

Abstract
Traumatic brain injury (TBI) is the most common cause of death and acquired disability among children and young adults in the developed countries. In clinical studies, the incidence of depression is high after TBI, and the mechanisms behind TBI-induced depression remain unclear. In the present study, we subjected rats to a moderate fluid percussion into the closed cranial cavity to induce TBI. After 3 days of recovery, injured rats were given a forced swim test (FST) and novelty-suppressed feeding tests. We found that TBI rats exhibited increased duration of immobility and longer latency to begin chewing food in a new environment compared with sham-operated rats. Western blot analysis showed that TBI led to a decrease in the phosphorylated levels of extracellular signal regulated kinases (ERK1/2) and p38 mitogen-activated protein kinase (p38 MAPK). Fluoxetine, a selective serotonin reuptake inhibitor (SSRI), significantly reduced the duration of immobility when administered once per day for 14 days. Consistent with behavioral tests, fluoxetine treatment reversed TBI-induced decrease in p-ERK1/2 and p-p38 MAPK levels. Pre-treatment with a selective tryptophan hydroxylase inhibitor para-chlorophenylalanine (PCPA) blocked the antidepressant effect of fluoxetine. PCPA also prevented the effect of fluoxetine on ERK1/2 phosphorylation without affecting p38 MAPK phosphorylation. Pre-treatment with ERK inhibitor SL327 but not p38 MAPK inhibitor SB203580 prevented the antidepressant effect of fluoxetine. These results suggest that ERK1/2 plays a critical role in TBI-induced depression.
Key words: antidepressant, depression, extracellular signal regulated kinases (ERK), hippocampus, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability in humans. According to the Centers for Disease Control and Prevention, the annual incidence of TBI in the United States is ∼1.7 million.1 Motor vehicle accidents are the major cause of fatal head injury. In addition, the use of improvised explosive devices in war zones has resulted in increasing numbers of blast-related head injuries in both military personnel and civilians.2,3 Concurrently, an increased frequency of concussive brain injury associated with athletics has been reported.4
Depression is the most frequently diagnosed psychiatric disorder after TBI.5,6 Nearly half of those with TBI experience major depressive disorder (MDD) in the first year.7 Despite the high prevalence of affective disorders after TBI, the pathophysiological mechanisms still remain to be defined. To better understand the pathophysiology of TBI and to evaluate potential therapeutic approaches, various animal models have been developed and characterized.8–11 Each is intended to mimic certain components of clinical TBI, although it is difficult to establish consistent models that include most or all of the factors that contribute to post-trauma tissue damage. These models include devices that accelerate or rotate the skull, impact injury to the intact and freely movable cranium, and rapid injection of fluid into a closed cranium (fluid percussion). Fluid percussion models produce brain injury by rapidly injecting fluid volumes into the cranial cavity,12 which produces pressure transients similar to those recorded in human cadaver skulls during sudden impact as well as neurological signs of behavioral suppression resembling signs of unconsciousness in humans.13
Prolonged neurological dysfunction that results from a brain insult is often attributed to irreversible structural damage such as loss of neurons or axonal degeneration. Pathological and immunohistological studies have reported that the hippocampus displayed vulnerability to traumatic insults. Autopsy reports and brain imaging studies of patients with depression pointed to abnormalities in several brain regions including prefrontal and cingulated cortex, hippocampus, striatum, amygdala, and thalamus.14–17 Further, the volume of hippocampus was smaller in patients with depression, which was associated with the duration of depression and the length of treatment.18–20
Using the fluid percussion model of brain injury, we found that TBI caused depression in rats assessed with the forced swim test. The purpose of this study was to delineate the cellular mechanisms responsible for TBI-induced depression and to develop better pharmacological strategies for treatment.
Methods
Animals
Adult male Sprague-Dawley rats weighing about 280–300 g were used in the experiments. Animals were kept under a 12-h/12-h light/dark cycle and allowed free access to food and water. All experimental procedures conformed to the National Institutes of Health guidelines and were approved by the National Cheng-Kung University Medical Center Animal Care and Use Committee to minimize discomfort to the animals during surgery and in the recovery period.
Fluid percussion injury (FPI)
Rats were anesthetized with sodium pentobarbital (25 mg/kg, intraperitoneally [IP]; Sigma Chemical Co, St Louis, MO) and a mixture containing ketamine (44 mg/kg, intramuscularly [IM]; Nan Kuang Pharmaceutical, Tainan, Taiwan), atropine (0.02633 mg/kg, IM; Sintong Chemical Industrial, Co, Ltd, Taoyuan, Taiwan), and xylazine (6.77 mg/kg, IM; Bayer, Leverkusen, Germany). The animals were placed in a stereotaxic frame, and the scalp was incised sagittally. Animals were subjected to a lateral FPI. After an incision in the scalp was made, a 4.8-mm circular craniotomy was performed midway between lambda and bregma 3.0 mm to the right of the central suture. A modified Leur-Lock connector (trauma cannula), 2.6 mm inner diameter, was secured into the craniotomy with cyanoacrylic adhesive and dental acrylic. A moderate FPI (2.2 atm) was produced by rapidly injecting a small volume of saline into the closed cranial cavity with a fluid percussion device (VCU Biomedical Engineering, Richmond, Va). The animal was removed from the device, the acrylic removed, and the incision sutured. Each injured and sham-injured animal for the fluid percussion model was closely evaluated immediately after TBI for behavioral recovery.
Triphenyltetrazolium chloride (TTC) staining
Rats were sacrificed at day 4 after TBI. Under deep anaesthesia (sodium pentobarbital, 100 mg/kg, IP) animals were perfused intracardially with saline. The brain tissue was then removed, immersed in cold saline for 5 min, and sliced into 2.0-mm sections with a tissue slicer. The brain slices were incubated in 2% TTC dissolved in phosphate buffered saline for 30 min at 37°C, and then transferred to 5% formaldehyde solution for fixation. The volume of infarction, as revealed by negative TTC stains indicating dehydrogenase-deficient tissue, was measured in each slice and summed using computerized planimetry (PC-based Image Tools software).
Open field test
The open field test, which is often used to assess anxiety, was performed as described previously.21 Rats were placed on the corner of a square box with dimensions, 40 cm×40 cm×40 cm (white floor and black wall) and allowed to explore for 10 min. The Noldus video tracking system was used to record the total distance as a measure of locomotor activity, the duration in the central zone, and the latency of the first entry into the central zone as the index of anxiety-like behaviors. Data were analyzed by EthoVison XT5.1 software. The box was cleaned with 75% ethanol to eliminate odors.
Forced swim test (FST)
The forced swim test, which was sensitive to conventional antidepressant treatment, was performed as described previously.22 Rats were gently placed in a large Perspex cylinder (height 40 cm, diameter 30 cm) with 30 cm of water at 24±1°C for 10 min. Water was changed between experiments. The duration of immobility, as the index of despaired behavior, was defined as no movement of limbs but respiration movement and was recorded by SONY digital camera from a horizontal angle.
Novelty suppressed feeding (NSF) test
The procedure was adapted from Stedenfeld and associates.23 At day 1, rats were subjected to a lateral FPI to induce TBI. At day 2 and day 3, food was removed from the cage, while water remained available ad libitum. At the day of testing, the rats were transferred to the testing room, and at the onset of the test, the rats were placed in an open field (80 cm×80 cm) with a pre-weighed food pellet in the center. For this experiment, latency to begin chewing food was measured.
Sucrose preference test
Rodents are born with an interest in sweet foods or solutions. Reduced preference for sweet solution in the sucrose preference test represents anhedonia.24 Rats were presented with two identical drinking bottles—one containing a 1% sucrose solution and the other normal tap water for a 24-h period. The position of the bottles was randomly assigned to prevent any influence of place preference. The bottles were carefully weighed before and after the 24-h period, and the percentage of the sucrose solution consumed was calculated.
Social interaction test
This experiment was conducted in an acrylic box (50×50×40 cm), and the testing room had a weak red light. The experimental rats and control rats dyed with black hair color were put in the box together, and allowed freedom of movement for 20 min. The time spent by the rats in active social interaction, such as sniffing, following, allogrooming, and mounting was recorded through computer-imaging recognition from the videotapes.
Elevated plus maze test (EPM)
EPM consisted of four arms (each arm 55 cm long×5 cm wide, white floor), which were elevated 50 cm above the floor. The black wall of closed arms was 25 cm in height. Two open and two closed arms formed a cross. At beginning of the test, rats were placed at the end of one of the closed arms, and subsequently the time spent in the open and closed arms was recorded for 10 min. In addition, the number of entries into either the open or closed arms was recorded by the Noldus video tracking system. Data were analyzed by EthoVison XT5.1 software. The maze was cleaned with 75% ethanol to eliminate odors between mice.
Western blot analysis
Brain tissue from the hippocampus was immediately removed and sonicated in a ice-cold homogenizing buffer containing 1% Triton X-100, 0.1% SDS, 50 mM Tris-HCl, pH 7.5 with complete preotease inhibitor cocktail (Roche, 11873580001). The lysates were centrifuged at 14,000 rpm for 30 min at 4°C. Supernatants were collected, and then protein concentrations in the supernatants were determined by taking 1μL of sample for protein assay. Samples (10 μg) were mixed with 4 μl of SDS sample buffer and placed in a boiling water bath for 10 min. Equal amounts of protein were loaded onto a 8.5% SDS-polyacrylamide gels and were run on 70 mV for 20 min and on 120 mV for 2 h. Separated proteins were transferred onto a polyvinylidene difluoride membrane at a constant ampere of 350 mA for 1.5 h.
The membrane was blocked in 5% nonfat milk in Tris buffered saline (TBS) for 1 h at room temperature and washed in Tris buffered saline with Tween (TBST) three times for 10 min in each. The membrane was incubated with either phospho-specific extracellular signal regulated kinases (ERK) 1 and ERK 2 antibodies (I :2000; Santa Cruz Biotechnology), ERK 1 and ERK 2 antibodies (1:2000; Cell Signaling), phospho-specific p38 antibody (I: 2500; BD Transduction Laboratories), p38 antibody (I: 2500; BD Transduction Laboratories), phospho-specific stress-activated protein kinase/Jun-amino-terminal kinase (SAPK/JNK) antibody (I: 1000; BD Transduction Laboratories), SAPK/JNK antibody (I: 1000; BD Transduction Laboratories), α-tublin (I: 100000; Sigma) in TBS overnight at 4°C with constant shaking. The membranes were washed with TBST in each, three times for 10 min, then incubated for 1 h with secondary antibody dissolved in blocking buffer in room temperature. Finally, the membranes were incubated in enhanced chemiluminescence detection reagent for detection of the labeled proteins. Western blots were developed in the linear range used for densitometry.
Statistical analysis
Data were expressed as mean±standard error of the mean. Behavioral data were analyzed by analysis of variance (ANOVA). Student t test or Bonferroni multiple comparison tests were used as post hoc comparisons. The level of significance was p<0.05.
Results
At day 1, we subjected rats to a lateral FPI by rapidly injecting a small volume of saline into the close cranial cavity. The levels of injury were evaluated by staining brain sections with TTC at day 4 (Fig. 1A). Figure 1B shows that larger cortical brain lesions developed in rats with TBI compared with those of sham-operated rats (Fig. 1C). In a separate group of rats, behavioral tests were performed at day 4. There were no differences on distance travelled in the inner area (t(32)=0,736, p>0.1), time spent in inner area (t(32)=0,435, p>0.1), total velocity (t(32)=0,01, p>0.5), and total distance travelled (t(32)=0,07, p>0.5) between sham-operated and TBI rats in an open field test. In the elevated plus maze (EPM), there was no difference in the time spent in the open arms between naïve (15.5±3.4%, n=12) and TBI (27.5±8.9%, n=12, p>0.1) rats. These results suggest that TBI rats did not exhibit anxiety-like behavior.
FIG. 1.
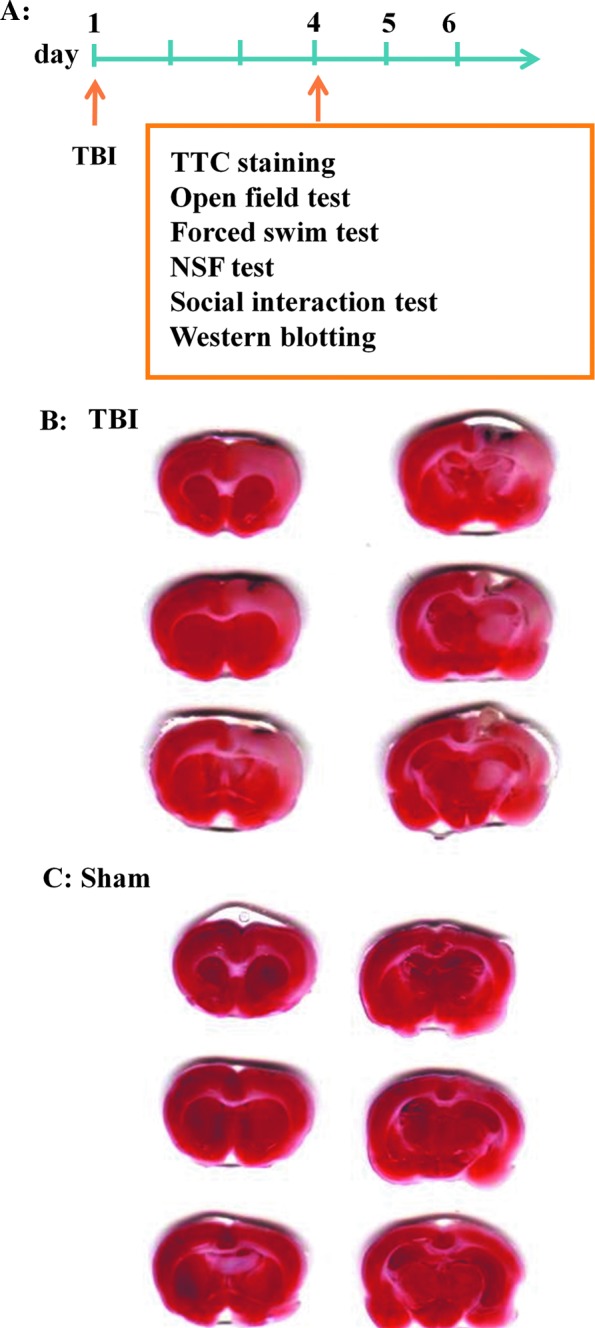
Traumatic brain injury (TBI) induces cortical brain lesions. (A) Time line of the experiments. (B,C) At day 1, rats were subjected to a lateral fluid percussion injury (B) or were sham-operated (C). The levels of injury were evaluated by staining brain sections with triphenyltetrazolium chloride (TTC) at day 4. NSF, novelty suppressed feeding. Color image is available online at www.liebertpub.com/neu
In the FST, TBI rats displayed despair behavior as indicated by increased time of immobility. One-way ANOVA showed that TBI rats exhibited significantly longer immobility time compared with sham-operated and naïve control rats (F(2,25)=3.406, p<0.05) (Fig. 2A). We performed the NSF test to assess depression by measuring the latency of an animal to approach and eat food in a novel environment. Figure 2B shows that the latency to begin chewing food was not different between sham-operated and TBI rats in the home cage (t(8)=1,194, p>0.1). In a new environment, however, TBI rats exhibited significantly longer latency to begin chewing food as compared with sham-operated rats (t(8)=2,336, p<0.05). Moreover, the duration (t(7)=3,06, p<0.05) (Fig. 2C) and frequency (t(7)=2,631, p<0.05) (Fig. 2D) of social interaction was less in the TBI rats than in the sham-operated rats. Thus, TBI exhibited depression-like behaviors.
FIG. 2.

Traumatic brain injury (TBI) induces depression-like behaviors in rats. (A) In the forced swim test, TBI rats (n=10) exhibited significantly longer immobility time compared with sham-operated (n=9) and naïve (n=9) rats. *p<0.05 vs. sham-operated. (B) In the novelty suppressed feeding test, the latency to begin chewing food was not different between sham-operated (n=5) and TBI (n=5) rats in the home cage. In a new environment, however, TBI rats exhibited significantly longer latency to begin chewing food compared with sham-operated rats. *p<0.05 vs. sham-operated. (C, D) In social interaction test, the duration and frequency were less in the TBI (n=4) rats than in the sham-operated (n=5) rats. *p<0.05 vs. sham-operated.
It has been shown that inhibition of MAPK signaling produced a depressive-like phenotype and blocked behavioral actions of antidepressants.25 We examined whether the increased immobility in TBI rats was mediated by MAPK. Rats were subjected to FPI on day 1, sacrificed on day 4, and hippocampus homogenate was immunoblotted with an antibodies that specifically recognizes dually phosphorylated ERK1/2 (both p44 and p42 isoforms), p38 MAPK and JUN NH2-terminal kinases (JNKs) (both p54 and p46 isoforms), as well as antibodies against total ERK1/2, pMAPK, and JNKs. As shown in Figure 3A and 3B, TBI led to a significant decrease in the phosphorylated levels of ERK1 (49.2±3.8%, n=6, p<0.01), ERK2 (56.6±2.7%, n=6, p<0.01) p38 MAPK (40.1±6.1%, n=6, p<0.01) but not JNK (p54 isoform F(2,14)=0.335, p>0.5; p46 isoform F(2,14)=3.043, p>0.05). In addition, no changes in the immunoreactivities against total ERK1/2 and p38 MAPK were detected. We also measured phosphorylated levels of ERK1/2, p38 MAPK, and JNK in the amygdala. In contrast to what was observed in the hippocampus, TBI did not influence the phosphorylated levels of ERK1/2, p38 MAPK, and JNK in the amygdala (Fig. 3C).
FIG. 3.

Traumatic brain injury (TBI) decreases phosphorylated levels of extracellular signal regulated kinases (ERK1/2) and p38 mitogen-activated protein kinase (MAPK) but not jun-amino-terminal kinase (JNK) in the hippocampus. Rats were subjected to fluid percussion injury on day 1, sacrificed on day 4. (A) Hippocampus and basolateral amygdala homogenates were immunoblotted with antibodies that specifically recognize dually phosphorylated ERK1/2 (both p44 and p42 isoforms), p38 MAPK, and JNK (both p54 and p46 isoforms), as well as antibodies against total ERK1/2, pMAPK, and JNK. (B) TBI led to a significant decrease in the phosphorylated levels of ERK1/2 and 38 MAPK but not JNK in the hippocampus. (C) TBI did not influence the phosphorylated levels of ERK1/2, p38 MAPK, and JNK in the amygdala.
Selective serotonin reuptake inhibitors (SSRIs) belong to one of the most effective and commonly used antidepressants. Therefore, we examined whether SSRI fluoxetine reversed TBI-induced increase in the immobility time in the FST. On day 1, rats were subjected to FPI and fluoxetine (2.5 mg/kg) or the vehicle was administered IP to the rats from day 4 to day 17. FST was performed on day 18. Figure 4A showed that TBI rats exhibited longer immobility time than those of sham-operated rats (t(10)=2,343, p<0.05) in vehicle treatment. Administration of fluoxetine had no effect on the sham-operated rats (t(9)=0,218, p>0.5) but significantly reduced immobility time in the TBI rats (t(12)=3,033, p<0.05). In the NSF test, although fluoxetine-treated rats showed a trend for decreased latency to begin chewing food compared with saline-treated rats, the difference did not reach statistical significance (p>0.05) (Fig. 4B). The sucrose preference test was regarded as a criterion of anhedonia when the preference score was less than 65%. We found that TBI rats treated with saline exhibited lower sucrose preference (50.0±2.4%, n=5) than those of treated with fluoxetine (66.4±5.6%, n=5) (t(8)=2.675, p<0.05) (Fig. 4C). Further, TBI rats treated with fluoxetine showed higher frequency (t(7)=4.519, p<0.01) (Fig. 4D) and longer duration (t(7)=4.195, p<0.01) (Fig. 4E) of social interaction compared with those saline-treated TBI rats.
FIG. 4.

Fluoxetine rescues traumatic brain injury (TBI)-induced depression-like behaviors. On day 1, rats were subjected to fluid percussion injury, and fluoxetine (2.5 mg/kg) or the vehicle was administered intraperitoneally to the rats from day 4 to day 17. Behavioral tests were performed on day 18. (A) In the forced swim test, TBI rats exhibited longer immobility time than those of sham-operated rats. Administration of fluoxetine had no effect on the sham-operated rats but significantly reduced immobility time in the TBI rats. *p<0.05 vs. sham-operated, #p<0.05 vs. saline. (B) In the novelty suppressed feeding test, TBI rats exhibited significantly longer latency to begin chewing food compared with sham-operated rats in a new environment. Although fluoxetine-treated TBI rats showed a trend for decreased latency to begin chewing food compared with saline-treated rats, the difference did not reach statistical significance (p>0.05). *p<0.05 vs. sham-operated. (C) In the sucrose preference test, TBI rats treated with saline exhibited lower sucrose preference than those treated with fluoxetine. *p<0.05 vs. saline. (D, E) In the social interaction test, the duration (D) and frequency (E) were restored by fluoxetine in the TBI rats. **p<0.01 vs. saline.
We determined whether TBI-mediated decreases in the phosphorylated levels of ERK1/2 and p38 MAPK could be reversed by fluoxetine. On day 1, rats were subjected to FPI, and fluoxetine (2.5 mg/kg) or saline was administered IP to the rats from day 4 to day 17. Western blotting analysis was performed on day 18. As illustrated in Figure 5, the phosphorylated levels of ERK1 (63.5±3.0% of naive, n=5, p<0.001), ERK2 (68.1±2.7% of naive, n=5, p<0.001) and p38MAPK (59.7±5.7% of naive, n=5, p<0.001) in the hippocampus were significantly lower in the saline-treated TBI rats compared with naïve control rats. Fluoxetine treatment reversed TBI-induced decreases in the phosphorylated levels of ERK1 (104.0±12.8% of naïve, n=5, p<0.05), ERK2 (98.9±8.6% of naïve, n=5, p<0.01) (Fig. 5A), and p38 MAPK (112.0±8.9% of naïve, n=6, p<0.01) (Fig. 5B) in the hippocampus. Interestingly, the phosphorylated state of ERK1/2 in the contralateral site of TBI rats was not altered by fluoxetine treatment (see online supplementary figure at ftp.liebertpub.com), a result in agreement with a previous report that ERK 1/2 phosphorylation was not changed after 2 weeks of treatment.26
FIG. 5.

Fluoxetine (Flu) reverses traumatic brain injury (TBI)-induced decrease in phosphorylated levels of extracellular signal regulated kinases (ERK1/2) and p38 mitogen-activated protein kinase (MAPK) in the hippocampus. On day 1, rats were subjected to fluid percussion injury and fluoxetine (2.5 mg/kg), or the vehicle was administered intraperitoneally to the rats from day 4 to day 17. On day 18, hippocampus homogenates of the ipsilateral site were immunoblotted with antibodies that specifically recognize dually phosphorylated ERK1/2 and p38 MAPK, as well as antibodies against total ERK1/2 and pMAPK. Fluoxetine treatment reversed TBI-induced decrease in the phosphorylated levels of ERK1/2 (A) and p38 MAPK (B) in the hippocampus. ###p<0.001 vs. naïve; *p<0.05, **p<0.01 vs. saline (Sal).
Para-chlorophenylalanine (PCPA) is a selective inhibitor of tryptophan hydroxylase, the rate-limiting enzyme in 5-HT biosynthesis.27 Previous studies showed that pre-treatment with PCPA blocked fluoxetine-induced reduction in immobility during FST.28 We examined whether pre-treatment with PCPA blocked the effect of fluoxetine on the immobility in TBI rats. PCPA was administered from day 14 to day 17, and FST was performed on day 18. As shown in Figure 6A, TBI rats treated with fluoxetine exhibited shorter immobility time than those treated with saline (t(12)=3.03, p<0.05). Pre-treatment with PCPA significantly reversed the effect of fluoxetine on the immobility time (t(10)=3,077, p<0.05 vs. fluoxetine). We also probed the phosphorylated levels of ERK1/2 and p38 MAPK in the hippocampus after treatment with PCPA. Figure 6B showed that fluoxetine treatment reversed TBI-induced decreases in the phosphorylated levels of ERK1 (139.5±19.7% of naïve, n=5), ERK2 (110.8±5.3% of naïve, n=5) (Fig. 6B), and p38 MAPK (112.0±8.9% of naive, n=6) (Fig. 6C) in the hippocampus. Pre-treatment with PCPA blocked the effect of fluoxetine on ERK1 (77.8±1.9% of naïve, n=4, p<0.05 vs. fluoxetine) and ERK2 (67.8±4.7% of naïve, n=4, p<0.001 vs. fluoxetine) phosphorylation. In contrast, pre-treatment with PCPA had no effect on the phosphorylated level of p38 MAPK (118.0±8.7% of naïve, n=5, p=0.64) (Fig. 6C). These results suggest that the ERK1/2 but not p38 MAPK signal pathway is involved in the antidepressant effect of fluoxetine on TBI.
FIG. 6.

Block of the effects of fluoxetine on traumatic brain injury (TBI)-induced depression-like behavior and decrease in phosphorylated levels of extracellular signal regulated kinases (ERK1/2) and p38 mitogen-activated protein kinase (MAPK) in the hippocampus. On day 1, rats were subjected to fluid percussion injury and fluoxetine or vehicle was administered (2.5 mg/kg, intraperitoneally [IP]) to the rats from day 4 to day 17. Para-chlorophenylalanine (PCPA) (250 mg/kg, IP) was administered 1 h before fluoxetine from day 14 to day 17. (A) The forced swim test was performed on day 18. TBI rats treated with fluoxetine exhibited shorter immobility time than those treated with saline. Pre-treatment with PCPA significantly reversed the effect of fluoxetine on the immobility time. *p<0.05 vs. saline, #p<0.05 vs. fluoxetine. (B) On day 18, Western blotting analysis showed that pre-treatment with PCPA blocked the effect of fluoxetine on the ERK1/2 phosphorylation. ###p<0.001 vs. naïve; *p<0.05, p<0.001 vs. TBI+fluoxetine. (C) PCPA had no effect on the phosphorylated level of p38MAPK. ###p<0.001 vs. naïve.
To confirm the involvement of ERK1/2 in the antidepressant effect of fluoxetine, a blood–brain barrier (BBB) penetrating ERK inhibitor α-[Amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)benzeneacetonitrile (SL327, 30 mg/kg, IP) was administered on day 17 and day 18 and FST was performed on day 18 (30 min after SL327 injection). Figure 7A shows that immobility time was not affected by fluoxetine treatment in the sham-operated rats but was significantly reduced in the TBI rats (t(12)=3.304, p<0.05). Fluoxetine's effect on immobility time was blocked in SL327-treated (n=7) but not in vehicle control (n=7) rats (t(12)=6.13, p<0.001). In addition, the immobility time of SL327-treated fluoxetine TBI mice was not different from those without SL327 and fluoxetine treatment mice (t(12)=0.301, p>0.5) indicating that SL327 completely blocked the effect of fluoxetine.
FIG. 7.

Effects of extracellular signal regulated kinases (ERK1/2) and p38 mitogen-activated protein kinase (MAPK) inhibitors on the antidepressant effect of fluoxetine. ERK1/2 inhibitor SL327 (30 mg/kg, n=7) (A) or p38 MAPK inhibitor SB203580 (0.1 mg/kg, n=3 or 2 mg/kg, n=3) (B) was administered intraperitoneally on day 17 and day 18 and the forced swim test was performed on day 18 (30 min after SL327 or SB203580 injection). SL327 completely blocked the effect of fluoxetine whereas SB203580 was ineffective. #p<0.05 vs. saline; ***p<0.001 vs. fluoxetine+dimethyl sulfoxide (DMSO).
We determined the effect of SB203580, a p38 MAPK inhibitor, on the action of fluoxetine. SB203580 at doses of 0.1 mg/kg to 2 mg/kg has been shown to block isoflurane preconditioning-induced neuroprotection against ischemia29 and inhibit inflammation-induced fibrosis in rats with myocardial ischemia.30 We found that 0.1 mg/kg (n=3) or 2 mg/kg (n=3) SB203580 was ineffective to affect the action of fluoxetine, and their effects were combined in Figure 7B. The immobility time of SB203580-treated fluoxetine TBI mice was not different from those vehicle-treated fluoxetine TBI mice (t(11)=0.936, p>0.1). Finally, we examined whether fluoxetine has a beneficial effect on TBI-induced brain damage. As depicted in Figure 8, infarction volume was not altered by fluoxetine treatment. Together, these data indicate that, although fluoxetine did not impact infarction volume, it did produce an antidepressant effect in TBI rats.
FIG. 8.
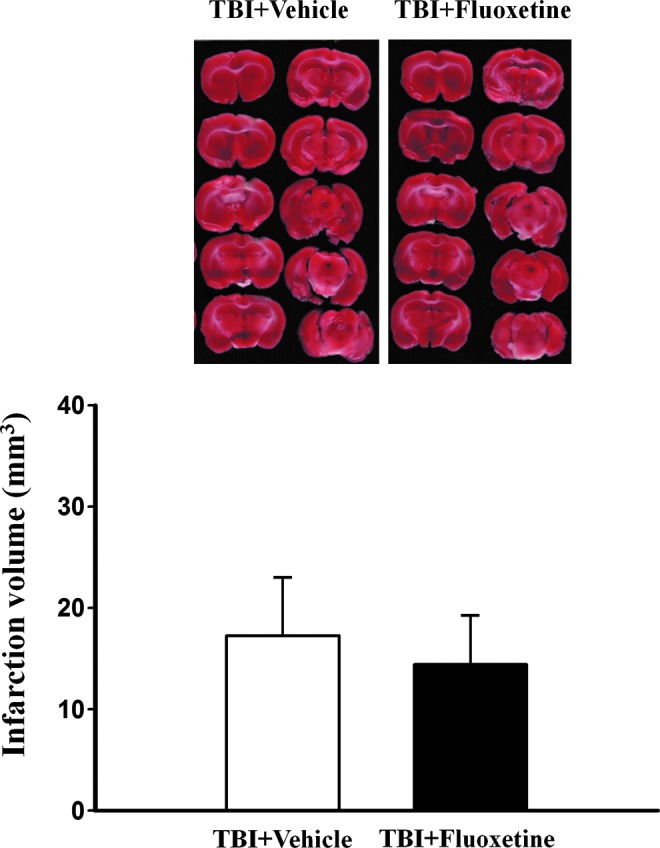
Fluoxetine does not affect traumatic brain injury (TBI)-induced brain damage. Rats were subjected to fluid percussion injury, and fluoxetine (2.5 mg/kg) or vehicle was administered intraperitoneally to the rats from day 4 to day 17. The levels of injury were evaluated by staining brain sections with triphenyltetrazolium chloride at day 18. Color image is available online at www.liebertpub.com/neu
Discussion
It has been shown that the incidence of patients with TBI and depression is high, ranging from 6% to 77%.31,32 The cellular processes underlying TBI-induced depression remain unclear, however. In the present study, using a fluid percussion model of brain injury, we have demonstrated that TBI caused depression-like behaviors in the FST, NSF test, and social interaction test in rats. TBI led to a significant decrease in the phosphorylated levels of ERK1/2 and p38 MAPK but not JNK in the hippocampus. The effects were restricted to the hippocampus because TBI had no effect on the phosphorylated levels of ERK1/2 and p38 MAPK in the amygdala. These results were in accordance with previous reports showing decreased phosphorylated levels of ERK1/2 in the post-mortem brain of depressed suicide subjects and in a rat model of depression induced by neonatal exposure of clomipramine.33,34
Anxiety is also common after TBI in humans. It has been shown that TBI rats displayed anxiety-like behaviors 1 and 3 months after injury.35 Using open fields test, however, we did not observe change in time spent in the inner area. The difference between these two studies could be because of the intensity of pressure pulse delivered by a fluid-percussion device (3.2–3.5 atmospheres vs. 2.2 atmospheres in the present study). Alternatively, the different species of rat used (Wistar vs. Sprague-Dawley rats in the present study) may account for the difference.
Fluoxetine is one of the most commonly used antidepressants in clinics. We found that chronic administration of fluoxetine significantly reduced immobility time in FST, increased sucrose preference, and improved social interaction of TBI rats, consistent with its antidepressant effect. In the NSF test, however, fluoxetine failed to decrease latency to begin chewing food. This may be explained by the anorexic effect of fluoxetine.36
MAPK phosphatase-1 (MKP-1) is a major negative regulator of ERK1/2. MKP-1 expression was increased in the hippocampal area of the post-mortem brain of patients with depression.32 MKP-1 was upregulated in the hippocampus in an animal model of chronic unpredictable stress-induced depression.37 In the present study, a serotonin synthesis inhibitor PCPA prevented the antidepressant effect of fluoxetine on TBI rats indicating the involvement of the serotonergic system in the behavioral effects of fluoxetine. Interestingly, PCPA prevented the reversal effect of fluoxetine on TBI-induced decrease in ERK1/2 phosphorylation without affecting the reversal effect of fluoxetine on TBI-induced decrease in p38 MAPK phosphorylation. These results suggest that the ERK1/2 signal pathway is responsible for both TBI-induced depression and antidepressant effect of fluoxetine. Consistent with this notion, pre-treatment with ERK inhibitor SL327 but not p38 MAPK inhibitor SB203580 blocked the antidepressant effect of fluoxetine (Fig. 7).
Prolonged neurological dysfunction that results from a brain insult is often attributed to irreversible structural damage such as loss of neurons or axonal degeneration. Here we showed that the hippocampal ERK1/2 signal pathway played a critical role in TBI-induced depression. These results suggest that the development of a positive regulator of ERK1/2 or negative regulator of MKP-1 may provide a new direction for the treatment of TBI-induced depression.
Supplementary Material
Acknowledgment
This study was supported by grants NHRI-EX101-10117NI from the National Health Research Institute, NSC101-2321-B-006-025 from the National Science Council and Aim for the Top University Project of the National Cheng-Kung University. We thank Dr Min-Der Lai for critical comments on thearticle.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Rutland-Brown W. Langlois J.A. Thomas K.E. Xi Y.L. Incidence of traumatic brain injury in the United States, 2003. J. Head Trauma Rehabil. 2006;21:544–548. doi: 10.1097/00001199-200611000-00009. [DOI] [PubMed] [Google Scholar]
- 2.French L.M. Parkinson G.W. Assessing and treating veterans with traumatic brain injury. J. Clin. Psychol. 2008;64:1004–1013. doi: 10.1002/jclp.20514. [DOI] [PubMed] [Google Scholar]
- 3.Hoge C.W. McGurk D. Thomas J.L. Cox A.L. Engel C.C. Castro C.A. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N. Engl. J. Med. 2008;358:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- 4.Ellemberg D. Henry L.C. Macciocchi S.N. Guskiewicz K.M. Broglio S.P. Advances in sport concussion assessment: from behavioral to brain imaging measures. J. Neurotrauma. 2009;26:2365–2382. doi: 10.1089/neu.2009.0906. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy R.E. Livingston L. Riddick A. Marwitz J.H. Kreutzer J.S. Zasler N.D. Evaluation of the Neurobehavioral Functioning Inventory as a depression screening tool after traumatic brain injury. J. Head Trauma Rehabil. 2005;20:512–526. doi: 10.1097/00001199-200511000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Hart T. Hoffman J.M. Pretz C. Kennedy R. Clark A.N. Brenner L.A. A longitudinal study of major and minor depression following traumatic brain injury. Arch. Phys. Med. Rehabil. 2012;93:1343–1349. doi: 10.1016/j.apmr.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 7.Bombardier C.H. Fann J.R. Temkin N.R. Esselman P.C. Barber J. Dikmen S.S. Rates of major depressive disorder and clinical outcomes following traumatic brain injury. JAMA. 2010;303:1938–1945. doi: 10.1001/jama.2010.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hicks R.R. Smith D.H. Lowenstein D.H. Saint Marie R. McIntosh T.K. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J. Neurotrauma. 1993;10:405–414. doi: 10.1089/neu.1993.10.405. [DOI] [PubMed] [Google Scholar]
- 9.Huber D. Petreanu L. Ghitani N. Ranade S. Hromadka T. Mainen Z. Svoboda K. Sparse optical microstimulation in barrel cortex drives learned behaviour in freely moving mice. Nature. 2008;451:61–64. doi: 10.1038/nature06445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iversen L. Cannabis and the brain. Brain. 2003;126:1252–1270. doi: 10.1093/brain/awg143. [DOI] [PubMed] [Google Scholar]
- 11.Pierce J.E. Smith D.H. Trojanowski J.Q. McIntosh T.K. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- 12.Thompson H.J. Lifshitz J. Marklund N. Grady M.S. Graham D.I. Hovda D.A. McIntosh T.K. Lateral fluid percussion brain injury: a 15-year review and evaluation. J. Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- 13.Sato M. Chang E. Igarashi T. Noble L.J. Neuronal injury and loss after traumatic brain injury: time course and regional variability. Brain Res. 2001;917:45–54. doi: 10.1016/s0006-8993(01)02905-5. [DOI] [PubMed] [Google Scholar]
- 14.Drevets W.C. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr. Opin. Neurobiol. 2001;11:240–249. doi: 10.1016/s0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- 15.Manji H.K. Drevets W.C. Charney D.S. The cellular neurobiology of depression. Nat. Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- 16.Harrison P.J. The neuropathology of primary mood disorder. Brain. 2002;125:1428–1449. doi: 10.1093/brain/awf149. [DOI] [PubMed] [Google Scholar]
- 17.Rajkowska G. Depression: what we can learn from postmortem studies. Neuroscientist. 2003;9:273–284. doi: 10.1177/1073858403252773. [DOI] [PubMed] [Google Scholar]
- 18.Sheline Y.I. Neuroimaging studies of mood disorder effects on the brain. Biol. Psychiatry. 2003;54:338–352. doi: 10.1016/s0006-3223(03)00347-0. [DOI] [PubMed] [Google Scholar]
- 19.MacQueen G.M. Yucel K. Taylor V.H. Macdonald K. Joffe R. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol. Psychiatry. 2008;64:880–883. doi: 10.1016/j.biopsych.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 20.Savitz J. Drevets W.C. Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci. Biobehav. Rev. 2009;33:699–771. doi: 10.1016/j.neubiorev.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prut L. Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur. J. Pharmacol. 2003;463:3–33. doi: 10.1016/s0014-2999(03)01272-x. [DOI] [PubMed] [Google Scholar]
- 22.Adachi M. Barrot M. Autry A.E. Theobald D. Monteggia L.M. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol. Psychiatry. 2008;63:642–649. doi: 10.1016/j.biopsych.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stedenfeld K.A. Clinton S.M. Kerman I.A. Akil H. Watson S.J. Sved A.F. Novelty-seeking behavior predicts vulnerability in a rodent model of depression. Physiol. Behav. 2011;103:210–216. doi: 10.1016/j.physbeh.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielsen C.K. Arnt J. Sánchez C. Intracranial self-stimulation and sucrose intake differ as hedonic measures following chronic mild stress: interstrain and interindividual differences. Behav. Brain Res. 2000;107:21–33. doi: 10.1016/s0166-4328(99)00110-2. [DOI] [PubMed] [Google Scholar]
- 25.Duman C.H. Schlesinger L. Kodama M. Russell D.S. Duman R.S. A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol. Psychiatry. 2007;61:661–670. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 26.Fumagalli F. Molteni I.R. Calabrese F. Frasca A. Racagni G. Riva M.A. Chronic fluoxetine administration inhibits extracellular signal-regulated kinase 1/2 phosphorylation in rat brain. J. Neurochem. 2005;93:1551–1560. doi: 10.1111/j.1471-4159.2005.03149.x. [DOI] [PubMed] [Google Scholar]
- 27.Koe B.K. Weissman A. p-Chlorophenylalanine: a specific depletor of brain serotonin. J. Pharmacol. Exp. Ther. 1966;154:499–516. [PubMed] [Google Scholar]
- 28.Page M.E. Detke M.J. Dalvi A. Kirby L.G. Lucki I. Serotonergic mediation of the effects of fluoxetine, but not desipramine, in the rat forced swimming test. Psychopharmacology (Berl) 1999;147:162–167. doi: 10.1007/s002130051156. [DOI] [PubMed] [Google Scholar]
- 29.Zheng S. Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of P38 mitogen-activated protein kinases. Mol. Pharmacol. 2004;65:1172–1180. doi: 10.1124/mol.65.5.1172. [DOI] [PubMed] [Google Scholar]
- 30.Yin H. Zhang J. Lin H. Wang R. Qiao Y. Wang B. Liu F. p38 mitogen-activated protein kinase inhibition decreases TNFalpha secretion and protects against left ventricular remodeling in rats with myocardial ischemia. Inflammation. 2008;31:65–73. doi: 10.1007/s10753-007-9050-2. [DOI] [PubMed] [Google Scholar]
- 31.Jorge R. Robinson R.G. Mood disorders following traumatic brain injury. NeuroRehabilitation. 2002;17:311–324. [PubMed] [Google Scholar]
- 32.van Reekum R. Bolago I. Finlayson M.A. Garner S. Links P.S. Psychiatric disorders after traumatic brain injury. Brain Injury. 1996;10:319–327. doi: 10.1080/026990596124340. [DOI] [PubMed] [Google Scholar]
- 33.Dwivedi Y. Rizavi H.S. Roberts R.C. Conley R.C. Tamminga C.A. Pandey G.N. Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J. Neurochem. 2001;77:916–928. doi: 10.1046/j.1471-4159.2001.00300.x. [DOI] [PubMed] [Google Scholar]
- 34.Feng P. Guan Z. Yang X. Fang J. Impairments of ERK signal transduction in the brain in a rat model of depression induced by neonatal exposure of clomipramine. Brain Res. 2003;991:195–205. doi: 10.1016/j.brainres.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 35.Jones N.C. Cardmone L. Williams J.P. Salsberg M.R. Myers D. O'Brien TJ. Experimental traumatic brain injury induces a pervasive hyperanxious phenotype in rats. J. Neurotrauma. 2008;25:1367–1374. doi: 10.1089/neu.2008.0641. [DOI] [PubMed] [Google Scholar]
- 36.Clifton P.G. Barnfield A.M. Philcox L. A behavioural profile of fluoxetine-induced anorexia. Psychopharmacology (Berl) 1989;97:89–95. doi: 10.1007/BF00443419. [DOI] [PubMed] [Google Scholar]
- 37.Duric V. Banasr M. Licznerski P. Schmidt H.D. Stockmeier C.A. Simen A.A. Newton S.S. Duman R.S. A negative regulator of MAP kinase causes depressive behavior. Nat. Med. 2010;16:1328–1332. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.