

Abstract
Coumadin (R/S-warfarin) is a highly efficacious and widely used anticoagulant; however, its highly variable metabolism remains an important contributor to uncertainties in therapeutic responses. Pharmacogenetic studies report conflicting findings on the clinical relevance of CYP2C19. A resolution to this controversy is impeded by a lack of detail on the potential role of CYP2C19 in warfarin metabolism. Consequently, we assessed the efficiency of CYP2C19 metabolism of R- and S-warfarin and explored possible contributions in the liver using in vitro methods. Recombinant CYP2C19 metabolized R- and S-warfarin mainly to 6-, 7-, and 8-hydroxywarfarin, while 4′-hydroxywarfarin was a minor metabolite. Overall R-warfarin metabolism was slightly more efficient than that for S-warfarin. Metabolic pathways that produce R-6-, 7-, and 8-hydroxywarfarin in human liver microsomal reactions correlated strongly with CYP2C19 S-mephenytoin hydroxylase activity. Similarly, CYP1A2 activity toward phenacetin correlated with formation of R-6 and 7-hydroxywarfarin such that R-8-hydroxywarfarin seems unique to CYP2C19 and possibly a biomarker. In following, CYP2C19 likely impacts R-warfarin metabolism and patient response to therapy. Intriguingly, CYP2C19 may contribute to S-warfarin metabolism in patients, especially when CYP2C9 activity is compromised due to drug interactions or genetic polymorphisms.
Keywords: Warfarin, CYP2C19, Cytochrome P450, metabolism, biomarker
BACKGROUND
Introduced into clinical practice in the 1950s, Coumadin (R/S-warfarin) has become one of the most widely used medications for the treatment and prevention of thromboembolic events [1]. Although highly effective [2-4], warfarin therapy poses risks for hemorrhaging (overdosing) or ischemic stroke (underdosing) due to a narrow therapeutic range and high inter-individual variability in response [1]. An important contributor to inter-patient variability in response to warfarin is the metabolism of the drug. Consequently, an understanding of metabolic pathways contributing to warfarin inactivation and clearance is critical for improving patient outcomes during anticoagulant therapy.
Warfarin is clinically available as an equal mixture of R and S enantiomers; metabolic clearances for both drug forms occur mainly through oxidative reactions catalyzed by cytochrome P450s (CYP for specific isoforms). As shown in Fig. (1), these enzymes demonstrate enantio- and regiospecificity when hydroxylating warfarin to 6-, 7-, 8-, 10-, and 4′-hydroxywarfarin [5, 6]. CYP2C9 efficiently metabolizes S-warfarin [7], while CYP2C19 [8] and CYP3A (CYP4/5/7) [9-11] may contribute to minor metabolic pathways. Although not studied as extensively, R-warfarin metabolism is potentially catalyzed mainly by CYP1A2, CYP2C19, and CYP3A [5, 9]. Collectively, these metabolic reactions transform pharmacologically active warfarin to relatively inactive hydroxywarfarins [5, 6], and thus understanding warfarin metabolism is necessary to assess the impact on active drug levels and the corresponding effect on patient response to therapy.
Fig. (1).
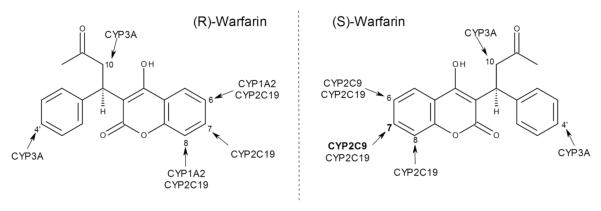
General metabolic pathways of R- and S-warfarin catalyzed by several cytochrome P450s.
Recent pharmacogenetic studies involving CYP2C19 have yielded findings that challenge the sole importance of CYP2C9 in patient response to warfarin therapy. An analysis of genetic and environmental effects on warfarin response with 311 patients (mostly Caucasian) identified significant associations between CYP2C19 polymorphisms and several clinical factors including stable dosage, warfarin sensitivity, and overdosing [12]. Similarly, CYP2C19 polymorphisms led to higher bleeding frequencies in Korean patients (n=66) possibly due to overdosing [13]. By contrast, two studies with Japanese patients (n=125 [14] and n=16 [15]) demonstrated that CYP2C19 polymorphisms did not impact the anticoagulant effect of warfarin treatment, even though variants inhibited R-warfarin but not S-warfarin clearance. An explanation for these observations would require CYP2C19 activity to impact R- or S-warfarin metabolism (or both) to influence drug response. Nevertheless, in vitro studies for CYP2C19 have been limited to identifying metabolites from these reactions [8] and establishing R-8-hydroxywarfarin as a biomarker for the enzyme [16]. The relative efficiencies of CYP2C19 metabolism of R- and S-warfarin and their overall contributions to drug clearance have not been reported. This information is potentially crucial for explaining variations in the significance of CYP2C19 in patient responses to therapy.
As a step toward resolving this controversy, we employed in vitro systems to assess the efficiencies for CYP2C19 metabolic pathways for R- and S-warfarin and determine their significance among all oxidative metabolic pathways present in the liver. We measured steady-state kinetic parameters for recombinant CYP2C19 toward R- and S-warfarin using an UPLC based method developed by our group [17]. We then assessed the importance of CYP2C19 in R- and S-warfarin metabolism relative to other hepatic P450s present through correlative activity studies using human liver microsomes. Based on previous studies by others [8, 18], some metabolic pathways for R-warfarin are likely shared between CYP2C19 and CYP1A2. Consequently, we employed common marker reactions to assess their respective activities in the human liver microsomes. For CYP2C19, we used 4′-hydroxylation of S-mephenytoin [19, 20] and for CYP1A2 O-deethylation of phenacetin [21, 22]. These P450-specific activities were correlated with the formation of each R- and S-warfarin hydroxywarfarin metabolite. Based on Pearson correlation analyses, it was possible to predict the relative significance of CYP2C19 and CYP1A2 contributions to each metabolic pathway for R- and S-warfarin in the liver.
RESULTS
Recombinant CYP2C19 Metabolism of R-warfarin
For R-warfarin, CYP2C19 produced four of the commonly observed hydroxywarfarin metabolites but not 10-hydroxywarfarin (Figs. 1 and 2, Panel A). The maximal rate of turnover (Vmax) for the R-7-hydroxywarfarin reaction was the highest, while the slowest rates were observed for the formation of R-8- and 4′-hydroxywarfarin, as shown in Table 1. The corresponding Km values were comparable for generating R-6-, 7-, and 8-hydroxywarfarin, but the one for 4′-hydroxywarfarin was about two-fold higher. Collectively, metabolic efficiencies (Vmax/Km) were in order of decreasing magnitude: R-7-hydroxywarfarin > 6-hydroxywarfarin = 8-hydroxywarfarin.
Fig. (2).

Steady-state metabolism of R- and S-warfarin by recombinant CYP2C19. Data are shown for reactions with R-warfarin (A) and S-warfarin (B) in which 50 nM CYP2C19 incubated with 7.8 to 500 μM substrate at 37°C and pH 7.4 as described in Materials and Methods. Kinetic profiles for the formation of each product of the reactions are as follows: squares (∎) for 6-hydroxywarfarin, triangles (▴) for 7-hydroxywarfarin, inverted triangles (▾) for 8-hydroxywarfarin, and diamonds (◆) for 4′-hydroxywarfarin. Reported values represent three experiments performed in duplicate. Data were fit to the Michaelis-Menten mechanism using GraphPad Prism 5® software (La Jolla, CA).
Table 1.
Kinetic Parameters of R- and S-warfarin Metabolism by Recombinant CYP2C19a
| Producta | R-warfarin | S-warfarin | ||||
|---|---|---|---|---|---|---|
| Vmaxb | Km (μM) |
Vmax/Km (× 103)c |
Vmaxb | Km (μM) |
Vmax/Km (× 103)c |
|
| 6-hydroxywarfarin | 0.34 (0.32-0.38) |
20 (12-27) |
17 | 0.40 (0.34-0.45) |
33 (17-49) |
12 |
| 7-hydroxywarfarin | 0.55 (0.49-0.60) |
14 (7.2-21) |
39 | 0.50 (0.47-0.53) |
32 (24-40) |
16 |
| 8-hydroxywarfarin | 0.21 (0.20-0.22) |
17 (13-21) |
12 | 0.31 (0.28-0.34) |
17 (9.0-26) |
18 |
| 4′-hydroxywarfarin | 0.18 (0.16-0.20) |
39 (23-56) |
4.6 | 0.17 (0.13-0.20) |
83 (31-135) |
2.0 |
95% confidence intervals are shown in parentheses
Units are nmol/min/nmole P450
Units are per min per μM
Recombinant CYP2C19 Metabolism of S-warfarin
CYP2C19 metabolized S-warfarin into the same four hydroxywarfarin metabolites as observed for R-warfarin (Figs 1 and 2, Panel B). Vmax was highest for formation of S-7-hydroxywarfarin followed by S-6-, 8-, and then 4′-hydroxywarfarin in decreasing order (Table 1). The corresponding Km values were lowest for the S-8-hydroxywarfarin reaction, intermediate for S-6- and 7-hydroxywarfarin, and highest for S-4′-hydroxywarfarin. These differences in kinetic parameters resulted in similar metabolic efficiencies (Vmax/Km) for S-7- and 8-hydroxywarfarin but lower values for S-6-hydroxywarfarin and especially S-4′-hydroxywarfarin. Compared to R-warfarin, overall efficiency of S-warfarin metabolism was only about two-fold less than that observed for R-warfarin.
Metabolism of R- and S-warfarin Metabolism by Human Liver Microsomes
For correlative studies, we determined the specific activity for nine human liver microsomes to oxidize R- and S-warfarin to the corresponding hydroxywarfarin metabolites. When either form of the drug served as a substrate, microsomal reactions generated 6-, 7-, 8-, 10-, and 4′-hydroxywarfarin, although there was significant variation in their rates of formation. For R-warfarin, the rates of R-10-hydroxywarfarin formation were the highest in most cases and those for R-8-hydroxywarfarin the lowest. The range and median rates in nmol/min/mg protein for R-warfarin metabolic pathways, respectively, were as follows: R-6-hydroxywarfarin, 0.028-0.067, 0.041; R-7-hydroxywarfarin, 0.016-0.057, 0.033; R-8-hydroxywarfarin, 0.0053-0.023, 0.0096; R-10-hydroxywarfarin, 0.055-0.19, 0.13; and R-4′-hydroxywarfarin, 0.028-0.057, 0.042. For S-warfarin reactions, rates of S-7-hydroxywarfarin formation exceeded those for other metabolites followed by S-10-hydroxywarfarin, while the lowest rates were observed for S-4′-hydroxywarfarin. The range and median rates in nmol/min/mg protein for S-warfarin metabolic pathways, respectively, were as follows: S-6-hydroxywarfarin, 0.11-0.20, 0.15; S-7-hydroxywarfarin, 0.51-0.77, 0.54; S-8-hydroxywarfarin, 0.085-0.27, 0.15; S-10-hydroxywarfarin, 0.086-1.3, 0.24; and S-4′-hydroxywarfarin, 0.051-0.12, 0.087. Overall metabolism of R-warfarin was less efficient than that for the S enantiomer.
Correlative Activity Studies Between S-mephenytoin and Warfarin as Substrates
We correlated rates of R- and S-warfarin metabolism to CYP2C19 S-mephenytoin activity present in human liver microsomes to assess the importance of CYP2C19 in warfarin metabolism relative to other hepatic P450s. For S-mephenytoin reactions, rates varied from no activity to 67 nmol/min/mg protein with a median value of 30. As shown in Table 2, S-mephenytoin activity positively correlated with formation of R-6-, 7-, and especially 8-hydroxywarfarin based on a significant Pearson Correlation P values, i.e. <0.007 in all cases. Even though CYP2C19 metabolizes S-warfarin, none of those metabolic pathways catalyzed by HLMs correlated with CYP2C19 S-mephenytoin activity. Consequently, CYP2C19 likely plays an important role in hepatic metabolism of R-warfarin and not S-warfarin.
Table 2.
Pearson Correlation Values Between R- and S-hydroxywarfarin Formation Rates and P450 Activities as Measured with a Bank of Nine Human Liver Microsomal Samples (See Figs. 3 and 4)a, b
| P450 isoform |
Probe | Drug | 6-Hydroxywarfarin | 7-Hydroxywarfarin | 8-Hydroxywarfarin | 10-Hydroxywarfarin | 4′-Hydroxywarfarin | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R value | P value | R value | P value | R value | P value | R value | P value | R value | P value | |||
| CYP1A2 | PHEN | R-WAR S-WAR |
0.66 0.66 |
0.056 0.054 |
0.67 0.50 |
0.049* 0.17 |
0.45 0.16 |
0.22 0.67 |
0.40 0.063 |
0.29 0.87 |
−0.44 −0.023 |
0.24 0.95 |
| CYP2C19 | MEPH | R-WAR S-WAR |
0.91 0.37 |
0.0006*** 0.32 |
0.82 0.28 |
0.0069** 0.46 |
0.90 −0.045 |
0.0009*** 0.91 |
−0.020 −0.98 |
0.96 0.80 |
−0.50 −0.46 |
0.17 0.22 |
PHEN, phenacetin; MEPH, S-mephenytoin; WAR, warfarin
Asterisks indicate statistically significant correlations
Correlative Activity Studies Between Phenacetin and Warfarin as Substrates
Based on our findings and those by others, CYP1A2 and CYP2C19 share similar metabolic pathways for R-warfarin, and thus we assessed their relative significance in R- and S-warfarin metabolism when both are present in HLMs. Specifically, we carried out correlative studies between warfarin metabolism and CYP1A2 phenacetin activity using human liver microsomes. For phenacetin reactions, rates varied from 14 to 82 pmol/min/mg protein with a median value of 46. As shown in, Table 2, CYP1A2 phenacetin activity correlated weakly with R-6-hydroxywarfarin (P 0.056) and 7-hydroxywarfarin (P 0.049). These findings indicate CYP1A2 is a major contributor to only two metabolic pathways for these drug enantiomers.
DISCUSSION
We assessed the efficiency of CYP2C19 metabolic pathways toward R- and S-warfarin and explored their possible contributions in the liver using in vitro methods. Based on steady-state studies with recombinant enzyme, CYP2C19 is a high affinity, but non-selective catalyst toward hydroxylation of R- and S-warfarin. Subsequent correlative activity studies with human liver microsomes indicated that these properties make CYP2C19 competitive in metabolizing R-warfarin and even S-warfarin under certain circumstances. Taken together, these findings advance an understanding of the potential role for CYP2C19 in warfarin metabolism and provide insights that may explain conflicting reports from genetic studies on the clinical relevance of CYP2C19.
Based on our steady-state studies, recombinant CYP2C19 demonstrated broad regiospecificity and low enantio-selectivity toward R- and S-warfarin. Both forms of the drug underwent oxidation at the 6, 7, 8, and 4′ positions (Figs. 1 and 2). Their relative levels at saturating substrate concentration (500 μM) yielded the same pattern observed in a previous study carried out a single warfarin concentration (2.9 mM) using recombinant CYP2C19 present in yeast microsomes [8]. Oxidation at positions 6, 7, and 8 on the coumarin ring requires only minor changes in the binding mode of the substrate, because these sites are proximal to one another. This is not the case for oxidation at the 4′ position, whereby warfarin must bind in the opposite orientation for catalysis to occur. The metabolic efficiencies, and hence underlying energetics, of these reactions are relatively similar, albeit slightly less so for the production of 4′-hydroxywarfarin. Nevertheless, the collective oxidation of coumarin ring far exceeds that for the phenyl ring of the warfarin molecule. The chiral center had minimal effect on the efficiency of warfarin oxidation. Metabolism of R-warfarin was only at best two-fold more efficient than that for S-warfarin. Taken together, CYP2C19 is not a very selective catalyst in warfarin metabolism.
The Km value for the R-8-hydroxywarfarin reaction contrasts significantly with that reported in the literature. An early effort to characterize CYP2C19 metabolism of R-warfarin to R-8-hydroxywarfarin employed human liver microsomes pre-treated with furafylline to inactivate CYP1A2 and thus minimize its contributions to the reactions [16]. Three different pre-treated human liver microsomal preparations yielded a Km for the R-8-hydroxywarfarin metabolic pathway ranging from 289 to 395 μM. These values are 20-fold higher than we report for recombinant CYP2C19, and thus implicate a much less efficient route of metabolism of R-warfarin by this enzyme than reported here. A source for these differences may lie in the design of the experiments. We employed supersomes containing only CYP2C19 as the P450 capable of metabolizing R-warfarin. While pre-treatment of microsomes with furafylline inactivates CYP1A2, other P450s, e.g. CYP1A1 [18], present in microsomal reactions may contribute to R-warfarin metabolism and thus compromise the ability to accurately estimate kinetic parameters for the CYP2C19 reaction. Furafylline pre-treatment of microsomes may also induce unexpected consequences that impact CYP2C19 activity, which was not explored in the original study. It is important to further note that the authors did not extend their study to other metabolic pathways for R-warfarin or any of those for S-warfarin. The limited scope of the original study was presumably due to the contributions of multiple other P450s to the reactions, which would confound those analyses. Consequently, the design of our study provides the first extensive characterization of the capacity of CYP2C19 to metabolize R- and S-warfarin.
CYP2C9 and CYP2C19 are members of the same P450 family and share 91% sequence identify; however, their limited sequence differences have significant consequences on warfarin metabolism. Whereas CYP2C19 slightly favors R-warfarin as a substrate, CYP2C9 demonstrates an almost 1000-fold higher metabolic efficiency toward S-warfarin than R-warfarin [23]. CYP2C9 is essentially only an S-warfarin hydroxylase and a more efficient one than CYP2C19. The CYP2C9 kinetic parameters toward S-warfarin reveal a higher affinity for substrate (Km about 2.8 μM) but somewhat lower overall turnover rate (0.056 nmol/min/nmol P450 for S-6-hydroxywarfarin and 0.068 nmol/min/nmol P450 for S-7-hydroxywarfarin, respectively) [24]. CYP2C9 metabolic efficiency toward S-warfarin is then higher than that for CYP2C19 and more important at low S-warfarin levels based on relative Km values.
Despite metabolism of both drug enantiomers, only contributions of CYP2C19 to R-warfarin metabolism seem significant in microsomal liver preparations. S-Warfarin metabolism is presumably dominated by CYP2C9 [25]. Rates of R-6-, 7-, and 8-hydroxywarfarin formation correlated significantly with CYP2C19 activity as measured by S-mephenytoin activity. A similar correlation was reported previously for R-8-hydroxywarfarin formation using human liver microsomes pre-treated with furafylline [16]. Modification of the microsomes was deemed necessary to inactivate CYP1A2 and thus prevent its contribution to the observed rate for R-8-hydroxywarfarin formation and facilitate correlative studies for CYP2C19. Our studies demonstrated that this step was not required to effectively show CYP2C19 is responsible for generation of R-8-hydroxywarfarin as well as R-6- and 7-hydroxywarfarin. CYP1A2 activity as measured by O-dealkylation also correlated, albeit weakly, with formation of R-6- and 7-hydroxywarfarin, indicating those metabolites are not suitable biomarkers for CYP2C19 activity due to ambiguity in their origination in R-warfarin metabolism. CYP2C19 activity correlated more strongly than CYP1A2 activity with R-7-hydroxywarfarin, suggesting CYP2C19 may be the more dominant enzyme for that pathway. Lack of a correlation for the rate of R-4′-hydroxywarfarin formation is likely due to the contribution of multiple P450s to this pathway, such as CYP2C8 [8] and CYP3A4 [9-11], which would confound this type of analysis.
Correlation between CYP1A2 activity and rates of R-6-hydroxywarfarin was expected based on reported in vitro studies; however, the findings for R-7-hydroxywarfarin were surprising. Recombinant CYP1A2 metabolized R-warfarin into R-6- and 8-hydroxywarfarin, but not R-7-hydroxywarfarin [18]. Nevertheless, anti-CYP1A2 antibodies inhibited the formation of R-7-hydroxywarfarin by human liver microsomes [26]. Other metabolic pathways were not explored in that study. The results from those studies as well as our observations suggest CYP1A2 activity toward R-warfarin may depend on whether it is in the recombinant form versus present in a microsomal fraction among multiple other P450s and proteins. Further studies are necessary to validate the possible dependency of CYP1A2 activity toward warfarin on the expression system.
Based on our findings, we can explore the possible impact of CYP2C19 on warfarin metabolism in humans. Bioavailability of warfarin will drive binding interactions to P450s responsible for its metabolism. The median plasma levels for R- and S-warfarin for 36 patients in one study were 0.48 mg/mL (1.6 μM) and 0.87 mg/mL (2.8 μM) respectively [27]. However, only 0.53% of the drugs were not bound to sera proteins, such that actual levels capable of participating in metabolism were 0.015 μM for R-warfarin and 0.0085 μM for S-warfarin. Under these low substrate concentrations, the highest affinity enzymes (i.e. lowest Km) will dominate metabolic pathways for the drugs. In following, the P450 with the lowest Km for S-warfarin, CYP2C9, is responsible for metabolizing about 80% of the drug [7] indicating a possible minor role for CYP2C19 in most patients. This difference in contribution between the enzymes may not be the same in patients possessing CYP2C9 polymorphisms. CYP2C9*2, *3, and *5 variants display higher Km and lower Vmax values relative to wild type CYP2C9*1, such that their metabolic properties are competitive or poorer than CYP2C19 [23, 28, 29]. Consequently, CYP2C19 may significantly contribute to metabolism of S-warfarin in those patients.
Although R-warfarin is a less potent anticoagulant than S-warfarin, its accumulation in patients may contribute to patient response during therapy [30] and thus its metabolism may have clinical relevance. R-10-Hydroxywarfarin is the major metabolite in clinical samples from patients undergoing warfarin therapy [17, 31] and studies described here using human liver microsomes. These findings support the role of CYP3A4, which is solely responsible for generating R-10-hydroxywarfarin, as a major metabolizer of R-warfarin. CYP3A4 displays a comparable affinity toward R-warfarin as CYP2C19 (Km ~30 μM versus Km ~20 μM) [9]. In following, it is conceivable that CYP2C19 and CYP3A4 compete with one another for R-warfarin during metabolism.
In sum, CYP2C19 can contribute to the metabolism of both R- and S-warfarin; however, the relative clinical importance of CYP2C19 and its polymorphisms in pharmacogenetic studies may depend on other factors, such as other P450 polymorphisms and/or competition with other enzymes. These complexities may underlie why CYP2C19 polymorphisms lead to warfarin sensitivity in some patients [12, 13], but not others [14, 15]. Advances in the profiling of R- and S-warfarin and their metabolites in patients undergoing anticoagulant therapy may directly assess drug metabolism [6, 17] and therefore provide further evidence to help resolve the controversial importance of CYP2C19 in patient response to anticoagulant therapy with warfarin.
MATERIALS AND METHODS
Materials
Ethyl alcohol, perchloric acid, and other general chemicals were purchased from Thermo Fisher Scientific (Waltham, MA). R- and S- warfarin, 7-hydroxycoumarin, mephenytoin, 4′-hydroxymephenytoin, 2,4-dimethylphenol, phenacetin, acetaminophen, and 2-acetamidophenol were obtained from Sigma (St. Louis, MO). All hydroxywarfarins were purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). The following human recombinant CYP2C19 and human liver microsomes were purchased from BD Biosciences (San Jose, CA): HH13, HG03, HG74, HG93, HK37, HLM150, CMV negative pooled, male pooled, female pooled.
Steady-state Metabolism of R- and S-warfarin
For these studies, we reconstituted 50 nM of human recombinant CYP2C19 (BD Biosciences) with 7.8 to 500 μM R-, S-warfarin in 50 mM potassium phosphate pH 7.4 at 37°C. The mixtures were preincubated for 5 min at the reaction temperature. Reactions were initiated by the addition of NADPH regenerating system (final concentration: 1 mM NADP+, 3 mM glucose-6-phosphate, 3 mM MgCl2, and 1 U glucose-6-phosphate dehydrogenase) and quenched after 15 min with an equal volume of 0.4 N perchloric acid containing the 20 μM of internal standard (7-hydroxycoumarin). The resulting quenched reactions were clarified by centrifugation and supernatants recovered for subsequent UPLC analyses. Each set of steady-state reactions was performed in duplicate and replicated over multiple days.
UPLC Analysis of Warfarin Reactions
We employed a rapid UPLC method for resolving and quantitating hydroxywarfarins generated by these experiments [17]. In brief, we employed an Acquity UPLC® (Waters, Milford, MA) system coupled to a TSQ-Quantum Ultra triple-quadrupole mass analyzer (ThermoFinnigan, San Jose, CA) that was equipped with a 2.1 mm × 150 mm Acquity BEH C18 1.7 μm particle column (Waters) operating at 40°C. A gradient method using H2O (0.01% formic acid v/v) (A) and methanol (B) was used to elute the warfarin and hydroxywarfarins at a flow rate of 250 μL/min. All compounds were detected in selected reaction monitoring mode (SRM) using the m/z transitions described [17].
Determination of Steady-state Kinetic Parameters
All metabolites were normalized relative to the internal standard and quantified relative to authentic standards. Kinetic parameters Km and Vmax were obtained by fitting the data to the Michaelis-Menten equation using GraphPad Prism 5® software (La Jolla, CA).
Assessing R- and S-warfarin Metabolism by Human Liver Microsomes
We assessed the activities of an array of human liver microsomes toward R- and S-warfarin for correlative studies determining the relative contribution of CYP2C19 to their metabolism. The reaction mixture contained 50 mM Tris-HCl (pH 7.7 at 25°C or pH 7.4 at 37°C), 8 mM MgCl2, 25 μg/mL alamethicin, 5 mM saccharolactone, 25 μM R- or S-warfarin, and 2 mg/mL human liver microsomes. Alamethicin and saccharolactone are reagents that facilitate UDP-glucuronosyltransferase (UGT) reactions and are included in a general buffer used by our group to phenotype activities for P450s, UGTs, and their combined activities. In preliminary studies, we demonstrated that metabolism of either 25 μM R- or S-warfarin by HLM150 was not affected by alamethicin and saccharolactone (Table 3), and thus their inclusion would not have impacted the results from the correlative studies. In addition, samples were not pre-reacted with furafylline to inactivate CYP1A2 as reported by others [16]. The final reaction mixtures were preincubated for 5 min at 37°C and then initiated upon addition of NADPH to a final concentration of 1 mM. The quench, work up and analysis of reaction samples were identical as that described previously for the reactions with recombinant enzyme. Experiments were carried out in triplicate for each HLM sample.
Table 3.
Impact of Alamethicin and Saccharolactone on HLM150 Specific Activity Toward R- and S-warfarin used in Correlative Studiesa
| Substrate | Reaction Adjunct | 6-Hydroxywarfarin | 7-Hydroxywarfarin | 8-Hydroxywarfarin | 10-Hydroxywarfarin | 4′-Hydroxywarfarin |
|---|---|---|---|---|---|---|
| R-warfarin | absence presence |
0.0362 (0.0017) 0.0342 (0.0022) |
0.0389 (0.0019) 0.0391 (0.0021) |
0.0105 (0.0023) 0.0095 (0.0013) |
0.113 (0.003) 0.121 (0.007) |
0.0444 (0.032) 0.0382 (0.027) |
| S-warfarin | absence presence |
0.183 (0.005) 0.179 (0.006) |
0.770 (0.018) 0.785 (0.015) |
0.269 (0.014) 0.271 (0.012) |
0.226 (0.010) 0.216 (0.013) |
0.0745 (0.0027) 0.0727 (0.0022) |
Units for observed rates are nmol/min/mg protein. Standard deviation for average of values are shown in parentheses.
Phenotyping CYP2C19 Activity using a S-mephenytoin Hydroxylation Assay
We measured rates of S-mephenytoin 4′-hydroxylation by human liver microsomes as a measure of CYP2C19 activity [19, 20]. The reaction mixture contained 50 mM Tris-HCl (pH 7.7 at 25°C or pH 7.4 at 37°C), 8 mM MgCl2, 25 μg/mL alamethicin, 5 mM saccharolactone, 40 μM Smephenytoin, and 2 mg/mL human liver microsomes. Preliminary studies demonstrated that metabolism of 40 μM S-mephenytoin by HLM150 was not affected by alamethicin and saccharolactone (17.7 ± 0.8 nmol/min/mg protein absence versus 16.5 ± 1.2 nmol/min/mg protein presence), and thus their inclusion would not have impacted the results from the correlative studies. Reaction mixtures were preincubated for 5 min at 37°C. Unlike efforts by others, [16] samples were not pre-reacted with furafylline to inactivate CYP1A2. The final reactions were initiated upon addition of NADPH to a final concentration of 1 mM. The reaction was incubated for 30 min at 37°C, and quenched with a solution containing 0.1 % formic acid in methanol and an internal standard (10 μM 2,4-dimethylphenol). The quenched reactions were centrifuged, and the supernatant transferred to a vial for analysis by HPLC.
Samples were injected onto a 4.6 × 150 mm Zorbax Eclipse 5 μm XDB-C18 column (Agilent) at 40°C using a Waters Breeze HPLC system. The mobile phase consisted of 0.1% acetic acid in water (A) and 100% methanol (B) and was delivered at a flow rate of 1.2 mL/min. A gradient elution program was used beginning with 60% A and 40% B held for the first 0.5 min followed by a linear gradient from 40% B to 100% B (0.5 - 5 min), 100% B maintained for 2 min, and 100% B to 40% B (7 – 10 min) with 40% B maintained for 1 min. The elution of mephenytoin, 4′-hydroxymephenytoin, and 2,4-dimethylphenol were monitored at 230 nm, and the amount of 4′-hydroxymephenytoin generated in the reaction determined relative to standards after correction with the internal standard. The rate for the corresponding activity was calculated as the amount of 4-hydroxymephenytoin produced during the 15 min reactions. Final activity values reflected the average of three experimental replicates.
Phenotyping CYP1A2 Activity using a Phenacetin Odealkylation Assay
We measured rates of phenacetin O-dealkylation by human liver microsomes as a measure of CYP1A2 activity [21]. The reaction included 50 mM Tris-HCl (pH 7.7 at 25°C or pH 7.4 at 37°C), 8 mM MgCl2, 25 μg/mL alamethicin, 5 mM saccharolactone, and 0.5 mg/mL HLM150s. A stock solution of phenacetin (160 μM) was prepared in methanol and diluted into the enzyme reaction (final 40 μM). This concentration of phenacetin was chosen to minimize contributions of lower affinity P450s to the reaction [21], and thus maximize CYP1A2 contributions to activity present in microsomes. Preliminary studies demonstrated that metabolism of 40 μM phenacetin by HLM150 was not affected by alamethicin and saccharolactone (0.0376 ± 0.0028 nmol/min/mg protein absence versus 0.0367 ± 0.0015 nmol/min/mg protein presence), and thus their inclusion would not have impacted the results from the correlative studies. The reaction mixtures were preincubated for 5 min at 37°C and reactions initiated upon addition of 1 mM NADPH (final). After 15 min, each reaction was quenched with a solution containing 0.1 % formic acid in methanol and an internal standard (4 μM 2-acetamidophenol). Quenched reactions were centrifuged, and the supernatant transferred to a vial for analysis by HPLC.
Samples were injected on 4.6 × 150 mm Zorbax Eclipse 5 μm XDB-C18 column (Agilent) at 40 °C using a Waters Breeze HPLC system. The mobile phase consisted of 0.1% acetic acid in water (A) and 100% methanol (B) was delivered at a flow rate of 1.2 mL/min. A gradient elution program was used beginning with 70% A and 30% B held for the first 0.5 min followed by a linear gradient from 30% B to 100% B (0.5 – 3.5 min), 100% B maintained for 1.5 min, and 100% B to 30% B (5 – 8 min) with 30% B maintained for 1 min. The elution of phenacetin, acetaminophen, and 2-acetamidophenol were monitored at 254 nm, and the amount of acetaminophen generated in the reaction determined relative to standards after correction with the internal standard. The rate for the corresponding activity was calculated as the amount of acetaminophen produced during the 15 min reactions. Final activity values reflected the average of three experimental replicates.
Correlation of Phenotyping Data and Statistical Analyses
Statistical analyses of the correlation data were conducted using GraphPad Prism 5® software (La Jolla, CA). A P value less than or equal to 0.05 was considered statistically significant. Phenotypic marker activities for CYP1A2 and CYP2C19 were correlated against formation rates of all five possible hydroxywarfarins derived from R- or S-warfarin using Pearson correlation analyses.
ACKNOWLEDGEMENTS
Grover P. Miller was supported by a pilot study grant from NIH UL1 TR000039 and a bridging grant from the University of Arkansas for Medical Sciences. So-Young Kim, Ji-Yeon Kang, and Sun-Ha Park were supported by Chonnam National University Second Stage BK21 Project from the Ministry of Education, Sciences, & Technology of the Republic of Korea. Gunnar Boysen was supported by NIH grant R21ES019684 and the Arkansas Tobacco Settlement Proceeds Act of 2000.
Footnotes
CONFLICT OF INTEREST The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- [1].Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2008;133:160S–98S. doi: 10.1378/chest.08-0670. [DOI] [PubMed] [Google Scholar]
- [2].Linkins L, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med. 2003;139:893–900. doi: 10.7326/0003-4819-139-11-200312020-00007. [DOI] [PubMed] [Google Scholar]
- [3].Schulman S. Care of Patients Receiving Long-Term Anticoagulant Therapy. N Engl J Med. 2003;349:675–683. doi: 10.1056/NEJMcp025373. [DOI] [PubMed] [Google Scholar]
- [4].Eikelboom J, Hankey GJ. The beginning of the end of warfarin? Med J Aust. 2004;180:548–51. doi: 10.5694/j.1326-5377.2004.tb06088.x. [DOI] [PubMed] [Google Scholar]
- [5].Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73:67–74. doi: 10.1016/s0163-7258(96)00140-4. [DOI] [PubMed] [Google Scholar]
- [6].Jones DR, Miller GP. Assays and applications in warfarin metabolism: what we know, how we know it and what we need to know. Expert Opin Drug Metab Toxicol. 2011;7:857–74. doi: 10.1517/17425255.2011.576247. [DOI] [PubMed] [Google Scholar]
- [7].Takahashi H, Kashima T, Nomoto S, Iwade K, Tainaka H, Shimizu T, Nomizo Y, Muramoto N, Kimura S, Echizen H. Comparisons between in-vitro and in-vivo metabolism of (S)-warfarin: catalytic activities of cDNA-expressed CYP2C9, its Leu359 variant and their mixture versus unbound clearance in patients with the corresponding CYP2C9 genotypes. Pharmacogenetics. 1998;8:365–373. doi: 10.1097/00008571-199810000-00001. [DOI] [PubMed] [Google Scholar]
- [8].Kaminsky L, de Morais S, Faletto M, Dunbar D, Goldstein J. Correlation of human cytochrome P4502C substrate specificities with primary structure: warfarin as a probe. Mol Pharmacol. 1993;43:234–239. [PubMed] [Google Scholar]
- [9].Jones DR, Kim SY, Boysen G, Yun CH, Miller GP. Contribution of Three CYP3A Isoforms to Metabolism of R- and S-Warfarin. Drug Metab Lett. 2010;4:213–9. doi: 10.2174/187231210792928242. [DOI] [PubMed] [Google Scholar]
- [10].Rettie A, Korzekwa KR, Kunze KL, Lawrence RF, Eddy AC, Aoyama T, Gelboin HV, Gonzalez FJ, Trager WF. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem Res Toxicol. 1992;5:54–9. doi: 10.1021/tx00025a009. [DOI] [PubMed] [Google Scholar]
- [11].Ngui JS, Chen Q, Shou M, Wang RW, Stearns RA, Baillie TA, Tang W. In Vitro Stimulation of Warfarin Metabolism by Quinidine: Increases in the Formation of 4′- and 10-Hydroxywarfarin. Drug Metab Dispos. 2001;29:877–886. [PubMed] [Google Scholar]
- [12].Jorgensen AL, Al-Zubiedi S, Zhang JE, Keniry A, Hanson A, Hughes DA, Eker D, Stevens L, Hawkins K, Toh CH, Kamali F, Daly AK, Fitzmaurice D, Coffey A, Williamson PR, Park BK, Deloukas P, Pirmohamed M. Genetic and environmental factors determining clinical outcomes and cost of warfarin therapy: a prospective study. Pharmacogenet Genomics. 2009;19:800–12. doi: 10.1097/FPC.0b013e3283317ab5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee S, Hwang HJ, Kim JM, Chung CS, Kim JH. CYP2C19 polymorphism in Korean patients on warfarin therapy. Arch Pharm Res. 2007;30:344–9. doi: 10.1007/BF02977616. [DOI] [PubMed] [Google Scholar]
- [14].Obayashi K, Nakamura K, Kawana J, Ogata H, Hanada K, Kurabayashi M, Hasegawa A, Yamamoto K, Horiuchi R. VKORC1 gene variations are the major contributors of variation in warfarin dose in Japanese patients. Clin Pharmacol Ther. 2006;80:169–78. doi: 10.1016/j.clpt.2006.04.010. [DOI] [PubMed] [Google Scholar]
- [15].Uno T, Sugimoto K, Sugawara K, Tateishi T. The effect of CYP2C19 genotypes on the pharmacokinetics of warfarin enantiomers. J Clin Pharm Ther. 2008;33:67–73. doi: 10.1111/j.1365-2710.2008.00887.x. [DOI] [PubMed] [Google Scholar]
- [16].Wienkers LC, Wurden CJ, Storch E, Kunze KL, Rettie AE, Trager WF. Formation of (R)-8-hydroxywarfarin in human liver microsomes: a new metabolic marker for the (S)-mephenytoin hydroxylase, P4502C19. Drug Metab Dispos. 1996;24:610–614. [PubMed] [Google Scholar]
- [17].Jones DR, Boysen G, Miller GP. Novel multi-mode ultra performance liquid chromatography-tandem mass spectrometry assay for profiling enantiomeric hydroxywarfarins and warfarin in human plasma. J Chromatogr B. 2011;879:1056–1062. doi: 10.1016/j.jchromb.2011.03.022. [DOI] [PubMed] [Google Scholar]
- [18].Zhang Z, Fasco MJ, Huang Z, Guengerich FP, Kaminsky LS. Human cytochromes P450 1A1 and 1A2: R-warfarin as a probe. Drug Metab Dispos. 1995;23:1339–1345. [PubMed] [Google Scholar]
- [19].Wrighton SA, Stevens JC, Becker GW, VandenBranden M. Isolation and characterization of human liver cytochrome P450 2C19: correlation between 2C19 and S-mephenytoin 4′-hydroxylation. Arch Biochem Biophys. 1993;306:240–245. doi: 10.1006/abbi.1993.1506. [DOI] [PubMed] [Google Scholar]
- [20].Goldstein JA, Faletto MB, Romkes-Sparks M, Sullivan T, Kitareewan S, Raucy JL, Lasker JM, Ghanayem BI. Evidence that CYP2C19 is the major (S)-mephenytoin 4′-hydroxylase in humans. Biochemistry. 1994;33:1743–1752. doi: 10.1021/bi00173a017. [DOI] [PubMed] [Google Scholar]
- [21].Distlerath LM, Reilly PEB, Martin MV, Davis GG, Wilkinson GR, Guengerich FP. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1985;260:9057–9067. [PubMed] [Google Scholar]
- [22].Pantuck EJ, Hsiao K-C, Maggio A, Nakamura K, Kuntzman R, Conney AH. Effect of cigarette smoking on phenacetin metabolism. Clin Pharmacol Ther. 1974;15:9–17. doi: 10.1002/cpt19741519. [DOI] [PubMed] [Google Scholar]
- [23].Sullivan-Klose TH, Ghanayem BI, Bell DA, Zhang ZY, Kaminsky LS, Shenfield GM, Miners JO, Birkett DJ, Goldstein JA. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics. 1996;6:341–349. doi: 10.1097/00008571-199608000-00007. [DOI] [PubMed] [Google Scholar]
- [24].Jones D, Kim SY, Guderyon M, Yun CH, Moran JH, Miller GP. Hydroxywarfarin Metabolites Potently Inhibit CYP2C9 Metabolism of S-Warfarin. Chem. Res. Toxicol. 2010 doi: 10.1021/tx1000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rettie AE, Korzekwa KR, Kunze KL, Lawrence RF, Eddy AC, Aoyama T, Gelboin HV, Gonzalez FJ, Trager WF. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem Res Toxicol. 1992;5:54–9. doi: 10.1021/tx00025a009. [DOI] [PubMed] [Google Scholar]
- [26].Yamazaki H, Shimada T. Human liver cytochrome P450 enzymes involved in the 7-Hydroxylation of R- and S-Warfarin Enantiomers. Biochem Pharmacol. 1997;54:1195–1203. doi: 10.1016/s0006-2952(97)00304-3. [DOI] [PubMed] [Google Scholar]
- [27].Chan E, McLachlan AJ, Pegg M, MacKay AD, Cole RB, Rowland M. Disposition of warfarin enantiomers and metabolites in patients during multiple dosing with rac-warfarin. Br J Clin Pharmacol. 1994;37:563–9. doi: 10.1111/j.1365-2125.1994.tb04305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Haining RL, Hunter AP, Veronese ME, Trager WF, Rettie AE. Allelic variants of human cytochrome P450 2C9: baculovirus-mediated expression, purification, structural characterization, substrate stereoselectivity, and prochiral selectivity of the wild-type and I359L mutant forms. Arch Biochem Biophys. 1996;333:447–458. doi: 10.1006/abbi.1996.0414. [DOI] [PubMed] [Google Scholar]
- [29].Dickmann LJ, Rettie AE, Kneller MB, Kim RB, Wood AJJ, Stein CM, Wilkinson GR, Schwarz UI. Identification and functional characterization of a new CYP2C9 variant (CYP2C9*5) expressed among African Americans. Mol Pharmacol. 2001;60:382–387. doi: 10.1124/mol.60.2.382. [DOI] [PubMed] [Google Scholar]
- [30].Breckenridge A, Orme M, Wesseling H, Lewis RJ, Gibbons R. Pharmacokinetics and pharmacodynamics of the enantiomers of warfarin in man. Clin Pharmacol Ther. 1974;15:424–30. doi: 10.1002/cpt1974154424. [DOI] [PubMed] [Google Scholar]
- [31].Locatelli I, Kmetec V, Mrhar A, Grabnar I. Determination of warfarin enantiomers and hydroxylated metabolites in human blood plasma by liquid chromatography with achiral and chiral separation. J Chromatogr B. 2005;818:191–198. doi: 10.1016/j.jchromb.2004.12.024. [DOI] [PubMed] [Google Scholar]