondary antibodies were diluted with the blocking buffer and incubated for 1 h each at room temperature. After the immunostaining, the coverslips were extensively rinsed with distilled water and mounted onto glass slides for observation. Neurons were viewed with an inverted light microscope (Zeiss Axiovert 200, Carl Zeiss MicroImaging, Inc.) equipped with epifluorescence optics. Images were captured with a CCD camera controlled by Axiovision software (Carl Zeiss MicroImaging, Inc.). For embryonic cortical neurons, the axons were stained with either the anti-βIII tubulin antibody (Tuj1) or anti-Tau1 antibody. The axons were then manually traced, and the axon lengths were recorded. For adult DRG neurons, after staining with Tuj1, the longest axon of each neuron was traced and measured. Axon length was measured with the “measure/curve” application of AxioVision software.
For quantification of axon length, we restricted the analysis to neurons with processes equal to or longer than two cell bodies in diameter. In each experiment, ∼100 neurons per condition were measured to calculate the mean value. The mean and SEM of neurite-bearing cells were calculated from at least three independent experiments. Thus, n values indicate the number of independent experiments performed.
In vivo electroporation of adult DRG neurons and quantification of axon regeneration
The in vivo electroporation of adult mouse DRGs was performed as described previously (Saijilafu et al. 2011). Briefly, under anesthesia induced by katamine (100 mg/kg) and xylazine (10 mg/kg), a small dorsolateral laminectomy was performed to expose the left L4–L5 DRGs. EGFP plasmid (3–4 μg/μL) or EGFP plus miR-138 RNA oligos (100 μM) were injected into the DRGs using pulled-glass capillaries (1.5 μL per ganglion). Immediately after injection, electroporation was performed by applying five pulses of current (35 V for 15 msec at 950-msec intervals) using a custom-made tweezer-like electrode powered by the Electro Square Porator ECM830 (BTX Genetronics). The wound was then closed, and the mice were allowed to recover. Two days after the electroporation, the sciatic nerves were crushed with fine forceps, and the crushed sites were marked with nylon epineural sutures. Three days later, the mice were perfused with 4% PFA in sodium phosphate buffer (pH 7.4). The whole nerve segment was then dissected out and further fixed in 4% PFA overnight at 4°C. Before whole-mount flattening, it was confirmed that the place of epineural suture matched the injury site, and experiments were included in the analysis only when the crush site was clearly identifiable. The same control group was used to determine how overexpression of the miR-138 mimics or knocking down of SIRT1 with siRNA affected axon regeneration.
For quantification of in vivo axon regeneration, the fluorescence images of flattened whole-mount nerves were first obtained. All identifiable EGFP-labeled axons in the sciatic nerve were then manually traced from the crush site to the distal growth cone to measure the length of axon regeneration. The n values indicate the number of mice.
qRT-PCR of SIRT1
Total RNA was isolated with the TRizol reagent (Invitrogen), and RNA was reverse-transcribed by using Moloney murine leukemia virus reverse transcriptase (Roche Applied Science). To quantify the mRNA levels with the RT-PCR, aliquots of single-stranded cDNA were amplified with gene-specific primers and Power SYBR Green PCR Master Mix (Invitrogen) using the CFX96 RT-PCR detection system (Bio-Rad). The PCR reactions contained 20–40 ng of cDNA, Universal Master Mix (Invitrogen), and 200 nM forward and reverse primers in a final reaction volume of 20 μL. The ratio of different samples was calculated by the data analysis software built in with the CFX96 RT-PCR system. The sequences of the SIRT1 primers used were forward, CGTCTCAGCGTCACTCCCAAGC; and reverse, ACGCAATCCTGCTCCCTCCC.
Quantification of mature microRNAs using RT-PCR
Individual reverse transcription and TaqMan microRNA assays were performed on a CFX96 RT-PCR detection system. The 15-μL reverse transcription reactions consisted of 10 ng of total RNA isolated with TRIzol (Invitrogen, 15596-026), 5 U of MultiScribe reverse transcriptase, 0.5 mM dNTPs, 1× reverse transcription buffer, 4 U of RNase inhibitor, and nuclease free water. The reverse transcription reactions were incubated for 30 min at 16°C, 30 min at 42°C, and 5 min at 85°C and then stored at 4°C until used in TaqMan assays. The 10-μL TaqMan RT-PCR reactions included 1× TaqMan universal PCR master mix, 1× TaqMan microRNA primers for miR-138 or RNU6B, 1.33 μL of undiluted cDNA, and nuclease free water. The reactions were run with the standard cycling protocol without the 50°C incubation stage. The reactions were incubated for 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. The fluorescence readings were collected during the 60°C step. Each TaqMan assay was done in either triplicate or quadruplicate for each sample tested. Relative quantities were calculated using the ΔΔCT method with RNU6B TaqMan microRNA control assay as the endogenous control and calibrated to the wild-type samples (Livak and Schmittgen 2001).
ChIP
ChIP was performed according to the published method (Liu et al. 2010a). Briefly, six to eight naïve or 10–15 axotomized L4 and L5 DRGs were collected and homogenized with 1% formaldehyde (Sigma-Aldrich) for 20 min on ice. The homogenized tissue was washed with cold PBS, suspended with 200 μL of cold cell lysis buffer (5 mM PIPES at pH 8.0, 85 mM KCl, 0.5% NP40, 1× complete proteinase inhibitor) and then incubated for 5 min on ice. The lysates were centrifuged at 3000 rpm for 5 min, and the pellets were resuspended in 200 μL of SDS lysis buffer (Millipore). After 10 min of incubation on ice, lysates were sonicated (six pulses, 10 sec each, at a power output of 40%, with 1-min incubations on ice in between each pulse) to shear the genomic DNA into 200- and 1000-base-pair (bp) fragments. To verify the size of the sheared chromatin (average size ∼500–600 bp), 5-μL aliquots of the lysates were treated with 1 μL of proteinase A (20 mg/mL) for 20 min at 50°C, and the sample was analyzed using a 1.5% agarose gel.
To perform the immunoprecipitation, the sonicated cell supernatant was diluted 10-fold in ChIP dilution buffer (Millipore), and the protease inhibitor was added. To reduce nonspecific background, the diluted cell supernatant was precleared with 80 μL of salmon sperm DNA/protein-A agarose–50% slurry (Millipore) for 30 min at 4°C with agitation. The agarose was then removed by brief centrifugation. The precleared chromatin was rotated overnight at 4°C with 10 μg of normal rabbit IgG (Upstate Biotechnology) or rabbit anti-SIRT1 (Millipore). Antibodies were pulled down with 70 μL of blocked protein-A agarose beads for 1 h at 4°C with rotation. The beads were then washed sequentially (twice for each solution) in immunoprecipitation dilution buffer, TSE-500 solution (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris at pH 8.1, 500 mM NaCl), freshly prepared LiCl washing solution (100 mM Tris at pH 8.1, 300 mM LiCl, 1% NP40, 1% deoxycholic acid), and 1× TE for 10 min at 4°C. Protein–DNA complexes were eluted from the protein-A agarose beads twice with 250 μL of immunoprecipitation elution buffer (50 mM NaHCO3, 1% SDS) for 15 min at 37°C with rotation. Formaldehyde-induced protein–DNA cross-linking was heat-reversed by incubating the protein–DNA complex overnight at 65°C. DNA was purified using phenol:chloroform:isoamyl alcohol (25:24:1) isolations and precipitated with 2 vol of 100% ethanol containing 10 μg of linear acrylamide overnight at −80°C. Immunoprecipitated and purified DNA fragments were resuspended in nuclease-free water. The DNA concentrations were determined, and each sample was diluted to 1 ng/μL.
Eight nanograms of DNA was used in 20-μL SYBR Green RT-PCR reactions, including 1× Power SYBR Green Master Mix and 0.5 μM forward and reverse primers. Reactions were run on a CFX96 RT-PCR system (Bio-Rad) using the standard default cycling protocol without the 50°C incubation: 10 min at 95°C and 40 cycles of 15 sec at 95°C and 1 min at 60°C. Primers sequences spaced at 1-kb intervals spanning 4 kb upstream of to 1 kb downstream from mmu-mir-138-1 on chromosome 8 were designed using the primer premier 5.0 software: (1) 4 kb upstream (FW, 5′-CTGAACCCAGGTACAAAGCAG-3′; RV, 5′-CAAGAACAGAAGGGAGAGGC-3′), (2) 3 kb upstream (FW, 5′-AGATGGGGTGTCTCTTGTTAAAG-3′; RV, 5′-CCTCTGTCTGCTTTCTCTTTGG-3′), (3) 2 kb upstream (FW, 5′-GCACCTCATACTGAAACCAAAGC-3′; RV, 5′-CCTATATCAAGCCCTGCCAAC-3′), (4) 1 kb upstream (FW, 5′-GCCTGTGCTGTCTTCCTCTC-3′; RV, 5′-TCCCATACCCTCGCTCTAAC-3′), and (5) 1 kb downstream (FW, 5′-TGGAACAGGAAGGAAAACGGA-3′; RV, 5′-GGAGGGTCCCCACAGAAAAC-3′).
Enrichment of DNA was calculated as the ratio of RT-PCR values between the SIRT1 immunoprecipitation sample and the rabbit IgG immunoprecipitation sample. All ChIP experiments were done from three independent chromatin preparation experiments, and all RT-PCR reactions were carried out in triplicate for each sample.
SIRT1 3′ UTR dual-luciferase assay
3′ UTR sequences of SIRT1 mRNA were PCR-amplified from mouse SIRT1 cDNA. The sequences of primers for Sirt1 were forward sequence, CCAGCTCGAGGGATTCAGGAATTGCTCCACCA; and reverse sequence, CCAGGCGGCCGCCTCCTCTGGCAGTAATGGTCCT.
The primers were designed to include XhoI and NotI restriction sites and a 4-bp extra random sequence. The PCR products were digested with XhoI and NotI and then cloned into the psiCHECK-2 dual-luciferase vector (Promega) digested with the same enzymes. The obtained constructs were cotransfected with miR-138 mimics or inhibitor into CAD cells using Lipofectine 2000 (Invitrogen). Luciferase expression was detected using the dual-luciferase reporter 1000 system (Promega) according to manufacturer's protocol. Briefly, 48 h after transfection, the cell culture medium was removed, and cells were lysed with 20 μL of 1× lysis buffer for 15 min at room temperature. One-hundred microliters of luciferase assay buffer II was added and mixed briefly. Firefly luciferase (F-luc) activity was immediately read using a microplate reader (Molecular Devices). One-hundred microliters of Stop & Glo buffer with Stop & Glo substrate was then added and mixed briefly, and R-luc activity was immediately read. R-luc activity was normalized to F-luc activity to normalize the variation in transfection efficiencies. All luciferase readings were taken from three or four individual wells for each psiCHECK-2-3′ UTR construct or control construct tested. The miR-138 target site in the Sirt1-3′ UTR was changed using the PCR method based on the study by Rivetti di Val Cervo et al. (2012).
Statistics
Data are presented as mean ± SEM. Two-tailed Student's t-test was used to determine the statistical significance between different experimental groups, which was set at a value of P < 0.05.
Acknowledgments
This work was supported by grants (to F.Q.Z) from the National Institutes of Health (R01NS064288) and The Craig H. Neilsen Foundation. The SIRT1 and SIRT1 (H363Y) plasmids were created by the Michael Greenberg laboratory and were purchased from Addgene. C.L. and F.Z. developed the project and designed the experiments. C.L. and R.W. performed the major experiments. S., Z.J., and B.Z. were involved in some experiments. F.Z. supervised the project and cowrote the manuscript with C.L.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.209619.112.
References
- Araki T, Sasaki Y, Milbrandt J 2004. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305: 1010–1013 [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015 [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Bonev B, Garcez P, Stanley P, Guillemot F, Papalopulu N 2012. MicroRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nat Neurosci 15: 697–699 [DOI] [PubMed] [Google Scholar]
- Ebert MS, Sharp PA 2012. Roles for microRNAs in conferring robustness to biological processes. Cell 149: 515–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA 2007. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4: 721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineberg SK, Kosik KS, Davidson BL 2009. MicroRNAs potentiate neural development. Neuron 64: 303–309 [DOI] [PubMed] [Google Scholar]
- Franke K, Otto W, Johannes S, Baumgart J, Nitsch R, Schumacher S 2012. miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling. EMBO J 31: 2908–2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L, Mak G, Kim D, Su SC, Tsai LH 2010. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466: 1105–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaub P, Tedeschi A, Puttagunta R, Nguyen T, Schmandke A, Di Giovanni S 2010. HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ 17: 1392–1408 [DOI] [PubMed] [Google Scholar]
- Gaub P, Joshi Y, Wuttke A, Naumann U, Schnichels S, Heiduschka P, Di Giovanni S 2011. The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 134: 2134–2148 [DOI] [PubMed] [Google Scholar]
- Guo W, Qian L, Zhang J, Zhang W, Morrison A, Hayes P, Wilson S, Chen T, Zhao J 2011. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J Neurosci Res 89: 1723–1736 [DOI] [PubMed] [Google Scholar]
- Hur EM, Saijilafu, Lee BD, Kim SJ, Xu WL, Zhou FQ 2011a. GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev 25: 1968–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Yang IH, Kim DH, Byun J, Saijilafu, Xu WL, Nicovich PR, Cheong R, Levchenko A, Thakor N, et al. 2011b. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc Natl Acad Sci 108: 5057–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisliouk T, Yosefi S, Meiri N 2011. miR-138 inhibits EZH2 methyltransferase expression and methylation of histone H3 at lysine 27, and affects thermotolerance acquisition. Eur J Neurosci 33: 224–235 [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20 [DOI] [PubMed] [Google Scholar]
- Li X, Jin P 2010. Roles of small regulatory RNAs in determining neuronal identity. Nat Rev Neurosci 11: 329–338 [DOI] [PubMed] [Google Scholar]
- Li XH, Chen C, Tu Y, Sun HT, Zhao ML, Cheng SX, Qu Y, Zhang S 2013. Sirt1 promotes axonogenesis by deacetylation of Akt and inactivation of GSK3. Mol Neurobiol doi: 10.1007/s12035-013-8437-3 [DOI] [PubMed] [Google Scholar]
- Liu C, Zhao X 2009. MicroRNAs in adult and embryonic neurogenesis. Neuromolecular Med 11: 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Teng ZQ, Santistevan NJ, Szulwach KE, Guo W, Jin P, Zhao X 2010a. Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell 6: 433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Tedeschi A, Park KK, He Z 2010b. Neuronal intrinsic mechanisms of axon regeneration. Annu Rev Neurosci 34: 131–152 [DOI] [PubMed] [Google Scholar]
- Liu CM, Hur EM, Zhou FQ 2012a. Coordinating gene expression and axon assembly to control axon growth: Potential role of GSK3 signaling. Front Mol Neurosci 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lv XB, Wang XP, Sang Y, Xu S, Hu K, Wu M, Liang Y, Liu P, Tang J, et al. 2012b. miR-138 suppressed nasopharyngeal carcinoma growth and tumorigenesis by targeting the CCND1 oncogene. Cell Cycle 11: 2495–2506 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL 2009. KLF family members regulate intrinsic axon regeneration ability. Science 326: 298–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernosterer G, Leuschner PJ, Alenius M, Martinez J 2006. Post-transcriptional regulation of microRNA expression. RNA 12: 1161–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, et al. 2008. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322: 963–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW 2010. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther 9: 844–855 [DOI] [PubMed] [Google Scholar]
- Rivetti di Val Cervo P, Lena AM, Nicoloso M, Rossi S, Mancini M, Zhou H, Saintigny G, Dellambra E, Odorisio T, Mahe C, et al. 2012. p63-microRNA feedback in keratinocyte senescence. Proc Natl Acad Sci 109: 1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijilafu, Zhou FQ 2012. Genetic study of axon regeneration with cultured adult dorsal root ganglion neurons. J Vis Exp 66: e4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijilafu, Hur EM, Zhou FQ 2011. Genetic dissection of axon regeneration via in vivo electroporation of adult mouse sensory neurons. Nat Commun 2: 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto J, Miura T, Shimamoto K, Horio Y 2004. Predominant expression of Sir2α, an NAD-dependent histone deacetylase, in the embryonic mouse heart and brain. FEBS Lett 556: 281–286 [DOI] [PubMed] [Google Scholar]
- Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, Khudayberdiev S, Leuschner PF, Busch CJ, Kane C, et al. 2009. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat Cell Biol 11: 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel G, Saba R, Schratt G 2011. MicroRNAs in neurons: Manifold regulatory roles at the synapse. Curr Opin Genet Dev 21: 491–497 [DOI] [PubMed] [Google Scholar]
- Smith DS, Skene JH 1997. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci 17: 646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland IT, Richards L, Holmes FE, Wynick D, Uney JB, Wong LF 2011. Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS ONE 6: e23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino T, Maruyama M, Tanno M, Kuno A, Houkin K, Horio Y 2010. Protein deacetylase SIRT1 in the cytoplasm promotes nerve growth factor-induced neurite outgrowth in PC12 cells. FEBS Lett 584: 2821–2826 [DOI] [PubMed] [Google Scholar]
- Vo NK, Cambronne XA, Goodman RH 2010. MicroRNA pathways in neural development and plasticity. Curr Opin Neurobiol 20: 457–465 [DOI] [PubMed] [Google Scholar]
- Wu D, Raafat A, Pak E, Clemens S, Murashov AK 2012. Dicer-microRNA pathway is critical for peripheral nerve regeneration and functional recovery in vivo and regenerative axonogenesis in vitro. Exp Neurol 233: 555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakuchi M 2012. MicroRNA regulation of SIRT1. Front Physiol 3: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D, Wang G, Liu Y, Huang W, Wu M, Zhu S, Jia W, Deng AM, Liu H, Kang J 2012. miR-138 promotes induced pluripotent stem cell generation through the regulation of the p53 signaling. Stem Cells 30: 1645–1654 [DOI] [PubMed] [Google Scholar]
- Zhang HY, Zheng SJ, Zhao JH, Zhao W, Zheng LF, Zhao D, Li JM, Zhang XF, Chen ZB, Yi XN 2011. MicroRNAs 144, 145, and 214 are down-regulated in primary neurons responding to sciatic nerve transection. Brain Res 1383: 62–70 [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Walzer M, Wu YH, Zhou J, Dedhar S, Snider WD 2006. Neurotrophins support regenerative axon assembly over CSPGs by an ECM-integrin-independent mechanism. J Cell Sci 119: 2787–2796 [DOI] [PubMed] [Google Scholar]
- Zhou S, Yu B, Qian T, Yao D, Wang Y, Ding F, Gu X 2011. Early changes of microRNAs expression in the dorsal root ganglia following rat sciatic nerve transection. Neurosci Lett 494: 89–93 [DOI] [PubMed] [Google Scholar]