

Abstract
Micro RNA-146a (miRNA-146a) is an inducible, 22 nucleotide, small RNA over-expressed in Alzheimer’s disease (AD) brain. Up-regulated miRNA-146a targets several inflammation-related and membrane-associated messenger RNAs (mRNAs), including those encoding complement factor-H (CFH) and the interleukin-1 receptor associated kinase-1 (IRAK-1), resulting in significant decreases in their expression (p < 0.05, ANOVA). In this study we assayed miRNA-146a, CFH, IRAK-1 and tetraspanin-12 (TSPAN12), abundances in primary human neuronal-glial (HNG) co-cultures, in human astroglial (HAG) and microglial (HMG) cells stressed with Aβ42 peptide and tumor necrosis factor alpha (TNFα). The results indicate a consistent inverse relationship between miRNA-146a and CFH, IRAK-1 and TSPAN12 expression levels, and indicate that HNG, HAG and HMG cell types each respond differently to Aβ42-peptide + TNFα-triggered stress. While the strongest miRNA-146a-IRAK-1 response was found in HAG cells, the largest miRNA-146a-TSPAN12 response was found in HNG cells, and the most significant miRNA-146a-CFH changes were found in HMG cells, the ‘resident scavenging macrophages’ of the brain.
Keywords: Brain gene expression, Complement factor H (CFH), Human astroglial cells, Microglial cells, Interleukin-1 receptor associated kinase 1, (IRAK-1), microRNA (miRNA), Neurodegeneration, Neuroinflammation, Post-transcriptional control, Small RNA, Tetraspanin 12 (TSPAN12)
Inflammatory neurodegeneration in Alzheimer’s disease (AD) appears to be orchestrated in part by a complex interplay of pro-inflammatory transcription factors, microRNAs (miRNAs), and specific innate immune and inflammatory messenger RNAs (mRNAs), resulting in the progressive development of pathogenic gene expression. In this signaling system, miRNAs represent a recently discovered family of small, evolutionarily conserved, non-coding regulatory RNAs that recognize the 3′ un-translated regions (UTRs) of specific mRNA populations, that through complementary base-pairing with target mRNA 3′ UTRs, function in translational inhibition in the expression of that particular target mRNA [32,16,29]. Specifically up-regulated miRNAs may target multiple mRNAs, and may explain in part the general down-regulation of genes expressed in AD-affected brain [14,4,15]. Originally described in mouse immune cells, an inducible, brain abundant miRNA-146a has been found to be significantly up-regulated (p < 0.05, ANOVA) in anatomical regions exhibiting AD neuropathology, is unchanged in brain regions spared of AD-type pathology, and is associated with the targeted down-regulation of specific inflammation-relevant messenger RNAs (mRNAs) [16,3,33,22]. These include mRNAs encoding complement factor H (CFH) [33,22,8,35] and the interleukin-1 receptor associated kinase 1 (IRAK-1) [12,6]. Decreased expression of CFH appears to contribute to dysregulated complement signaling with significant pro-inflammatory consequences as AD progresses, and decreases in IRAK-1 are associated with a compensatory surge in IRAK-2 expression and increases in pathogenic transcription from NF-κB-sensitive pro-inflammatory genes [14,4,15,12,6,36,30,17,7,26,27]. Interestingly, increases in miRNA-146a have also been shown in 5 different transgenic mouse models of AD and in virus, neurotoxic metal- and oxidation-stressed human brain cells [1,9–11,13,18–20,24; unpublished observations; discussed further below]. It is not well understood, however, what brain cell types contribute to this altered miRNA-146a-mediated progression of pathogenic events. These studies were undertaken to further our understanding of miRNA-146a-mediated alterations in CFH and IRAK-1 gene expression in Aβ42- and TNFα-stressed human neuronal-glial (HNG), human astroglial (HAG) and a human microglial (HMG) cells in primary culture. Previous data have shown that in AD, increased abundance of Aβ42 peptides and tumor necrosis factor alpha (TNFα) are important participants in driving pro-inflammatory signaling [15,22,8,35]. We also studied expression of the transmembrane spanning tetraspanin 12 (TSPAN12); besides roles in membrane signal transduction, TSPAN12 also serves as an essential partner for ADAM10, thus facilitating ADAM10-dependent proteolysis of beta-amyloid precursor protein (βAPP) into non-amyloidogenic signaling pathways [36,26,27,20]. The data suggest that miRNA-146a, CFH, IRAK-1 and TSPAN12 are part of a pathogenetic system variably expressed in different brain cell types, and suggest that miRNA-146a mediated effects on gene expression in HNG, HAG or HMG cells may differentially contribute to inflammatory aspects of brain cell degeneration.
The primary culture of human neuronal-glial (HNG; also referred to as human neural cells) and human astroglial (HAG) cells, including glial-specific glial fibrillary acidic protein (GFAP), neuron-specific beta-tubulin III (βTUBIII), and Hoescht 33258 staining have been previously described [1,5–7,9–11,13,17–24,26–28,33]. Human microglial (HMG) cells were cultured according to established protocols (Clonexpress, Gaithersburg, MD) and stained with an antibody that recognizes a microglial-specific Iba1 (sc-32725, Santa Cruz) (Fig. 1). After 1 week of culture brain cells to be stressed received at each change of medium human recombinant tumor necrosis factor alpha (TNFα; 10 nM; T0157, Sigma–Aldrich Chemical, St. Louis, MO) plus Aβ42 peptide (5 μM; Sigma–Aldrich). Aβ42 peptides were prepared using the hexafluoroisopropanol (HFIP) evaporation-dimethyl sulfoxide-solubilization method as previously described [21,38,34]. Control primary cells received cell culture grade human serum albumin (HSA; Sigma–Aldrich). After Aβ42 peptide + TNFα addition cells were cultured for 1.0 additional week after which total RNA and protein fractions were analyzed using equivalent numbers of brain cells [6,24,21]. A guanidine isothiocyanate- and silica gel-based membrane total RNA purification system and miRNA isolation kit (PureLink™ Invitrogen, Carlsbad, CA) were used to isolate total RNA; total RNA concentrations were quantified using RNA 6000 Nano LabChips and a 2100 Bioanalyzer (Caliper Technologies, Mountainview, CA; Agilent Technologies, Palo Alto, CA). In previous studies specific small RNAs including 5S RNA, miRNA-132 and miRNA-146a were initially analyzed and quantified using miRNA arrays (LC Sciences, Houston, TX) or Northern dot blots as previously described [33,21–24,6,13,9,28,5]. For real-time quantitative PCR (qPCR) analysis of miRNAs, total RNA fractions were prepared from HNG, HAG or HMG cells using an miRNeasy Mini Kit (Qiagen, Valencia, CA), and qPCR analysis was performed using individual TaqMan miRNA assays using a TaqMan MiRNA Reverse Transcription Kit, TaqMan Universal PCR Master Mix, and an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems; Carlsbad, CA) [15,22,18,1,11]. Altered small RNA levels were further verified using a quantitative Northern dot blot ‘focusing’ assay that utilizes a T4 PNK kinase radiolabel system employing [α-32P]-dATP (6000 Ci/mmol; Invitrogen, Carlsbad, CA) that significantly concentrates small RNA and miRNA signals [33,22,6,26,27,20]. In all past and present studies undertaken all 3 techniques yielded quantitatively similar results; Northern dot blot and qPCR data for miRNA-132 and miRNA-146a are shown in Fig. 2. 5S RNA and miRNA-132 were used as endogenous controls as their levels have been previously shown not to change under control or stressed physiological conditions in any human brain cell type studied [33,11,28,23]. Expression of CFH, IRAK-1, TSPAN12 and β-actin (as an internal control) at the level of mRNA and protein were performed as previously described [33,21–23,6,30,28]. TSPAN12 protein was analyzed using a rabbit polyclonal antibody to TSPAN12 (ab90091; Abcam, Cambridge, MA) [21]. Relative 5S RNA, miRNA-132 and miRNA-146a, CFH, IRAK-1 or TSPAN12 mRNA and protein signal strengths were quantified against 5SRNA (for miRNA) or β-actin (for protein) in each sample using data-acquisition software provided with a GS250 molecular imager (Bio-Rad, Hercules, CA) [22,17,7,26,27,20,19]. Graphic presentations were performed using Excel algorithms (Microsoft, Seattle, WA) and Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA). Statistical significance was analyzed using a two-way factorial analysis of variance (p, ANOVA; SAS Institute, Cary, NC). A ‘p’ value of <0.05 was deemed as statistically significant; experimental values in the Figures are expressed as means ± one standard deviation (SD) of that mean.
Fig. 1.

Primary cultures of human neuronal-glial [HNG] cells (A), human astroglia [HAG] cells (B), and human microglial [HMG] cells (C) are currently used to study human brain aging- and AD-relevant disease mechanisms [15,33,22,6,27,20,13,18,1,11,24,5,38]. HNG and HAG cells are stained using antibody to glial fibrillary acidic protein (GFAP), a glial-specific marker (green fluorescence; λmax = 556 nm); HMG cells are stained with fluorescent antibody to a human microglial-specific Iba1 (green fluorescence; λmax = 504 nm; sc-32725, Santa Cruz); in addition to GFAP, HNG cells are stained with βTUBIII, a neuron-specific marker (red; λmax = 702 nm) and Hoescht 33258 to highlight apoptotic features of cell nuclei (blue; λmax = 461 nm) (C). Brain cell types and numbers can be electronically quantified according to differential nuclear and cytosol staining and λmax [1,21,38]. Using the λmax index after 2 weeks in culture HNG cells Exhibit 50/50 neuronal/astroglial cells, HAG cultures exhibit at least 95% of cells staining with a λmax = 556 nm and HMG cultures exhibit at least 95% of cells staining with a λmax = 504 nm [20,21; unpublished]. Interestingly, highly purified monocultures of HMG cells begin expressing GFAP mRNA and react with antibody to GFAP after 2 or more weeks in culture [37]; each cell type shown here are 2 weeks in culture; HNG and HAG cells are about 50% confluent; HMG cells are about 25% confluent; all magnifications 20× [21,38].
Fig. 2.
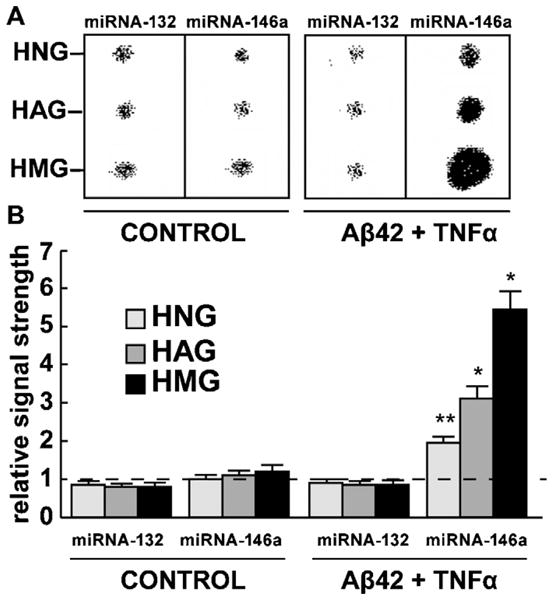
Analysis of a control miRNA-132 and an inducible miRNA-146a in HNG, HAG and HMG cells both in untreated controls and in Aβ42 peptide + TNFα-stressed cells. Northern dot blot analysis (A) and signal quantitation using qPCR analysis (B); similar results were obtained using either miRNA array, Northern dot blot or qPCR analysis. While the levels of miRNA-132 were found to not change under any treatment condition, miRNA-146a was induced 2.1-, 3.2- and 5.4-fold over controls in HNG, HAG and HMG cells, respectively after the stress treatments indicated. Signals were quantified against internal 5SRNA levels; for ease of comparison a dashed horizontal line at 1.0 indicates miRNA-146a levels in control HNG cells; N = 5 for each cell type determination; significance over controls *p < 0.01; **p < 0.05 (ANOVA).
The primary culture of HNG, HAG, and HMG cells provide a useful platform in the study of brain cell aging and neurodegenerative disease-relevant mechanisms (Fig. 1) [1,5,6,11,13,18–24,26–28,33,38]. These primary human brain cells are typically stained differentially using primary antibodies to GFAP, a human microglial-specific Iba1; in addition to GFAP staining HNG cells are stained with βTUBIII, a neuron-specific marker. Hoescht 33258 staining specifically highlights cellular nuclei, and in combination with brain cell specificity-staining and morphological features the actual cell number can be quantified for individual brain cell types [20,1,21,38]. Human neurons do not grow well in primary culture by themselves without the support of astroglial cells, therefore only human neural (neuronal-glial co-cultures; 50/50 neurons/astroglial cells) were used in these experiments [1,21,38]. Interestingly, and in agreement with a previous report, aged or cell culture medium-augmented HMG cells were observed to begin expressing GFAP and stain with GFAP antibody after about 2 weeks in primary culture [1,38]. While 5S RNA and miRNA-132 exhibited no significant change (p > 0.05) in relative abundance in any of the samples tested under any treatment conditions, miRNA-146a was found to be increased approximately 2.1-, 3.2-and 5.4-fold, respectively, in HNG, HAG and HMG primary brain cell lines (Fig. 2).
We further observed a consistent inverse correlation between miRNA-146a and CFH, IRAK-1 or TSPAN12 expression in the 3 brain cell types examined in this study, except for TSPAN12 expression in HMG cells which did not change [22,6,13] (Fig. 3). Indeed, the magnitude of decreased CFH, IRAK-1 or TSPAN12 expression varied amongst different cell types. For example the greatest repression of CFH mRNA and protein was observed to be down-regulated to 0.23- and 0.14-fold of controls in HMG cells (Fig. 3B); the greatest repression of IRAK-1 mRNA and protein was observed to be down-regulated to 0.21- and 0.15-fold of controls in HAG cells (Fig. 3C), and the greatest repression of TSPAN12 mRNA and protein was observed to be down-regulated to 0.51- and 0.27-fold of controls in HNG cells (Fig. 3D), and in these cases the results were highly significant (p < 0.05, ANOVA).
Fig. 3.

Levels of complement factor H (CFH), interleukin-1 receptor associated kinase 1 (IRAK-1) and TSPAN12 protein in control and in stressed (Aβ42 peptide + TNFα) HNG, HAG and HMG cells compared to β-actin levels within the same sample (A). Relative mRNA and protein levels (as determined by qPCR or Western analysis, respectively) are quantified for CFH (B), IRAK-1 (C) or TSPAN12 (D). For ease of comparison a dashed horizontal line in (B–D) at 1.0 indicates relative control levels. Interestingly, TSPAN12 mRNA and protein signals remained unchanged in HMG cells after any treatment condition studied here (D); N = 8 for each cell type; significance compared to control levels *p < 0.01; **p < 0.05 (ANOVA).
First described as an endotoxin- and pro-inflammatory cytokine-responsive small inducible RNA in murine monocytes [33] miRNA-146a has since been shown to be significantly up-regulated in inflammatory central nervous system (CNS) disorders associated with AD [22,6,17], viral infection [11,10,9,24], prion-induced inflammatory neurodegeneration [31] and in inflammation associated with experimental and human temporal lobe epilepsy [2]. Interestingly, up-regulated miRNA-146a has also been shown to negatively regulate IL-1β-induced inflammation in human lung epithelial cells, hence providing a novel cell-specific mechanism for the regulation of inflammation and the innate immune response [25]. Significant up-regulation of miRNA-146a is also observed in interleukin-1beta- (IL-1β-), Aβ42 peptide- or neurotoxic metal sulfate-mediated oxidation-stressed HNG brain cell cultures [27,20,19], and in at least 5 different transgenic mouse models of AD that over-express beta-amyloid peptides (p < 0.05, ANOVA) [13]. These observations, and the immune and inflammatory genetic signaling associated with miRNA-146a, including a cell-specific and inducible CFH and IRAK-1 down-regulation [3,33,22,8,6,31], prompted us to further examine miRNA-146a-CFH, miRNA-146a-IRAK-1 and miRNA-146a-TSPAN12 signaling circuits in three primary cell types of the human brain.
HMG cells, sometimes referred to as the ‘resident macrophages’ of the CNS are an extremely plastic, scavenging cell type that are the first and main form of active innate immune defense in the CNS [22,8,35]. HMG cells constitute 10–20% of total brain cell populations, and while they arise from a hematopoietic lineage it has been intriguingly proposed that these cells possess stem cell properties and, depending on their environment, may be an important precursor to other major brain cell types including HAG and other non-HMG brain cell types [35,21,38]. Indeed highly purified monocultures of primary HMG cells cultured for less than 2 weeks specifically stain with the human microglial-specific marker Iba1 (also known as allograft inflammatory factor 1; Santa Cruz), while more aged HMG cells also stain with antibodies to GFAP [37; unpublished observations]. HMG cells are also known to express high levels of CFH, a soluble glycoprotein inhibitor of both the immune system’s classical and alternate complement activation system which critically mediates the inflammatory response to infection, and further contributes to micoglial cell recruitment at inflammatory sites [35,12,27]. CFH (also known as factor H, H factor, HF, H factor-1, HF1, β1H globulin, C3b inactivator accelerator, A-C3bINA, AMBP-1, adrenomedullin binding protein-1, and AM binding protein-1) normally acts as a complement system repressor as a specific inhibitor of the C3–C3b transition; hence miRNA-146a-mediated CFH deficits are conducive to excessive complement pathway activation associated with autoimmunity and a sustained inflammatory response [22,8,27,20,19]. CFH is not only significantly reduced in AD and in cytokine and Aβ42-peptide stressed brain cells in vitro [22,8,35,12,6], but approximately 50% of patients with the retinal inflammatory disorder age-related macular degeneration possess specific sequence variants in the CFH gene that essentially diminish the effectiveness of CFH protein as the primary inhibitor of complement activation [35]. HMG that normally ‘scavenge’ the CNS for damaged neurons, amyloid plaques and infectious or neurotoxic agents as mobile pathogen-sensing macrophages appear to also advance the spreading of inflammatory signals [8,27,18]. MiRNA-146a-mediated CFH deficits may promote microglial activation, wherein maximally immune responsive forms of microglia further secrete a host of pro-inflammatory cytokines including IL-1α, IL-1β, TNFα, cathepsins, metalloproteinases and lipophylic amines that directly contribute to neuronal damage and the progression of degenerative inflammatory neuropathology [35,12,17]. HAG cells are an immune-responsive cell type involved in the neuropathogenesis of inflammatory diseases of the brain, where again the activation of inflammatory mediators and cytokines play an important roles [22,8,35,12,6]. Previous studies report a significant miRNA-146a-mediated depletion of IRAK-1 in HAG cells when compared to HNG cells receiving identical experimental stress protocols, and this HAG cell depletion of IRAK-1 is linked to a compensatory up-regulation of IRAK-2 and the sustained activation of the transcription factor NF-κB, and of transcription from NF-κB-sensitive pro-inflammatory genes [33,6].
Interestingly, using miRBASE algorithms (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/) we note that TSPAN1, TSPAN14 and TSPAN18 mRNA 3′-UTRs have been identified as miRNA-146a targets; we further note that TSPAN12 appears to be targeted by miRNA-146a, in part because many TSPAN mRNA 3′-UTRs contain highly homologous RNA sequences (unpublished observations). βAPP, TSPAN12, and βAPP processing enzymes such as ADAM10 (a disintegrin and metalloprotease) are membrane-associated components of βAPP-mediated signaling enriched in neuronal cell membranes and were found to be most abundant in HNG cells in these studies (data not shown) [12,36,17]. While TSPAN12 over-expression enhances ADAM10-dependent processing of βAPP in neuroblastoma cells, deficits in TSPAN12 may shunt βAPP catabolism into amyloidogenic pathways resulting in the increased generation of pro-inflammatory Aβ42 peptides [36,30,17,21].
Brain gene expression is regulated by a rich variety of chromatin structural, transcriptional, post-transcriptional and epigenetic controls that coordinate gene product abundance through variations in mRNA speciation and complexity, and the neurobiology of miR-NAs and their interactive roles in human brain function are only beginning to become understood. Each brain cell type expresses not only variation in what kinds of mRNA are expressed but also variable abundances of inducible miRNAs and their target mRNAs (Fig. 2) [32,16,29,14,4,15,3,22]. As brain tissues are composed of multiple neuronal, astroglial, microglial, and other cell types, the contributions of each brain cell type to miRNA and mRNA speciation and gene expression remain to be further elucidated. Distinctly different molecular-genetic responses to miRNA-146a-mediated CFH, IRAK-1 and TSPAN12 expression are apparent even in juxtaposed brain cell types (Figs. 1 and 3). Investigations involving systematic analysis and bioinformatics combined with studies in control versus stressed primary brain cells and affected regions of AD brains should provide further insight into pathogenic spreading mechanisms and the role of intrinsically specific miRNA-mRNA interactions in both the onset and propagation of the AD process.
In summary, these data are the first to show differential regulation of miRNA-146a, CFH, IRAK-1 and TSPAN12 in three different primary brain cell cultures including HNG, HAG and HMG cells in response to Aβ42-peptide and TNFα-induced stress. While HNG cell cultures contain approximately 50/50 populations of signaling neurons and astroglial support cells, HAG and HMG cells are immunological-responsive cell types of the CNS and important mediators of pro-inflammatory and pathogenic signaling in neurodegeneration. As similar miRNA-146a up-regulation is also observed in AD affected brain regions, the specific mis-regulation of miRNA-146a-CHF, miRNA-146a-IRAK-1 or miRNA-146a-TSPAN12 circuits confined to different cell types of the brain has important implications in our understanding of the extra-neuronal acquisition, transmission and ‘spreading’ of inflammatory aspects of neurological disorders. These findings further support the idea that a directed modification of specific miRNA signaling in selected brain cell types will provide novel therapeutic strategies of benefit in the clinical management of AD.
Acknowledgments
Thanks are extended to Drs. P.N. Alexandrov, S.O. Gettner, J.M. Hill, D. Guillot and Y. Zhao for expert advice, statistical analysis and technical assistance. This work was presented in part at the International Conference on Alzheimer’s disease (ICAD) 10th annual meeting 10–15 July 2010, Honolulu, HI. These studies were supported in part by a Translational Research Initiative grant from Louisiana State University Board of Regents (W.J.L.), by an Alzheimer Association Investigator-Initiated Research Grant IIRG-09-131729 (W.J.L.), and by National Institutes of Health Research Grant NIA AG038834 (W.J.L.).
References
- 1.Alexandrov PN, Zhao Y, Pogue AI, Tarr MA, Kruck TPA, Percy ME, Lukiw WJ. Synergistic effects of iron and aluminum on stress-related gene expression in primary human neural cells. J Alzheimers Dis. 2005;8:117–127. doi: 10.3233/jad-2005-8204. [DOI] [PubMed] [Google Scholar]
- 2.Aronica E, Fluiter K, Iyer A, Zurolo E, Vreijling J, van Vliet EA, Baayen JC, Gorter JA. Expression pattern of miRNA-146a, an inflammation-associated miRNA, in experimental and human temporal lobe epilepsy. Eur J Neurosci. 2010;31:1100–1107. doi: 10.1111/j.1460-9568.2010.07122.x. [DOI] [PubMed] [Google Scholar]
- 3.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MiRNAs:, new regulators of immune cell development and function. Nat Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 4.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002;70:462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 5.Cui JG, Kuroda H, Chandrasekharan NV, Pelaez RP, Simmons DL, Bazan NG, Lukiw WJ. Cyclooxygenase-3 gene expression in Alzheimer hippocampus and in stressed human neural cells. Neurochem Res. 2004;29:1731–1737. doi: 10.1023/b:nere.0000035809.70905.8a. [DOI] [PubMed] [Google Scholar]
- 6.Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ. Differential regulation of interleukin-1 receptor-associated Kinase-1 (IRAK-1) and IRAK-2 by miRNA-146a and NF-kB in stressed human astroglial cells and in Alzheimer’s disease. J Biol Chem. 2010;285:38951–38960. doi: 10.1074/jbc.M110.178848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ershov AV, Lukiw WJ, Bazan NG. Selective transcription factor induction in retinal pigment epithelial cells during photoreceptor phagocytosis. J Biol Chem. 1996;271:28458–28462. doi: 10.1074/jbc.271.45.28458. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths MR, Neal JW, Fontaine M, Das T, Gasque P. Complement factor H protects against experimental autoimmune encephalomyelitis. J Immunol. 2009;182:4368–4377. doi: 10.4049/jimmunol.0800205. [DOI] [PubMed] [Google Scholar]
- 9.Higaki S, Gebhardt BM, Lukiw WJ, Thompson HW, Hill JM. Effect of immunosuppression on gene expression in the HSV-1 latently infected mouse trigeminal ganglion. Invest Ophthalmol Vis Sci. 2002;43:1862–1869. [PubMed] [Google Scholar]
- 10.Hill JM, Lukiw WJ, Gebhardt BM, Higaki S, Loutsch JM, Myles ME, Thompson HW, Kwon BS, Bazan NG, Kaufman HE. Gene expression analyzed by microarrays in HSV-1 latent mouse trigeminal ganglion following heat stress. Virus Genes. 2001;23:273–280. doi: 10.1023/a:1012517221937. [DOI] [PubMed] [Google Scholar]
- 11.Hill JM, Zhao Y, Clement C, Neumann DM, Lukiw WJ. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport. 2009;20:1500–1505. doi: 10.1097/WNR.0b013e3283329c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol. 2008;9:684–691. doi: 10.1038/ni.1606. [DOI] [PubMed] [Google Scholar]
- 13.Li YY, Cui JG, Hill JM, Bhattacharjee S, Zhao Y, Lukiw WJ. Increased expression of miRNA-146a in Alzheimer’s disease transgenic models. Neurosci Lett. 2011;487:94–98. doi: 10.1016/j.neulet.2010.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loring JF, Wen X, Lee JM, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer’s disease. DNA Cell Biol. 2001;20:683–695. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- 15.Lukiw WJ. Gene expression profiling in fetal, aged, and Alzheimer hippocampus: a continuum of stress-related signaling. Neurochem Res. 2004;29:1287–1297. doi: 10.1023/b:nere.0000023615.89699.63. [DOI] [PubMed] [Google Scholar]
- 16.Lukiw WJ. MiRNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- 17.Lukiw WJ, Bazan NG. Survival signaling in Alzheimer’s disease. Biochem Soc Trans. 2006;34:1277–1282. doi: 10.1042/BST0341277. [DOI] [PubMed] [Google Scholar]
- 18.Lukiw WJ, Pogue AI. Induction of specific miRNA species by ROS-generating metal sulfates in primary human brain cells. J Inorg Biochem. 2007;101:1265–1269. doi: 10.1016/j.jinorgbio.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lukiw WJ, LeBlanc HJ, Carver LA, McLachlan DR, Bazan NG. Run-on gene transcription in human neocortical nuclei Inhibition by nanomolar aluminum and implications for neurodegenerative disease. J Mol Neurosci. 1998;11:67–78. doi: 10.1385/JMN:11:1:67. [DOI] [PubMed] [Google Scholar]
- 20.Lukiw WJ, Percy ME, Kruck TPA. Nanomolar aluminum induces pro-inflammatory and apoptotic gene expression in human brain cells. J Inorg Biochem. 2005;99:1895–1898. doi: 10.1016/j.jinorgbio.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 21.Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer’s disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukiw WJ, Zhao Y, Cui JG. A NF-κB-sensitive miRNA-146a-mediated inflammatory circuit in AD and in stressed human brain cells. J Biol Chem. 2008;283:31315–31322. doi: 10.1074/jbc.M805371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukiw WJ, Cui JG, Li YY, Culicchia F. Up-regulation of miRNA-221 (chr Xp11.3) and caspase-3 accompanies down-regulation of the survivin-1 homolog BIRC1 (NAIP) in glioblastoma multiforme (GBM) J Neurooncol. 2009;91:27–32. doi: 10.1007/s11060-008-9688-0. [DOI] [PubMed] [Google Scholar]
- 24.Lukiw WJ, Cui JG, Yuan YY, Bhattacharjee PS, Corkern M, Clement C, Hill JM. Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport. 2010;21:922–927. doi: 10.1097/WNR.0b013e32833da51a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry M, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in microRNA-146a expression negatively regulate the IL-1β-induced inflammatory response in human lung alveolar epithelial cells. J Immunol. 2008;(180):5689–5698. doi: 10.4049/jimmunol.180.8.5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer’s disease. Neuroreport. 2004;15:1507–1510. doi: 10.1097/01.wnr.0000130539.39937.1d. [DOI] [PubMed] [Google Scholar]
- 27.Pogue AI, Li YY, Cui JG, Zhao Y, Kruck TP, Percy ME, Tarr MA, Lukiw WJ. Characterization of an NF-kB-regulated miRNA-146a in metal-sulfate-stressed human brain cells. J Inorg Biochem. 2009;11:156–164. doi: 10.1016/j.jinorgbio.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Pogue AI, Cui JG, Li YY, Zhao Y, Culicchia F, Lukiw WJ. MiRNA-125b (miRNA-125b) function in astrogliosis and glial cell proliferation. Neurosci Lett. 2010;476:18–22. doi: 10.1016/j.neulet.2010.03.054. [DOI] [PubMed] [Google Scholar]
- 29.Provost P. MiRNAs as a molecular basis for mental retardation, Alzheimer’s and prion diseases. Brain Res. 2010;1338:58–66. doi: 10.1016/j.brainres.2010.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riazanskaia N, Lukiw WJ, Grigorenko A, Korovaitseva G, Dvoryanchikov G, Moliaka Y, Nicolaou M, Farrer L, Bazan NG, Rogaev E. Regulatory region variability in the human presenilin-2 (PSEN2) gene: potential contribution to the gene activity and risk for AD. Mol Psychiatr. 2002;7:891–898. doi: 10.1038/sj.mp.4001101. [DOI] [PubMed] [Google Scholar]
- 31.Saba R, Goodman CD, Huzarewich RL, Robertson C, Booth SA. A miRNA signature of prion induced neurodegeneration. PLoS One. 2008;3:e3652. doi: 10.1371/journal.pone.0003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satoh J. MiRNAs and their therapeutic potential for human diseases: aberrant miRNA expression in Alzheimer’s disease brains. J Pharmacol Sci. 2010;114:269–275. doi: 10.1254/jphs.10r11fm. [DOI] [PubMed] [Google Scholar]
- 33.Sethi P, Lukiw WJ. MiRNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci Lett. 2009;459:100–104. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- 34.Stine WB, Jungbauer L, Yu C, LaDu MJ. Preparing synthetic Aβ in different aggregation states. Methods Mol Biol. 2011;670:13–32. doi: 10.1007/978-1-60761-744-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weisman D, Hakimian E, Ho GJ. Interleukins, inflammation, and mechanisms of Alzheimer’s disease. Vitam Horm. 2006;74:505–530. doi: 10.1016/S0083-6729(06)74020-1. [DOI] [PubMed] [Google Scholar]
- 36.Xu D, Sharma C, Hemler ME. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 2009;23:3674–3681. doi: 10.1096/fj.09-133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokoyama A, Yang L, Itoh S, Mori K, Tanaka J. Microglia, a potential source of neurons, astrocytes, and oligodendrocytes. Glia. 2004;45:96–104. doi: 10.1002/glia.10306. [DOI] [PubMed] [Google Scholar]
- 38.Zhao Y, Calon F, Julien C, Winkler JW, Petasis NA, Lukiw WJ, Bazan NG. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer models. PLoS One. 2011;6:e15816. doi: 10.1371/journal.pone.0015816. [DOI] [PMC free article] [PubMed] [Google Scholar]