

Abstract
Plasmacytoid dendritic cells (pDCs) are innate immune cells that are specialized to produce interferon-alpha (IFNα) and participate in activating adaptive immune responses. Although IFNα inhibits HIV-1 (HIV) replication in vitro, pDCs may act as inflammatory and immunosuppressive dendritic cells (DCs) rather than classical antigen-presenting cells during chronic HIV infection in vivo, contributing more to HIV pathogenesis than to protection. Improved understanding of HIV–pDC interactions may yield potential new avenues of discovery to prevent HIV transmission, to blunt chronic immune activation and exhaustion, and to enhance beneficial adaptive immune responses. In this chapter we discuss pDC biology, including pDC development from progenitors, trafficking and localization of pDCs in the body, and signaling pathways involved in pDC activation. We focus on the role of pDCs in HIV transmission, chronic disease progression and immune activation, and immu-nosuppression through regulatory T cell development. Lastly, we discuss potential future directions for the field which are needed to strengthen our current understanding of the role of pDCs in HIV transmission and pathogenesis.
3.1 Introduction
Dendritic cells (DCs) are innate immune cells that play a critical role in the host response to infection, as they routinely patrol mucosal and lymph tissue and blood, are recruited to inflamed tissues, and are among the first cells to sense and respond to microbes (Steinman and Hemmi 2006). When DCs encounter pathogens, they recognize conserved structures of the microbe termed pathogen-associated molecular patterns (PAMPs). DCs recognize PAMPs by means of germline-encoded pattern-recognition receptors (PRRs). The interaction of microbial PAMPs with DC PRRs, including Toll-like receptors (TLRs) and NOD-like receptors, activates specific intracellular signaling pathways which mediate rapid antimicrobial effector functions at the site of pathogen sensing (Medzhitov 2001; Fritz et al. 2005; Tada et al. 2005). Additionally, DCs process and present microbial antigens to adaptive immune cells to program specific T and B cell responses (Guermonprez et al. 2002; Pulendran et al. 2010). DCs prime expansion of antigen-specific T cells, polarize CD4+ T cells, establish memory, regulate T cell exhaustion, and influence antibody affinity maturation and isotype switching. The specificity of the adaptive immune responses depends on the Major Histocompatability Complex (MHC) class molecule in which the antigen is presented, the concurrent combination of cytokines released, and the co-stimulatory molecules that are expressed by the DCs. Signaling pathways elicited upon PRR sensing by DCs and signals received from the tissue microenvironment ensure tailoring of an immune response to the type of pathogen (extracellular, vacuolar, intracellular) by dictating a cell-mediated vs. humoral immunity. DCs not only dictate the type of immune response acutely, but also help program the type of immune memory and prevent immunopathology through induction of regulatory mechanisms.
The two major subsets of DCs in human blood, myeloid DCs (mDCs—also referred to as conventional DC) and plasmacytoid DCs (pDCs), differ in morphology, phenotype, and function. mDCs and pDCs express different but complementary TLRs, which allow them to respond to different types of pathogens. mDCs recognize diverse pathogens due to their broad TLR expression, and display a flexible program of cytokine secretion influencing Th1, Th2, Th17, or regulatory T cell responses (Treg). While pDCs do not secrete the Th1 skewing cytokine IL-12 in humans, mDCs secrete high amounts of IL-12 in response to some bacterial or viral pathogens. pDCs specifically recognize pathogens containing ssRNA by TLR7 and unmethylated CpG DNA motifs via TLR9 and produce up to 1,000-fold more interferon-alpha (IFNα) than other types of blood cells in response to viruses (McKenna et al. 2005). Like mDCs, pDCs also display a differential response towards different microbes, varying from secretion of type I IFN to maturation and antigen presentation for T helper and T regulatory cell responses.
In this review, we focus on what is known about pDCs in HIV infection. We discuss data gathered from cell biology and immunological experiments, as well as data derived from infected humans and nonhuman primates (NHP), to demonstrate the complexity of pDC functions during acute and chronic HIV infection. In doing so, we argue that pDCs often effect conflicting functions in antiviral defense and immunopathology. Although much remains to be learned, we propose that pDCs play a crucial role both early during infection and during the chronic phase, contributing to immune activation and eventual disease progression.
Thus, while lack of activation of mDCs by HIV impairs the development of adaptive immune responses (Lore et al. 2002; Granelli-Piperno et al. 2004), rapid activation of pDCs by HIV to produce inflammatory cytokines and chemokines at mucosal sites of transmission may enhance initial infection. At the same time, persistent activation of pDCs to produce IFNα during chronic infection may contribute to immune activation and inflammation (Fernandez et al. 2011; Jacquelin et al. 2009; Bosinger et al. 2009), which are associated with disease progression to AIDS and with the development of co-morbidities such as cardiovascular disease, kidney and liver disease, and development of non-AIDS malignancies (Baker and Duprez 2010; Lekakis and Ikonomidis 2010; Ho et al. 2010; Lichtenstein et al. 2010; El-Sadr et al. 2006). In general, during acute viral infections, type I IFN produced by pDCs act as immunostimulatory cytokines favoring mDC maturation (Fonteneau et al. 2004), and as antiviral agents, activating intracellular restriction factors, inhibiting proliferation, and inducing apoptosis of infected target cells, such as T cells (Barber 2001). In contrast, chronic and systemic activation of pDCs during persistent infection, such as HIV, can paradoxically lead to deleterious inflammation and perturbation of T cell proliferation, homeostasis, and cell death (Heikenwalder et al. 2004). Additionally, it has been shown that HIV-activated pDCs produce the immu-nosuppressive enzyme indoleamine (2,3)-dioxygenase (IDO), favoring the development of regulatory T cells (Manches et al. 2008; Fallarino et al. 2007; Munn and Mellor 2004; Honda et al. 2005).
Altogether, pDCs are now implicated in the following aspects of HIV pathogenesis: (1) inciting recruitment of CCR5 + CD4+ T cells to mucosal sites of HIV inoculation during transmission (Haase 2010); (2) inducing apoptosis of CD4+ T cells through their persistent production of type I IFN (Stary et al. 2009; Fonteneau et al. 2004; Boasso et al. 2008; Herbeuval et al. 2005b); (3) increased immune activation of T cell subsets through type I IFN production; and (4) favoring generation of immunosuppressive regulatory T cells over immunostimulatory Th17 cells through their production of IDO.
3.2 Plasmacytoid Dendritic Cell Biology
The discovery and identification of pDCs came from the convergence of studies of rare blood and lymphoid tissue cell populations. Early studies in 1980s identified a specific cell type based on their characteristic plasma cell morphology and location in the T cell areas of reactive human lymphoid tissue (Vollenweider and Lennert 1983). They were later shown to express the T cell-associated marker CD4, but lacked CD3 and B cell markers, while expressing MHC-II and some myeloid markers. Based on this phenotype, the cells were termed “plasmacytoid monocytes” by Facchetti et al. (1988). In 1991, O’Doherty et al. identified a subset of blood CD4+ CD3− CD11c− immature DC that developed into mature DCs in the presence of monocyte-derived cytokines (O’Doherty et al. 1994, 1993) and in 1993 Grouard et al. also identified a subset of CD4+ CD3− CD11c− cells with a plasmacytoid morphology in the T cell area of human tonsils, which could differentiate into mature DC upon culture with IL-3 or IL-3 and CD40L. These cells were found to display an identical morphology and phenotype to plasmacytoid monocytes and to immature HLA-DR + CD11c− blood DC. Because these cells could differentiate into mature DC, they were termed pDC precursors (Grouard et al. 1997).
The existence of a rare blood cell capable of secreting extremely high amounts of IFNα had been established in the 1970s, when these cells were labeled as “natural IFN-producing cells” (NIPC). Like plasmacytoid monocytes, they did not express markers of T, B, monocyte, or NK cells, expressed MHC-II, and, importantly, secreted massive amounts of type I IFN in response to select viruses. The identical phenotype and morphology led Siegal et al. to finally identify the NIPC with pDCs (Siegal et al. 1999).
Aside from their lack of lineage markers (CD3, CD19, CD14, CD11c) and expression of MHC-II and co-stimulatory molecules (in the activated state), pDCs express several selective markers, such as sialic acid-binding immunoglobulin-like lectin H (SIGLEC-H) and bone marrow stromal antigen 2 (BST2) in mice and blood DC antigen 2 (BDCA2; also known as CLEC4C), BDCA4, leukocyte immunoglob-ulin-like receptor, subfamily A, member 4 (LILRA4; also known as ILT7) in humans, and high expression of the IL-3 receptor alpha (CD123).
3.2.1 Development
Data regarding pDC development have been garnered from mouse studies, although the recent identification of a genetic DC deficiency in humans has allowed identification of the relevant molecular players and will further help delineate the exact role of pDCs during viral and bacterial infections. Differentiation of DCs from hematopoietic progenitors relies on the activity of cytokines, such as FMS-related tyrosine kinase 3 ligand (Flt3L) and GM-CSF. As such, the Flt3 receptor is expressed in a fraction of hematopoietic stem cells and is maintained in a subpopulation of common myeloid progenitors (CMPs) (D’Amico and Wu 2003). CMPs give rise to macrophage and DC progenitors (MDP), a direct precursor of the Common DC progenitors (CDP). Flt3 and GM-CSFR expression is regulated by the transcription factor PU.1, a member of the ETS family of transcription factors, which regulate the development of monocytes and DCs (Carotta et al. 2010).
As stated above, mDCs and pDCs display complementary roles in immunity, as evidenced by their pattern of PRR, migration, cytokine secretion, and antigen presentation. The development program directing pDC vs. mDC differentiation starts at the CDP stage, through two developmental systems. pDC differentiation requires the expression of the E protein E2-2 (Cisse et al. 2008), in the absence of E protein antagonist of DNA binding (ID2). The basic helix-loop-helix E2-2 transcription factor directly binds to the promoters of several pDC selective genes such as BDCA2, LILRA4, IRF7, the pre-TCRa chain gene, IRF8, and SPIB (a relative of PU.1). E proteins are crucial for lymphopoiesis, which may explain the importance of E2-2 for expression of lymphoid-related genes in pDC (SPIB, RAG1, IL7R, TDT). Deletion of murine E2-2 blocked the development of pDCs, and haploinsufficiency in humans affected by Pitt–Hopkins syndrome yields an aberrant pDC profile and impaired IFN type I responses (Cisse et al. 2008). ID proteins directly inhibit binding of E proteins, and ID2 expression is extremely low in CDP, pre-pDC, and pDC, in contrast to conventional myeloid DCs which express high levels of ID2.
pDC differentiation also requires a high concentration of IRF8 and low levels of PU.1, and IRF8 and PU.1 may act as a complex on composite DNA elements (Carotta et al. 2010; Schiavoni et al. 2002; Tsujimura et al. 2003). The requirement of PU.1 and IRF8 has been confirmed on PU.1- or IRF8-deficient mice, and in humans deficient in IRF8, resulting in inhibition of pDC differentiation (this may be due to alteration of CDP formation, rather than a specific pDC defect). PU.1 and IRF8 share target genes in pDCs, such as Ciita94, Tlr9, and IFNα. SPIB is the closest homolog of PU.1 in mammals, and inhibiting the expression of either transcription factor in human hematopoietic progenitors inhibits pDC differentiation.
3.2.2 IFN Secretion
One of the primary functions of pDCs is the secretion of large amounts of type I IFN in response to viruses and bacterial DNA. Thus, upon recognition of some viruses, pDCs start transcribing type I IFN genes within a few hours. A major part of the pDC transcriptome is then devoted to type I IFN genes, and pDCs secrete up to 10 pg/cell type I IFN within 24 h, which represents up to 1,000 times the amount produced by any other blood cell type (Liu 2005). Human pDCs can secrete all the subtypes of type I IFNs, IFNα, IFNβ, IFNλ, IFNω, and IFNτ.
Secretion of type I IFN is one functional outcome of recognition of nucleic acids by pDC. Receptors for the sensing of microbial genetic material exist both in the endosomal and cytosolic compartment of the cell, although it seems that pDCs rely on endosomal TLR sensing system for recognition of viruses (Kato et al. 2005). Endosomal TLRs, i.e., TLR7 and TLR9, in pDCs have extracellular domains comprising multiple repeats of Leucine-Rich Repeats which mediate recognition, linked to a cytosolic Toll/IL-1 receptor homology (TIR) domain responsible for initiation of signal transduction (Akira and Takeda 2004).
TLR9 recognizes unmethylated DNA containing Cytosine–Guanosine dinucleotides (CpG), which allows preferential sensing of bacterial genomes, which contain a much higher fraction of unmethylated DNA than mammalian genomes. Based on the functional response of pDCs and B cells to CpG DNA, several classes of CpG motifs have been described. The prototypical type A CpG (CpGA) comprises oligonucleotides with a phosphodiester backbone and nuclease-resistant phosphorothioate ends. They also contain a poly-G tail, allowing aggregation of oligonucleotides into large complexes. CpGA oligonucleotides induce the secretion of high amounts of type I IFN, but limited phenotypic maturation of pDC (up-regulation of MHC and co-stimulatory molecules). CpG type B (CpGB) oligonucleotides only comprise phos-phorothioate links, and do not contain a poly-G tail. CpGB induces limited amounts of Type I IFN, but a strong maturation of pDC accompanied by secretion of TNFα and other inflammatory cytokines (Krieg 2002). TLR7recognizesguanosine–uridine-rich single-stranded RNA (ssRNA), as well as the potent synthetic antiviral imidazo-quinolines. Although Uridine tetramers may be the minimal motifs recognized by TLR7, GU-rich sequences preferentially induce IFNα by pDCs, and there is evidence that CpG motifs in viral RNA can also contribute to RNA recognition (Greenbaum et al. 2009; Jimenez-Baranda et al. 2011). Although guanosine- and uridine-rich regions in the HIV LTR were known to stimulate TLR7 (Heil et al. 2004), the molecular determinant of HIV virion recognition in pDC remained unknown, until it was proven that endosomal delivery of viral RNA, rather than early retrotranscipts, stimulates pDC through TLR7 (Beignon et al. 2005).
The endosomal and lysosomal localization of TLR7 and TLR9 in activated pDCs allow sampling of internalized material while preventing inappropriate activation by self-DNA or -RNA. Further restriction of TLR activation is ensured by processing of TLR in the endo-lysosomal compartments. Delivery of TLR7 and TLR9 to the endosomes is dependent on the RE-resident protein Unc93b1, and activation of TLR9 requires its cleavage by acidic proteases in endo-lysosomal compartments (Barbalat et al. 2011). Asparagine endopeptidase is a key protease allowing N-terminal processing of TLR9 (Sepulveda et al. 2009), in conjunction with other acidic proteases, and only the cleaved form of TLR9 can associate with the signaling adaptor MyD88. The same form of receptor processing likely exists for TLR7. The preprocessing and endosomal localization of TLR7 and TLR9 allows recognition of foreign ligands such as internalized viruses without actual infection of pDC. Many inactivated viruses, including chemically inactivated HIV, can trigger IFNα secretion with the same potency as live viruses (Beignon et al. 2005). Some live cytosolic viruses may still gain access to the endosomal compartments through autophagy-mediated delivery of cytosolic content to the endolysosomes (Lee et al. 2007).
TLR7 and TLR9 both signal through the TIR-containing adaptor MyD88, which leads to activation of IRF7 for type I IFN secretion. The engagement of TLR-MyD88 leads to assembly of a multiprotein complex in the cytoplasmic tail of TLR, comprising IRAK4, Bruton’s tyrosine kinase (BTK), and IRF7. IRF7 is ubiquitylated by the ubiquitin E3 ligase activity of TRAF6 and further phosphorylated by IRAK4. Activated IRF7 then interacts with TRAF3, IKKα, IRAK1, and osteopontin, translocating to the nucleus for type I IFN gene transcription. TRAF6 also ubiquitylates the protein kinase transforming growth factor-β (TGFβ)-activated kinase 1 (TAK1) for activation of NF-κB and MAPK, leading to transcription of inflammatory cytok-ines, chemokines, and co-stimulatory molecules (Akira and Takeda 2004). IRF7 expression in pDCs is constitutively high, in part due to continuous autocrine feedback by low levels of type I IFN (O’Brien et al. 2011), and by low expression of the translational repressors 4e-BPs (Colina et al. 2008). Upon TLR stimulation, a first wave of type I IFN can also positively feedback to amplify the IFN response, and activation of IRF8 further amplifies IFN secretion by pDCs.
TLR sensing of HIV by pDCs mostly occurs through endosomal recognition of viral RNA by TLR7. Although free virus triggers IFN secretion by pDCs, it is likely that cell-associated virus also contributes to pDC activation, possibly through the uptake of HIV-containing apoptotic cell vesicles (Lepelley et al. 2011), enhancing endosomal delivery of HIV RNA to the TLR-containing endosomes. Uptake of HIV-containing apoptotic vesicles may also potentiate delivery of HIV antigens for cross-presentation (see below). Cytosolic sensing of HIV has not been shown to significantly contribute to IFNα secretion in pDCs. The cytosolic exonuclease TREX1 suppresses cytosolic innate sensing in CD4+ T cells and macrophages (Lepelley et al. 2011), and the HIV Tat gene product can inhibit Protein Kinase R (PKR) (Cai et al. 2000), which may partly account for the predominance of TLR recognition of HIV for IFN secretion in pDCs.
3.2.3 Antigen Presentation
Another major function of pDCs is the presentation of virus antigens to CD4+ and CD8+ T cells. Although pDCs are generally thought to be weaker antigen-presenting cells (APCs) than mDCs, activated pDCs can efficiently activate memory CD4+ and CD8+ responses, and in some instances can prime naïve T cells. Initial characterization of pDCs in terms of T cell stimulation was based on allogeneic mixed lymphocyte reactions. It was shown that pDCs stimulated by influenza virus and CD40L can induce strong allogeneic Th1 cultured with IL-3 or IL-3 + CD40L can prime allogeneic CD4+ T cells to differentiate into IL-4-secreting Th2 cells or IL-10-secreting Tregs (Rissoan et al. 1999). The Th1 polarization induced by Flu-activated pDCs may reflect their high expression of MHC and co-stimulatory molecules, as well as secretion of type I IFN (Cella et al. 2000; Huber and Farrar 2011).
Upon viral infection, pDCs can potently present viral antigens and stimulate expansion of anti-viral CD4+ and CD8+ T cells (Fonteneau et al. 2003). However, in HIV infection, pDCs are poorly infected, and it is unclear to what extent they can present endogenous viral antigens. However, pDCs can acquire exogenous antigens in the form of soluble proteins or cell-derived material to cross-present to CD4+ and CD8+ T cells. Early work suggested that pDCs are not able to cross-present exogenous antigens to CD8+ T cells, even upon activation by CpG, but could present endogenous antigens. These experiments involved loading of pDCs with model antigen with concomitant CpG activation, and injection of antigen-loaded pDC in mice (Salio et al. 2004). However, in other studies, pDC could present exogenous ovalbumin (OVA) to naïve CD8 T cells in vitro, but only when pDCs were activated by CpG. In this study, they could not present OVA to OVA-specific CD4+ T cells, unless the soluble protein was complexed with specific antibodies (Kool et al. 2011). OVA-immune complexes enhanced antigen uptake and induced its rerouting to acidic MHC-II+ compartments (Benitez-Ribas et al. 2006). More recent work has examined the cell biology of viral antigen cross-presentation in mouse and human pDCs. DiPucchio et al. showed that influenza virus stimulation induces routing of MHC-I molecules to the cell surface within 30 min of exposure, and cross-presentation of Flu antigen to CD8+ T cells within 4 h. Infection of pDCs was probably not required, as cross-presentation was dependent on endocytic recycling and independent of the proteasome. Cross-presentation by pDCs to memory CD8+ T cells was more potent than cross-presentation by mDCs, suggesting that pDCs may have evolved a rapid mechanism of viral antigen presentation for activation of memory responses (Di Pucchio et al. 2008). Other reports have shown that pDCs can cross-prime naïve CD8+ T cells after TLR activation (Mouries et al. 2008). The requirement for TLR activation for cross-presentation may be due to TLR-MyD88 recruitment of TAP molecules to nonacidic early endosomal compartment in some forms of cross-presentation (Burgdorf et al. 2008). Finally, it has been shown that type I IFN can enhance cross-presentation of viral antigens by dendritic cells in vivo, possibly through enhanced delivery of co-stimulatory signals (Le Bon et al. 2003).
Cross-presentation of HIV antigen by pDCs has now been conclusively demonstrated in several in vitro studies. Uptake of HIV lipopeptides (Hoeffel et al. 2007), uptake of noninfectious HIV particles, and exposure to HIV-infected apoptotic cells (Larsson et al. 2002) lead to cross-presentation of HIV antigen to specific CD8+ T cells in a proteasome-dependent manner. These studies highlight the dual role of pDCs during HIV infection, where they can secrete high amounts of type I IFN, but also present viral antigens to CD4+ and CD8+ T cells. Furthermore, cytokines secreted by pDC can also induce maturation of bystander APCs in a TNFα- and IFNα-dependent way (Fonteneau et al. 2004).
3.2.4 Migration
Unlike mDCs, pDCs are found in rare numbers in peripheral tissues under steady-state conditions. pDCs circulate through the body via the bloodstream, and enter secondary lymphoid tissues via High Endothelial venules (HEV) (Cella et al. 1999; Liu 2005). In inflammatory conditions, pDCs leave the bloodstream and accumulate at the site of infection, where they can secrete IFNα, take up antigens, and migrate to draining lymph nodes for antigen presentation. pDC homing to HEV is facilitated by its expression of L-selectin and PSGL1, the counter ligands of E and P selectins, respectively. Furthermore, pDCs express CXCR4, a receptor for the homeostatic chemokine CXCL12 expressed by HEV. Under reactive conditions, pDCs express additional molecules involved in homing to secondary lymphoid organs, such as CCR5 and CXCR3 (Diacovo et al. 2005; Yoneyama et al. 2004). pDCs can also express the chemokine receptor CCR9 and migrate to its ligand CCL25 to home into the small intestine (Wendland et al. 2007). pDCs purified from blood express an array of chemokine receptors. However, pDCs do not migrate in vitro towards the respective ligands (with the exception of CXCR4), indicating that these chemokine receptors are not functional in the steady state (Penna et al. 2001). Although CXCR3 is unable to induce migration of pDCs, it potentiates the chemotactic response to CXCL12, and mediates adhesion and migration to heparan sulfates expressed by endothelial cells (Krug et al. 2002). In addition, pDCs exposed to IFNα acquire the ability to migrate in response to CCR2, CCR5, and CXCR3 ligands (Cicinnati et al. 2008).
In addition to chemokines, pDCs can respond to two agonists released by damaged tissues at the site of inflammation: Adenosine can engage the Adenosine Receptor A1, and F2L can trigger the formyl peptide receptor FPR3 (Devosse et al. 2009; Schnurr et al. 2004). pDCs express functional receptor for the anaphylatoxins C3a and C5a, and can also migrate in response to IL-18, an inflammasome-generated inflammatory mediator (Gutzmer et al. 2006; Kaser et al. 2004). Thus, in addition to chemotactic chemokines, pDCs can be recruited through signals associated with inflammation and tissue damage (Jimenez-Baranda et al. 2012). We discuss below the crucial role of pDC recruitment at the site of HIV entry. Viruses can activate pDCs and direct their secretion of T and NK cell chemotactic chemokines, such as CXCL9, CXCL10, and CCL4 (Piqueras et al. 2006; Megjugorac et al. 2004; Bendriss-Vermare et al. 2005). Although it is possible that secretion of the CCR5 ligands CCL3 and CCL4 by HIV-activated pDCs could limit viral spreading, the presence of inflammatory pDCs at the site of viral transmission seems to fuel HIV spread rather than limit it.
3.3 HIV–Plasmacytoid Dendritic Cell Interactions
3.3.1 Entry
HIV enters susceptible cells either through direct fusion with the cell membrane or through receptor-mediated endocytosis. Direct fusion of HIV occurs following a series of interactions between the heterotrimeric HIV glycoprotein gp120/gp41 and cell-surface CD4 receptor and a co-receptor, CXCR4 or CCR5. HIV fusion is pH independent and results in insertion of HIV cores into the cytoplasm with subsequent reverse transcription, integration, and productive infection (Stein et al. 1987). HIV enters mDCs either through CD4 receptor-mediated endocytosis and/or through C-type lectin receptors such as DC-specific ICAM-3-grabbing nonintegrin (DC-SIGN) (Geijtenbeek et al. 2000). mDCs do not become activated by HIV to produce cytokines or to mature for antigen presentation (Fonteneau et al. 2004). mDCs are minimally infected by HIV but coculture of HIV-infected mDCs with T cells results in explosive infection (Frank et al. 1999). The block to HIV replication/infection in mDC is likely due to the high expression of the restriction factor SAMHD1 (Laguette et al. 2011; Goldstone et al. 2011; Hrecka et al. 2011), a deoxy-nucleoside triphosphate triphosphohydrolase. It is thought that SAMHD1, which is highly expressed in mDCs, restricts HIV replication by hydrolyzing the majority of dNTPs, thus inhibiting reverse transcription and viral complementary DNA (cDNA) synthesis. SAMHD1 has only been analyzed in monocyte-derived dendritic cells and macrophages, but has not yet been investigated in primary mDCs, pDCs, or monocytes; therefore it is unclear whether SAMHD1 function in these cell types contributes to inhibition of HIV replication.
pDCs are also minimally infected by HIV (Fong et al. 2002), but in contrast to mDCs are highly activated to produce IFNα by both live and inactivated but fusion-competent HIV (Beignon et al. 2005; Fonteneau et al. 2004). pDCs express CD4, CXCR4, and CCR5, but not DC-SIGN. HIV entry and activation of pDCs to produce IFNα require HIV envelope glycoprotein (Beignon et al. 2005). When pDCs are incubated with HIV and b12, a human neutralizing anti-gp 120 mAb, or 17b, a human neutralizing anti-gp120 mAb that binds to an epitope induced by binding gp120 to CD4, IFNα production is inhibited. HIV entry and activation of pDCs to produce IFNα also require cell surface CD4. In pDCs incubated with HIV in the presence of an anti-CD4 antibody, IFNα is markedly inhibited. However, co-receptor usage does not seem necessary for pDC activation by HIV (Beignon et al. 2005; Herbeuval et al. 2005a; Schmidt et al. 2005; Haupt et al. 2008). Further support of the necessity of CD4-gp120 binding to pDC to induce IFNα production has been supported by the finding that the degree of IFNα induction is correlated with the affinity of the virus to CD4 (Haupt et al. 2008). pDCs are not activated by non-virion-associated HIV envelope glycoprotein, however, and may actually inhibit pDC activation (Beignon et al. 2005; Martinelli et al. 2007).
3.3.2 Activation
pDCs are activated by the interaction of ssRNA or unmethylated CpG DNA (CpG) with TLRs 7 and 9, respectively, in endosomal compartments of the cell. The source of ssRNA is usually viral and the source of CpG DNA may be bacterial products or self-DNA, as seen in systemic lupus erythematosis (SLE) (Boule et al. 2004). Depending on the activating stimuli, pDCs can be functionally dichotomous. pDCs can either become IFNα-producing cells (IPCs) that minimally upregulate maturation molecules but produce substantial amounts of IFNα (over 1,000× other cell types) (McKenna et al. 2005) or become potent APCs that do not produce IFNα but rather produce NF-κB-dependent inflammatory cytokines such as TNFα, IL-6, and IL-8, and upregulate co-stimulatory molecules for potent presentation of antigen to T cells (O’Brien et al. 2011).
Whether pDCs become IPCs or APCs seems related to the endosomal compartments to which the stimulating ligands traffic. Much of the work on understanding how trafficking of TLR ligands determines phenotype and function in pDCs has been done by studying CpGs in pDCs. It is well established that different CpGs predominantly induce IFNα- or NF-κB-dependent responses, based primarily on their sequence and/or secondary structure. For example, it has been shown that CpGA has poly G sequences at its ends as well as internal palindromic sequences containing CG dinucleotides that form multimolecular complexes. A large fraction of internalized CpGA traffics to and is retained in early endosomal compartments of pDCs, and results in IFNα signaling. In contrast, CpGB, which comprises one or more unmethylated 6mer CpG motifs in a fully phosphorothioated backbone, remains monomeric and traffics rapidly to lysosomes to stimulate NF-κB-dependent responses (Honda et al. 2005).
Compartmental localization of TLR agonists within pDCs determines whether pDCs can act as effective APCs. CpGB and influenza stimulate pDCs to form NF-κB-dependent intracellular pools of MHC II molecules that are persistently neosynthe-sized and accumulate in antigen-loading compartments. In contrast, CpGA stimulation of pDCs does not lead to the formation of MHC II intracellular clusters (Sadaka et al. 2009). CpGB-stimulated pDCs efficiently process and present CMV antigens and are capable of stimulating CMV-specific effector memory T helper cells. CpGA-stimulated pDCs produce large amounts of type I IFNs, but fail to induce CMV-specific CD4+ effector memory T cells to produce IFN-γ (Jaehn et al. 2008). Interestingly, influenza virus induces IFNα production but also matures pDCs fully, in comparison to CpGB, which stimulates minimal IFNα production. Agonists such as influenza virus or CpGC can both stimulate IFNα production and antigen-presenting capacity in pDCs; however, these pDCs that obtain the mature antigen-presenting phenotype become refractory to IFNα production (Kerkmann et al. 2003). Thus, TLR agonists which strongly activate NF-κB signaling pathways to fully mature pDCs to gain an APC phenotype result in a pDC refractory state to further cytokine production.
Mechanisms by which IFNα signaling pathways are turned off in TLR-activated pDCs remain to be elucidated. Potential mechanisms for inhibited IFNα production after strong maturation include changes at the transcriptional and posttranscriptional level. At the transcriptional level, it has been shown that IRF7 mRNA is downregulated after a strong maturational stimulus (O’Brien et al. 2011). At the posttranscriptional level, it has previously been shown that strong activation of the NF-κB signaling pathway causes ubiquitination and proteasomal degradation of IRAK1, a necessary component of the transductional transcriptional processor complex necessary for IRF7 phosphorylation and nuclear translocation (Kubo-Murai et al. 2008). In contrast, IRF7 is upregulated in human pDCs both in vitro and in vivo, possibly contributing to persistence of IFN signaling in HIV disease (O’Brien et al. 2011; Sabado et al. 2010).
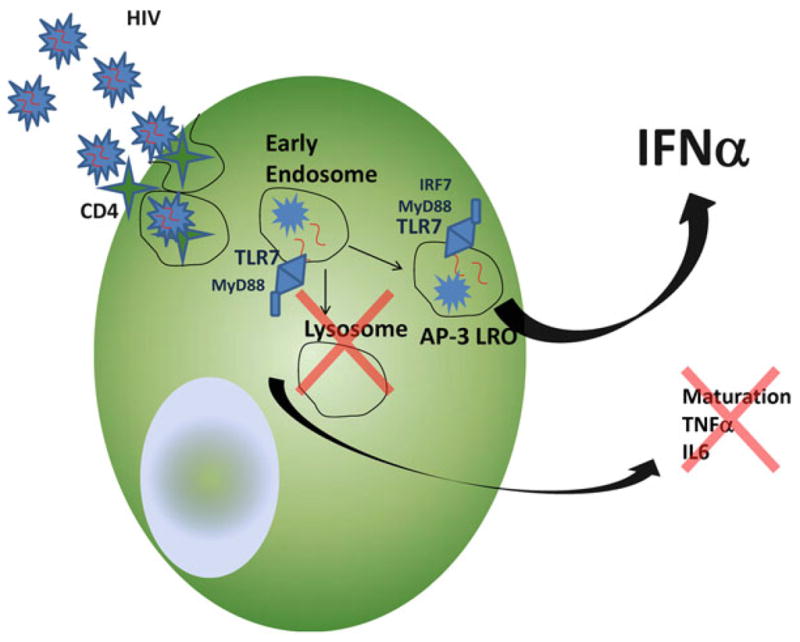
New data also implicate adaptor-protein 3 (AP-3) as a critical endosomal protein complex involved in IFNα signaling in pDCs (Sasai et al. 2010). A newer model of pDC spatiotemporal trafficking is emerging, whereby stimulatory agonists first traffic to a common early endosomal compartment to trigger NF-κB-dependent responses, including IL-6 and TNFα production and expression of co-stimulatory molecules. CpGB, which strongly stimulates NF-κB pathways but does not stimulate IFNα, is rapidly found in lysosomal compartments. Alternatively, CpGA, which weakly stimulates NF-κB pathways but stimulates substantial IFNα production, traffics from the common early endosomal compartment to an AP-3 lysosome-related organelle (LRO) compartment, where IFNα signaling pathways are triggered (Sasai et al. 2010).
The activation of pDCs by HIV has been studied in detail. HIV activates pDCs to produce high levels of IFNα and lower levels of TNFα, and minimally upregulates co-stimulatory molecules, regardless of the HIV CXCR4 or CCR5 strain used. Endocytosis, but not fusion, is required for pDC activation by HIV. C34, a peptide that inhibits post-CD4 binding conformational changes in the transmembrane gp41 that is required for fusion, does not inhibit HIV activation of pDCs. However, inhibitors of endocytosis such as dimethyl amiloride, cytochalasin D, and chlorpromaz-ine inhibit IFNα production by HIV-activated pDCs. Additionally, inhibitors of endosomal acidification, such as chloroquine, quinacrine, ammonium chloride, and bafilomycin, inhibit IFNα production by HIV-activated pDCs. Altogether, CD4-mediated endocytosis and endosomal acidification seem to be required for pDCs to become activated and to produce IFNα in response to HIV (Beignon et al. 2005).
Studies have been performed to understand whether HIV RNA or DNA is responsible for pDC activation. pDCs cultured with recombinant virions deficient in viral RNA do not become stimulated to produce IFNα or TNFα. Additionally, pDCs cultured with plasmid DNA encoding HIV, circular or linear, and formulated in cationic lipids, do not become stimulated to produce IFNα. In contrast to viral DNA, pDCs cultured with purified HIV RNA preincubated with cationic lipids produce high levels of IFNα and TNFα, and RNAse treatment inhibits this effect (Beignon et al. 2005). Additionally, pDCs are activated to produce IFNα by guanosine- and uridine-rich ssRNA motifs derived from the 5′ untranslated region of HIV-1, which have been reported to interact with TLR7 and TLR8 (Heil et al. 2004). HIV pseudotyped with vesicular stomatitis virus envelope (VSV-G), which is pantropic and allows for infection of all cell types tested, activates pDCs to produce IFNα and TNFα but does not activate mDCs (Beignon et al. 2005).
pDCs are likely activated by HIV through TLR7, as TLR7 oligonucleotide inhibitors are much more potent than TLR9 inhibitors in blocking IFNα production by HIV-exposed pDCs in human pDCs (Beignon et al. 2005; Mandl et al. 2008). Furthermore, silencing of TLR7 by siRNA in a pDC line inhibited the majority of IFNα production by HIV-1-infected cells (Lepelley et al. 2011). It remains to be elucidated whether incomplete inhibition of IFNα in HIV-activated pDCs by TLR7 oligonucleotide inhibitors and siRNA occurs because of limited specificity of these reagents, or whether additional and as yet undefined pDC PRRs sense HIV.
Classically, dendritic cells that have been activated by a microbial pathogen become refractory to subsequent activation. This phenomenon, commonly referred to as TLR tolerance, is thought to be a protective mechanism against unopposed and deleterious immune activation and inflammation (Geisel et al. 2007). In pDCs, however, certain viral and synthetic TLR ligands do not conform to this phenomenon and do not make pDCs refractory to subsequent activation. Importantly, both CpGA and HIV have been shown to activate the type I IFN receptor-mediated auto-crine feedback loop and to persistently activate pDCs to produce IFNα. pDCs stimulated with CpGA or HIV can be restimulated to produce IFNα whereas CpGB- and influenza-activated pDCs become refractory to stimulation and cannot secrete IFNα. Both HIV and CpGA traffic to “early” EEA-1 and “recycling” transferrin receptor endosomal compartments. This results in limited NF-κB-dependent responses and weak T cell immunity but strong and persistent IFNα secretion. In contrast, influenza virus strongly activates NF-κB-dependent responses like CpGB to mature pDC for strong antigen presentation to T cells and to inhibit ongoing IFNα production (O’Brien et al. 2011).
Altogether, the data support a model (Fig. 3.1) in which HIV enters pDCs through CD4-mediated endocytosis to traffic predominantly to early and recycling endo-somes to stimulate weak NF-κB-dependent responses but strong and persistent IFNα responses. Both NF-κB and IFNα responses are pH dependent, and HIV RNA predominantly stimulates these responses through endosomal TLR7. It remains unclear whether HIV activation of pDCs to become IPCs instead of APCs plays a role in HIV transmission, persistent inflammation and immune activation, or suboptimal adaptive immune response development during HIV infection.
Fig. 3.1.
pDC entry and activation by HIV
3.4 The Role of Plasmacytoid Dendritic Cells in HIV Transmission and Primary Infection
3.4.1 Transmission
Although lentiviral infections such as HIV are characterized by long incubation periods and a protracted clinical course, the initial transmission event which leads to systemic infection is rapid and explosive (Geisel et al. 2007). Much of the research that informs this knowledge comes from the simian Immunodeficiency Virus (SIV) rhesus macaque NHP model of mucosal transmission. The rhesus macaque (RM) model of pathogenic SIV closely parallels that of HIV, in that the animals experience high levels of viral replication, progressive CD4+ T cell depletion, chronic immune activation, and mucosal and systemic immunodeficiency leading to AIDS (Paiardini et al. 2009; Brenchley et al. 2010).
The transmission events that transpire during the first 2 weeks of HIV infection are logistically and ethically difficult to study in humans because HIV is asymptomatic during this time period. RM studies have proven invaluable to investigate these critical early time points of infection. From these studies, we now understand that virions cross mucosal epithelia within hours of inoculation and comprise a small homogeneous founder population. Genetic studies of transmitted virus support the concept of a small infected founder population, as infection in humans is acquired from a single virus genotype in 80% of cases (Keele et al. 2008). This small founder population then undergoes a local expansion during the first week of infection to establish an irreversible, systemic, and self-propagating chronic infection. During the second week of infection the virus can be detected in lymph nodes where latent infection is thought to persist. At this time gut-associated lymphoid tissue (GALT) is also highly infected, with resultant massive loss of CD4+ T cells (Li et al. 2009; Brenchley and Douek 2008).
A new model of HIV mucosal transmission events has emerged, based upon detailed immunohistochemistry studies of cervical and vaginal tissues taken from RMs that were intravaginally inoculated with SIV in vivo. Upon exposure to SIV, endocervical epithelial cells produce macrophage inflammatory protein 3 (MIP-3α) which then attracts substantial numbers of pDCs. pDCs are the first predominant cell type to arrive to infected mucosal sites and are activated by HIV to produce MIP1β and other chemokines and cytokines which attract CD4+ T cells. While CD4+ T cells are present in small numbers in normal endocervical mucosal tissue, release of cytokines and chemokines by HIV-activated pDCs in locally HIV-infected tissue recruits high numbers of CD4+ T cells for explosive and sustained infection. Furthermore, it has been shown that topical vaginal application of glycerol monolaurate, which inhibits the production of MIP-3α, prevents local and systemic SIV infection, therefore suggesting that pDC recruitment to mucosal sites of HIV inoculation is critical to transmission and subsequent systemic infection (Li et al. 2009). Altogether, this emerging model of initial SIV transmission events suggests that innate signaling, including pDC activation, during HIV mucosal inoculation paradoxically facilitates rather than restricts viral replication and expansion of infection.
3.5 pDC Function in Primary Infection
In chronic HIV infection, pDC and mDC are lost from the blood and this depletion correlates with high plasma viral load and low CD4+ T cell counts (Barron et al. 2003; Donaghy et al. 2001, 2003). Similarly, blood mDC and pDC numbers are markedly reduced in primary infection and are dysfunctional. Sabado et al. found that the ability of mDC and pDC to stimulate T cells, as measured by a mixed lymphocyte reaction (MLR), was not de fi cient (Sabado et al. 2010), whereas Huang et al. found that mDC’s ability to stimulate T cells in MLR was diminished (pDC MLRs were not tested) (Huang et al. 2011). Differences in study results were thought due to different techniques used for performing the MLR. Sabado et al. tested MLR using radioactive incorporation of tritium on day 5 of T-cell DC cocul-ture, whereas Huang et al. used a CFSE-based assay that allows for measuring proliferative responses over the entire period of culture. Additionally, MLRs in the Huang study were performed in more patients with earlier acute HIV-1 infection. Notably in the Sabado study, pDC from acutely HIV-infected subjects with the highest HIV RNA viral loads had the lowest ability to stimulate T cells.
In the Sabado study, both mDC and pDC from primary HIV-infected subjects (mostly with later acute HIV infection, as characterized by Fiebig stage III–VI) were low in number but were hyperfunctional as compared to uninfected control subjects. In response to overnight TLR7 agonist stimulation with R848 (resiqui-mod), mDCs and pDCs from HIV-infected subjects produced more inflammatory cytokines in culture supernatants than normal control DCs. mDC produced significantly more IL-6, TNFα, IP-10, MIP1α, MIP1β, RANTES, and IL-12p70 in response to R848, while pDC produced significantly higher levels of IFNα, IL-6, TNFα, MIP1α, MIP1β, and RANTES in response to R848. pDCs from primary HIV-infected subjects also produced cytokines/chemokines in response to stimulation with HIV. In contrast, in the Huang study, which used intracellular cytokine staining to study cytokine production in response to CLO97, which is a synthetic TLR7/8 agonist very similar to R848, results were mixed, with mDC IL-12p70 or TNF-α production being significantly reduced during acute- and early-stage HIV-1 infection, whereas frequencies of IL-6-secreting mDCs were similar between the patient groups. They showed that IFNα-producing pDCs, as well as the per-cell intensity of IFNα production by pDCs, were substantially decreased in subjects with acute- and early-stage HIV-1 infection. Proportions of IL6- and TNFα-secreting pDCs were similar between patients from primary HIV-1 infection and HIV-1-negative controls, but per-cell intensities of cytokine production tended to be higher in patients with primary HIV-1 infection, specifically with regard to IL-6. Future studies may elucidate differing results between these two studies, but it seems apparent that DC dysfunction during the earliest stages of HIV infection likely contributes to inadequate adaptive immune response development.
In AIDS-susceptible NHP (RMs) that have been infected intravenously with SIV, pDCs are mobilized in the blood within 3 days after infection (Barratt-Boyes et al. 2010) to increase three- to sevenfold in blood and 10- to 20-fold in lymphoid tissue (Brown et al. 2009). Subsequently, by day 14 pDCs are depleted in blood but are highly activated, highly infected, and apoptotic in lymphoid tissue. Nevertheless, remaining pDCs had essentially normal functional responses to stimulation through TLR 7, with half of lymph node pDCs producing both TNFα and IFNα (Brown et al. 2009). These findings reveal that cell migration and death both contribute to pDC depletion in acute SIV infection. Lymph node pDCs during acute SIV are highly activated, infected, and apoptotic, which likely influences CD4+ T cell activation and infection.
3.6 pDC Function in Chronic Infection
HIV-1 disease progression is associated with a decline of circulating pDCs in blood, which correlates with high viral load and reduced CD4 counts. Chronic production of IFNα has been observed in HIV patients and is associated with disease progression. Increased IFNα expression in circulating peripheral blood mononuclear cells (PBMCs) correlates with HIV/SIV disease progression (Barron et al. 2003; Donaghy et al. 2001, 2003). In particular, a specific subtype of IFNα, namely, IFNα2b, is preferentially upregulated in HIV-1 patients throughout the course of the disease (Lehmann et al. 2009). In SIV models, persistent decline of circulating pDCs and chronic over-expression of IFNα are also observed during pathogenic SIV infection of nonnatural hosts, but not during nonpathogenic SIV infection of natural hosts.
Although pDCs are depleted in blood during chronic HIV infection, they accumulate in lymph nodes. The frequency of pDCs in lymph nodes is higher in HIV-infected patients than in HIV-uninfected controls, and highest in patients with the highest viral loads (Lehmann et al. 2010). Additionally, pDCs from lymph nodes of HIV-infected patients secrete higher amounts of IFNα spontaneously (in the absence of exogenous stimuli) but do not express higher levels of co-stimulatory molecules, suggesting that these pDCs are activated in vivo to produce IFNα but are immature IPCs, not APCs. pDCs in lymph nodes of HIV-infected patients, although higher in frequency than in uninfected controls, also exhibit increased apoptosis, as evidenced by increased staining of Annexin V, a marker for cells undergoing apoptosis. Circulating pDCs in the blood of HIV-infected patients express higher levels of lymph-node homing markers CCR7 and CD62L. Additionally, pDCs from HIV-infected patients respond more potently to CCR7 ligands, CCL19 and CCL21, in migration assays, suggesting that pDCs in chronically HIV-infected patients are poised to migrate more readily to lymphoid tissues. Therefore pDC depletion in the blood of HIV-infected subjects is likely due to redistribution of pDCs to lymphoid compartments where they ultimately die. These results have been confirmed in chronically SIV-infected macaques, which evidenced four times more pDC in GALT than uninfected control macaques, and the pDCs in chronically SIV-infected RMs were more stimulatory to produce IFNα than uninfected control pDCs (Reeves et al. 2012). SIV-infected macaques also have depleted pDCs in blood but elevated pDCs in lymphoid compartments during acute infection, which decline in AIDS, likely due to apoptosis. The acute phase of nonpathogenic SIV infection in natural hosts (e.g., sooty mangabeys (SMs) and African green monkeys (AGMs)) also involves a decline of circulating pDCs, which initially relocate to lymph nodes, and then return to the circulation following the onset of the chronic phase, therefore implicating a role for pDC dynamics in pathogenic lentiviral infection in humans and animal models (Diop et al. 2008).
During chronic HIV infection, circulating blood pDCs have diminished functionality in response to TLR7 and TLR9 stimulation (Finke et al. 2004; Tilton et al. 2008; Kamga et al. 2005). This compromised functionality is thought to occur because pDCs are exposed to HIV, CpGs from bacterial translocation from the gut, and inflammatory cytokines in vivo; therefore they are more refractory to stimulation. Notably, CD4 is expressed at lower levels on the surface of pDCs in viremic HIV patients, but is more highly expressed on the surface of pDCs from uninfected controls and elite controllers. However, the intracellular CD4 is higher in HIV viremic patients than uninfected controls and elite controllers, suggesting that pDC activation in vivo causes CD4 internalization by pDCs. Decreased surface CD4 expression correlated with decreased IFN production in response to stimulation of pDCs with HIV (Machmach et al. 2012). It is unclear why pDCs in lymphoid tissues are hyperstimulatory during chronic HIV or SIV infection (Lehmann et al. 2010) while circulating blood pDCs are hypostimulatory. One possible explanation for these divergent responses between lymphoid and blood pDCs is that pDCs were stimulated with different reagents. In the blood pDC studies, pDCs were stimulated with synthetic TLR7 and TLR9 agonists. In the human lymphoid tissue pDC study, spontaneous activation was measured and in the SIV GALT study, pDCs within PBMCs were stimulated with PMA and/or poly:IC.
3.7 The Role of pDCs in Immune Activation During Chronic HIV Infection
HIV infection is marked by aberrant immune activation, which is a better correlate of disease progression to AIDS than viremia (Hazenberg et al. 2003; Deeks et al. 2004; Papagno et al. 2004; Benito et al. 2005; Giorgi et al. 1999; Bofill et al. 1996). Chronic immune activation and inflammation also persist in HIV infection despite antiretroviral therapy (ART) (Hunt et al. 2003; El-Sadr et al. 2006) and contribute to increased risk of serious non-AIDS conditions such as cardiovascular disease, kidney disease, liver disease, and non-AIDS-defining malignancies (Baker and Duprez 2010; Lekakis and Ikonomidis 2010; Ho et al. 2010; Lichtenstein et al. 2010; El-Sadr et al. 2006). Immune activation is characterized by increased expression of HLADR, CD38, and Ki67 on CD4+ and CD8+ T cells. The cause of immune activation in AIDS is unknown, but stimulation of innate immune cells directly by HIV and indirectly by products of bacterial translocation may be major contributors. Thus chronic injection of TLR7 or TLR9 agonists in mice induces characteristic lymphoid tissue disruption and immune deficiency, a syndrome resembling HIV-induced immunodeficiency (Heikenwalder et al. 2004; Baenziger et al. 2009).
Both human and animal studies support a role for pDCs and IFNα in the pathogenesis of HIV immune activation and inflammation. High plasma titers of IFNα during acute- and late-stage disease have been shown to correlate with disease progression (von Sydow et al. 1991). When matched for blood HIV RNA levels, women progress to AIDS more rapidly than men, express higher markers of immune activation, and produce more IFNα per pDC when challenged with HIV ex vivo, thought related to effects of female sex hormones on pDC functionality (Meier et al. 2009). Lymphoid tissue and circulating PBMCs derived from HIV-infected subjects with progressive disease express much higher levels of IFNα and related inducible genes as compared to uninfected controls (Herbeuval et al. 2006; Lehmann et al. 2008).
Primate studies have contributed critical insight into potential mechanisms of immune activation in chronic HIV infection. In contrast to the RM model where SIV infection results in AIDS, nonpathogenic or natural SIV hosts exist, including the SM and the AGM among others. Natural hosts are endemically infected with SIV with high levels of viral replication and yet they have a life span similar to uninfected animals, as they do not develop immunode fi ciency, immune activation, or AIDS (Paiardini et al. 2009; Brenchley et al. 2010; Jacquelin et al. 2009). Transcriptional pro fi ling in pathogenic and nonpathogenic SIV-infected primates reveals differences in IFNα responses, where both hosts have strong IFNα response signatures during acute infection, but only the pathogenically infected animals that go on to develop AIDS maintain elevated IFNα response signatures over the course of chronic infection (Jacquelin et al. 2009; Manches and Bhardwaj 2009). Currently, it remains unresolved whether the lack of sustained immune activation in natural SIV infection is due to a general attenuated response to infection, or due to induction of regulatory mechanisms that suppress immune responses generated during the acute infection. While one report claimed that pDCs from natural SIV hosts produced deficient IFNα responses to SIV because of perturbation of IFNα signaling pathways, other groups have not found differences in pDC responsiveness between pathogenic and non-pathogenic (natural) SIV hosts. Studies are underway to closely examine whether there are true differences between pDC antigen-presentation and IFNα responses between pathogenic and nonpathogenic SIV models (Fig. 3.2).
Fig. 3.2.

Differential transcriptomes in nonpathogenic vs. pathogenic SIV infection
In pathogenic SIV/HIV infection, GALT is a critical site of mucosal CD4 depletion and immune activation-induced tissue pathology (Brenchley and Douek 2008). In AIDS-susceptible RMs, pDCs in the blood upregulate β7-integrin and are rapidly recruited to the colorectum after intravenous SIV infection. In vivo blockade of α4 β7-integrin inhibited pDC recruitment to the colorectum and reduced immune activation (Ansari et al. 2011). The up-regulation of β7-integrin expression on pDCs in the blood also was observed in HIV-infected humans but not in chronically SIV-infected SMs (Kwa et al. 2011). These studies further implicate a role for activated pDC in the pathophysiology of immune activation in HIV.
Although IFNα is directly antiviral because it programs cell death and reduces viral replication in HIV-infected cells (Karpov 2001), it likely contributes to inhibition of T cell differentiation and death of HIV-uninfected bystander cells in HIV infection (Demoulins et al. 2008). IFNα induces TNF-related apoptosis-inducing ligand (TRAIL) and its death receptor (DR) 5 on CD4+ T lymphocytes in peripheral blood and in secondary lymphatic tissue, leading to the apoptosis of uninfected CD4+ T cells, likely contributing to the characteristic destruction of lymph node architecture in advanced stages of HIV-1 infection (Herbeuval et al. 2005a). Type I IFN can upregulate transcription of p53 and integrates with the p53 pathway to regulate apoptosis of virally infected cells (Takaoka et al. 2003). It has been observed that type I IFN-regulated genes, including cell cycle-associated genes, are upregu-lated in activated CD4+ T cells of HIV-infected patients (Sedaghat et al. 2008). Additionally, IFNα administration significantly enhances CD8+ T cell activation in chronically HIV-infected subjects (Manion et al. 2012). Besides IFN-induced TRAIL-mediated apoptosis of CD4+ T cells, recent models argue for a dynamic regulation of memory T cell homeostasis by inflammatory cytokines and type I IFN. First, chronic TLR stimulation can induce reversible blockade of thymic output, in a type I IFN-dependent way (Demoulins et al. 2008). Second, type I IFN is known to enhance bystander T cell proliferation (Tough et al. 1996), and constitutive proliferation of memory T cells due to specific or bystander activation could contribute to exhaustion of the memory CD4+ T cell reservoir. In addition, IFNα has been shown to inhibit telomerase activity and contribute to telomere shortening in human T cells (Reed et al. 2004). Depletion of self-renewing central memory CD4+ T cells by chronic stimulation has been shown to be a tipping point in SIV-infected macaques, upon which insufficient central memory cells are available to generate CD4+ effector memory and immunodeficiency ensues (Okoye et al. 2007). The hyperproliferative state modulated by chronic IFNα secretion may drive memory T cell exhaustion in the long term (Sedaghat et al. 2008).
Because stimulation of innate immune cells, including pDCs, by HIV and circulating bacterial products from gut translocation is thought to be a major cause of immune activation and inflammation in HIV infection, clinical trials have tested whether IFNα inhibitors and inhibitors of innate immune TLR signaling in HIV infection blunt immune activation. In older studies, HIV-infected subjects were vaccinated against IFN-alpha-2b in a phase I/II study and then a double-blind placebo-controlled phase II/III clinical trial, respectively (Gringeri et al. 1996, 1999). Although the immunogenicity of the vaccine was low, subjects who responded to vaccination had a lower rate of disease progression, supporting the evidence that IFNα plays a role in driving immune activation and disease progression. Chloroquine has also been studied as an immunomodulatory agent in HIV infection as it has been shown to reduce endosomal TLR signaling through inhibition of acidification in endosomal compartments where TLR ligands bind TLRs. Endosomal acidification is necessary for cleavage of TLRs for activation of inflammatory signaling pathways. In vitro, chloroquine inhibits IFNα production by HIV-activated pDCs. Chloroquine also decreases CD8+ T cell activation and inhibits indoleamine 2,3 dioxygenase (IDO) and programmed death ligand 1 (PDL-1), two negative regulators of T cell responses (Martinson et al. 2010). When mice are treated with chloroquine, the production of proinflammatory cytokines upon stimulation with LPS is reduced (Hong et al. 2004).
Clinical trials have supported the use of chloroquine to reduce immune activation in HIV-infected patients. When chloroquine was administered to ART-naïve subjects for 2 months, patients showed immunological improvement, as the frequency of CD38+ HLA-DR + CD8+ T cells, proliferation of T cells, and circulating LPS levels were significantly reduced (Murray et al. 2010). Six months of chloroquine administration to HIV-infected clinical nonresponders, who had inadequate reconstitution of CD4+ T cells despite suppressive ART, had decreased frequency of activated T cells, decreased circulating LPS, decreased production of inflammatory cytokines (IL-6, TNF-alpha) in response to ex vivo stimulation of TLR ligands, and evidenced improved CD4+ T cell counts and pDC numbers (Piconi et al. 2011). Larger placebo-controlled trials are under way to fully elucidate whether chloro-quine is effective at immunomodulating innate immune signaling to curb immune activation and T cell depletion in HIV infection.
3.8 Contribution of pDCs to Th17/Treg Balance During HIV Infection
In addition to the role of pDCs in inflammation-associated decline of the CD4+ T cell compartment, pDCs can potentially affect the balance of CD4+ T cell subsets in the periphery and in the gastrointestinal tract of infected individuals. Recent studies have focused on specific populations of CD4+ T cells that seem to share a common developmental pathway, and may be important in controlling HIV-associated immune activation: Th17 and Treg. Th17 cells are thought to be important for host defense against microbes, and in particular may be crucial for preservation of the integrity of the gut-associated mucosa and prevention of bacterial translocation (Kanwar et al. 2010). On the other hand, Tregs are potent suppressors of T cells and DC activation, and they may play a dual role in HIV infection, by suppressing HIV-specific T cell responses, but also by dampening the damaging immune activation during chronic disease. Th17 and Treg differentiation is reciprocally regulated in vitro upon stimulation of naïve CD4+ T cells, in the presence of TGFβ and the inflammatory cytokines IL-6, IL-1β, and IL-23. While TGFβ upregulates the expression of the master transcription factors RORγt and Foxp3 for Th17 and Treg development, respectively, signaling through IL-6 skews Treg development towards Th17 commitment (Veldhoen et al. 2006; Bettelli et al. 2006; Zhou et al. 2008). Treg and Th17 cells also share common chemokine receptors (CCR4, CCR6) and homing properties towards CCL20 (Acosta-Rodriguez et al. 2007).
Early studies of Treg dynamics during HIV infection reported decreased numbers of Treg in the blood of chronically infected patients, a concept compatible with the fact that Treg can be infected by HIV (El Hed et al. 2010). The interpretation of those data is complicated by the fact that there is no unambiguous marker of Treg in humans, as Foxp3 can also be expressed by recently activated CD4+ effector cells. However, Tregs can be identified with a good probability using a combination of markers, including high expression of Foxp3, CD25, low expression of the IL-7Rα (CD127), and expression of inhibitory surface molecules, such as CTLA-4 and GITR (Miyara et al. 2011). The emerging picture is that despite a relative disappearance of Tregs in the circulation, both data in humans and in NHP show an increased frequency of Foxp3 CTLA4+ Tregs in the secondary lymphoid tissue of chronically infected individuals (Boasso et al. 2006). Furthermore, SIV infection in pathogenic and nonpathogenic NHP models showed that disease progression is associated with the relative loss of Th17 cells and increase in the frequency of CD4+ Foxp3+ Tregs in blood and lymphoid mucosal tissue (Kanwar et al. 2010). Infection and loss of Th17 in acute SIV infection (Kader et al. 2009) may contribute to altered Treg/Th17 ratios in later phases. In addition, even though massive depletion of CD4+ T cells occurs in the gastrointestinal tract of all SIV-infected monkeys, bacterial translocation is not observed in nonpathogenic infections, suggesting the crucial role of the preservation of a normal Treg/T17 balance in the gut.
As mentioned, the relative expansion of Tregs in infected individuals plays a dual role. In vitro removal of CD4+ CD25+ cells from peripheral leukocytes of HIV-infected patients or SIV-infected macaques enhances HIV- or SIV-specific immune responses (Kinter et al. 2004;Aandahl et al. 2004). Furthermore, the frequency of Tregs inversely correlates with the magnitude of SIV-specific CTL in infected RMs (Estes et al. 2006). On the other hand, decreased frequency of circulating Tregs correlates with hyperacti-vation in HIV-infected patients, and early induction of TGFβ, Foxp3, and IL-10 in nonpathogenic models of SIV infection is associated with early resolution of inflammation (Bosinger et al. 2009; Jacquelin et al. 2009; Ploquin et al. 2006).
The mechanisms of Treg/Th17 deregulation are not known. However, besides direct infection of Th17 cells, there is evidence that DC-derived cytokines and immunoregulatory enzymes can play an important role in regulating Treg/Th17 balance. Thus, type I IFN constrains the development of Th17 cells (Guo et al. 2008). Type I IFN induces up-regulation of SOCS3 and downregulation of IL-1β and IL-23 in DC, while naïve T cells cultured in Th17 polarizing condition in the presence of type I IFN downregulate RORγt, IL-17A, and IL23R in CD4+ T cells (Ramgolam et al. 2009). Another mechanism controlling peripheral Treg differentiation and affecting Treg/Th17 ratios is expression of immunoregulatory enzymes in dendritic cells. The enzyme IDO that catalyzes the rate-limiting step in tryptophan catabolism has a well-identified role in dampening T cell activation, through depletion of tryp-tophan, an amino acid essential for proliferation of effector cells. IDO also plays a role in the generation and activation of Treg, through activation of the amino-acid starvation response gene GCN2 in developing CD4+ T cells, but also through the generation of soluble tryptophan catabolites, which can contribute to Foxp3+ Treg differentiation (Munn et al. 2005; Fallarino et al. 2006; Favre et al. 2010). Importantly, IDO activity is upregulated in HIV-infected patients, and is associated with disease progression and a deregulated Treg/Th17 ratio (Favre et al. 2010). pDCs activated by HIV and other TLR agonists express IDO, and induce the generation of Treg from naïve CD4+ T cells (Manches et al. 2008; Moseman et al. 2004). In addition, IDO expression by pDC has been shown to prevent conversion of Treg into Th17 cells and IDO functioned as a molecular switch to maintain the stability of Treg at the detriment of Th17 differentiation in an inflammatory environment (Baban et al. 2009; Sharma et al. 2009). Inhibition of IDO by 1-methyl tryptophan showed enhancement of HIV-specific CTL responses and clearance of HIV-infected macrophages, in an in vivo model of HIV infection in humanized mice (Potula et al. 2005). Furthermore, Tregs have been shown to induce IDO in DCs through CTLA-4 engagement, and CTLA-4 blockade in RMs inhibits IDO expression, enhances CD8+ T-cell responses, and contributes to lower viral load (Hryniewicz et al. 2006).
New mouse models of conditional pDC knockout have recently been developed, and provide remarkable insights into the role of pDC during acute and chronic viral infections. Thus, conditional knockout of BDCA-2 depletes mouse pDC, which impairs generation of anti-vesicular stomatitis virus CD8+ T cell responses, but had no effect in a model of acute lymphocytic choriomeningitis virus (LCMV) infection (Swieki, Immunol Rev 2010). Similarly, conditional knockout of E2-2 preventing the development and maintenance of pDC did not affect T cell responses and clearance of LCMV Armstrong strain. However, pDC-deficient animals failed to clear LCMV Docile strain, resulting in chronic infection (Cervantes-Barragan et al. 2012). pDC-deficient mice displayed reduced numbers of LCMV-specific CD4+ T cell and impaired CD8+ T cell responses. Enhancement of CD4+ T cell responses was nevertheless independent of antigen presentation by pDC and likely due to decreased IFN in pDC-deficient mice. Interestingly, in yet another conditional knockout, Siglec-H deletion leads to specific pDC depletion, but it was shown that pDC promoted the induction of peripheral tolerance by generating antigen-specific CD4+ Foxp3+ Treg. Importantly, pDC maintained higher number of CD4+ Foxp3+ Treg in the lamina propria of the small intestine, but not in the periphery, while pDC-depleted mice had enhanced number of Th17 cells in the lamina propria, indicating that pDCs are involved in the homeostasis of Th17/Treg ratios in the gastrointestinal tract (Takagi et al. 2011). Thus, although the role of pDC-derived IFN and IDO expression may be part of a complex immunoregulatory network, the high levels of type I IFN and possibly the antigen-presenting capabilities of pDCs likely affect the balance of beneficial and detrimental Th17 and Treg responses in chronically infected individuals.
3.9 Conclusions and Future Directions
The role of pDCs in HIV immunopathogenesis remains incompletely characterized. Much of the evidence linking pDCs and IFNα to HIV transmission and disease progression is correlative, but causative data is emerging. Although pDCs have the capacity to mature into APCs to stimulate potent adaptive immune responses, in HIV infection pDCs more likely effect a cytokine- and chemokine-producing phenotype, which may paradoxically fuel viral transmission and immune activation. Further research is needed to clarify the role of pDCs in HIV transmission, disease progression, and chronic inflammation (Fig. 3.3).
Fig. 3.3.

Dual role of pDC in HIV infection
3.9.1 Future Directions: Transmission
Through careful analysis of mucosal tissue samples from macaques infected in vivo with SIV, it has been shown that pDCs arrive first to produce inflammatory cytok-ines and T-cell-attracting chemokines (Li et al. 2009). Inhibition of pDC trafficking to mucosal tissues using topical glycerol monolaurate actually prevented SIV transmission and systemic infection. Based on this report it seems that inhibition of innate immune responses and inflammation during HIV mucosal exposure actually prevent transmission, whereas earlier studies which tested immune-activating topical microbicides, like imiquimod and CpG, actually enhanced infection and viremia (Wang et al. 2005). Additionally, it has been shown that exposed, uninfected sex workers actually have depressed mucosal immunity as compared to uninfected controls (Lajoie et al. 2012), supporting an immune quiescence model of protection, whereby lower T-cell activation/recruitment at the mucosal compartment reduces HIV target cell numbers and is an important component of protection from HIV infection.
Topical and oral ART as pre-exposure prophylaxis has proven to be a highly efficacious strategy to prevent HIV transmission (Grant et al. 2010; Abdool Karim et al. 2010), but this approach has had various limitations. Some of these studies were stopped prematurely due to futility (van der Straten et al. 2012) and others have raised concerns about systemic toxicity like bone loss with oral tenofovir use (Liu et al. 2011). Strategies that employ daily oral ART pre-exposure prophylaxis also may not be cost-effective (Keller and Smith 2011). Therefore, integrating immunomodulating microbicides like glycerol monolaurate into the armamentarium of prevention strategies should be considered. Additionally, specific inhibitors of pDC activation could be explored as topical microbicides. In vaginal and foreskin tissue explants, Phosphorothioate 2′ deoxyribose oligomers, when applied topically, have been shown to both inhibit HIV infection and dampen HIV-associated local inflammation, including IFNα production (Fraietta et al. 2010). Cross-linking CD4, BDCA-2, BDCA-4, or CD-123 on pDCs has been shown to inhibit IFNα production both by inhibiting translocation of IRF7 to the nucleus, therefore inhibiting IFNα signaling pathways, and by maturing the pDCs so that IFN signaling is inhibited (Fanning et al. 2006). Immunoproteasome inhibitors (Ichikawa et al. 2012) have also been used to inhibit pathogenic IFNα signaling in pDCs.
3.9.2 Future Directions: Chronic Immune Activation
The cause of immune activation and inflammation in HIV infection remains unknown, but candidate mechanisms include the stimulation of innate immune cells by replicating and non-replicating HIV and by circulating bacterial products that have translocated from damaged gut mucosa. Critical insight into the pathogenesis of immune activation in HIV infection has been gained by studying pathogenic as compared to nonpathogenic SIV infection. Although both RMs and SMs or AGMs are highly viremic with SIV, only RMs develop AIDS while both SMs and AGMs have a normal life span. Only RMs have heightened immune activation, inflammation, and CD4 T cell decline. A major clue to the etiology of immune activation in chronic pathogenic SIV and HIV is that type I IFN-stimulated genes (ISGs) are highly upregulated in RMs but not so in SMs or AGMs. Although one study suggested that pDC responses in natural SIV infection are blunted due to defects in IFNα signaling pathways (Mandl et al. 2008), subsequent studies have not supported these findings (Harris et al. 2010) but rather showed that ISGs are highly upregulated in RMs, SMs, and AGMs during acute infection, but this transcription signal normalizes in SMs and AGMs after 4–6 weeks of SIV infection (Manches and Bhardwaj 2009). Similarly, a rare group of HIV-infected humans who maintain a significant viremia but do not experience a decline in CD4+ T cells also have transcriptomes which are characterized by low ISGs as compared to HIV-infected progressors (Rotger et al. 2011). These studies support a model of active immune suppression of IFN responses in nonpathogenic as compared to pathogenic SIV infection, rather than any specific deficit in the ability of pDCs to produce type I IFN in response to SIV.
Understanding the cause of these persistent IFN responses during chronic HIV and pathogenic SIV infection remains an important unanswered question. Additionally, understanding how IFN-responses are resolved despite persistent viral replication in nonpathogenic SIV infection will provide potential targets for immune therapy in HIV-infected humans. Because pDCs are considered to be the main source of type I IFN, more detailed studies are needed to dissect any differences in SIV–pDC interactions between RMs and SMs or AGMs. For example, differential responses to TLR ligands predict disease transmission and progression in HIV-infected subjects (Bochud et al. 2007; Pine et al. 2009; Rotger et al. 2011; Freguja et al. 2012; Ricci et al. 2010; Soriano-Sarabia et al. 2008); therefore there may be functional differences in the response of innate immune cells, including pDCs, to HIV and to circulating bacterial products from gut translocation. It has been shown that HIV stimulates pDCs to produce IFNα instead of maturing to become an APC, while other ssRNA viruses like influenza both stimulate IFNα production and mature the cells, ultimately inhibiting persistent IFNα production. It is unknown whether trafficking and stimulation of TLR7 agonists, including SIV, or TLR9 agonists, including bacterial DNA (CpG), differ between RM and SM or AGM pDCs. If SIV or CpGs mature SM or AGM pDCs but do not mature RM pDCs, this would be suggestive that differential trafficking and/or stimulation of signaling pathways could account for differential systemic responses to SIV in vitro. Another intriguing possibility is that there is inhibited infection of SIV in lymphoid tissues of SMs, such that there would be a lack of stimulatory ligands for pDCs to produce IFNα (Paiardini et al. 2011). Also, it has been shown that there is increased trafficking of pDCs to GALT (Kwa et al. 2011; Reeves et al. 2012) in RM, which produce inflammatory cytokines, including IFNα.
Non-pDC IFN responses may also contribute to chronic immune activation. Monocyte-derived macrophages derived from PBMCs of macaques upregulated ISGs in response to SIV infection, but ISG expression was inhibited after silencing the expression of MDA-5, a cytosolic RNA receptor that activates IFN signaling pathways. However, when chloroquine was added to the experiment, there was further reduction in ISG expression, indicating that both an endosomal TLR pathway and a cytosolic MDA-5 pathway are implicated in ISG expression (Co et al. 2011).
From the transcriptome studies of PBMCs from blood and lymphoid tissues of pathogenic vs. nonpathogenic PMBCs, immune regulatory genes such as IDO and ADAR were more highly expressed in nonpathogenic SIV disease (Jacquelin et al. 2009). It is not known which cell types are responsible for increased expression of these immunoregulatory molecules. The interplay between innate immune cells and adaptive immune cells to achieve this more immunoregulatory state is also unknown. For example, SIV might induce pDCs in SM and AGMs to become predominantly IDO-producing cells instead of IFNα-producing cells in vivo, as compared to RMs. In addition to IDO and ADAR, the differential expression of other immunoregula-tory molecules on innate and adaptive immune cells, both in vivo and ex vivo in AGMs and SMs as compared to RMs, would be informative.
Contributor Information
Meagan O’Brien, Division of Infectious Diseases, Department of Medicine, NYU Cancer Institute, New York University School of Medicine, New York, NY, USA
Olivier Manches, NYU Cancer Institute, New York University School of Medicine, New York, NY, USA
Nina Bhardwaj, Email: Nina.Bhardwaj@nyumc.org, Department of Medicine, Dermatology, and Pathology, New York University School of Medicine, NYU Cancer Institute, 522 First Avenue, Smilow 1303, New York, NY 10016, USA
References
- Aandahl EM, Michaelsson J, Moretto WJ, Hecht FM, Nixon DF. Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalo-virus antigens. J Virol. 2004;78(5):2454–2459. doi: 10.1128/JVI.78.5.2454-2459.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdool Karim Q, Abdool Karim SS, Frohlich JA, Grobler AC, Baxter C, Mansoor LE, Kharsany AB, Sibeko S, Mlisana KP, Omar Z, Gengiah TN, Maarschalk S, Arulappan N, Mlotshwa M, Morris L, Taylor D. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010;329(5996):1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8(6):639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Ansari AA, Reimann KA, Mayne AE, Takahashi Y, Stephenson ST, Wang R, Wang X, Li J, Price AA, Little DM, Zaidi M, Lyles R, Villinger F. Blocking of alpha4beta7 gut-homing integrin during acute infection leads to decreased plasma and gastrointestinal tissue viral loads in simian immunodeficiency virus-infected rhesus macaques. J Immunol. 2011;186(2):1044–1059. doi: 10.4049/jimmunol.1003052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183(4):2475–2483. doi: 10.4049/jimmunol.0900986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenziger S, Heikenwalder M, Johansen P, Schlaepfer E, Hofer U, Miller RC, Diemand S, Honda K, Kundig TM, Aguzzi A, Speck RF. Triggering TLR7 in mice induces immune activation and lymphoid system disruption, resembling HIV-mediated pathology. Blood. 2009;113(2):377–388. doi: 10.1182/blood-2008-04-151712. [DOI] [PubMed] [Google Scholar]
- Baker JV, Duprez D. Biomarkers and HIV-associated cardiovascular disease. Curr Opin HIV AIDS. 2010;5(6):511–516. doi: 10.1097/COH.0b013e32833ed7ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- Barber GN. Host defense, viruses and apoptosis. Cell Death Differ. 2001;8(2):113–126. doi: 10.1038/sj.cdd.4400823. [DOI] [PubMed] [Google Scholar]
- Barratt-Boyes SM, Wijewardana V, Brown KN. In acute pathogenic SIV infection plasma-cytoid dendritic cells are depleted from blood and lymph nodes despite mobilization. J Med Primatol. 2010;39(4):235–242. doi: 10.1111/j.1600-0684.2010.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron MA, Blyveis N, Palmer BE, MaWhinney S, Wilson CC. Influence of plasma viremia on defects in number and immunophenotype of blood dendritic cell subsets in human immunodeficiency virus 1-infected individuals. J Infect Dis. 2003;187(1):26–37. doi: 10.1086/345957. [DOI] [PubMed] [Google Scholar]
- Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, Larsson M, Gorelick RJ, Lifson JD, Bhardwaj N. Endocytosis of HIV-1 activates plasmacytoid den-dritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115(11):3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendriss-Vermare N, Burg S, Kanzler H, Chaperot L, Duhen T, de Bouteiller O, D’Agostini M, Bridon JM, Durand I, Sederstrom JM, Chen W, Plumas J, Jacob MC, Liu YJ, Garrone P, Trinchieri G, Caux C, Briere F. Virus overrides the propensity of human CD40L-activated plasmacytoid dendritic cells to produce Th2 mediators through synergistic induction of IFN-{gamma} and Th1 chemokine production. J Leukoc Biol. 2005;78(4):954–966. doi: 10.1189/jlb.0704383. [DOI] [PubMed] [Google Scholar]
- Benitez-Ribas D, Adema GJ, Winkels G, Klasen IS, Punt CJ, Figdor CG, de Vries IJ. Plasmacytoid dendritic cells of melanoma patients present exogenous proteins to CD4+ T cells after Fc gamma RII-mediated uptake. J Exp Med. 2006;203(7):1629–1635. doi: 10.1084/jem.20052364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito JM, Lopez M, Lozano S, Ballesteros C, Martinez P, Gonzalez-Lahoz J, Soriano V. Differential upregulation of CD38 on different T-cell subsets may influence the ability to reconstitute CD4+ T cells under successful highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2005;38(4):373–381. doi: 10.1097/01.qai.0000153105.42455.c2. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Boasso A, Vaccari M, Nilsson J, Shearer GM, Andersson J, Cecchinato V, Chougnet C, Franchini G. Do regulatory T-cells play a role in AIDS pathogenesis? AIDS Rev. 2006;8(3):141–147. [PubMed] [Google Scholar]
- Boasso A, Hardy AW, Landay AL, Martinson JL, Anderson SA, Dolan MJ, Clerici M, Shearer GM. PDL-1 upregulation on monocytes and T cells by HIV via type I interferon: restricted expression of type I interferon receptor by CCR5-expressing leukocytes. Clin Immunol. 2008;129(1):132–144. doi: 10.1016/j.clim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochud PY, Hersberger M, Taffe P, Bochud M, Stein CM, Rodrigues SD, Calandra T, Francioli P, Telenti A, Speck RF, Aderem A. Polymorphisms in Toll-like receptor 9 influence the clinical course of HIV-1 infection. AIDS. 2007;21(4):441–446. doi: 10.1097/QAD.0b013e328012b8ac. [DOI] [PubMed] [Google Scholar]
- Bofill M, Mocroft A, Lipman M, Medina E, Borthwick NJ, Sabin CA, Timms A, Winter M, Baptista L, Johnson MA, Lee CA, Phillips AN, Janossy G. Increased numbers of primed activated CD8 + CD38 + CD45RO + T cells predict the decline of CD4+ T cells in HIV-1-infected patients. AIDS. 1996;10(8):827–834. doi: 10.1097/00002030-199607000-00005. [DOI] [PubMed] [Google Scholar]
- Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, Francella N, Sidahmed A, Smith AJ, Cramer EM, Zeng M, Masopust D, Carlis JV, Ran L, Vanderford TH, Paiardini M, Isett RB, Baldwin DA, Else JG, Staprans SI, Silvestri G, Haase AT, Kelvin DJ. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest. 2009;119(12):3556–3572. doi: 10.1172/JCI40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med. 2004;199(12):1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008;1(1):23–30. doi: 10.1038/mi.2007.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Silvestri G, Douek DC. Nonprogressive and progressive primate immunodeficiency lentivirus infections. Immunity. 2010;32(6):737–742. doi: 10.1016/j.immuni.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KN, Wijewardana V, Liu X, Barratt-Boyes SM. Rapid influx and death of plasmacy-toid dendritic cells in lymph nodes mediate depletion in acute simian immunodeficiency virus infection. PLoS Pathog. 2009;5(5):e1000413. doi: 10.1371/journal.ppat.1000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol. 2008;9(5):558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- Cai R, Carpick B, Chun RF, Jeang KT, Williams BR. HIV-I TAT inhibits PKR activity by both RNA-dependent and RNA-independent mechanisms. Arch Biochem Biophys. 2000;373(2):361–367. doi: 10.1006/abbi.1999.1583. [DOI] [PubMed] [Google Scholar]
- Carotta S, Dakic A, D’Amico A, Pang SH, Greig KT, Nutt SL, Wu L. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity. 2010;32(5):628–641. doi: 10.1016/j.immuni.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5(8):919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. 2000;1(4):305–310. doi: 10.1038/79747. [DOI] [PubMed] [Google Scholar]
- Cervantes-Barragan L, Lewis KL, Firner S, Thiel V, Hugues S, Reith W, Ludewig B, Reizis B. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc Natl Acad Sci U S A. 2012;109(8):3012–3017. doi: 10.1073/pnas.1117359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicinnati VR, Kang J, Sotiropoulos GC, Hilgard P, Frilling A, Broelsch CE, Gerken G, Beckebaum S. Altered chemotactic response of myeloid and plasmacytoid dendritic cells from patients with chronic hepatitis C: role of alpha interferon. J Gen Virol. 2008;89(Pt 5):1243–1253. doi: 10.1099/vir.0.83517-0. [DOI] [PubMed] [Google Scholar]
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, Holter W, Rauch A, Zhuang Y, Reizis B. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135(1):37–48. doi: 10.1016/j.cell.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Co JG, Witwer KW, Gama L, Zink MC, Clements JE. Induction of innate immune responses by SIV in vivo and in vitro: differential expression and function of RIG-I and MDA5. J Infect Dis. 2011;204(7):1104–1114. doi: 10.1093/infdis/jir469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colina R, Costa-Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, Makrigiannis AP, Bell JC, Sonenberg N. Translational control of the innate immune response through IRF-7. Nature. 2008;452(7185):323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- D’Amico A, Wu L. The early progenitors of mouse dendritic cells and plasmacytoid predendritic cells are within the bone marrow hemopoietic precursors expressing Flt3. J Exp Med. 2003;198(2):293–303. doi: 10.1084/jem.20030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, Hunt P, Martin JN, Kahn JO, Levy J, McGrath MS, Hecht FM. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. 2004;104(4):942–947. doi: 10.1182/blood-2003-09-3333. [DOI] [PubMed] [Google Scholar]
- Demoulins T, Abdallah A, Kettaf N, Baron ML, Gerarduzzi C, Gauchat D, Gratton S, Sekaly RP. Reversible blockade of thymic output: an inherent part of TLR ligand-mediated immune response. J Immunol. 2008;181(10):6757–6769. doi: 10.4049/jimmunol.181.10.6757. [DOI] [PubMed] [Google Scholar]
- Devosse T, Guillabert A, D’Haene N, Berton A, De Nadai P, Noel S, Brait M, Franssen JD, Sozzani S, Salmon I, Parmentier M. Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J Immunol. 2009;182(8):4974–4984. doi: 10.4049/jimmunol.0803128. [DOI] [PubMed] [Google Scholar]
- Di Pucchio T, Chatterjee B, Smed-Sorensen A, Clayton S, Palazzo A, Montes M, Xue Y, Mellman I, Banchereau J, Connolly JE. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol. 2008;9(5):551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacovo TG, Blasius AL, Mak TW, Cella M, Colonna M. Adhesive mechanisms governing interferon-producing cell recruitment into lymph nodes. J Exp Med. 2005;202(5):687–696. doi: 10.1084/jem.20051035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diop OM, Ploquin MJ, Mortara L, Faye A, Jacquelin B, Kunkel D, Lebon P, Butor C, Hosmalin A, Barre-Sinoussi F, Muller-Trutwin MC. Plasmacytoid dendritic cell dynamics and alpha interferon production during Simian immunodeficiency virus infection with a nonpathogenic outcome. J Virol. 2008;82(11):5145–5152. doi: 10.1128/JVI.02433-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghy H, Pozniak A, Gazzard B, Qazi N, Gilmour J, Gotch F, Patterson S. Loss of blood CD11c(+) myeloid and CD11c(-) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood. 2001;98(8):2574–2576. doi: 10.1182/blood.v98.8.2574. [DOI] [PubMed] [Google Scholar]
- Donaghy H, Gazzard B, Gotch F, Patterson S. Dysfunction and infection of freshly isolated blood myeloid and plasmacytoid dendritic cells in patients infected with HIV-1. Blood. 2003;101(11):4505–4511. doi: 10.1182/blood-2002-10-3189. [DOI] [PubMed] [Google Scholar]
- El Hed A, Khaitan A, Kozhaya L, Manel N, Daskalakis D, Borkowsky W, Valentine F, Littman DR, Unutmaz D. Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. J Infect Dis. 2010;201(6):843–854. doi: 10.1086/651021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sadr WM, Lundgren JD, Neaton JD, Gordin F, Abrams D, Arduino RC, Babiker A, Burman W, Clumeck N, Cohen CJ, Cohn D, Cooper D, Darbyshire J, Emery S, Fatkenheuer G, Gazzard B, Grund B, Hoy J, Klingman K, Losso M, Markowitz N, Neuhaus J, Phillips A, Rappoport C. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355(22):2283–2296. doi: 10.1056/NEJMoa062360. [DOI] [PubMed] [Google Scholar]
- Estes JD, Li Q, Reynolds MR, Wietgrefe S, Duan L, Schacker T, Picker LJ, Watkins DI, Lifson JD, Reilly C, Carlis J, Haase AT. Premature induction of an immunosuppressive regulatory T cell response during acute simian immunodeficiency virus infection. J Infect Dis. 2006;193(5):703–712. doi: 10.1086/500368. [DOI] [PubMed] [Google Scholar]
- Facchetti F, de Wolf-Peeters C, Mason DY, Pulford K, van den Oord JJ, Desmet VJ. Plasmacytoid T cells. Immunohistochemical evidence for their monocyte/macrophage origin. Am J Pathol. 1988;133(1):15–21. [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C, Santamaria P, Fioretti MC, Puccetti P. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176(11):6752–6761. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- Fallarino F, Gizzi S, Mosci P, Grohmann U, Puccetti P. Tryptophan catabolism in IDO + plas-macytoid dendritic cells. Curr Drug Metab. 2007;8(3):209–216. doi: 10.2174/138920007780362581. [DOI] [PubMed] [Google Scholar]
- Fanning SL, George TC, Feng D, Feldman SB, Megjugorac NJ, Izaguirre AG, Fitzgerald-Bocarsly P. Receptor cross-linking on human plasmacytoid dendritic cells leads to the regulation of IFN-alpha production. J Immunol. 2006;177(9):5829–5839. doi: 10.4049/jimmunol.177.9.5829. [DOI] [PubMed] [Google Scholar]
- Favre D, Mold J, Hunt PW, Kanwar B, Loke P, Seu L, Barbour JD, Lowe MM, Jayawardene A, Aweeka F, Huang Y, Douek DC, Brenchley JM, Martin JN, Hecht FM, Deeks SG, McCune JM. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. 2010;2(32):32ra36. doi: 10.1126/scitranslmed.3000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez S, Tanaskovic S, Helbig K, Rajasuriar R, Kramski M, Murray JM, Beard M, Purcell D, Lewin SR, Price P, French MA. CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J Infect Dis. 2011;204(12):1927–1935. doi: 10.1093/infdis/jir659. [DOI] [PubMed] [Google Scholar]
- Finke JS, Shodell M, Shah K, Siegal FP, Steinman RM. Dendritic cell numbers in the blood of HIV-1 infected patients before and after changes in antiretroviral therapy. J Clin Immunol. 2004;24(6):647–652. doi: 10.1007/s10875-004-6250-5. [DOI] [PubMed] [Google Scholar]
- Fong L, Mengozzi M, Abbey NW, Herndier BG, Engleman EG. Productive infection of plasmacytoid dendritic cells with human immunodeficiency virus type 1 is triggered by CD40 ligation. J Virol. 2002;76(21):11033–11041. doi: 10.1128/JVI.76.21.11033-11041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonteneau JF, Gilliet M, Larsson M, Dasilva I, Munz C, Liu YJ, Bhardwaj N. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 2003;101(9):3520–3526. doi: 10.1182/blood-2002-10-3063. [DOI] [PubMed] [Google Scholar]
- Fonteneau JF, Larsson M, Beignon AS, McKenna K, Dasilva I, Amara A, Liu YJ, Lifson JD, Littman DR, Bhardwaj N. Human immunodeficiency virus type 1 activates plasmacy-toid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J Virol. 2004;78(10):5223–5232. doi: 10.1128/JVI.78.10.5223-5232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Mueller YM, Do DH, Holmes VM, Howett MK, Lewis MG, Boesteanu AC, Alkan SS, Katsikis PD. Phosphorothioate 2′deoxyribose oligomers as microbicides that inhibit human immunodeficiency virus type 1 (HIV-1) infection and block Toll-like receptor 7 (TLR7) and TLR9 triggering by HIV-1. Antimicrob Agents Chemother. 2010;54(10):4064–4073. doi: 10.1128/AAC.00367-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank I, Kacani L, Stoiber H, Stossel H, Spruth M, Steindl F, Romani N, Dierich MP. Human immunodeficiency virus type 1 derived from cocultures of immature dendritic cells with autologous T cells carries T-cell-specific molecules on its surface and is highly infectious. J Virol. 1999;73(4):3449–3454. doi: 10.1128/jvi.73.4.3449-3454.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freguja R, Gianesin K, Del Bianco P, Malacrida S, Rampon O, Zanchetta M, Giaquinto C, De Rossi A. Polymorphisms of innate immunity genes influence disease progression in HIV-1 infected children. AIDS. 2012;26(6):765–768. doi: 10.1097/QAD.0b013e3283514350. [DOI] [PubMed] [Google Scholar]
- Fritz JH, Girardin SE, Fitting C, Werts C, Mengin-Lecreulx D, Caroff M, Cavaillon JM, Philpott DJ, Adib-Conquy M. Synergistic stimulation of human monocytes and dendritic cells by Toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur J Immunol. 2005;35(8):2459–2470. doi: 10.1002/eji.200526286. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, Cornelissen IL, Nottet HS, KewalRamani VN, Littman DR, Figdor CG, van Kooyk Y. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100(5):587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- Geisel J, Kahl F, Muller M, Wagner H, Kirschning CJ, Autenrieth IB, Frick JS. IL-6 and maturation govern TLR2 and TLR4 induced TLR agonist tolerance and cross-tolerance in den-dritic cells. J Immunol. 2007;179(9):5811–5818. doi: 10.4049/jimmunol.179.9.5811. [DOI] [PubMed] [Google Scholar]
- Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, Shih R, Lewis J, Wiley DJ, Phair JP, Wolinsky SM, Detels R. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179(4):859–870. doi: 10.1086/314660. [DOI] [PubMed] [Google Scholar]
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, Webb M. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480(7377):379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- Granelli-Piperno A, Golebiowska A, Trumpfheller C, Siegal FP, Steinman RM. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc Natl Acad Sci U S A. 2004;101(20):7669–7674. doi: 10.1073/pnas.0402431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapia M, Guanira-Carranza JV, Ramirez-Cardich ME, Montoya-Herrera O, Fernandez T, Veloso VG, Buchbinder SP, Chariyalertsak S, Schechter M, Bekker LG, Mayer KH, Kallas EG, Amico KR, Mulligan K, Bushman LR, Hance RJ, Ganoza C, Defechereux P, Postle B, Wang F, McConnell JJ, Zheng JH, Lee J, Rooney JF, Jaffe HS, Martinez AI, Burns DN, Glidden DV. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363(27):2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum BD, Rabadan R, Levine AJ. Patterns of oligonucleotide sequences in viral and host cell RNA identify mediators of the host innate immune system. PLoS One. 2009;4(6):e5969. doi: 10.1371/journal.pone.0005969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gringeri A, Santagostino E, Cusini M, Muca-Perja M, Marinoni A, Mannucci PM, Burny A, Criscuolo M, Lu W, Andrieru JM, Mbika JP, Lachgar A, Fall LS, Chams V, Feldman M, Hermans P, Zagury JF, Bizzini B, Musicco M, Zagury D. Absence of clinical, virological, and immunological signs of progression in HIV-1-infected patients receiving active anti-interferon-alpha immunization: a 30-month follow-up report. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;13(1):55–67. doi: 10.1097/00042560-199609000-00009. [DOI] [PubMed] [Google Scholar]
- Gringeri A, Musicco M, Hermans P, Bentwich Z, Cusini M, Bergamasco A, Santagostino E, Burny A, Bizzini B, Zagury D. Active anti-interferon-alpha immunization: a European-Israeli, randomized, double-blind, placebo-controlled clinical trial in 242 HIV-1-infected patients (the EURIS study) J Acquir Immune Defic Syndr Hum Retrovirol. 1999;20(4):358–370. doi: 10.1097/00042560-199904010-00006. [DOI] [PubMed] [Google Scholar]
- Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plas-macytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med. 1997;185(6):1101–1111. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118(5):1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzmer R, Kother B, Zwirner J, Dijkstra D, Purwar R, Wittmann M, Werfel T. Human plasmacytoid dendritic cells express receptors for anaphylatoxins C3a and C5a and are chemoattracted to C3a and C5a. J Invest Dermatol. 2006;126(11):2422–2429. doi: 10.1038/sj.jid.5700416. [DOI] [PubMed] [Google Scholar]
- Haase AT. Targeting early infection to prevent HIV-1 mucosal transmission. Nature. 2010;464(7286):217–223. doi: 10.1038/nature08757. [DOI] [PubMed] [Google Scholar]
- Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, Silvestri G, Muller-Trutwin M, Vasile-Pandrea I, Apetrei C, Hirsch V, Lifson J, Brenchley JM, Estes JD. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol. 2010;84(15):7886–7891. doi: 10.1128/JVI.02612-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Donhauser N, Chaipan C, Schuster P, Puffer B, Daniels RS, Greenough TC, Kirchhoff F, Schmidt B. CD4 binding affinity determines human immunodeficiency virus type 1-induced alpha interferon production in plasmacytoid dendritic cells. J Virol. 2008;82(17):8900–8905. doi: 10.1128/JVI.00196-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazenberg MD, Otto SA, van Benthem BH, Roos MT, Coutinho RA, Lange JM, Hamann D, Prins M, Miedema F. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS. 2003;17(13):1881–1888. doi: 10.1097/00002030-200309050-00006. [DOI] [PubMed] [Google Scholar]
- Heikenwalder M, Polymenidou M, Junt T, Sigurdson C, Wagner H, Akira S, Zinkernagel R, Aguzzi A. Lymphoid follicle destruction and immunosuppression after repeated CpG oligode-oxynucleotide administration. Nat Med. 2004;10(2):187–192. doi: 10.1038/nm987. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, Dolan MJ, Yagita H, Lifson JD, Shearer GM. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood. 2005a;106(10):3524–3531. doi: 10.1182/blood-2005-03-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbeuval JP, Hardy AW, Boasso A, Anderson SA, Dolan MJ, Dy M, Shearer GM. Regulation of TNF-related apoptosis-inducing ligand on primary CD4+ T cells by HIV-1: role of type I IFN-producing plasmacytoid dendritic cells. Proc Natl Acad Sci U S A. 2005b;102(39):13974–13979. doi: 10.1073/pnas.0505251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Kruhlak MJ, Anderson SA, Dolan MJ, Dy M, Andersson J, Shearer GM. Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc Natl Acad Sci U S A. 2006;103(18):7000–7005. doi: 10.1073/pnas.0600363103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JE, Deeks SG, Hecht FM, Xie Y, Schnell A, Martin JN, Ganz P, Hsue PY. Initiation of antiretroviral therapy at higher nadir CD4+ T-cell counts is associated with reduced arterial stiffness in HIV-infected individuals. AIDS. 2010;24(12):1897–1905. doi: 10.1097/QAD.0b013e32833bee44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Ripoche AC, Matheoud D, Nascimbeni M, Escriou N, Lebon P, Heshmati F, Guillet JG, Gannage M, Caillat-Zucman S, Casartelli N, Schwartz O, De la Salle H, Hanau D, Hosmalin A, Maranon C. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity. 2007;27(3):481–492. doi: 10.1016/j.immuni.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434(7036):1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- Hong Z, Jiang Z, Liangxi W, Guofu D, Ping L, Yongling L, Wendong P, Minghai W. Chloroquine protects mice from challenge with CpG ODN and LPS by decreasing proinflammatory cytokine release. Int Immunopharmacol. 2004;4(2):223–234. doi: 10.1016/j.intimp.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474(7353):658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryniewicz A, Boasso A, Edghill-Smith Y, Vaccari M, Fuchs D, Venzon D, Nacsa J, Betts MR, Tsai WP, Heraud JM, Beer B, Blanset D, Chougnet C, Lowy I, Shearer GM, Franchini G. CTLA-4 blockade decreases TGF-beta, IDO, and viral RNA expression in tissues of SIVmac251-infected macaques. Blood. 2006;108(12):3834–3842. doi: 10.1182/blood-2006-04-010637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yang Y, Al-Mozaini M, Burke PS, Beamon J, Carrington MF, Seiss K, Rychert J, Rosenberg ES, Lichterfeld M, Yu XG. Dendritic cell dysfunction during primary HIV-1 infection. J Infect Dis. 2011;204(10):1557–1562. doi: 10.1093/infdis/jir616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber JP, Farrar JD. Regulation of effector and memory T-cell functions by type I inter-feron. Immunology. 2011;132(4):466–474. doi: 10.1111/j.1365-2567.2011.03412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, Deeks SG. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis. 2003;187(10):1534–1543. doi: 10.1086/374786. [DOI] [PubMed] [Google Scholar]
- Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, Barnard J, Nevarez S, Goldman BI, Kirk CJ, Looney RJ, Anolik JH. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012;64(2):493–503. doi: 10.1002/art.33333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, Dillies MA, Roques P, Butor C, Silvestri G, Giavedoni LD, Lebon P, Barre-Sinoussi F, Benecke A, Muller-Trutwin MC. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest. 2009;119(12):3544–3555. doi: 10.1172/JCI40093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaehn PS, Zaenker KS, Schmitz J, Dzionek A. Functional dichotomy of plasmacytoid den-dritic cells: antigen-specific activation of T cells versus production of type I interferon. Eur J Immunol. 2008;38(7):1822–1832. doi: 10.1002/eji.200737552. [DOI] [PubMed] [Google Scholar]
- Jimenez-Baranda S, Greenbaum B, Manches O, Handler J, Rabadan R, Levine A, Bhardwaj N. Oligonucleotide motifs that disappear during the evolution of influenza virus in humans increase alpha interferon secretion by plasmacytoid dendritic cells. J Virol. 2011;85(8):3893–3904. doi: 10.1128/JVI.01908-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Baranda S, Silva IP, Bhardwaj N. Plasmacytoid dendritic cells lead the charge against tumors. J Clin Invest. 2012;122(2):481–484. doi: 10.1172/JCI61345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kader M, Wang X, Piatak M, Lifson J, Roederer M, Veazey R, Mattapallil JJ. Alpha4(+) beta7(hi)CD4(+) memory T cells harbor most Th-17 cells and are preferentially infected during acute SIV infection. Mucosal Immunol. 2009;2(5):439–449. doi: 10.1038/mi.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamga I, Kahi S, Develioglu L, Lichtner M, Maranon C, Deveau C, Meyer L, Goujard C, Lebon P, Sinet M, Hosmalin A. Type I interferon production is profoundly and transiently impaired in primary HIV-1 infection. J Infect Dis. 2005;192(2):303–310. doi: 10.1086/430931. [DOI] [PubMed] [Google Scholar]
- Kanwar B, Favre D, McCune JM. Th17 and regulatory T cells: implications for AIDS pathogenesis. Curr Opin HIV AIDS. 2010;5(2):151–157. doi: 10.1097/COH.0b013e328335c0c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpov AV. Endogenous and exogenous interferons in HIV-infection. Eur J Med Res. 2001;6(12):507–524. [PubMed] [Google Scholar]
- Kaser A, Kaser S, Kaneider NC, Enrich B, Wiedermann CJ, Tilg H. Interleukin-18 attracts plasmacytoid dendritic cells (DC2s) and promotes Th1 induction by DC2s through IL-18 receptor expression. Blood. 2004;103(2):648–655. doi: 10.1182/blood-2002-07-2322. [DOI] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23(1):19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A. 2008;105(21):7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SB, Smith DM. The price of tenofovir-emtricitabine undermines the cost-effectiveness and advancement of pre-exposure prophylaxis. AIDS. 2011;25(18):2308–2310. doi: 10.1097/QAD.0b013e32834d3cab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkmann M, Rothenfusser S, Hornung V, Towarowski A, Wagner M, Sarris A, Giese T, Endres S, Hartmann G. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J Immunol. 2003;170(9):4465–4474. doi: 10.4049/jimmunol.170.9.4465. [DOI] [PubMed] [Google Scholar]
- Kinter AL, Hennessey M, Bell A, Kern S, Lin Y, Daucher M, Planta M, McGlaughlin M, Jackson R, Ziegler SF, Fauci AS. CD25(+)CD4(+) regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4(+) and CD8(+) HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J Exp Med. 2004;200(3):331–343. doi: 10.1084/jem.20032069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool M, Geurtsvankessel C, Muskens F, Madeira FB, van Nimwegen M, Kuipers H, Thielemans K, Hoogsteden HC, Hammad H, Lambrecht BN. Facilitated antigen uptake and timed exposure to TLR ligands dictate the antigen-presenting potential of plasmacytoid DCs. J Leukoc Biol. 2011;90(6):1177–1190. doi: 10.1189/jlb.0610342. [DOI] [PubMed] [Google Scholar]
- Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- Krug A, Uppaluri R, Facchetti F, Dorner BG, Sheehan KC, Schreiber RD, Cella M, Colonna M. IFN-producing cells respond to CXCR3 ligands in the presence of CXCL12 and secrete inflammatory chemokines upon activation. J Immunol. 2002;169(11):6079–6083. doi: 10.4049/jimmunol.169.11.6079. [DOI] [PubMed] [Google Scholar]
- Kubo-Murai M, Hazeki K, Nigorikawa K, Omoto T, Inoue N, Hazeki O. IRAK-4-dependent degradation of IRAK-1 is a negative feedback signal for TLR-mediated NF-kappaB activation. J Biochem. 2008;143(3):295–302. doi: 10.1093/jb/mvm234. [DOI] [PubMed] [Google Scholar]
- Kwa S, Kannanganat S, Nigam P, Siddiqui M, Shetty RD, Armstrong W, Ansari A, Bosinger SE, Silvestri G, Amara RR. Plasmacytoid dendritic cells are recruited to the colorectum and contribute to immune activation during pathogenic SIV infection in rhesus macaques. Blood. 2011;118(10):2763–2773. doi: 10.1182/blood-2011-02-339515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474(7353):654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie J, Juno J, Burgener A, Rahman S, Mogk K, Wachihi C, Mwanjewe J, Plummer FA, Kimani J, Ball TB, Fowke KR. A distinct cytokine and chemokine profile at the genital mucosa is associated with HIV-1 protection among HIV-exposed seronegative commercial sex workers. Mucosal Immunol. 2012;5(3):277–287. doi: 10.1038/mi.2012.7. [DOI] [PubMed] [Google Scholar]
- Larsson M, Fonteneau JF, Lirvall M, Haslett P, Lifson JD, Bhardwaj N. Activation of HIV-1 specific CD4 and CD8 T cells by human dendritic cells: roles for cross-presentation and non-infectious HIV-1 virus. AIDS. 2002;16(10):1319–1329. doi: 10.1097/00002030-200207050-00003. [DOI] [PubMed] [Google Scholar]
- Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4(10):1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315(5817):1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Lehmann C, Harper JM, Taubert D, Hartmann P, Fatkenheuer G, Jung N, van Lunzen J, Stellbrink HJ, Gallo RC, Romerio F. Increased interferon alpha expression in circulating plasmacy-toid dendritic cells of HIV-1-infected patients. J Acquir Immune De fi c Syndr. 2008;48(5):522–530. doi: 10.1097/QAI.0b013e31817f97cf. [DOI] [PubMed] [Google Scholar]
- Lehmann C, Taubert D, Jung N, Fatkenheuer G, van Lunzen J, Hartmann P, Romerio F. Preferential upregulation of interferon-alpha subtype 2 expression in HIV-1 patients. AIDS Res Hum Retroviruses. 2009;25(6):577–581. doi: 10.1089/aid.2008.0238. [DOI] [PubMed] [Google Scholar]
- Lehmann C, Lafferty M, Garzino-Demo A, Jung N, Hartmann P, Fatkenheuer G, Wolf JS, van Lunzen J, Romerio F. Plasmacytoid dendritic cells accumulate and secrete interferon alpha in lymph nodes of HIV-1 patients. PLoS One. 2010;5(6):e11110. doi: 10.1371/journal.pone.0011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekakis J, Ikonomidis I. Cardiovascular complications of AIDS. Curr Opin Crit Care. 2010;16(5):408–412. doi: 10.1097/MCC.0b013e32833e10a9. [DOI] [PubMed] [Google Scholar]
- Lepelley A, Louis S, Sourisseau M, Law HK, Pothlichet J, Schilte C, Chaperot L, Plumas J, Randall RE, Si-Tahar M, Mammano F, Albert ML, Schwartz O. Innate sensing of HIV-infected cells. PLoS Pathog. 2011;7(2):e1001284. doi: 10.1371/journal.ppat.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, Reilly CS, Peterson ML, Schultz-Darken N, Brunner KG, Nephew KR, Pambuccian S, Lifson JD, Carlis JV, Haase AT. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458(7241):1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein KA, Armon C, Buchacz K, Chmiel JS, Buckner K, Tedaldi EM, Wood K, Holmberg SD, Brooks JT. Low CD4+ T cell count is a risk factor for cardiovascular disease events in the HIV outpatient study. Clin Infect Dis. 2010;51(4):435–447. doi: 10.1086/655144. [DOI] [PubMed] [Google Scholar]
- Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- Liu AY, Vittinghoff E, Sellmeyer DE, Irvin R, Mulligan K, Mayer K, Thompson M, Grant R, Pathak S, O’Hara B, Gvetadze R, Chillag K, Grohskopf L, Buchbinder SP. Bone mineral density in HIV-negative men participating in a tenofovir pre-exposure prophylaxis randomized clinical trial in San Francisco. PLoS One. 2011;6(8):e23688. doi: 10.1371/journal.pone.0023688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lore K, Sonnerborg A, Brostrom C, Goh LE, Perrin L, McDade H, Stellbrink HJ, Gazzard B, Weber R, Napolitano LA, van Kooyk Y, Andersson J. Accumulation of DC-SIGN + CD40+ dendritic cells with reduced CD80 and CD86 expression in lymphoid tissue during acute HIV-1 infection. AIDS. 2002;16(5):683–692. doi: 10.1097/00002030-200203290-00003. [DOI] [PubMed] [Google Scholar]
- Machmach K, Leal M, Gras C, Viciana P, Genebat M, Franco E, Boufassa F, Lambotte O, Herbeuval J, Ruiz-Mateos E. Plasmacytoid dendritic cells reduce HIV production in elite controllers. J Virol. 2012;86(8):4245–4252. doi: 10.1128/JVI.07114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manches O, Bhardwaj N. Resolution of immune activation defines nonpathogenic SIV infection. J Clin Invest. 2009;119(12):3512–3515. doi: 10.1172/JCI41509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manches O, Munn D, Fallahi A, Lifson J, Chaperot L, Plumas J, Bhardwaj N. HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J Clin Invest. 2008;118(10):3431–3439. doi: 10.1172/JCI34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl JN, Barry AP, Vanderford TH, Kozyr N, Chavan R, Klucking S, Barrat FJ, Coffman RL, Staprans SI, Feinberg MB. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. 2008;14(10):1077–1087. doi: 10.1038/nm.1871. [DOI] [PubMed] [Google Scholar]
- Manion M, Rodriguez B, Medvik K, Hardy G, Harding CV, Schooley RT, Pollard R, Asmuth D, Murphy R, Barker E, Brady KE, Landay A, Funderburg N, Sieg SF, Lederman MM. Interferon-alpha administration enhances CD8+ T cell activation in HIV infection. PLoS One. 2012;7(1):e30306. doi: 10.1371/journal.pone.0030306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli E, Cicala C, Van Ryk D, Goode DJ, Macleod K, Arthos J, Fauci AS. HIV-1 gp120 inhibits TLR9-mediated activation and IFN-{alpha} secretion in plasmacytoid dendritic cells. Proc Natl Acad Sci U S A. 2007;104(9):3396–3401. doi: 10.1073/pnas.0611353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinson JA, Montoya CJ, Usuga X, Ronquillo R, Landay AL, Desai SN. Chloroquine modulates HIV-1-induced plasmacytoid dendritic cell alpha interferon: implication for T-cell activation. Antimicrob Agents Chemother. 2010;54(2):871–881. doi: 10.1128/AAC.01246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. 2005;79(1):17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Megjugorac NJ, Young HA, Amrute SB, Olshalsky SL, Fitzgerald-Bocarsly P. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. J Leukoc Biol. 2004;75(3):504–514. doi: 10.1189/jlb.0603291. [DOI] [PubMed] [Google Scholar]
- Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, Wen TF, Lindsay RJ, Orellana L, Mildvan D, Bazner S, Streeck H, Alter G, Lifson JD, Carrington M, Bosch RJ, Robbins GK, Altfeld M. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med. 2009;15(8):955–959. doi: 10.1038/nm.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, Amoura Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev. 2011;10(12):744–755. doi: 10.1016/j.autrev.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Moseman EA, Liang X, Dawson AJ, Panoskaltsis-Mortari A, Krieg AM, Liu YJ, Blazar BR, Chen W. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4 + CD25+ regulatory T cells. J Immunol. 2004;173(7):4433–4442. doi: 10.4049/jimmunol.173.7.4433. [DOI] [PubMed] [Google Scholar]
- Mouries J, Moron G, Schlecht G, Escriou N, Dadaglio G, Leclerc C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood. 2008;112(9):3713–3722. doi: 10.1182/blood-2008-03-146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med. 2004;10(1):15–18. doi: 10.1016/j.molmed.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Murray SM, Down CM, Boulware DR, Stauffer WM, Cavert WP, Schacker TW, Brenchley JM, Douek DC. Reduction of immune activation with chloroquine therapy during chronic HIV infection. J Virol. 2010;84(22):12082–12086. doi: 10.1128/JVI.01466-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien M, Manches O, Sabado RL, Jimenez Baranda S, Wang Y, Marie I, Rolnitzky L, Markowitz M, Margolis DM, Levy D, Bhardwaj N. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-alpha-producing and partially matured phenotype. J Clin Invest. 2011;121(3):1088–1101. doi: 10.1172/JCI44960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty U, Steinman RM, Peng M, Cameron PU, Gezelter S, Kopeloff I, Swiggard WJ, Pope M, Bhardwaj N. Dendritic cells freshly isolated from human blood express CD4 and mature into typical immunostimulatory dendritic cells after culture in monocyte-conditioned medium. J Exp Med. 1993;178(3):1067–1076. doi: 10.1084/jem.178.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty U, Peng M, Gezelter S, Swiggard WJ, Betjes M, Bhardwaj N, Steinman RM. Human blood contains two subsets of dendritic cells, one immunologically mature and the other immature. Immunology. 1994;82(3):487–493. [PMC free article] [PubMed] [Google Scholar]
- Okoye A, Meier-Schellersheim M, Brenchley JM, Hagen SI, Walker JM, Rohankhedkar M, Lum R, Edgar JB, Planer SL, Legasse A, Sylwester AW, Piatak M, Jr, Lifson JD, Maino VC, Sodora DL, Douek DC, Axthelm MK, Grossman Z, Picker LJ. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. J Exp Med. 2007;204(9):2171–2185. doi: 10.1084/jem.20070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiardini M, Pandrea I, Apetrei C, Silvestri G. Lessons learned from the natural hosts of HIV-related viruses. Annu Rev Med. 2009;60:485–495. doi: 10.1146/annurev.med.60.041807.123753. [DOI] [PubMed] [Google Scholar]
- Paiardini M, Cervasi B, Reyes-Aviles E, Micci L, Ortiz AM, Chahroudi A, Vinton C, Gordon SN, Bosinger SE, Francella N, Hallberg PL, Cramer E, Schlub T, Chan ML, Riddick NE, Collman RG, Apetrei C, Pandrea I, Else J, Munch J, Kirchhoff F, Davenport MP, Brenchley JM, Silvestri G. Low levels of SIV infection in sooty mangabey central memory CD T cells are associated with limited CCR5 expression. Nat Med. 2011;17(7):830–836. doi: 10.1038/nm.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagno L, Spina CA, Marchant A, Salio M, Rufer N, Little S, Dong T, Chesney G, Waters A, Easterbrook P, Dunbar PR, Shepherd D, Cerundolo V, Emery V, Griffiths P, Conlon C, McMichael AJ, Richman DD, Rowland-Jones SL, Appay V. Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 infection. PLoS Biol. 2004;2(2):E20. doi: 10.1371/journal.pbio.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna G, Sozzani S, Adorini L. Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. J Immunol. 2001;167(4):1862–1866. doi: 10.4049/jimmunol.167.4.1862. [DOI] [PubMed] [Google Scholar]
- Piconi S, Parisotto S, Rizzardini G, Passerini S, Terzi R, Argenteri B, Meraviglia P, Capetti A, Biasin M, Trabattoni D, Clerici M. Hydroxychloroquine drastically reduces immune activation in HIV-infected, antiretroviral therapy-treated immunologic nonresponders. Blood. 2011;118(12):3263–3272. doi: 10.1182/blood-2011-01-329060. [DOI] [PubMed] [Google Scholar]
- Pine SO, McElrath MJ, Bochud PY. Polymorphisms in Toll-like receptor 4 and Toll-like receptor 9 influence viral load in a seroincident cohort of HIV-1-infected individuals. AIDS. 2009;23(18):2387–2395. doi: 10.1097/QAD.0b013e328330b489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J. Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood. 2006;107(7):2613–2618. doi: 10.1182/blood-2005-07-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploquin MJ, Desoutter JF, Santos PR, Pandrea I, Diop OM, Hosmalin A, Butor C, Barre-Sinoussi F, Muller-Trutwin MC. Distinct expression profiles of TGF-beta1 signaling mediators in pathogenic SIVmac and non-pathogenic SIVagm infections. Retrovirology. 2006;3:37. doi: 10.1186/1742-4690-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, Takikawa O, Munn DH, Gendelman HE, Persidsky Y. Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood. 2005;106(7):2382–2390. doi: 10.1182/blood-2005-04-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol. 2010;11(8):647–655. doi: 10.1038/ni.1894. [DOI] [PubMed] [Google Scholar]
- Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-beta inhibits human Th17 cell differentiation. J Immunol. 2009;183(8):5418–5427. doi: 10.4049/jimmunol.0803227. [DOI] [PubMed] [Google Scholar]
- Reed JR, Vukmanovic-Stejic M, Fletcher JM, Soares MV, Cook JE, Orteu CH, Jackson SE, Birch KE, Foster GR, Salmon M, Beverley PC, Rustin MH, Akbar AN. Telomere erosion in memory T cells induced by telomerase inhibition at the site of antigenic challenge in vivo. J Exp Med. 2004;199(10):1433–1443. doi: 10.1084/jem.20040178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves RK, Evans TI, Gillis J, Wong FE, Kang G, Li Q, Johnson RP. Infection induces accumulation of plasmacytoid dendritic cells in the gut mucosa. J Infect Dis. 2012 doi: 10.1093/infdis/jis408. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci E, Malacrida S, Zanchetta M, Mosconi I, Montagna M, Giaquinto C, De Rossi A. Toll-like receptor 9 polymorphisms influence mother-to-child transmission of human immunodeficiency virus type 1. J Transl Med. 2010;8:49. doi: 10.1186/1479-5876-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal Malefyt R, Liu YJ. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999;283(5405):1183–1186. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, Martinez R, Sandler NG, Roque A, Liebner J, Battegay M, Bernasconi E, Descombes P, Erkizia I, Fellay J, Hirschel B, Miro JM, Palou E, Hoffmann M, Massanella M, Blanco J, Woods M, Gunthard HF, de Bakker P, Douek DC, Silvestri G, Martinez-Picado J, Telenti A. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Invest. 2011;121(6):2391–2400. doi: 10.1172/JCI45235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabado RL, O’Brien M, Subedi A, Qin L, Hu N, Taylor E, Dibben O, Stacey A, Fellay J, Shianna KV, Siegal F, Shodell M, Shah K, Larsson M, Lifson J, Nadas A, Marmor M, Hutt R, Margolis D, Garmon D, Markowitz M, Valentine F, Borrow P, Bhardwaj N. Evidence of dysregu-lation of dendritic cells in primary HIV infection. Blood. 2010;116(19):3839–3852. doi: 10.1182/blood-2010-03-273763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaka C, Marloie-Provost MA, Soumelis V, Benaroch P. Developmental regulation of MHC II expression and transport in human plasmacytoid-derived dendritic cells. Blood. 2009;113(10):2127–2135. doi: 10.1182/blood-2008-10-178152. [DOI] [PubMed] [Google Scholar]
- Salio M, Palmowski MJ, Atzberger A, Hermans IF, Cerundolo V. CpG-matured murine plasmacytoid dendritic cells are capable of in vivo priming of functional CD8 T cell responses to endogenous but not exogenous antigens. J Exp Med. 2004;199(4):567–579. doi: 10.1084/jem.20031059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science. 2010;329(5998):1530–1534. doi: 10.1126/science.1187029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC, 3rd, Belardelli F, Gabriele L. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med. 2002;196(11):1415–1425. doi: 10.1084/jem.20021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology. 2005;343(2):256–266. doi: 10.1016/j.virol.2005.09.059. [DOI] [PubMed] [Google Scholar]
- Schnurr M, Toy T, Shin A, Hartmann G, Rothenfusser S, Soellner J, Davis ID, Cebon J, Maraskovsky E. Role of adenosine receptors in regulating chemotaxis and cytokine production of plasmacytoid dendritic cells. Blood. 2004;103(4):1391–1397. doi: 10.1182/blood-2003-06-1959. [DOI] [PubMed] [Google Scholar]
- Sedaghat AR, German J, Teslovich TM, Cofrancesco J, Jr, Jie CC, Talbot CC, Jr, Siliciano RF. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. J Virol. 2008;82(4):1870–1883. doi: 10.1128/JVI.02228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda FE, Maschalidi S, Colisson R, Heslop L, Ghirelli C, Sakka E, Lennon-Dumenil AM, Amigorena S, Cabanie L, Manoury B. Critical role for asparagine endopeptidase in endocytic Toll-like receptor signaling in dendritic cells. Immunity. 2009;31(5):737–748. doi: 10.1016/j.immuni.2009.09.013. [DOI] [PubMed] [Google Scholar]
- Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, Mellor AL, He Y, Munn DH. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009;113(24):6102–6111. doi: 10.1182/blood-2008-12-195354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284(5421):1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- Soriano-Sarabia N, Vallejo A, Ramirez-Lorca R, Rodriguez Mdel M, Salinas A, Pulido I, Saez ME, Leal M. Influence of the Toll-like receptor 9 1635A/G polymorphism on the CD4 count, HIV viral load, and clinical progression. J Acquir Immune Defic Syndr. 2008;49(2):128–135. doi: 10.1097/QAI.0b013e318184fb41. [DOI] [PubMed] [Google Scholar]
- Stary G, Klein I, Kohlhofer S, Koszik F, Scherzer T, Mullauer L, Quendler H, Kohrgruber N, Stingl G. Plasmacytoid dendritic cells express TRAIL and induce CD4+ T-cell apoptosis in HIV-1 viremic patients. Blood. 2009;114(18):3854–3863. doi: 10.1182/blood-2009-04-217927. [DOI] [PubMed] [Google Scholar]
- Stein BS, Gowda SD, Lifson JD, Penhallow RC, Bensch KG, Engleman EG. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell. 1987;49(5):659–668. doi: 10.1016/0092-8674(87)90542-3. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- Swiecki M, Colonna M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol Rev. 2010;234(1):142–62. doi: 10.1111/j.0105-2896.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with Toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun. 2005;73(12):7967–7976. doi: 10.1128/IAI.73.12.7967-7976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H, Fukaya T, Eizumi K, Sato Y, Sato K, Shibazaki A, Otsuka H, Hijikata A, Watanabe T, Ohara O, Kaisho T, Malissen B. Plasmacytoid dendritic cells are crucial for the initiation of inflammation and T cell immunity in vivo. Immunity. 2011;35(6):958–971. doi: 10.1016/j.immuni.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, Taniguchi T. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- Tilton JC, Manion MM, Luskin MR, Johnson AJ, Patamawenu AA, Hallahan CW, Cogliano-Shutta NA, Mican JM, Davey RT, Jr, Kottilil S, Lifson JD, Metcalf JA, Lempicki RA, Connors M. Human immunodeficiency virus viremia induces plasmacytoid dendritic cell activation in vivo and diminished alpha interferon production in vitro. J Virol. 2008;82(8):3997–4006. doi: 10.1128/JVI.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272(5270):1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- Tsujimura H, Tamura T, Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J Immunol. 2003;170(3):1131–1135. doi: 10.4049/jimmunol.170.3.1131. [DOI] [PubMed] [Google Scholar]
- van der Straten A, van Damme L, Haberer JE, Bangsberg DR. How well does PREP work? Unraveling the divergent results of PrEP trials for HIV prevention. AIDS. 2012;26(7):F13–F19. doi: 10.1097/QAD.0b013e3283522272. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Vollenweider R, Lennert K. Plasmacytoid T-cell clusters in non-specific lymphadenitis. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;44(1):1–14. doi: 10.1007/BF02890155. [DOI] [PubMed] [Google Scholar]
- von Sydow M, Sonnerborg A, Gaines H, Strannegard O. Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res Hum Retroviruses. 1991;7(4):375–380. doi: 10.1089/aid.1991.7.375. [DOI] [PubMed] [Google Scholar]
- Wang Y, Abel K, Lantz K, Krieg AM, McChesney MB, Miller CJ. The Toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J Virol. 2005;79(22):14355–14370. doi: 10.1128/JVI.79.22.14355-14370.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland M, Czeloth N, Mach N, Malissen B, Kremmer E, Pabst O, Forster R. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc Natl Acad Sci U S A. 2007;104(15):6347–6352. doi: 10.1073/pnas.0609180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama H, Matsuno K, Zhang Y, Nishiwaki T, Kitabatake M, Ueha S, Narumi S, Morikawa S, Ezaki T, Lu B, Gerard C, Ishikawa S, Matsushima K. Evidence for recruitment of plas-macytoid dendritic cell precursors to inflamed lymph nodes through high endothelial venules. Int Immunol. 2004;16(7):915–928. doi: 10.1093/intimm/dxh093. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453(7192):236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]