

Bunyaviruses are a group of medically important viruses, many of which are insect-borne. Using genome-wide screening in Drosophila with the bunyavirus Rift Valley fever virus (RVFV), Hopkins et al. now identify mRNA decapping as a control point for viral RNA transcription. Depletion of the mRNA decapping enzyme Dcp2 enhances viral transcription, whereas ectopic expression impedes it. This study suggests a model whereby RVFV competes for the Dcp2-accessible mRNA pool, which can present a bottleneck for viral replication.
Keywords: Dcp2, RNA decapping, RVFV, bunyavirus, cap-snatching, RNAi screen
Abstract
Bunyaviruses are an emerging group of medically important viruses, many of which are transmitted from insects to mammals. To identify host factors that impact infection, we performed a genome-wide RNAi screen in Drosophila and identified 131 genes that impacted infection of the mosquito-transmitted bunyavirus Rift Valley fever virus (RVFV). Dcp2, the catalytic component of the mRNA decapping machinery, and two decapping activators, DDX6 and LSM7, were antiviral against disparate bunyaviruses in both insect cells and adult flies. Bunyaviruses 5′ cap their mRNAs by “cap-snatching” the 5′ ends of poorly defined host mRNAs. We found that RVFV cap-snatches the 5′ ends of Dcp2 targeted mRNAs, including cell cycle-related genes. Loss of Dcp2 allows increased viral transcription without impacting viral mRNA stability, while ectopic expression of Dcp2 impedes viral transcription. Furthermore, arresting cells in late S/early G2 led to increased Dcp2 mRNA targets and increased RVFV replication. Therefore, RVFV competes for the Dcp2-accessible mRNA pool, which is dynamically regulated and can present a bottleneck for viral replication.
RNA stability is a key factor in the regulation of eukaryotic gene expression. Within the RNA moiety, cis elements, including the 5′ 7mG cap and the 3′ poly-A tail, play dual roles in protecting the mRNA from exonuclease-mediated degradation and promoting translation. RNA degradation is both actively regulated and an essential part of normal RNA turnover (Tucker and Parker 2000). Two strategies account for the majority of mRNA turnover: 3′-to-5′-mediated decay via the exosome and 5′-to-3′ degradation by the exonuclease Xrn1. Both strategies are dependent on loss of protective cis elements; initial deadenylation of the poly-A tail signals for both exosome-dependent targeting and removal of the 5′ 7mG cap by the canonical decapping enzyme Dcp2 (Tucker and Parker 2000). Dcp2 cleavage of the cap exposes a 5′ monophosphate that is the substrate for Xrn1 (Muhlrad et al. 1994). Furthermore, perhaps as a regulatory mechanism, the RNA degradation machinery is largely compartmentalized within the cytoplasm. The decapping machinery and the 5′-to-3′ exonuclease are localized to processing (P) bodies (Ingelfinger et al. 2002; Van Dijk et al. 2002; Sheth and Parker 2003). P bodies are granules of ribonucleoproteins (RNPs), microscopically visible, and dynamic in their size and number. Additionally, P bodies act as storage depots; some RNAs targeted to the P body are degraded, while others may be released (Parker and Sheth 2007). Thus, the dynamic control of mRNA stability and turnover can be regulated by P-body biology. This is consistent with the fact that cellular conditions, including stress and translational inhibition, alter the visible morphology of P bodies within the cytoplasm (Eulalio et al. 2007b). Interestingly, however, microscopically visible P-body punctae are dispensable for the function of multiple mRNA decay pathways, suggesting that their structure is a marker for increased pools of accumulating mRNAs (Eulalio et al. 2007b).
As obligate intracellular pathogens with limited coding capacity, viral RNAs must replicate to high levels and hijack the translation apparatus while simultaneously avoiding the host's degradation machinery. Furthermore, RNA viruses must also maintain the stability of different RNA species, including the genome, anti-genome, and mRNA. Viruses have evolved complex strategies to protect their 5′ ends from exonucleases while facilitating translation. Some viruses that replicate in the nucleus hijack the endogenous capping machinery (e.g., retroviruses), while viruses that replicate in the cytoplasm cannot. To overcome this barrier, some cytoplasmic viruses encode their own capping machinery and generate mRNAs that resemble endogenous mRNAs (e.g., rhabdoviruses) (Li et al. 2008a). Other viruses protect the 5′ end from degradation by covalently attaching a protein to the 5′ end that prevents targeting by exonucleases (e.g., picornaviruses). However, this prevents canonical translation, and thus these viruses use internal ribosome entry sites to engage the translation machinery (Scotti et al. 1981). Another group of viruses “cap-snatch;” that is, they steal the 5′ end of host mRNAs using a virally encoded endonuclease, generating primers that are used by the viral RNA-dependent RNA polymerase to generate viral mRNAs (Garcin et al. 1995). The 5′ end of the viral mRNA is therefore indistinguishable from endogenous mRNAs and thus is both protected from degradation and able to recruit host ribosomes. All negative-sense segmented RNA viruses (orthomyxoviruses, arenaviruses, and bunyaviruses) cap-snatch. Of these, influenza A virus, an orthomyxovirus, is the best studied and snatches the 5′ end of pre-mRNAs in the nucleus (Herz et al. 1981; Plotch et al. 1981). Since bunyaviruses and arenaviruses replicate in the cytoplasm, they must use a distinct pool of mRNAs; however, while our mechanistic understanding of bunyaviral cap-snatching is increasing (van Poelwijk et al. 1996; van Knippenberg et al. 2005; Mir et al. 2010; Morin et al. 2010; Reguera et al. 2010; Cheng and Mir 2012), little is known about whether the host can combat this replication step or what pool of endogenous mRNAs are being targeted for this process.
Bunyaviruses are an emerging group of medically and agriculturally important viruses, many of which are insect-borne. Rift Valley fever virus (RVFV) is a mosquito-borne emerging bunyavirus in Africa that can cause encephalitic or hemorrhagic symptoms in infected humans, leads to spontaneous miscarriage in pregnant livestock, and causes high rates of mortality in young animals (Pepin et al. 2010; Boshra et al. 2011). Currently, there are no therapeutics or FDA-approved vaccines to combat bunyaviral infection. This is in part due to a lack of understanding of the molecular interactions occurring between bunyaviruses and host cells. We set out to identify host factors that restrict RVFV infection in insects using Drosophila as our model insect due to the ease of genetic manipulation both in vitro and in vivo. RNAi in Drosophila cells is robust, and conserved immune biology with humans has been demonstrated (e.g., Toll) (Lemaitre et al. 1996), suggesting that we can use Drosophila to probe both insect and human antiviral factors (Cherry 2005; Hao et al. 2008; Sabin et al. 2009; Sessions et al. 2009; Moser et al. 2010; Rose et al. 2011). Furthermore, Drosophila has been used as a model to study arboviral infection, including RVFV (Sabin et al. 2009; Sessions et al. 2009; Filone et al. 2010; Moser et al. 2010; Rose et al. 2011; Nakamoto et al. 2012).
Using genome-wide RNAi screening in Drosophila cells, we identified 131 genes that impact RVFV (strain MP12) infection, including Dcp2, the P-body-resident mRNA decapping enzyme. Dcp2 restricts RVFV in Drosophila and mosquito cells and also in adult flies. This restriction is likely general to bunyaviruses, since the distantly related bunyavirus La Crosse virus (LACV) is also restricted by Dcp2. Mechanistically, we found that the viral nucleocapsid (N) is localized to P bodies, and RVFV competes with the RNA degradation machinery for target mRNAs. Increasing the pool of mRNAs targeted for degradation via the depletion of Dcp2 or cell cycle arrest in late S/early G2 led to increased RVFV replication, while decreasing targets via ectopic expression of Dcp2 restricted infection. Therefore, our data point to a model in which the pool of Dcp2-accessible mRNAs is dynamically regulated and presents a bottleneck for RVFV replication.
Results
Genome-wide screening implicates the mRNA decapping machinery as a restriction factor for RVFV
In order to identify host factors that restrict RVFV, we performed a high-content genome-wide RNAi screen in Drosophila cells. Briefly, 384-well plates were arrayed with dsRNAs targeting ∼13,000 genes in the Drosophila genome (Ambion); Drosophila cells were seeded, and knockdown was allowed to proceed for 3 d. Cells were then infected with the MP12 strain of RVFV (multiplicity of infection [MOI] = 0.25) (Caplen et al. 1985), which differs by only 11 amino acids from the wild-type strain ZH548, making it likely that cellular factors that impact MP12 replication will also impact wild-type strains (Vialat et al. 1997). Thirty hours post-infection (hpi), cells were stained for total nuclei and RVFV N. Automated microscopy followed by automated image analysis was used to calculate the average percent infection per well (RVFV N-positive cells/total cells) from four sites per well, and the screen was performed in duplicate. Genes with a robust Z-score of ≥1.3 or less than or equal to−1.3 in duplicate (P < 0.05) that were nontoxic (robust Z-score greater than or equal to−2 in duplicate) were considered hits (Fig. 1A). One-hundred-seventy-nine genes were identified, among which 56 were part of multisubunit complexes (e.g., ribosome and proteasome). Therefore, we chose only one or two genes per complex to verify as a representative, leaving 143 genes to validate. We generated independent dsRNAs targeting unique regions of 143 genes from the initial set and validated 85 genes. Including the genes identified in the primary screen that were validated by another member of their complex, this comes to 131 genes. One-hundred-twenty-four genes restricted infection, while seven promoted infection (Fig. 1B; Supplemental Fig. S1; Supplemental Table S1). In addition, gene ontology (GO) enrichment analysis indicated that these candidates were significantly enriched for genes involved in DNA replication, the cell cycle, and mRNA metabolic processes (Fig. 1C). Importantly, our gene list was also enriched for genes conserved with humans and mosquitoes; 124 were conserved with mosquitoes and humans, four were conserved in mosquitoes, and only three did not have identified orthologs in these groups. This suggests that the factors and pathways identified may have conserved interactions with RVFV replication across multiple relevant hosts.
Figure 1.
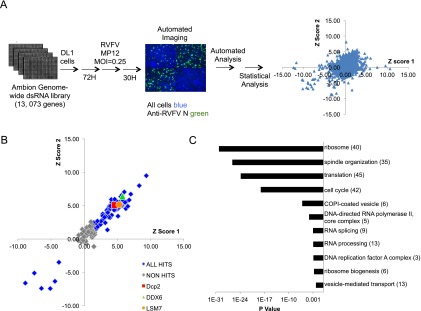
Genome-wide RNAi screen for host factors that impact RVFV in Drosophila. (A) Genome-wide RNAi screen pipeline. Cells were plated onto 384-well plates preplated with dsRNAs targeting the Drosophila genome. Three days later, cells were infected with RVFV MP12 (MOI = 0.25) for 30 h and processed for immunofluorescence. (Green) RVFV N; (blue) nuclei. Automated microscopy followed by image analysis was used to calculate robust Z-scores, which are shown for each replicate of the screen. (B) Primary candidates were validated using independent dsRNAs; for complexes with multiple candidates represented in the primary pool, a selection of genes was validated as representative of the complex. Robust Z-scores from 143 genes shown with validated genes in blue and genes that did not validate in gray. Dcp2, DDX6, and LSM7 are shown in red, green, and orange, respectively. (C) GO term enrichment for validated genes.
Additionally, we validated three genes that reside in P bodies (Sheth and Parker 2003): the canonical mRNA decapping enzyme Dcp2, LSM7 (part of the heptameric LSM1–7 complex that participates in decapping activation) (Nissan et al. 2010), and the Drosophila homolog of human DDX6/Rck/p54 (Me31B), which has been characterized as an activator of decapping in yeast (Coller et al. 2001; Fischer and Weis 2002), although its mechanism is unclear (Nakamura et al. 2001; Weston 2006; Sweet et al. 2012) (Figs. 1B, 2A–C; Supplemental Figs. S1, S2; Supplemental Table S1). P-body components participate in multiple mRNA degradation pathways, including silencing and decapping (Eulalio et al. 2007a), and P-body morphology can be altered by the loss of specific P-body components. Loss of Dcp2 leads to increased P-body size, while the loss of DDX6 leads to the dispersal of P bodies in Drosophila (Eulalio et al. 2007b; data not shown). Furthermore, visible P bodies are not necessarily required for functional activity of P-body-resident proteins, including RNA silencing, nonsense-mediated decay (NMD), and mRNA decay (Eulalio et al. 2007b). Therefore, we set out to elucidate whether P bodies themselves or particular functions within P bodies, such as decapping, were specifically involved in RVFV restriction. We screened seven other canonical P-body-resident proteins—including HPat (Patr-1), GW182 (gw), Dcp1 (a binding partner for Dcp2), EDC4 (Ge-1), staufen (stau), LSM14A (tral; not part of the LSM1–7 complex), and EDC3—and found that none of them impacted RVFV replication greater than twofold, although loss of LSM14A or EDC3 led to modest but significantly increased levels of RVFV infection (Fig. 2C). Interestingly, LSM14A and EDC3 are mRNA decapping activators in yeast (Nissan et al. 2010) and are required for DDX6 recruitment to P bodies in Drosophila (Tritschler et al. 2009), suggesting that their effects may be through this mechanism. Our data suggest that while RVFV replication is restricted by mRNA decapping (Dcp2 and four decapping activators), P-body integrity is not essential for this restriction or RVFV replication.
Figure 2.

Decapping restricts RVFV replication in Drosophila cells. (A) Drosophila cells were treated with the indicated dsRNA for 3 d, then infected with RVFV (MOI 0.01), and processed for immunofluorescence. (Green) RVFV N protein; (blue) nuclei. (B) Cells were treated as in A and infected with the indicated virus. dsRNA targeting each virus was used as a positive control (virus). Quantification of mean fold change in the percentage of cells infected with the indicated virus is shown. (Blue) DCV; (purple) SIN; (red) VSV; (green) RVFV (MP12 strain). Mean ± SD of three or more independent experiments. (**) P < 0.01. (C) Cells treated as in A with P-body component dsRNAs were quantified for the mean fold change in the percentage of RVFV-infected cells. Decapping activators are indicated, and genes significantly affecting RVFV replication are highlighted in blue. Mean ± SD of three or more independent experiments. (*) P < 0.05. (D) Representative deconvolved plane of a Drosophila cell expressing myc-Dcp2 (green) and infected with RVFV for 30 h. (Red) RVFV N; (blue) nuclei. (E) Quantification of RVFV N and myc-Dcp2 punctae colocalization events per cell (>150 cells from three independent experiments). The majority of infected cells (∼90%) have at least one colocalization between Dcp2 and RVFV N.
Dcp2 restricts the bunyavirus RVFV but not other families of RNA viruses
In order to determine the specificity of this restriction, we tested whether Dcp2 impacts the replication of RNA viruses from three disparate families: Drosophila C virus (DCV), Sindbis virus (SINV), and vesicular stomatitis virus (VSV). DCV is a positive-sense RNA picorna-like virus and natural pathogen of Drosophila (Johnson and Christian 1998; Cherry and Perrimon 2004). DCV does not use a 5′ cap for translation but rather couples a protein to the 5′ end for protection and uses internal ribosome entry sites for translation (Scotti et al. 1981; Wilson et al. 2000). SINV is a human arbovirus that is a positive-sense RNA alphavirus, and VSV is an arbovirus that is a negative-sense ssRNA rhabdovirus; both of these viruses encode their own 5′ capping machinery (Ahola and Kääriäinen 1995; Li et al. 2008a). We found that Dcp2 specifically restricts RVFV, since depletion of Dcp2 has no impact on the level of infection by DCV, SINV, or VSV (Fig. 2B). Since the only known role for Dcp2 is in decapping and it selectively restricted RVFV, we hypothesized that decapping per se specifically limits RVFV replication through this biological function.
RVFV N associates with P bodies
Since we identified P-body-resident proteins as antiviral against RVFV, we tested whether the viral replication machinery and P bodies interact during RVFV infection. Previous studies of the distantly related bunyavirus Sin Nombre virus found that the N protein, necessary for cap-snatching, forms visible punctae in mammalian cells that colocalize with the P-body-resident protein DCP1a (Mir et al. 2008). We generated cells expressing Dcp1-GFP, which labels P bodies (Eulalio et al. 2007b), and infected them with RVFV for 30 h. Analysis of these cells demonstrated that N punctae partially overlapped with P-body punctae (Supplemental Fig. S3A). More than half of the infected cells presented with at least one colocalization (Supplemental Fig. S3B). Furthermore, the majority of these events presented with partial overlap rather than complete colocalization (only three colocalizations were found to be coincident), resembling previous reports showing that P bodies may contain distinct compartments (Sen and Blau 2005; Weil et al. 2012). Additionally, colocalization studies examining P bodies and the Ty3 retrotransposon in yeast have shown similar patterns of partial overlap (Beliakova-Bethell et al. 2006). Since Dcp2, but not Dcp1, is antiviral (Fig. 2C), we next examined whether Dcp2 colocalized with RVFV N. We generated cells expressing myc-Dcp2 (Jäger and Dorner 2010) and infected them with RVFV for 30 h. We found that coincident colocalization occurred in ∼90% of infected cells (Fig. 2D, E), while only a small subset of cells showed instances of partial overlap between RVFV N and Dcp2 punctae (∼2%). Interestingly, when we coexpressed Dcp1-GFP and myc-Dcp2, we saw a spectrum of overlap where the large majority of punctae had substantial overlap of Dcp1 and Dcp2, while others showed partial overlap or no overlap (Supplemental Fig. S3C). This suggests complexity to these compartments. Taken together, our data suggest that RVFV (and perhaps bunyaviruses in general) interacts with P bodies or P-body-resident proteins.
Dcp2 restricts bunyavirus infection in adult flies
We set out to determine the role of Dcp2 in antiviral defense against RVFV at the organismal level in adult flies (Filone et al. 2010). Arboviral infection of the insect host is controlled by the innate immune system. If compromised, an otherwise nonlethal infection can become pathogenic and potentially fatal (van Rij et al. 2006; Wang et al. 2006; Sabin et al. 2009). Since Dcp2 is an essential gene (Lin et al. 2006; Pressman et al. 2012), we used in vivo RNAi technology to deplete Dcp2 post-developmentally in adult flies to determine the impact of the decapping machinery on RVFV replication. Briefly, transgenic flies bearing a UAS element driving the expression of an inverted repeat (IR) against Dcp2 were crossed to flies expressing the Gal4 protein under the control of a heat-shock-inducible promoter. Adult flies were subjected to heat shock, driving expression of the snap-back transgene and mRNA depletion, which had no impact on survival (Supplemental Fig. S3A,B). As expected, control flies infected with RVFV presented with little mortality; however, Dcp2-depleted flies succumbed to RVFV infection (Fig. 3A). This increase in mortality was accompanied by increased levels of viral replication as measured by Northern blot at 6 d post-infection (dpi) (Fig. 3B, C). We also examined viral RNA levels at 20 dpi, when flies are dying from infection, and found that while we see less of an increase in Dcp2-depleted flies compared with day 6, increased replication continues late into infection (Supplemental Fig. S4C,D). We hypothesize that the differences we see are greater at day 6 due to highly infected flies in the population succumbing to infection by 20 dpi. Additionally, we tested whether decapping restricted another distantly related bunyavirus, LACV, an orthobunyavirus transmitted by mosquitoes to humans (Gerhardt et al. 2001; Haddow and Odoi 2009). Control flies challenged with LACV presented with little mortality, while Dcp2-depleted flies succumbed to LACV infection (Fig. 3D). Additionally, we found increased viral RNA levels in flies infected with LACV as measured by quantitative RT–PCR 6 dpi (Supplemental Fig. S4E). Thus, Dcp2 restricts bunyaviruses both in cell culture and in vivo in adult flies.
Figure 3.
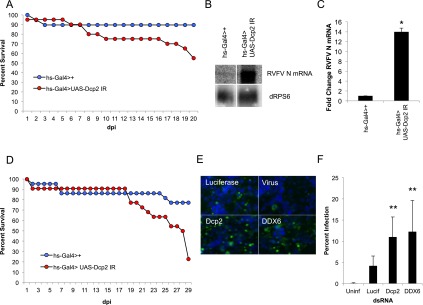
Dcp2 restricts bunyaviruses in flies and mosquitoes. (A) Adult flies carrying heat-shock-inducible Gal4 were crossed to flies that inducibly express dsRNA against Dcp2 (hs-Gal4>UAS-Dcp2 IR [red]), or controls (hs-Gal4>+ [blue]) were challenged with RVFV, and the percentage of survival is graphed as a function of days post infection (dpi). A representative of at least three experiments is shown. P < 0.001 log-rank test. (B) Northern blot analysis of RVFV-infected control flies and Dcp2-deficient flies probed for RVFV N mRNA or dRPS6 (cellular loading control) 6 dpi. (C) Quantification of three experiments as shown in B. Mean ± SD. (*) P < 0.05. (D) Flies as described in A were challenged with LACV. Percentage of survival is graphed as a function of time. A representative of at least three experiments is shown. P < 0.001 log-rank test. (E) Representative immunofluorescence of A. aegypti Aag-2 cells treated with the indicated dsRNA and subsequently infected with RVFV. (F) Quantification of mean fold change in percentage of infection with mean ± SD shown. (**) P < 0.01.
Decapping restricts RVFV in Aedes aegypti mosquito cells
RVFV is unusual among arboviruses in that it has been isolated in nature from a large number of mosquito species, and numerous mosquitoes can experimentally transmit this virus, including A. aegypti, the major vector for Dengue virus (Turell et al. 2010). Since there are no cell lines available for many of the more common vector mosquitoes that transmit RVFV and their genomes have not been sequenced, we took advantage of the fact that A. aegypti is a sequenced vector mosquito. Aag-2 cells, derived from these mosquitoes, are permissive to RVFV and amenable to robust RNAi (Fig. 3E; Campbell et al. 2008; Moutailler et al. 2011). There are two annotated Dcp2 orthologs in the A. aegypti genome (AAEL015607 and AAEL000783) with 99% amino acid sequence identity, allowing us to design a single dsRNA targeting a conserved region of both Dcp2 genes to deplete both simultaneously. Aag-2 cells were subjected to RNAi using the same method as with Drosophila cells. Three days post-RNAi, cells were challenged with RVFV (MOI of 0.06) for 24 h, fixed, and stained for total nuclei (Fig. 3E, blue) and RVFV N protein (Fig. 3E, green). Automated microscopy and automated image analysis revealed a significant increase in the percentage of RVFV-infected Dcp2-depleted mosquito cells (Fig. 3E,F), suggesting that decapping is a conserved mechanism of bunyaviral restriction among insects.
Dcp2 does not restrict RVFV by directly decapping viral mRNAs
One major difference between the replication strategies of segmented negative-sense viruses (RVFV and LACV) and nonsegmented negative-sense viruses (VSV) is the mechanism by which they cap their viral mRNAs. While VSV encodes its own 5′ capping machinery (VSV L) (Li et al. 2008a), RVFV does not. Rather, all segmented negative-sense RNA viruses (bunyaviruses, arenaviruses, and orthomyxoviruses) “cap-snatch” the 5′ ends of host mRNAs, simultaneously defending the 5′ end of their mRNAs from exonucleases and facilitating translation. Bunyavirus-encoded N protein specifically binds the 5′ caps of host mRNAs (Mir et al. 2010). The viral RNA-dependent RNA polymerase (L), which has endonucleolytic activity (Patterson et al. 1984; Reguera et al. 2010), then cleaves host mRNAs 10–18 nucleotides (nt) downstream from the 5′ cap and uses this primer as a template for viral mRNA transcription (Patterson et al. 1984; Bouloy et al. 1990; Simons and Pettersson 1991). Thus, all viral mRNAs from this family (RVFV encodes four mRNAs) (Supplemental Fig. S5) begin with a short sequence of nucleotides of nonviral origin.
We reasoned that if Dcp2 restricts RVFV replication at the step of cap-snatching, there are three likely mechanisms: (1) Dcp2 may be restricting the pool of available host mRNAs through normal metabolic turnover (indirect mechanism); (2) following cleavage by the viral L protein, short host-derived primers may be decapped prior to viral RNA transcriptional elongation; and (3) Dcp2 may be directly decapping host mRNA–viral mRNA conjugates following or concomitant to viral transcription (direct mechanism). The second mechanism is highly unlikely; following cap-binding and cleavage, the 5′ cap is bound by viral proteins and thus is likely occluded from Dcp2 recognition (Mir and Panganiban 2008), and in yeast, Dcp2 has been shown to be largely inactive on mRNAs <30 base pairs (bp) (Steiger et al. 2003).
In order to clarify which of the other mechanisms restricts RVFV, we took two approaches: We first assayed the cap status of viral mRNAs in the presence or absence of Dcp2, and second, we examined the decay rate of viral mRNA species in the presence or absence of Dcp2. First, we developed an assay to distinguish capped mRNAs from those that have been decapped by Dcp2, which leaves a 5′ monophosphate on mRNAs following cap cleavage (Muhlrad et al. 1994). Terminator exonuclease degrades 5′ monophosphate-bearing RNAs, including those that are the product of Dcp2-mediated decapping. Cellular 28S rRNA, which natively has a 5′ monophosphate, was completely digested by this enzyme (Fig. 4A,B), while the capped mRNA dRPS6 remained largely intact (95% protected) (Fig. 4A,C,D). We also analyzed the level of background digestion that occurs in this assay by radiolabeling the cap of GFP mRNA transcribed in vitro and then subjecting these capped mRNAs to digestion in the presence or absence of cellular mRNA, as this is the context of our experimental samples. We found that without cellular RNA, ∼18% of the GFP signal was lost in digested samples compared with mock controls. Interestingly, this level of digestion was reduced to ∼12% with the addition of cellular RNA, presumably due to the abundance of rRNA competing for digestion (Supplemental Fig. S6A).
Figure 4.

Dcp2 does not directly degrade viral mRNA. (A) Drosophila cells treated with the indicated dsRNA and infected with RVFV; total RNA was either mock-treated or digested with terminator exonuclease as indicated. Northern blots were probed for RVFV N mRNA (capped transcript), 28S rRNA (5′ monophosphate-bearing control transcript), or the cellular capped dRPS6 mRNA. (B) Quantification of 28S rRNA as shown in A, normalized to luciferase mock-treated. Mean ± SD for three or more independent experiments. (*) P < 0.05. (C) Quantification of RVFV N mRNA in mock-digested samples normalized to luciferase control. Mean ± SD for three or more independent experiments. (*) P < 0.05. (D) Ratio of digested RVFV N mRNA divided by the mock-treated sample, yielding the ratio of mRNA that is refractory to digestion. Mean ± SD for three or more independent experiments. (n.s.) Not significant. (E) Quantification of dRPS6 transcript in mock-digested samples normalized to luciferase control. Mean ± SD for three or more independent experiments. (n.s.) Not significant. (F) Ratio of digested dRPS6 mRNA transcript divided by the mock-treated sample, yielding the ratio of mRNA that is refractory to digestion. Mean ± SD for three or more independent experiments. (n.s.) Not significant. (G) Drosophila cells were treated with the indicated dsRNA and infected with RVFV. Twenty-eight hours post-infection, cells were treated with CHX, and total RNA was collected at the indicated time after CHX treatment. Northern blots were probed for RVFV N mRNA, the S-segment genome, and 28S rRNA as a loading control. (H) Quantification of RVFV N transcript in CHX-treated samples as shown in G. Mean ± SD for three or more independent experiments; no time points were significantly different between control and Dcp2-depleted cells. (I) Quantification of the RVFV S-segment genome in CHX-treated samples as shown in G. Mean ± SD for three or more independent experiments; no time points were significantly different.
If Dcp2 were directly decapping viral mRNAs post-cap-snatching, loss of Dcp2 would increase the proportion of undigested viral mRNA (capped) present in the viral RNA pool. Conversely, if Dcp2 were decapping the mRNA substrates available for RVFV to snatch from, there would be no change in the relative proportion of mRNA that was digested, even though there would be an increase in overall viral mRNA levels.
Since the N and nonstructural protein (NSs) mRNAs are significantly different in size from their genomic segment, this allows us to distinguish them from the small (S) genomic RNA. In contrast, the other viral mRNAs are not sufficiently different in size from the genomic RNA to distinguish by Northern blot (Supplemental Fig. S5). Thus, we chose to examine the N transcript, since it is essential; NSs is dispensable for replication in cell culture (Bouloy and Flick 2009). We found that ∼75% of the viral N mRNA was protected from digestion in control cells, presumably by a 5′ cap (Fig. 4F); accounting for background levels of digestion, this suggests that anywhere from 13% to 25% of the viral mRNA is uncapped natively. Moreover, we observed that Dcp2 knockdown leads to an overall increase in viral N mRNA levels (Fig. 4A,E); however, Dcp2 knockdown did not significantly alter the proportion of protected N mRNA (Fig. 4A,F). This suggests that Dcp2 does not directly decap viral mRNAs. Furthermore, we found that Dcp2 does not seem to regulate the steady-state levels of the housekeeping mRNA dRPS6, as there is no change in dRPS6 mRNA in the presence or absence of Dcp2 with or without digestion (Fig. 4A,C,D). These findings suggest that Dcp2 restricts the RNA substrates available for RVFV-mediated cap-snatching and does not globally affect the cap status of mRNAs within the cell. This is consistent with the finding that Dcp2 does not globally regulate mRNA turnover but impacts the stability of only small subsets of mRNAs from yeast to humans (Li et al. 2008b, 2012; Yoon et al. 2010). Furthermore, depletion of the P-body-resident 5′-to-3′ exonuclease Xrn1 does not impact viral infection levels, N transcript levels, or digestion assay results, suggesting that viral mRNAs themselves are not under considerable pressure from the 5′-to-3′ degradation pathway (Supplemental Fig. S6B–D).
Since there was a degree of background digestion occurring with the exonuclease assay, we set out to directly determine whether Dcp2 impacted viral mRNA stability. Unlike cellular mRNAs, whose decay can be measured by treatment with actinomycin D and subsequent monitoring for loss of mRNA, viral RNA-dependent RNA polymerases are refractory to actinomycin D, preventing the use of this approach. However, a unique feature of bunyaviruses is that transcription of mRNAs is coupled to translation (Barr 2007); therefore, treatment with cycloheximide (CHX), which inhibits translational elongation, prevents viral mRNA transcription, allowing us to examine the rate of viral mRNA decay.
For these studies, we infected dsRNA-treated Drosophila cells with RVFV for 28 h and then treated cells with CHX. We collected total RNA every 30 min and examined viral mRNA levels via Northern blot (Fig. 4G). RVFV N mRNA has a half-life of ∼30 min (Fig. 4H). Furthermore, RVFV N mRNA decayed at the same rate in Dcp2-depleted and control cells (luciferase) (Fig. 4H), suggesting that Dcp2 is not affecting viral mRNA stability. Furthermore, we observed no changes in viral genome levels over time with CHX treatment in the presence or absence of Dcp2, indicating that short-term disruption of translation does not globally impact the stability of other RNA species (Fig. 4I). Altogether, these data suggest that Dcp2 is not directly decapping viral mRNAs but rather decaps specific pools of mRNA that are the preferential targets of RVFV cap-snatching.
RVFV selectively snatches cell cycle-related mRNAs
As our results suggest that Dcp2 and RVFV compete for the same pool of mRNA targets, we set out to determine which mRNAs were being snatched. This may also reveal the particular mRNAs that are regulated by Dcp2-dependent decapping. Thus, we performed 5′ RNA ligase-mediated (RLM)-RACE and sequenced the 5′ ends of viral N mRNA transcripts from RVFV-infected Drosophila cells. Of the 40 sequenced reads, we were able to align 33 to the 5′ end of endogenous RNAs (Fig. 5A; Supplemental Fig. S5). Of these 33, we found four instances in which we found the same gene being snatched in independent experiments, leaving 29 independent genes. There were no obvious consensus sequence motifs within this gene set, suggesting that if there are structural or sequence-specific motifs targeted by Dcp2, they are not contained within the first 15 nt. We found that while nine of the 29 genes had no annotated GO terms, half of the remaining 20 genes (10) had terms associated with the cell cycle and mitosis (Fig. 5A; Supplemental Fig. S7).
Figure 5.
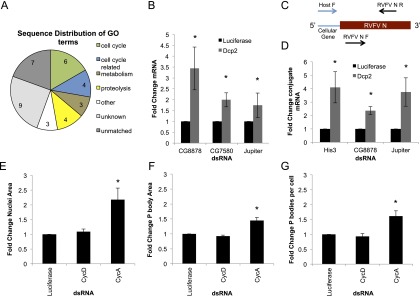
Cell cycle RNAs are an enriched substrate for RVFV cap-snatching. (A) Pie chart of annotated GO function of sequences from the 5′ end of RVFV mRNAs mapped to Drosophila. (B) Quantitative RT–PCR of levels of indicated target mRNAs normalized to RP49 as a cellular loading control and shown as fold change over nontargeting dsRNA (luciferase). Mean ± SD for three or more independent experiments. (*) P < 0.05. (C) Schematic representing the position of RT–PCR primers used in D. Forward primers recognizing the first 11 nt of the host gene 5′ untranslated region (UTR) were used amplify host-RVFV fusion mRNA, and internal primers were used to amplify total RVFV N mRNA. (D) Quantitative RT–PCR of the indicated host-RVFV N mRNA conjugates normalized to RP49 as a loading control and shown as fold change over nontargeting dsRNA (luciferase). Mean ± SD for three or more independent experiments. (*) P < 0.05. (E–G) Cells expressing Dcp1-GFP to monitor P bodies were treated with the indicated dsRNA for 3 d, fixed, imaged, and analyzed using MetaXpress software. Mean ± SD for three or more independent experiments. n > 500 cells; (*) P < 0.05. (E) Average nuclear area shown relative to luciferase control. (F) Average Dcp1-GFP foci size relative to luciferase control. (G) Average number of Dcp1-GFP foci per cell relative to luciferase control.
To determine whether the stability of these target mRNAs is impacted by Dcp2, we performed RNAi in Drosophila cells and assayed host mRNA levels by quantitative RT–PCR. We found that, indeed, mRNA levels for three genes tested (CG8878, CG7580, and Jupiter) were increased upon Dcp2 depletion (Fig. 5B). To determine whether these genes were used as a target for RVFV snatching, we used a forward primer containing the first 11 bp of either CG8878 or Jupiter in addition to another gene identified (Histone 3 [His3], a replication-dependent histone) and a reverse primer in the N transcript. Using this assay, we found that the viral host-N fusion mRNAs were increased upon Dcp2 knockdown as compared with the control (Fig. 5D). These data suggest that Dcp2 affects RVFV replication primarily by restricting the substrate mRNAs available for cap-snatching and that cell cycle mRNAs are targeted by both decapping and cap-snatching.
P-body morphology is regulated by the cell cycle
Interestingly, previous studies in human HeLa cells suggested that the rapid turnover of replicating histone mRNAs (such as His3) at the end of S phase is dependent on Dcp2 (Mullen and Marzluff 2008; Su et al. 2013). This observation, along with our identification of a large number of cell cycle-related mRNAs as targets of RVFV-dependent cap-snatching, suggests that cell cycle-related mRNAs are degraded by Dcp2 in Drosophila. Emerging data also suggest that P bodies are regulated by a number of different biological inputs, including the cell cycle. P-body size and number increase as mammalian cells exit S phase and enter G2 (Yang et al. 2004). This may be due to the influx of mRNAs encoding DNA replication machinery and histones that need to be degraded, and this accumulation may result in increased granule assembly, leading to increased P-body size. We examined whether P-body size or number was influenced by the cell cycle in Drosophila. We used RNAi against cyclins to arrest the cells in G1 (CycD knockdown) or late S/G2 (CycA knockdown) (Björklund et al. 2006). As expected, we observed an increase in nuclear size upon S/G2 arrest (Fig. 5E). This was concomitant with a significant increase in P-body area and number (Fig. 5F,G), suggesting that cell cycle-dependent regulation of P bodies is deeply conserved and that P-body size during the cell cycle may serve as a marker for the load of mRNAs destined for degradation.
Cell cycle arrest in late S/early G2 enhances RVFV replication
Since P-body size is regulated by the cell cycle with the apex at late S/early G2, which is likely due to increased targeting of RNAs for degradation, and mRNA target levels are seemingly a bottleneck for RVFV replication, we hypothesized that RVFV may replicate most efficiently when P bodies are at their largest and P-body mRNAs are in high abundance. Indeed, analysis of our validated RNAi screen gene set revealed 39 genes that had the GO term “cell cycle” and were antiviral (Fig. 1C; Supplemental Fig. S1). Furthermore, the entire DNA replication factor A complex (RPA2, RpA-70, and CG15220), whose depletion results in S-phase arrest, was also antiviral in our screen (Supplemental Fig. S1; Supplemental Table S1). Moreover, we found that 28 genes impacted the cell cycle arresting at S/G2, as measured by increased nuclear area upon depletion (Supplemental Fig. S1; Supplemental Table S1), of which 26 genes were antiviral, and 15 genes did not have an annotated GO term associated or literature reference to the cell cycle, suggesting that they may have a previously unknown role in the cell cycle. To validate this, we performed RNAi against a panel of genes that arrest the cell cycle at specific stages. Treatment of cells with dsRNA to arrest in S/G2 (CycA, cdc2, and RnRs) led to increased levels of infection, while arrest in G1 (CycD, cdc2c, and CycE) had no impact (Fig. 6A,B), consistent with the fact that in log phase, >80% of Drosophila cells are in G1 (Boutros et al. 2004). Furthermore, we observed increases in RVFV mRNA upon loss of CycA, but not CycD, as measured by Northern blot (Fig. 6C; Supplemental Fig. S8A), along with increased His3-N mRNA accumulation (Fig. 6D). To determine whether this was specific for RVFV infection, we challenged arrested cells with VSV. We found that VSV replication was unaffected by S/G2 arrest, while G1 arrest by cdc2 modestly promoted infection (Fig. 6B). Altogether, these data suggest that cell cycle arrest in late S/early G2 is advantageous for RVFV replication, and this enhancement is specific to bunyaviruses.
Figure 6.
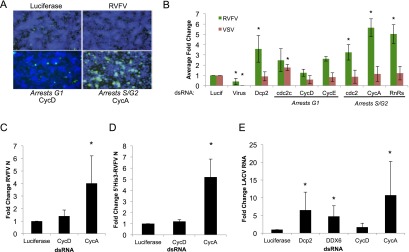
Cell cycle arrest in late S/early G2 phase enhances bunyavirus replication. (A) Representative images of DL1 cells treated with the indicated dsRNA, infected with RVFV for 30 h, fixed, and stained for RVFV N protein (green) and total nuclei (blue). (B) Quantification of cells treated with the indicated dsRNA and then infected with either RVFV or VSV for 30 or 24 h, respectively. Mean ± SD for three or more experiments shown. (*) P < 0.05. (C) Cells were treated with the indicated cell cycle gene dsRNA. Quantification of Northern blot analysis of RVFV N mRNA normalized to dRPS6 with the mean ± SD for three or more experiments shown. (*) P < 0.05. (D) Quantitative RT–PCR for 5′ His3-RVFV N fusion mRNA in cells treated for the indicated dsRNA for 3 d and then infected with RVFV for 30 h. Mean ± SD for three or more experiments shown. (*) P < 0.05. (E) DL1 cells treated with the indicated dsRNA and infected with LACV for 72 h. Quantification of Northern blot analysis of LACV N mRNA and the S-segment genome/anti-genome normalized to dRPS6, with the mean ± SD for three or more experiments shown. (*) P < 0.05.
Diverse bunyaviruses are restricted by similar mechanisms in cells
Our finding that Dcp2 restricts LACV in adult flies prompted us to test this in cell culture. Depletion of Dcp2 in Drosophila cells led to increased levels of LACV replication as measured by viral RNA levels; similar results were seen with DDX6 depletion (Fig. 6E; Supplemental Fig. S8B). Next, we tested whether LACV, like RVFV, also replicated more efficiently in late S/early G2, a time when Dcp2 targeted mRNAs should be abundant. Indeed, we found that S/G2 arrest led to increased LACV RNA replication (Fig. 6E; Supplemental Fig. S8B). These data suggest that cap-snatching is a bottleneck in the bunyaviral life cycle and that modulation of target mRNA levels impacts the replication of diverse bunyaviruses.
Dcp2 is limiting: Ectopic expression restricts RVFV replication
Since loss of Dcp2 leads to increased replication, we hypothesized that enforced expression of Dcp2 may restrict RVFV replication by decapping—and thereby limiting—the pool of available mRNAs. First, we confirmed that RNAi against Dcp2 substantially depleted myc-Dcp2 in our Dcp2-expressing cells by immunoblot (Fig. 7A). Next, wild-type or Dcp2-expressing cells were infected with RVFV, and immunoblot analysis of RVFV glycoprotein (RVFV Gn) revealed that Dcp2 knockdown increases RVFV infection (Fig. 7A), consistent with our findings measured by microscopy (Fig. 2A) or Northern blot (Fig. 4E). Additionally, we found that ectopic Dcp2 expression significantly restricted RVFV infection as measured by both immunoblot (Fig. 7A) and microscopy (Fig. 7B). Furthermore, Dcp2 knockdown restored infection in ectopically Dcp2-expressing cells to wild-type levels (compare with luciferase-treated infected cells) (Fig. 7A,B). These treatments had no effect on VSV infection (Fig. 7C), demonstrating specificity. Therefore, Dcp2 levels define a set point for RVFV infection.
Figure 7.
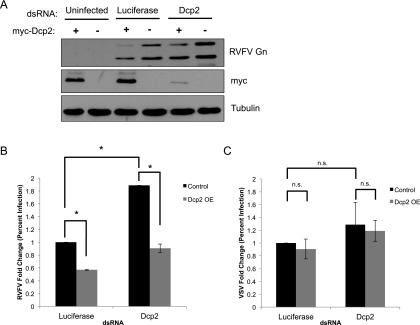
Ectopic expression of Dcp2 restricts RVFV replication. (A) Control cells or cells expressing myc-Dcp2 were treated with the indicated dsRNA for 3 d, infected with RVFV for 30 h, and analyzed by immunoblot. (B) Cells were treated and infected as in A and processed for immunofluorescence and quantification of the percentage of infection, with the mean ± SD for three or more experiments shown. (*) P < 0.05. (C) Cells were treated as described in A and infected with VSV for 24 h. Cells were processed for microscopy and quantification of the percentage of infection, with the mean ± SD for three or more experiments shown. (n.s.) Not significant.
Discussion
Our genome-wide RNAi screen identified and validated a large number of genes that restrict RVFV replication. Among this gene set was the canonical mRNA decapping enzyme Dcp2 and two decapping activators. The interactions between mRNA decay and viral infection are an area of burgeoning study (Gaglia and Glaunsinger 2010; Dickson and Wilusz 2011; Moon et al. 2012); however, there is little known about the intersection of the RNA degradation machinery and bunyaviral infection, and so we explored this biology. Future studies will reveal the mechanistic roles that the other validated factors play in viral infection. We focused on Dcp2, which specifically restricts the replication of two diverse bunyaviruses (RVFV and LACV) but not other RNA viruses in insects both in vitro and in vivo. Bunyaviruses, unlike the other RNA viruses tested, use cap-snatching to generate the 5′ end of viral mRNAs. This is in part mediated by the bunyaviral N protein, which binds to 5′ capped mRNAs (Mir et al. 2010). This led us to postulate that RVFV cap-snatching competes with decapping and suggests a model in which the availability of mRNA substrates is rate-limiting for RVFV infection. Thus, modulation of these targets can create or eliminate a bottleneck in viral replication.
Indeed, we found that Dcp2 does not directly decap viral transcripts after snatching and transcription have occurred, as knockdown does not impact the cap status of viral mRNAs (Fig. 4F) or their stability (Fig. 4G,H), and, furthermore, that Xrn1 knockdown does not increase viral mRNA stability (Supplemental Fig. S6B–D). Rather, mRNA decapping during normal RNA turnover keeps the pool of available mRNA targets from which bunyaviruses can snatch at a low level. Thus, loss of the decapping enzyme Dcp2 leads to increased bunyaviral replication, and ectopic expression of Dcp2 restricts infection. Furthermore, we found that many mRNA targets that are snatched and incorporated into viral transcripts are cell cycle-related. Indeed, mRNA levels in P bodies are cyclically altered in phase with the cell cycle. As cells transit into late S/early G2, P bodies enlarge to accommodate mRNA degradation of mRNAs required for DNA replication, and these cells support higher levels of bunyaviral replication. Our genome-wide RNAi screen identified a large number of cell cycle genes—including all three subunits of the DNA replication factor A complex—that arrest the cell cycle at this time as antiviral. Interestingly, however, we found that P-body dispersion through knockdown of components known to be required for P-body integrity does not affect RVFV replication; this is consistent with data showing that microscopically visible P bodies are not required per se for P-body-associated functions, such as miRNA silencing and NMD. Indeed, our data strengthen previous findings that P-body morphology may be a marker for the accumulation of mRNAs destined for degradation within the cell.
We found that by modulating the pool of host mRNAs targeted for decapping, either through changes in the expression level of Dcp2 or by arresting cells in late S/early G2 phase, bunyavirus replication is affected in insects. Interestingly, mRNAs carrying NMD signals were incorporated at an increased rate into Sin Nombre transcripts in human cells, and Sin Nombre N localizes with P bodies (Mir et al. 2008). This suggests potential conservation of decapping as antiviral against bunyaviruses in mammals. In further support of this, Dcp2 is inducible by type I interferons (Samarajiwa et al. 2009; Li et al. 2012). While Drosophila only encode one characterized decapping enzyme, a second decapping enzyme (NUDT16) has been recently characterized in both mice and humans (Song et al. 2010), and an additional six nudix domain-containing proteins in mice and one in yeast have been shown to have various degrees of decapping activity (Song et al. 2013). Drosophila encodes no NUDT16 ortholog; however, it does encode for 14 other nudix domain containing proteins, none of which were identified as antiviral in our genome-wide screen. Recent studies have identified a novel decapper in bacteria, which do not cap their RNA, suggesting that decapping activity is ancient and preceded mRNA capping in evolution (Song et al. 2013). This may further support a role for decapping in antiviral defense. Interestingly, comparisons have revealed that DCP2 and NUDT16 have both redundant and specific targets (Li et al. 2011), suggesting that in eukaryotes, decapping may be far more complex than first thought.
One interesting question raised by these observations is why bunyaviruses transcribe their viral mRNA in an area rife with mRNA degradation machinery. This is particularly perplexing when considering the fact that viral RNA transcription is dependent on translation, and P bodies are ribosome-free. We have four hypotheses that might explain why bunyaviruses use mRNAs destined for degradation as the target for cap-snatching. First, these targets are largely present in a spatially concentrated area and are destined for degradation. Thus, snatching caps from these mRNAs should not negatively impact cell viability. Second, cytoplasmically replicating RNA viruses must compartmentalize their replication steps in order to enhance efficiency; segregating RNA transcription to these areas (P bodies) may prevent competition between cap-snatching, genome replication, and viral RNA packaging into virions. Third, microscopy studies indicate that P bodies, and perhaps the RNA degradation machinery in general, are surrounded by ribosomes (Cougot et al. 2012; Weil et al. 2012). Furthermore, recent studies in Drosophila have shown that during oogenesis and early egg activation, translation of mRNAs necessary for proper axis formation depends on RNA localization within subcompartments of P-body-like RNPs (Weil et al. 2012). RNAs located deep within the P-body core are associated with decapping activators and are translationally repressed, while those located toward the edge of the P body are able to interact with ribosomes associated with the periphery of the P body and can initiate translation. This pool of readily available ribosomes may be optimal for initiation of translation of viral mRNAs, since bunyaviral mRNA transcriptional elongation requires translation to be occurring concomitantly (Barr 2007). Thus, it is possible that these ribosomes serve this function. Finally, we speculate that the 5′ end of the host mRNA molecule, once cleaved, may represent an abnormal or “foreign” RNA structure to the host cell (5′ monophosphate on an RNA with a 3′ poly-A tail). While cellular RNA molecules exist with 5′ monophosphates, such as rRNA, these RNAs are heavily associated with proteins (which may occlude recognition of their 5′ end structure) and are not polyadenylated. Snatching in these localized environments may ensure that mRNAs that are targeted for snatching can be rapidly degraded by the P-body-resident processive exonuclease Xrn1. Indeed, there may be parallels between bunyaviral snatching of Dcp2 targeted pools in the cytoplasm and nuclear pre-mRNAs by orthomyxoviruses. Many pre-mRNAs are aberrantly synthesized, and therefore the nucleus has surveillance machinery that includes an Xrn1 homolog Rat1 that targets uncapped mRNAs for degradation. Therefore, in both cases, the uncapped host mRNA is under close surveillance and thus has a very short half-life.
Altogether, we explored the host factor dependencies of RVFV, leading to the finding that two diverse bunyaviruses are restricted by mRNA decapping. Furthermore, these studies have revealed new aspects of RNA decay and the regulation of these compartments; our results indicate that alteration of P-body morphology during cell cycle progression is a deeply conserved process from insects to mammals (Yang et al. 2004). We also provide evidence that in Drosophila, Dcp2 decaps mRNAs involved in cell cycle progression and DNA replication in addition to previously established roles in regulating histone mRNA levels in human cells (Mullen and Marzluff 2008). This suggests that decapping in Drosophila more generally targets mRNAs undergoing rapid turnover. RNA profiling studies in murine cells depleted of Dcp2 or NUDT16 suggest that in higher organisms, the decapping of mRNAs is specialized (Li et al. 2012). Thus, selective activation of decapping may potentially be a viable therapeutic approach to degrade RVFV-accessible mRNAs. Indeed, Dcp2 is potentially regulated: In yeast, phosphorylation of Dcp2 by Ste20 is necessary for Dcp2 recruitment to P bodies (Yoon et al. 2010), and vertebrate Dcp2 has a number of conserved uncharacterized phosphorylation sites (Hornbeck et al. 2011). Further studies will reveal whether decapping of specific cargo can be selectively regulated in insects and mammals.
Materials and methods
Cells, viruses, antibodies, and reagents
Drosophila DL1 cells were grown and maintained as previously described (Cherry and Perrimon 2004). VSV-eGFP (gift from J. Rose) was grown in BHK cells as described (Ramsburg et al. 2005). Sindbis/GFP (gift from R. Hardy) was grown in C636 cells (Burnham et al. 2007). DCV was grown and purified as described (Cherry and Perrimon 2004). The MP12 strain of RVFV was grown in Vero cells as described (Filone et al. 2010). An original strain of LACV was prepared as described previously (Janssen et al. 1984). Antibodies were obtained from the following sources: anti-GFP (Invitrogen), anti-tubulin (Sigma), anti-DCV (Cherry and Perrimon 2004), and anti-RVFV N ID8 and anti-RVFV Gn 4D4 (gifts from R. Doms). Fluorescent secondary antibodies were obtained from Invitrogen, and HRP-conjugated antibodies were from Amersham. Additional chemicals were obtained from Sigma.
RNAi
dsRNAs were generated as described (Boutros et al. 2004). For RNAi, DL1 or Aag2 cells were passaged into serum-free medium and plated into wells containing dsRNA. One hour later, complete medium was added, and cells were incubated for 3 d.
Viral infections
Three days post-RNAi, cells were infected with the indicated viral inoculum. VSV (MOI = 0.01) and DCV (MOI = 0.4) were processed at 24 hpi. SIN (MOI = 2.5) and RVFV (DL1-MOI = 0.01; Aag-2-MOI = 0.06) were spinoculated at 1200 rpm for 2 h and processed at 36 and 30 hpi, respectively. For RNA collection, cells were infected with either RVFV (MOI = 0.1) or LACV (MOI = 2.5), spinoculated at 1200 rpm for 2 h, and collected at either 30 or 72 h, respectively.
Immunofluorescence
Cells were fixed, processed, imaged by automated microscopy, and subjected to automated image analysis as described (Rose et al. 2011). For colocalization studies, z-stacks of 20 planes were taken at 63×, deconvolved, and scored.
RNA analysis
Total RNA was extracted, Northern-blotted, and quantified as previously described (Cherry et al. 2005). RT-qPCR was performed as previously described (Xu et al. 2012). Primer sequences are described in the Supplemental Material.
CHX treatment
Drosophila cells were treated with the indicated dsRNA and infected with RVFV. Twenty-four h post-infection, cells were treated with 10 μg/mL CHX, and total RNA was collected every 30 min.
5′ RACE and cloning
5′ RACE was performed using the FirstChoice RLM-RACE kit from Ambion according to the manufacturer's instruction. RT–PCR was performed as described (Xu et al. 2012) using primers specific for the 5′ RACE adaptor (forward) and RVFV N transcript (reverse) and gel-purified (Qiagen) prior to ligation using the TOPO TA cloning system (Invitrogen). Sequences were blasted against the Drosophila genome; those that matched within 40 bp of the annotated 5′ end of a transcript and containing less than or equal to one end mismatch were considered hits. Bowtie analysis yielded the same results.
Exonuclease digest assay
Ten micrograms of total RNA was denatured for 2 min at 95°C, then either mock-treated or digested using terminator exonuclease (Epicentre) as per the manufacturer's instructions, and evaluated by Northern blot.
Adult fly infections
Flies were obtained from the Vienna Drosophila RNAi Center (VDRC) or Bloomington Stock Center. Flies (4–7 d old) carrying UAS-Dcp2 IR (VDRC transformant: 22272) or control (w1118) were crossed to hs-Gal4 and challenged and heat-shocked every 2 d (RVFV) or maintained at 29°C throughout the experiment (LACV) (Cherry and Perrimon 2004). Flies were monitored daily for mortality (analyzed with a log-rank test), or 15 flies per condition were processed at the indicated time point post-infection for RNA analysis as described (Xu et al. 2012).
Acknowledgments
We thank L. Sabin for BOWTIE sequence matching; K. Lynch and G. Blakqori for critical reading of the manuscript; A. Yasunaga, P. Rose, S. Hanna, and other members of the Cherry laboratory for technical advice and helpful discussions; S. Solden for LACV and technical advice; R. Doms and C. Schmaljohn for RVFV antibodies; J. Rose for VSV-eGFP; R. Hardy for SlN/GFP; and VDRC and The Bloomington Stock Center for transgenic flies. Dcp1-GFP was a gift from E. Izaurralde, and Myc-Dcp2 was a gift from S. Dorner. S.C. is a recipient of the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award. This work was supported by grants from the National Institutes of Health (R01AI074951,U54AI057168, and RO1AI095500 to S.C., and T32 NS007180 to K.C.H.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.215384.113.
References
- Ahola TT, Kääriäinen LL 1995. Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc Natl Acad Sci 92: 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr JN 2007. Bunyavirus mRNA synthesis is coupled to translation to prevent premature transcription termination. RNA 13: 731–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliakova-Bethell N, Beckham C, Giddings TH Jr, Winey M, Parker R, Sandmeyer S 2006. Virus-like particles of the Ty3 retrotransposon assemble in association with P-body components. RNA 12: 94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björklund M, Taipale M, Varjosalo M, Saharinen J, Lahdenperä J, Taipale J 2006. Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nat Cell Biol 439: 1009–1013 [DOI] [PubMed] [Google Scholar]
- Boshra H, Lorenzo G, Busquets N, Brun A 2011. Rift Valley fever: Recent insights into pathogenesis and prevention. J Virol 85: 6098–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouloy M, Flick R 2009. Reverse genetics technology for Rift Valley fever virus: Current and future applications for the development of therapeutics and vaccines. Antiviral Res 84: 101–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouloy MM, Pardigon NN, Vialat PP, Gerbaud SS, Girard MM 1990. Characterization of the 5′ and 3′ ends of viral messenger RNAs isolated from BHK21 cells infected with Germiston virus (bunyavirus). Virology 175: 50–58 [DOI] [PubMed] [Google Scholar]
- Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N 2004. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303: 832–835 [DOI] [PubMed] [Google Scholar]
- Burnham AJ, Gong L, Hardy RW 2007. Heterogeneous nuclear ribonuclear protein K interacts with Sindbis virus nonstructural proteins and viral subgenomic mRNA. Virology 367: 212–221 [DOI] [PubMed] [Google Scholar]
- Campbell CL, Keene KM, Brackney DE, Olson KE, Blair CD, Wilusz J, Foy BD 2008. Aedes aegypti uses RNA interference in defense against Sindbis virus infection. BMC Microbiol 8: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplen H, Peters CJ, Bishop DHL 1985. Mutagen-directed attenuation of Rift Valley fever virus as a method for vaccine development. J Gen Virol 66: 2271–2277 [DOI] [PubMed] [Google Scholar]
- Cheng E, Mir MA 2012. Signatures of Host mRNA 5′ terminus for efficient hantavirus cap snatching. J Virol 86: 10173–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry S 2005. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Gen Dev 19: 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry S, Perrimon N 2004. Entry is a rate-limiting step for viral infection in a Drosophila melanogaster model of pathogenesis. Nat Immunol 5: 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry S, Doukas T, Armknecht S, Whelan S, Wang H, Sarnow P, Perrimon N 2005. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev 19: 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller JMJ, Tucker MM, Sheth UU, Valencia-Sanchez MAM, Parker RR 2001. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA 7: 1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougot N, Cavalier A, Thomas D, Gillet R 2012. The dual organization of P-bodies revealed by immunoelectron microscopy and electron tomography. J Mol Biol 420: 17–28 [DOI] [PubMed] [Google Scholar]
- Dickson AM, Wilusz J 2011. Strategies for viral RNA stability: Live long and prosper. Trends Genet 27: 286–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E 2007a. P bodies: At the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 8: 9–22 [DOI] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Schweizer D, Izaurralde E 2007b. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol 27: 3970–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filone CM, Hanna SL, Caino MC, Bambina S, Doms RW, Cherry S 2010. Rift Valley fever virus infection of human cells and insect hosts is promoted by protein kinase C ɛ. PLoS ONE 5: e15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N, Weis K 2002. The DEAD box protein Dhh1 stimulates the decapping enzyme Dcp1. EMBO J 21: 2788–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia MMM, Glaunsinger BAB 2010. Viruses and the cellular RNA decay machinery. Wiley Interdiscip Rev RNA 1: 47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcin D, Lezzi M, Dobbs M, Elliott RM, Schmaljohn C, Kang CY, Kolakofsky D 1995. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J Virol 69: 5754–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt RRR, Gottfried KLK, Apperson CSC, Davis BSB, Erwin PCP, Smith ABA, Panella NAN, Powell EEE, Nasci RSR 2001. First isolation of La Crosse virus from naturally infected Aedes albopictus. Emerg Infect Dis 7: 807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddow AD, Odoi A 2009. The incidence risk, clustering, and clinical presentation of La Crosse virus infections in the Eastern United States, 2003–2007. PLoS ONE 4: e6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y 2008. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454: 890–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz C, Stavnezer E, Krug R, Gurney T 1981. Influenza virus, an RNA virus, synthesizes its messenger RNA in the nucleus of infected cells. Cell 26: 391–400 [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M 2011. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40: D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelfinger DD, Arndt-Jovin DJD, Lührmann RR, Achsel TT 2002. The human LSm1–7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA 8: 1489–1501 [PMC free article] [PubMed] [Google Scholar]
- Jäger EE, Dorner SS 2010. The decapping activator HPat a novel factor co-purifying with GW182 from Drosophila cells. RNA Biol 7: 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen R, Gonzalez-Scarano F, Nathanson N 1984. Mechanisms of bunyavirus virulence. Comparative pathogenesis of a virulent strain of La Crosse and an avirulent strain of Tahyna virus. Lab Invest 50: 447–455 [PubMed] [Google Scholar]
- Johnson KN, Christian PD 1998. The novel genome organization of the insect picorna-like virus Drosophila C virus suggests this virus belongs to a previously undescribed virus family. J Gen Virol 79: 191–203 [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA 1996. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86: 973–983 [DOI] [PubMed] [Google Scholar]
- Li J, Rahmeh A, Morelli M, Whelan SPJ 2008a. A conserved motif in region V of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J Virol 82: 775–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Song MG, Kiledjian M 2008b. Transcript-specific decapping and regulated stability by the human Dcp2 decapping protein. Mol Cell Biol 28: 939–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Song M, Kiledjian M 2011. Differential utilization of decapping enzymes in mammalian mRNA decay pathways. RNA 17: 419–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Dai J, Song M, Fitzgerald-Bocarsly P, Kiledjian M 2012. Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7. Mol Cell Biol 32: 1164–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M-D, Fan S-J, Hsu W-S, Chou T-B 2006. Drosophila decapping protein 1, dDcp1, is a component of the oskar mRNP complex and directs its posterior localization in the oocyte. Dev Cell 10: 601–613 [DOI] [PubMed] [Google Scholar]
- Mir MA, Panganiban AT 2008. A protein that replaces the entire cellular eIF4F complex. EMBO J 27: 3129–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir MA, Duran WA, Hjelle BL, Ye C, Panganiban AT 2008. Storage of cellular 5′ mRNA caps in P bodies for viral cap-snatching. Proc Natl Acad Sci 105: 19294–19299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir MA, Sheema S, Haseeb A, Haque A 2010. Hantavirus nucleocapsid protein has distinct m7G cap- and RNA-binding sites. J Biol Chem 285: 11357–11368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SL, Barnhart MD, Wilusz J 2012. Inhibition and avoidance of mRNA degradation by RNA viruses. Curr Opin Microbiol 15: 500–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin B, Coutard B, Lelke M, Ferron F, Kerber R, Jamal S, Frangeul A, Baronti C, Charrel R, de Lamballerie X, et al. 2010. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog 6: e1001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser TS, Jones RG, Thompson CB, Coyne CB, Cherry S 2010. A kinome RNAi screen identified AMPK as promoting poxvirus entry through the control of actin dynamics. PLoS Pathog 6: e1000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutailler S, Roche B, Thiberge J-M, Caro V, Rougeon F, Failloux A-B 2011. Host alternation is necessary to maintain the genome stability of Rift Valley fever virus. PLoS Negl Trop Dis 5: e1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Decker CJ, Parker R 1994. Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′→3′ digestion of the transcript. Gene Dev 8: 855–866 [DOI] [PubMed] [Google Scholar]
- Mullen TE, Marzluff WF 2008. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Gene Dev 22: 50–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto M, Moy RH, Xu J, Bambina S, Yasunaga A, Shelly SS, Gold B, Cherry S 2012. Virus recognition by Toll-7 activates antiviral autophagy in Drosophila. Immunity 36: 658–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura AA, Amikura RR, Hanyu KK, Kobayashi SS 2001. Me31B silences translation of oocyte-localizing RNAs through the formation of cytoplasmic RNP complex during Drosophila oogenesis. Development 128: 3233–3242 [DOI] [PubMed] [Google Scholar]
- Nissan T, Rajyaguru P, She M, Song H, Parker R 2010. Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol Cell 39: 773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Sheth U 2007. P bodies and the control of mRNA translation and degradation. Mol Cell 25: 635–646 [DOI] [PubMed] [Google Scholar]
- Patterson JLJ, Holloway BB, Kolakofsky DD 1984. La Crosse virions contain a primer-stimulated RNA polymerase and a methylated cap-dependent endonuclease. J Virol 52: 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin M, Bouloy M, Bird BH, Kemp A, Paweska J 2010. Rift Valley fever virus (Bunyaviridae: phlebovirus): An update on pathogenesis, molecular epidemiology, vectors, diagnostics and prevention. Vet Res 41: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotch SJ, Bouloy M, Ulmanen I, Krug RM 1981. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23: 847–858 [DOI] [PubMed] [Google Scholar]
- Pressman S, Reinke CA, Wang X, CARTHEW RW 2012. A systematic genetic screen to dissect the microRNA pathway in Drosophila. G3 2: 437–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsburg E, Publicover J, Buonocore L, Poholek A, Robek M, Palin A, Rose JK 2005. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J Virol 79: 15043–15053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera J, Weber F, Cusack S 2010. Bunyaviridae RNA polymerases (L-Protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog 6: e1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose PP, Hanna SL, Spiridigliozzi A, Wannissorn N, Beiting DP, Ross SR, Hardy RW, Bambina SA, Heise MT, Cherry S 2011. Natural resistance-associated macrophage protein is a cellular receptor for Sindbis virus in both insect and mammalian hosts. Cell Host Microbe 10: 97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin LR, Zhou R, Gruber JJ, Lukinova N, Bambina S, Berman A, Lau C-K, Thompson CB, Cherry S 2009. Ars2 regulates both miRNA- and siRNA-dependent silencing and suppresses RNA virus infection in Drosophila. Cell 138: 340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ 2009. INTERFEROME: The database of interferon regulated genes. Nucleic Acids Res 37: D852–D857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti PD, Longworth JF, Plus N, Croizier G, Reinganum C 1981. The biology and ecology of strains of an insect small RNA virus complex. Adv Virus Res 26: 117–143 [DOI] [PubMed] [Google Scholar]
- Sen GL, Blau HM 2005. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat Cell Biol 7: 633–636 [DOI] [PubMed] [Google Scholar]
- Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, Ramirez JL, Dimopoulos G, Yang PL, Pearson JL, et al. 2009. Discovery of insect and human dengue virus host factors. Nature 458: 1047–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300: 805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons JFJ, Pettersson RFR 1991. Host-derived 5′ ends and overlapping complementary 3′ ends of the two mRNAs transcribed from the ambisense S segment of Uukuniemi virus. J Virol 65: 4741–4748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M-G, Li Y, Kiledjian M 2010. Multiple mRNA decapping enzymes in mammalian cells. Mol Cell 40: 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MG, Bail S, Kiledjian M 2013. Multiple Nudix family proteins possess mRNA decapping activity. RNA 19: 390–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiger M, Carr-Schmid A, Schwartz DC, Kiledjian M, Parker R 2003. Analysis of recombinant yeast decapping enzyme. RNA 9: 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Slepenkov SV, Slevin MK, Lyons SM, Ziemniak M, Kowalska J, Darzynkiewicz E, Jemielity J, Marzluff WF, Rhoads RE 2013. mRNAs containing the histone 3′ stem-loop are degraded primarily by decapping mediated by oligouridylation of the 3′ end. RNA 19: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet T, Kovalak C, Coller J 2012. The DEAD-box protein Dhh1 promotes decapping by slowing ribosome movement. PLoS Biol 10: e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler F, Braun JE, Eulalio A, Truffault V, Izaurralde E, Weichenrieder O 2009. Structural basis for the mutually exclusive anchoring of P body components EDC3 and Tral to the DEAD box protein DDX6/Me31B. Mol Cell 33: 661–668 [DOI] [PubMed] [Google Scholar]
- Tucker MM, Parker RR 2000. Mechanisms and control of mRNA decapping in Saccharomyces cerevisiae. Biochemistry 69: 571–595 [DOI] [PubMed] [Google Scholar]
- Turell MJ, Wilson WC, Bennett KE 2010. Potential for North American mosquitoes (Diptera: Culicidae) to transmit Rift Valley fever virus. J Med Entomol 47: 884–889 [DOI] [PubMed] [Google Scholar]
- Van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Séraphin B 2002. Human Dcp2: A catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J 21: 6915–6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Knippenberg I, Lamine M, Goldbach R, Kormelink R 2005. Tomato spotted wilt virus transcriptase in vitro displays a preference for cap donors with multiple base complementarity to the viral template. Virology 335: 122–130 [DOI] [PubMed] [Google Scholar]
- van Poelwijk F, Kolkman J, Goldbach R 1996. Sequence analysis of the 5′ ends of tomato spotted wilt virus N mRNAs. Arch Virol 141: 177–184 [DOI] [PubMed] [Google Scholar]
- van Rij RP, Saleh MC, Berry B, Foo C, Houk A, Antoniewski C, Andino R 2006. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Gene Dev 20: 2985–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialat P, Muller R, Vu TH, Prehaud C, Bouloy M 1997. Mapping of the mutations present in the genome of the Rift Valley fever virus attenuated MP12 strain and their putative role in attenuation. Virus Res 52: 43–50 [DOI] [PubMed] [Google Scholar]
- Wang X-H, Aliyari R, Li W-X, Li H-W, Kim K, Carthew R, Atkinson P, Ding S-W 2006. RNA interference directs innate immunity against viruses in adult Drosophila. Science 312: 452–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil TT, Parton RM, Herpers B, Soetaert J, Veenendaal T, Xanthakis D, Dobbie IM, Halstead JM, Hayashi R, Rabouille C, et al. 2012. Drosophila patterning is established by differential association of mRNAs with P bodies. Nat Cell Biol 14: 1305–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston A 2006. Xp54 and related (DDX6-like) RNA helicases: Roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res 34: 3082–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JEJ, Powell MJM, Hoover SES, Sarnow PP 2000. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol Cell Biol 20: 4990–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Grant G, Sabin LR, Gordesky-Gold B, Yasunaga A, Tudor M, Cherry S 2012. Transcriptional pausing controls a rapid antiviral innate immune response in Drosophila. Cell Host Microbe 12: 531–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Jakymiw A, Wood MR, Eystathioy T, Rubin RL, Fritzler MJ, Chan EK 2004. GW182 is critical for the stability of GW bodies expressed during the cell cycle and cell proliferation. J Cell Sci 117: 5567–5578 [DOI] [PubMed] [Google Scholar]
- Yoon JH, Choi EJ, Parker R 2010. Dcp2 phosphorylation by Ste20 modulates stress granule assembly and mRNA decay in Saccharomyces cerevisiae. J Cell Biol 189: 813–827 [DOI] [PMC free article] [PubMed] [Google Scholar]