

Abstract
Ndfip1 is an adaptor for the E3 ubiquitin ligase Itch. Both Ndfip1- and Itch-deficient T cells are biased towards TH2 cytokine production. Here we demonstrate that lungs from Ndfip1−/− mice showed increased numbers of neutrophils and TH17 cells. This was not because Ndfip1−/− T cells are biased towards Th17 differentiation. In fact, fewer Ndfip1−/− T cells differentiated into TH17 cells in vitro due to high IL-4 production. Rather, TH17 differentiation was increased in Ndfip1−/− mice due to increased numbers of IL-6 producing eosinophils. IL-6 levels in mice that lacked both Ndfip1 and IL-4 were similar to WT controls and these mice had fewer TH17 cells in their lungs. These results indicate that TH2 inflammation, such as that observed in Ndfip1−/− mice, can increase TH17 differentiation by recruiting IL-6 producing eosinophils into secondary lymphoid organs and tissues. This may explain why TH17 cells develop within an ongoing TH2 inflammatory response.
INTRODUCTION
CD4 T cells can differentiate into different helper (TH) subsets according to cues from their immediate cytokine environment. TH17 cells are a distinct group of CD4 TH cells that have been shown to protect against specific bacterial and fungal pathogens by producing pro-inflammatory cytokines that mediate neutrophil recruitment.1 While they can play protective roles, TH17 cells can also worsen the pathogenesis of inflammatory diseases such as multiple sclerosis, arthritis, inflammatory bowel disease (IBD), and asthma.2,3
TH17 differentiation can be either promoted or prevented by various cytokines. For example, CD4 TH cells can differentiate into TH17 cells in response to transforming growth factor- β (TGF-β) and the pro-inflammatory cytokine IL-6.4,5 While TGF- β and IL-6 are sufficient for their differentiation, IL-23 signaling promotes long-term maintenance of the TH17 cell lineage allowing TH17 cells to fully differentiate and proliferate.6 Additionally, IL-1 together with IL-6 can facilitate TH17 differentiation by increasing expression of retinoic-related orphan receptor γt (RORγt) and interferon response factor (IRF-4) and inducing higher levels of IL-17 production.7 In contrast, TH17 differentiation is inhibited by IL-2 as well as the TH2 and TH1-specific cytokines IL-4 and IFN-γ.8–10 Although cytokines made by TH1 and TH2 cells can directly inhibit TH17 differentiation, TH17 cells commonly occur alongside TH1 and TH2 cells in inflammatory settings such as in inflammatory bowel disease (IBD) and asthma.
Atopic asthma is characterized by TH2-mediated inflammation in the lung, and is often accompanied by eosinophilia, high serum IgE levels, and airway hyper-reactivity.11 While this disease is defined by the hallmark TH2-mediated inflammation, high levels of IL-17, as well as TH17 cells, are present in the lungs of asthmatic patients.12–14 Furthermore, high levels of IL-17 and neutrophil accumulation are normally seen in more severe cases of asthma15, 16, including steroid-resistant asthma17,18. TH17 cells have been shown to contribute to lung inflammation in mouse models of asthma, through the recruitment of neutrophils 19–22 that can promote tissue damage, and by inducing lung stromal cell and epithelial cells to produce pro-inflammatory cytokines and chemokines23–26.
Nedd4 family interacting protein 1 (Ndfip1) is an adaptor protein that binds to and augments the function of the E3 ligase Itch.27 Itch has been shown to ubiquitylate, and cause the degradation of JunB28, a transcription factor that promotes the expression of IL-2, IL-4 and IL-529. In the absence of Ndfip1, activated CD4 T cells accumulate high levels of JunB and become TH2 polarized.27 We recently reported that Ndfip1−/− mice naturally develop TH2-mediated inflammation in the skin, gastrointestinal (GI) tract, and lungs, and die prematurely.27, 30 These mice have increased percentages of activated CD4 T cells in their spleens and lymph nodes (LN) that are TH2-biased, as well as eosinophilia. Additionally, they have high circulating levels of IgE and IL-5 in the serum. Histological analysis revealed infiltrating inflammatory cells in the perivascular regions in the lung and goblet cell hyperplasia.27 Importantly, neutrophils were also evident in the perivascular regions of the lung, as well as within the alveolar space. Given the reported role of TH17 cells in lung inflammation and in the recruitment of neutrophils into the airways, we sought to determine whether Ndifp1−/− regulates TH17 differentiation.
In this report we show that mice lacking Ndfip1 have increased numbers of TH17 cells. This finding suggested a scenario in which Ndfip1 acts to prevent TH17 differentiation, much like it does for TH2 differentiation. On the contrary, in vitro, Ndfip1−/− T cells were defective in becoming TH17 cells. This is because increased IL-4 production by Ndfip1−/− T cells inhibit TH17 differentiation. Thus, Ndfip1 promotes TH17 differentiation by inhibiting production of IL-4. In addition, our data show that while IL-4 directly inhibits TH17 differentiation in vitro, the TH2 inflammatory environment in Ndfip1−/− mice promotes the differentiation of TH17 in vivo by increasing the numbers of IL-6-producing eosinophils. Together, these data suggest that TH2 responses can promote TH17 responses in vivo. This might help to explain why TH17 cells develop in the setting of inflammatory diseases such as asthma.
METHODS
Mice
Ndfip1−/− mice have been previously described.27 They have been backcrossed to the C57BL6 background for more than 9 generations. All mice were kept in a semi-barrier facility at the Children’s Hospital of Philadelphia. All experimentation was approved and followed guidelines established by the Children’s Hospital of Philadelphia Institutional Animal Care and Use Committee. Ndfip1−/− mice were bred from heterozygous parents and their WT littermates were used as controls. The IL-4−/− mice31 were purchased from the Jackson Laboratories and bred with Ndfip1−/− mice in our mouse facility. Mice 4–8 weeks old were used for experiments.
Analysis of lung bronchial alveolar lavage (BAL) fluids and tissue homogenates
Lung tissue homogenates were prepared by perfusing the lungs and removing either a piece or a whole lung, which was then digested with DNase (20µg/ml), collagenase 1 (0.9 mg/ml) and collagenase 1A (0.8 mg/ml) (Sigma) in 20–25 ml of DMEM, shaking for 1 hour at room temperature. The cell suspension was filtered through a 100µm, then 40µm mesh and 10% FCS was added. The cells were then washed and either cultured with PMA, ionomycin, and Golgi-stop (BD Bioseciences) for intracellular cytokine staining, or directly prepared for flow cytometry. Neutrophils in the BAL fluids were collected as previously described32 and analyzed by flow cytometry using antibodies against CD11b (M1/70) and Ly6G (1A8) from BD Biosciences.
Collection of BAL
At designated time points, the animals were euthanized, the diaphragm was incised, and the trachea isolated and cannulated with a 20-gauge catheter, which was immobilized with 4-0 silk suture. BAL fluid was collected from the whole lung using 4 instillation/withdrawal passes of 0.8 ml PBS containing 100 nM Diethylenetriaminepentaacetic acid (DTPA; Sigma), as previously described.32 BAL fluid (0.17 ml) was centrifuged and placed on glass cytospin slides, which were then stained by Diff-Quick reagents (Fisher Scientific) to enumerate leukocyte subtypes based on their cellular and nuclear morphological features.
Flow cytometry and antibodies
Unstimulated cells were harvested from lung tissue homogenates (described above) and spleens and then stained with LIVE/DEAD Fixable Dead Cell Blue stain as per manufacturer’s instructions (Invitrogen). Cells were then pre-incubated with Fc Block (anti-CD16/32, 2.4G2, BD Biosciences) prior to surface staining. Cells were stained for surface markers for 30 minutes at 4°C, washed and then fixed with the Foxp3 Fixation/Permeabilization Kit from eBioscience. Samples were analyzed the next day on an LSRFortessa (BD Biosciences). The antibodies used to stain for leukocyte cell subsets in the lung tissue homogenates and spleen samples were anti-CD11c (N418), anti-CD4 (GK1.5), anti-Ly6G (1A8), anti-CD11b (M1/70), and anti-c-Kit (2B8) from BioLegend; anti-Foxp3 (FJK-16s) and anti-FcεRIα (MAR-1) from eBioscience; and anti-SiglecF (E502440) from BD Biosciences.
TH17 differentiation
Naïve CD4+ T cells were sorted by gating out the CD25+ cells and selecting the CD44low CD62Lhigh population of CD4+ T cells. Cells were cultured in 48 well plates using 5×105 cells per well, and activated with plate-bound anti-CD3 (5µg/ml) and anti-CD28 (5µg/ml) (BD Pharmingen). TH17 differentiating conditions included TGF- β (5ng/ml) (PreproTech) and IL-6 (20ng/ml) (R and D systems), or TGF- β (0.5ng/ml), IL-6 (20ng/ml), IL-1 (20ng/ml) (PreproTech), and IL-23 (50ng/ml) (R and D Systems). Cells were cultured for 5 days and analyzed for IL-17 and IL-4 production by flow cytometry by intracellular cytokine staining. Supernatants were collected at 20–24 hours to measure IL-2 production by ELISA. In some of the experiments, IL-4 (Biolegend) and IL-2 (BD Biosciences) blocking antibodies were added at the initiation of the cultures (20µg/ml).
Measurement of cytokines by ELISA
ELISA was performed to measure cytokines from either serum or spleen cultures. For serum, mice were bled and serum was collected using minicollect tubes from Greiner bio-one, and stored at −80°C until used for ELISA. Spleen cultures were set up using 4×106 splenocytes per well in a round-bottom 96 well plate. Soluble anti-CD3 (5µg/ml) was used to activate T cells. Supernatants were collected at 24 hours after stimulation and placed at −80°C until used. For IL-6 production from spleen cultures, splenocytes were stimulated with PMA (1µM) and ionomycin (30µg/ml) for 4.5–5 hours and then supernatants were collected and placed at −80°C until used. ELISA was performed using the eBioscience kit for the respective cytokine. Based on the manufacturers protocol, the limit of sensitivity for the ELISA measuring IL-6 was 4pg/ml, however, our extrapolated results showed linear detection of positive controls as low as 2pg/ml. All results shown are within the linear range of extrapolated data.
Measurement of Serum IgE by ELISA
Serum was collected as described above. The amount of IgE from the serum of the indicated mice was analyzed using the Mouse IgE ELISA kit from BioLegend as per the instructions provided in the kit. Plates were analyzed using a Synergy HT Microplate Reader.
Intracellular cytokine staining
For intracellular cytokine staining, the cells from lung tissue homogenates or TH17 differentiation cultures were re-stimulated with PMA (1µM) and ionomycin (30µg/ml), in the presence of Golgi-stop for 4.5 hours. Cells were fixed either using the BD Biosciences Kit for intracellular cytokine staining or the Foxp3 Fixation/Permeabilization Kit from eBioscience. For TH17 differentiation cultures, cells were stained with anti-IL-17 (eBio17B7, eBioscience) and anti-IL-4 (11B11, BD Biosciences), and analyzed by flow cytometry. For analysis of T helper subsets from lung and spleen samples, cells were stained with the LIVE/DEAD Fixable Cell stain and blocked with Fc Block as described above. The cells were stained for intracellular cytokines using the following antibodies: IL-4 (11B11, BioLegend), IL-9 (RM9A4, BioLegend), IFN-γ (XMG1.2, BioLegend), IL-5 (TRFK5, BD Biosciences), IL-6 (MP5-20F3, BD Biosciences), and IL-10 (JES5-16E3, eBioscience).
Anti-IL-6 antibody treatment
Ndfip1−/− mice were treated with IL-6 blocking antibody (R&D Systems), using 1mg in 250µl of PBS, or with 1mg/250µl isotype control antibodies, per injection. Intraperitoneal injections were performed weekly, at 4 and 5 weeks of age, and mice were analyzed at 6 weeks of age.
Statistics
All statistical analysis was performed by Student’s T-tests. A P value ≤ 0.05 was considered to determine statistical significance. Error bars represent standard deviation of the mean.
RESULTS
Ndfip1−/− mice have an increase in lung neutrophils and TH17 cells
Previous work from our group has shown that Ndfip1−/− mice develop TH2-mediated pulmonary inflammation characterized by goblet cell hyperplasia and increased numbers of perivascular lymphocytes and eosinophils. Neutrophils, leukocytes that are associated with TH17 rather than TH2-mediated inflammation, were also evident within the perivascular space. To quantify the neutrophils from Ndfip1−/− and Ndfip1+/+ littermates, we analyzed cells from bronchial alveolar lavage (BAL) by both morphological and flow cytometric approaches. Examination of cytospin preparations revealed high numbers of neutrophils among cells isolated from BAL of Ndfip1−/− mice, but not Ndfip1+/+ littermate controls (Figure 1A and Supplementary Figure 1A). This result was confirmed using flow cytometry of cells isolated from BAL. Antibodies against Ly6G and CD11b, markers used to identify neutrophils (Figure 1B), revealed a significant increase in the number of neutrophils in the airspace of lungs from Ndfip1−/− animals. This increase in neutrophils in the lungs of Ndfip1−/− mice suggested that TH17 cells might be present amongst the TH2 cells that have been reported previously.
Figure 1. Ndfip1−/− mice have increased lung neutrophils and TH17 cells.

(A) Number of neutrophils in the BAL fluids form Ndfip1+/+ or Ndfip1−/− mice as determined by cytospin. This graph includes the average and standard deviation from 3 different mice of each genotype. (B) Number of neutrophils (Ly6G+ CD11b+) from the BAL fluids of these mice, analyzed by flow cytometry, using antibodies against Ly6G and CD11b, including data from 5 different mice of each genotype. (C) The percentage of CD4 T cells that are IL-17+ in lung tissue homogenates from Ndfip1−/− vs. Ndfip1+/+ mice. (D) The percentage of lung cells that are CD4+ IL-17+ (TH17) in lung tissue homogenates from Ndfip1+/+ or Ndfip1−/− mice, including the average and standard deviation from 7 different mice of each genotype. * refers to a significant difference between Ndfip1+/+ and Ndfip1−/−, p<0.01; *** refers to p<0.0001.
TH17 cells are known to recruit neutrophils into the lung during inflammatory conditions.33 To test whether Ndfip1−/− mice contained increased numbers of TH17 cells, we isolated cells from the lungs of Ndfip1−/− and Ndfip1+/+ littermates and used flow cytometry to determine the percentage of CD4+ T cells that expressed intracellular IL-17. Analysis of lung tissue homogenates from 6 week old Ndfip1−/− mice or Ndfip1+/+ littermates showed a significant increase in the percentage of TH17 cells in the lungs of Ndfip1−/− mice (Figure 1C). As shown in figure 1D, the percentage of cells in the lung tissue homogenates that were CD4+ and IL-17+ was increased approximately 7 fold in Ndfip1−/− mice compared to Ndfip1+/+ littermate controls.
We next sought to analyze the percentages of TH2 cytokine producing cells in the lung and spleen of Ndfip1−/− and Ndfip1+/+ mice, as well as with other cytokine producing CD4+ subsets. The most frequent cytokine producing subsets in both lung and spleens of Ndfip1−/− animals were those producing IL-4 or IL-5 (Supplementary Figure 1B). The percentages of these TH2 cytokine-producing cells in Ndfip1−/− mice were, on average, 12 fold higher than those in Ndfip1+/+ littermates. Other cytokine producing subsets were also increased, but to a lesser extent.
Together, these data show that Ndfip1−/− mice have an ongoing TH17 immune response in addition to their previously described TH2 mediated inflammation.
Ndfip1−/− T cells have a defect in TH17 differentiation in vitro
Ndfip1−/− T cells are more likely to become TH2 cytokine producers in vivo and in vitro.27 This is because Ndfip1 is required for Itch-mediated degradation of JunB, a transcription factor that promotes IL-4 and IL-5 transcription.29 Thus, one function of Ndfip1 is to prevent IL-4 production and TH2 differentiation. Given the increased percentages of TH17 cells in the lungs of Ndfip1-deficient mice, we hypothesized that Ndfip1 might also inhibit TH17 differentiation. To test this, we analyzed TH17 differentiation of Ndfip1+/+ and Ndfip1−/− T cells in vitro. Naïve, sorted CD4 T cells from Ndfip1−/− mice or Ndfip1+/+ littermate controls were activated with anti-CD3 and anti-CD28 in the presence of TGF-β and IL-6, conditions shown previously to induce TH17 differentiation.4,5 Consistent with these reports, approximately 11% of the Ndfip1+/+ T cells became IL-17 producing cells under these conditions. Contrary to our expectations, Ndfip1−/− CD4 T cells showed a reduced capacity to differentiate into TH17 cells (Figure 2A and B), as fewer than 4% of the cells were IL-17 producers. Thus, after 5 days in culture, Ndfip1−/− CD4 T cells showed a significant decrease in IL-17 production under these conditions. It has been shown that low levels of TGF-β in conjunction with IL-6, IL-1, and IL-23 induce higher percentages of IL-17 producing T cells in culture than TGF-β and IL-6 alone.7 Supporting this, using WT cells, we obtained the highest level of TH17 differentiation using 0.5ug/mL of TGF-β together with IL-6, IL-1, and IL-23 (data not shown). We therefore tested the ability of naïve Ndfip1−/− CD4 T cells to differentiate into TH17 cells under these conditions. After five days of culture, naïve Ndfip1−/− CD4+ T cells again showed a reduced capacity for IL-17 production compared to CD4+ T cells isolated from Ndfip1+/+ littermate controls (Figure 2C and D). Approximately 40% of Ndfip1+/+ T cells differentiated into TH17 cells, while fewer than 10% of the Ndfip1−/− T cells became IL-17 producers. The combined data from 5–6 separate experiments is shown in Figure 2 B and D. Taken together these data show that Ndfip1−/− T cells are defective at differentiating into TH17 cells in vitro.
Figure 2. Ndfip1−/− CD4 T cells are defective in differentiating into TH17 cells.
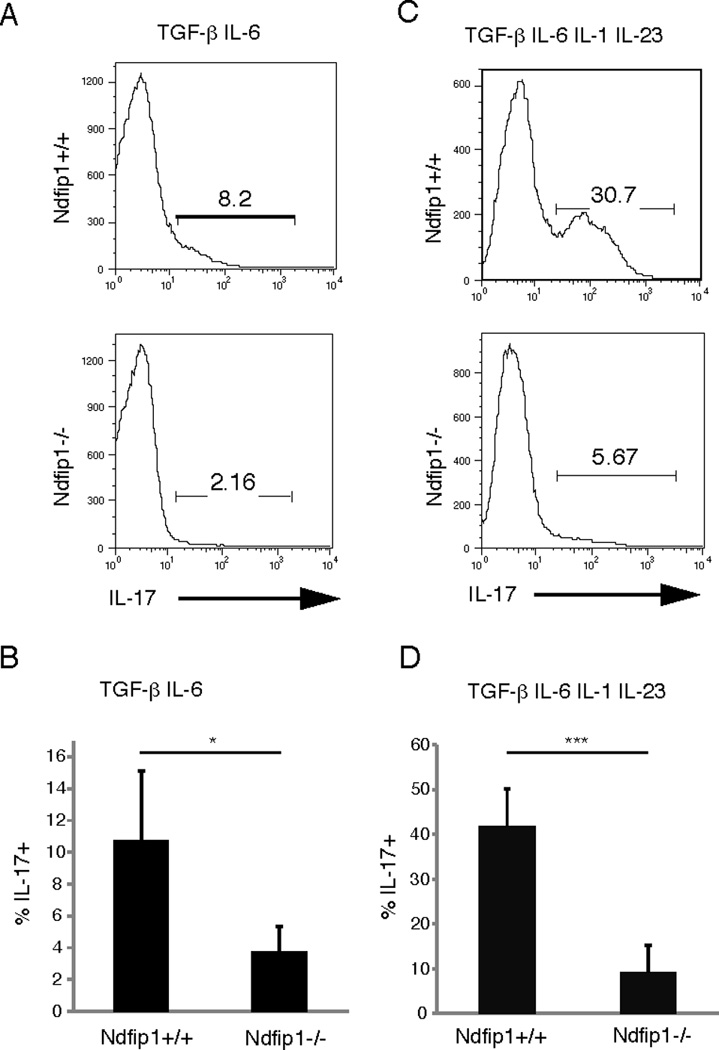
(A) Naïve sorted CD4 T cells were activated with plate-bound anti-CD3 and anti-CD28 in the presence of TGF-β and IL-6. The percentage of IL-17+ cells was analyzed by flow cytometry after 5 days of culture. (B) Same as A, shown as the combined data from 5 different experiments. (C) Naïve sorted CD4 T cells were activated with CD3 and CD28 in the presence of TGF-β, IL-6, IL-1, and IL-23. The percentage of IL-17+ cells was analyzed by flow cytometry after 5 days of culture. (D) Same as B, shown as the combined data from 6 different experiments. * refers to p<0.01; *** refers to p<0.001.
IL-4 and IL-2 are cytokines that have been previously shown to inhibit TH17 differentiation.8–10 Ndfip1−/− T cells can produce increased levels of IL-2 and IL-4 under certain conditions.27, 34 To test whether Ndfip1−/− T cells produce higher levels of IL-2 than their Ndfip1+/+ counterparts, we measured IL-2 produced by Ndfip1−/− or Ndfip1+/+ T cells after activation with anti-CD3 and anti-CD28 in the presence or absence of TH17 polarizing conditions (Supplementary Figure 2A). When cultured in the presence of anti-CD3 and anti-CD28, Ndfip1−/− T cells produced a similar amount of IL-2 compared to Ndfip1+/+ T cells. Furthermore, under TH17 polarizing conditions, IL-2 production was also similar between the two genotypes. These data suggest that differences in IL-2 production are unlikely to account for the defect in TH17 differentiation in Ndfip1−/− T cells. Thus, we next considered a possible role for IL-4.
A previous report from our lab has shown that Ndfip1−/− CD4 T cells are biased towards the TH2 subtype and make more IL-4 than Ndfip1+/+ T cells when cultured under TH2 differentiating conditions.27 Importantly, when these cells were cultured under neutral conditions, in the absence of polarizing cytokines, higher percentages of Ndfip1−/− CD4 T cells produced IL-4 after 5 days in culture than their WT counterparts (Supplementary Figure 2B). Given the increased capacity of Ndfip1−/− T cells to become IL-4 producing cells in the absence of TH2 polarizing conditions, we sought to test whether these cells make IL-4 when activated under TH17 polarizing conditions. Based on our previous results (Figure 2) we tested IL-4 production under conditions that produce the highest percentages of TH17 cells, namely with TGF-β, IL-6, IL-1 and IL-23. Under these conditions, fewer than 1% of WT T cells produced IL-4. In WT cells, these percentages were similar to WT cells cultured under neutral conditions (Supplementary Figure 2B). Ndfip1−/− T cells, on the other hand, continued to produce IL-4 even under TH17 polarizing conditions. However, the percentages of IL-4 producing T cells were lower in Ndfip1−/− T cells cultured under TH17 conditions than under neutral conditions. Thus, TGF-β likely prevents some of the IL-4 production in these cells. Importantly, Ndfip1−/− T cells that produced IL-4 were not also producing IL-17 (Figure 3). These data suggest that Ndfip1−/− T cells can become TH2 cytokine producing cells even under TH17 polarizing conditions. TH17 differentiation is unlikely to occur in these IL-4 producing cells. Furthermore, IL-4 production by these cells could prevent TH17 differentiation by other cells in the cultures.
Figure 3. Blocking of IL-4 restores the defect in TH17 differentiation.
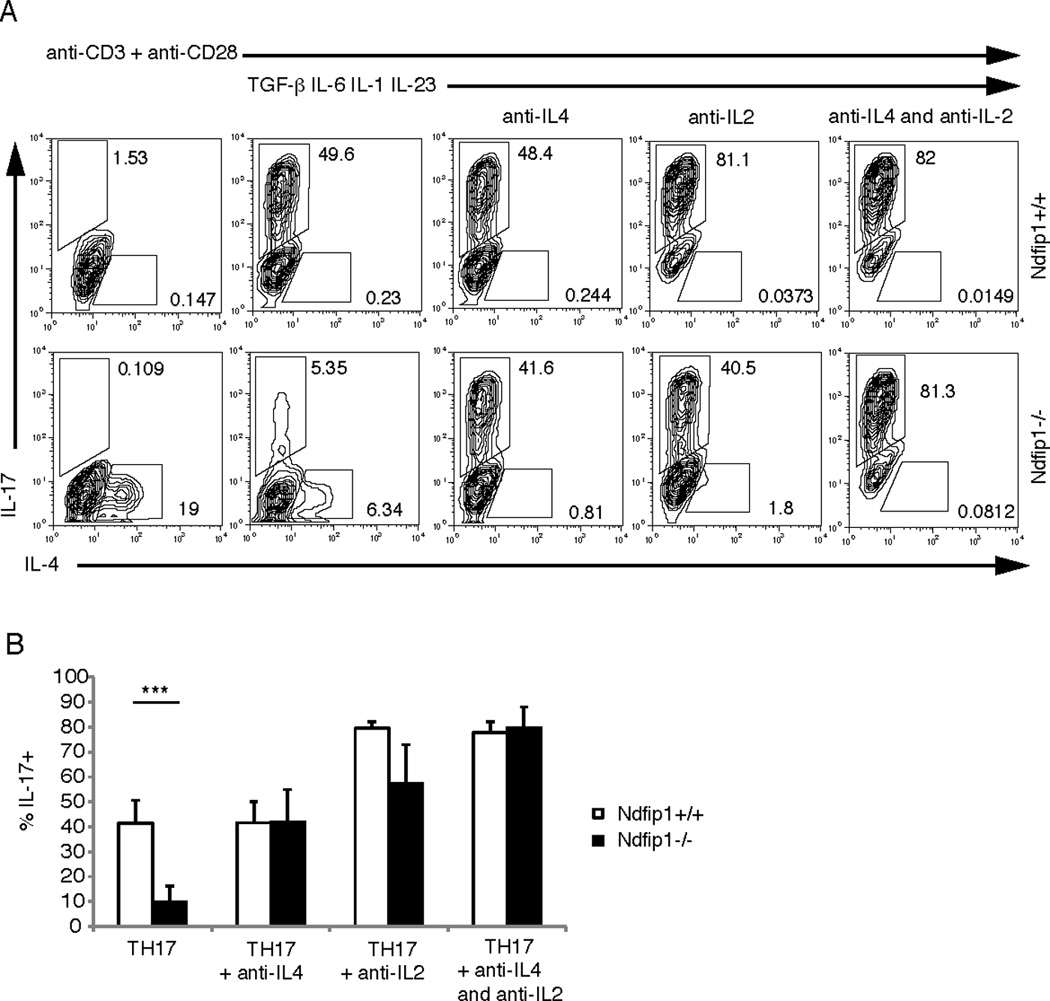
(A) Sorted naïve CD4 T cells were activated with plate-bound anti-CD3 and anti-CD28 in the presence of TGF-β, IL-6, IL-1, and IL-23 and blocking antibodies against IL-4 and/or IL-2 as specified. The percentage of IL-17 and IL-4 producers was analyzed using flow cytometry on day 5 of culture. This data is representative of 5 different experiments. (B) Combined data from 5 experiments. Open bars represent Ndfip1+/+ and black bars represent Ndfip1−/− T cells. *** p< 0.005.
Blocking IL-4 restores TH17 differentiation of Ndfip1−/− T cells
Given the capacity of IL-4 and IL-2 to inhibit TH17 differentiation, we next tested whether IL-4 production or a combination of both IL-4 and IL-2 production by Ndfip1−/− T cells could prevent TH17 differentiation in vitro. To test this, we cultured naïve Ndfip1+/+ and Ndfip1−/− CD4+ T cells under TH17 polarizing conditions in vitro in the presence or absence of IL-4 and IL-2 blocking antibodies. We then measured the percentages of these CD4+ T cells that differentiated into TH17 cells. As shown above, when activated in the presence of TGF-β, IL6, IL-1, and IL-23, Ndfip1−/− CD4 T cells were defective in differentiating into TH17 cells when compared to Ndfip1+/+ CD4 T cells. Importantly, in the presence of IL-4 blocking antibodies, the ability of Ndfip1−/− T cells to differentiate into TH17 cells was restored (Figure 3A and B). Both the percentage of cells making IL-17 and the amount of intracellular IL-17 being made by the TH17 cells reached levels similar to those of Ndfip1+/+ CD4 T cells. Furthermore, T cells from Ndfip1−/− IL-4−/− mice do not have a defect in TH17 differentiation (Supplementary Figure 2C). These data indicate that IL-4 produced by Ndfip1−/− CD4 T cells prevents their differentiation into TH17 cells.
It has been shown previously that blocking IL-2 can enhance TH17 differentiation.8,35 Consistent with these observations, we saw an increase in the percentage of Ndfip1+/+ cells that produced IL-17 in the presence of IL-2 blocking antibodies. Ndfip1−/− CD4 T cells also showed an increased percentage of TH17 producing cells in the presence of the IL-2 blocking antibody. Ndfip1+/+ cells showed an average of a 2 fold increase, compared to a 6 fold increase for Ndfip1−/− cells. However, in the presence of IL-2 blocking antibodies, the levels of IL-4 produced by Ndfip1−/− T cells, and to some extent Ndfip1+/+ T cells, also decreased significantly (Figure 3A). This suggests that in these cultures blocking IL-2 is promoting TH17 differentiation of Ndfip1−/− T cells by reducing IL-4 production. Furthermore, when used together, anti-IL4 and anti-IL2 blocking antibodies restored TH17 differentiation to levels seen in Ndfip1+/+ cultures (Figure 3 A and B). Taken together these data indicate that IL-4 produced by Ndfip1−/− T cells prevents their differentiation into TH17 cells. Furthermore, these results show that Ndfip1 facilitates TH17 differentiation by inhibiting the production of IL-4.
Ndfip1−/− mice have elevated levels of IL-6
Our results show that Ndfip1−/− mice have an increase in TH17 cells in the lung. Surprisingly, however, Ndfip1−/− CD4 T cells are less likely to differentiate into TH17 cells in vitro. This suggests that conditions exist in Ndfip1−/− mice that support TH17 differentiation. One possible factor required for TH17 differentiation in vivo is IL-6. Thus, we analyzed the levels of IL-6 in the serum of Ndfip1+/+ and Ndfip1−/− mice. Analysis of IL-6 levels in the serum of 6 week old mice by ELISA showed higher amounts of IL-6 in Ndfip1−/− mice than Ndfip1+/+ littermate controls (Figure 4A). Thus, increased IL-6 in vivo could promote the TH17 differentiation of CD4+ T cells observed in Ndfip1−/− mice. To test whether the increased level of IL-6 seen in Ndfip1−/− mice could account for the increased percentages of TH17 cells in the lungs, we treated Ndfip1−/− mice with an anti-IL-6 blocking antibody. Starting at 4 weeks of age, IL-6 blocking antibody was administered for two weeks. Following this treatment, we analyzed BAL fluid for the numbers of neutrophils and lung tissue homogenates for the presence of TH17 cells. Ndfip1−/− mice treated with isotype control showed similar numbers of neutrophils in their BAL fluid compared to untreated Ndfip1−/− mice (Figure 4B versus Supplementary Figure 1A), however treatment with IL-6 blocking antibodies led to a significant reduction in neutrophil numbers (Figure 4C). Eosinophil numbers were also reduced (Figure 4C). While isotype control-treated controls showed similar percentages of TH17 cells to untreated mice, Ndfip1−/− mice that received IL-6 blocking antibody had significantly fewer TH17 cells than isotype control-treated animals (Figure 4D). While the percentages of TH17 cells were reduced, the percentages of other cytokine producing CD4+ T cells were less affected (Supplementary Figure 3). Importantly, the reduced percentages of TH17 cells correlated with decreased lung inflammation (Figure 4E). These results indicate that mice treated with anti-IL-6 antibodies have lower levels of TH17 cells in the lungs, supporting our hypothesis that the high levels of IL-6 facilitate TH17 differentiation in Ndfip1−/− mice.
Figure 4. Ndfip1−/− mice have elevated levels of IL-6 that promotes TH17 differentiation.
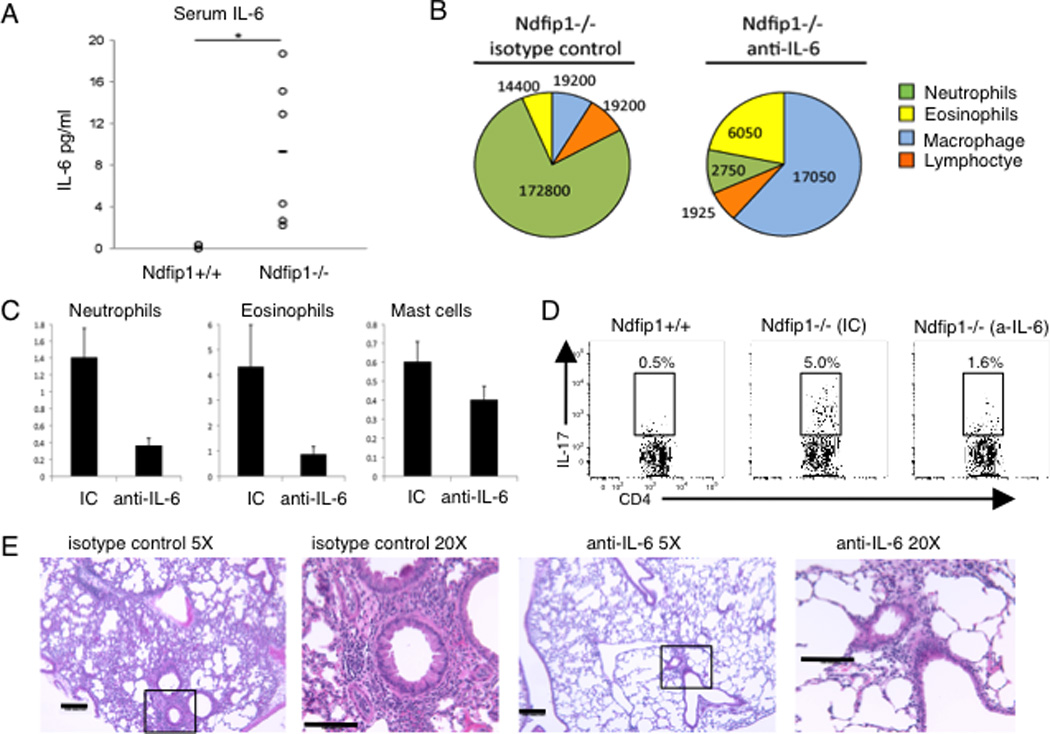
(A) IL-6 levels in the serum from Ndfip1−/− or Ndfip1+/+ littermate controls measured by ELISA. Each circle represents a different mouse and there are 6 mice of each phenotype included. *p<0.05. (B–D) Ndfip1−/− mice were treated with anti-IL6 or isotype control (IC) on weeks 4 and 5 and analyzed on week six of life. (B) BAL fluid from Ndfip1−/− mice treated either with anti-IL-6 or IC was collected and the number of the indicated cells was determined by cytospin. A representative pie chart is shown for each treatment group, n=2 (IC) and n=3 (anti-IL-6). (C) The number of neutrophils (Ly6G+ CD11b+), eosinophils (SiglecF+, Ly6G−) and Mast cells (FcεRIα+ SiglecF−) in the lung tissue homogenate was determined by flow cytometry. The homogenate was from half of the lung tissue. Graphs show the mean + s. d. (D) The percentage of lung cells from tissue homogenates that were CD4+ and IL-17+ were analyzed by flow cytometry. The plots shown are representative of 2–3 mice of the indicated genotypes and treatments. (E) Lungs from Ndfip1−/− mice treated with isotype control (IC) or anti-IL-6 were perfused, inflated with fixative and sectioned. H and E stained sections were analyzed at 5X and 20X. Bar shows 100 microns.
Together our data show that Ndfip1−/− CD4 T cells are partially defective in TH17 differentiation. This is because these cells produce too much IL-4, even in the presence of TGF-β. Furthermore, we propose that the high levels of IL-6 in Ndfip1−/− animals helps to promote TH17 differentiation in vivo, and explains why we observe increased percentages of the TH17 effector cell subtype in Ndfip1−/− mice compared to healthy Ndfip1+/+ littermates.
Eosinophil numbers are increased in the spleens of Ndfip1−/− mice and these cells are producing IL-6
As noted above we detect more IL-6 in the serum of mice lacking Ndfip1. It is known that CD4+ T cells, macrophages, eosinophils, mast cells and neutrophils can make IL-636, 37. To determine which cells in Ndfip1−/− mice were making IL-6 and thus driving TH17 differentiation, we restimulated cells from the lungs and spleens and assessed IL-6 production by intracellular cytokine staining. We detected more cells producing IL-6 in the spleens than in the lungs of Ndfip1−/− mice (Figure 5A and 5B and data not shown). While we could detect IL-6 production in CD4+ cells, the majority (about 86%) of the splenocytes making IL-6 were CD4 negative cells (Figure 5B). Amongst the CD4 negative cells, we did not detect a significant frequency of IL-6+ cells in the Ly6G+ neutrophil or c-kit+ mast cell populations (data not shown). In contrast, most of the IL-6+ cells were found among the SiglecF+ (Figure 5C) population suggesting that the cells making IL-6 in Ndfip1−/− mice were eosinophils. We have previously shown that mice lacking Ndfip1 have increased numbers of eosinophils in the esophagus30, however the spleen was not investigated in this previous report. Therefore, we assessed the frequency of eosinophils in the spleens of mice lacking Ndfip1. We found that Ndfip1−/− have increased percentages and numbers of eosinophils in the spleen (Figure 5D and 5E). Moreover, more of the eosinophils in Ndfip1−/− mice are activated as indicated by FcεRIα expression and are making IL-6 (Figure 5F and 5G). Based on these data, eosinophils are the source of IL-6 that promotes TH17 differentiation in vivo in mice lacking Ndfip1.
Figure 5. Eosinophils are significant producers of IL-6 in Ndfip1−/− mice.
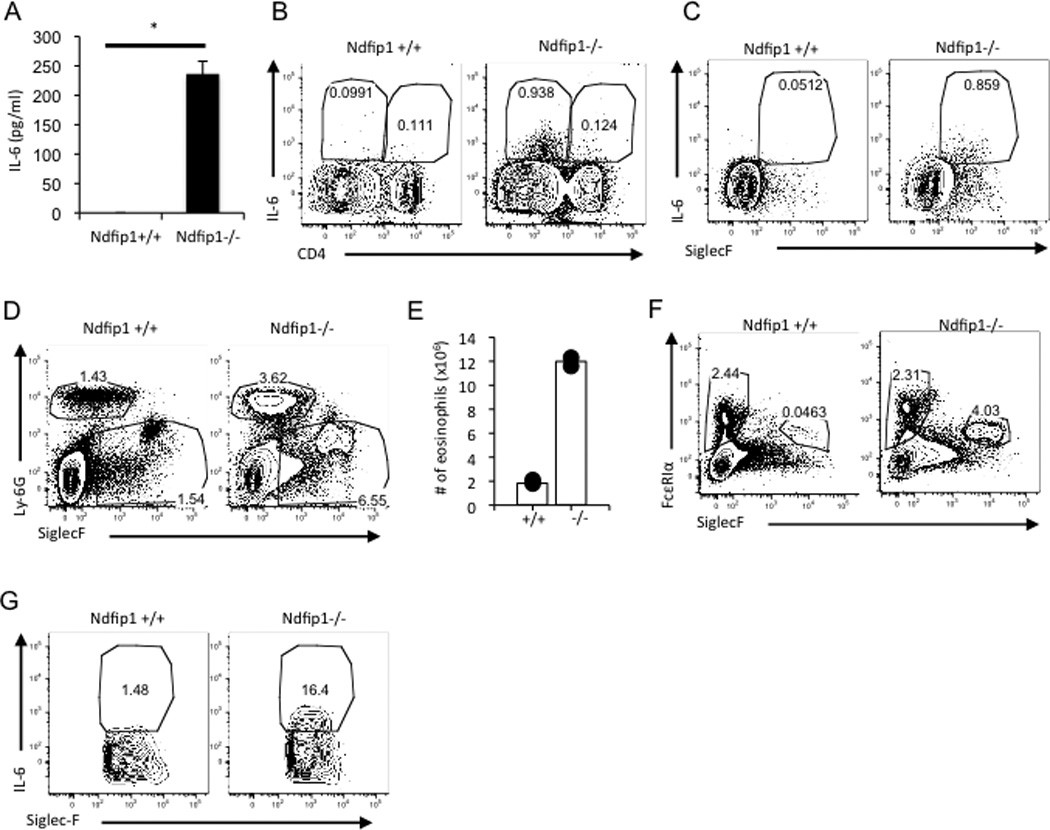
(A) The amount of IL-6 in the supernatants of splenocytes restimulated for 4.5–5 hours with PMA and ionomycin was measured by ELISA. Bar graph represents the mean + s.d. of triplicate samples from one mouse for each genotype. The results are representative of 3–4 mice per genotype. *p<0.00005. (B and C) Splenocytes were restimulated with PMA and ionomycin and then stained and analyzed by flow cytometry as described in materials and methods for surface markers and intracellular IL-6. (B) Representative flow plots gated on live cells are shown, n=3 mice per genotype. (C) Representative flow plots gated on live CD4− cells are shown, n=3 mice per genotype. (D) Representative facs plots of flow cytometric analysis from the spleens of indicated mice. Plots are gated on live cells and show the Ly6Ghigh neutrophil population and Ly6Glow/neg SiglecF+ eosinophil population, n=2 mice per genotype. (E) The numbers of eosinophils (Ly6Glow/neg SiglecF+) from the mice in D are shown. (F) Splenocytes were analyzed for the percentage of FcεRIα+ SiglecF− mast cells and FcεRIα+ SiglecF+ activated eosinophils. Representative plots are shown. n=2–3 mice per genotype. (G) Cells were stimulated as in B and C. IL-6 production by the eosinophil population is shown in the plots (Gated on CD4− Ly6Glow/neg SiglecF+ cells), n=3 mice per genotype.
The TH2 inflammatory environment in Ndfip1-deficient mice promotes TH17 differentiation
Our in vitro data shows that IL-4 can directly decrease TH17 differentiation, while our in vivo data suggests that TH2-mediated inflammation may promote TH17 differentiation indirectly by increasing the levels of IL-6. It is possible that even in the presence of IL-4-producing TH2 cells, the inflammatory conditions in Ndfip1−/− mice promote the differentiation of TH17 cells. To test this, we crossed Ndfip1−/− mice to IL-4−/− animals, and analyzed the percentages of TH17 cells in the lungs, IgE levels in the serum (Supplementary Figure 4A) and IL-6 production from splenocytes in the resulting Ndfip1−/−, IL-4−/− and Ndfip1−/−IL-4−/− progeny.
As illustrated in figure 6, Ndfip1−/− IL-4−/− double knockout (DKO) mice showed a lower level of IL-6 in the serum compared to Ndfip1−/− IL-4+/+ littermate controls (Figure 6A) as well a decreased IL-6 production by ex-vivo splenocytes (Figure 6B), indicating that the TH2 response in Ndfip1−/− mice is contributing to the high levels of IL-6 in these mice. Supporting this, eosinophils in the spleens of Ndfip1−/− IL-4−/− DKO mice showed reduced intracellular IL-6 (Figure 6C). Furthermore, Ndfip1−/− IL-4−/− DKO mice contained fewer neutrophils in the BAL fluid (Supplementary Figure 4B) and, in addition to the decrease in TH2 cytokine producing cells (Supplementary Figure 4C), also showed lower percentages of TH17 cells in the lungs compared to Ndfip1−/− IL-4+/+ controls (Figure 6D). Additionally, there is much less inflammation in the lungs of Ndfip1−/− IL-4−/− DKO mice compared to Ndfip1−/− mice (unpublished results Natalia Ramos and Allison Beal). These results indicate that the TH2 inflammatory environment seen in Ndfip1−/− mice contributes to the differentiation of TH17 cells. We propose that this is, at least in part, due to an increase in production of IL-6, which occurs as a consequence of the TH2-mediated inflammation.
Figure 6. The TH2 environment in Ndfip1−/− mice contributes to IL-6 expression and TH17 differentiation.
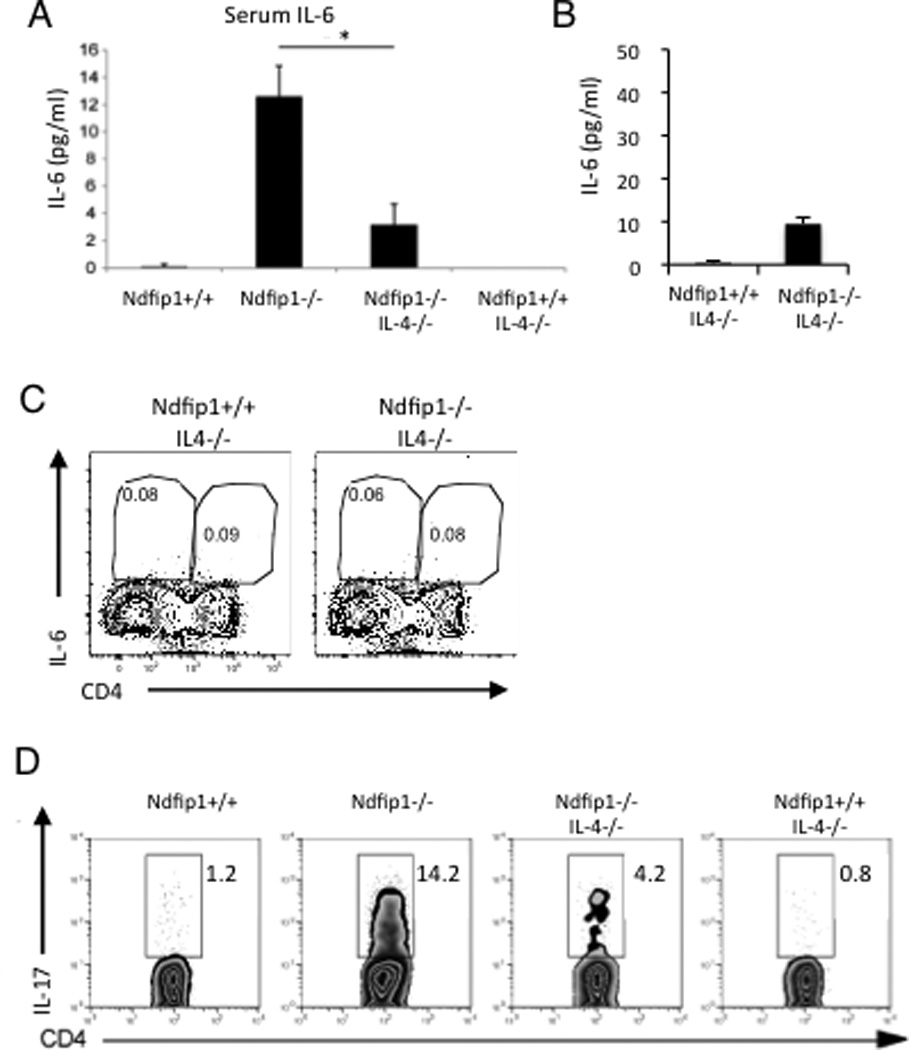
(A) IL-6 from the serum of the indicated mouse phenotypes analyzed through ELISA. *p<0.05. (B) The amount of IL-6 in the supernatants of splenocytes restimulated with PMA and ionomycin was measured by ELISA. Bar graph represents the mean + s.d. of triplicate samples from one mouse for each of the indicated genotype. The results are representative of 3 mice per genotype. **p<0.001. (C) Splenocytes were restimulated as indicated in the materials and methods and then analyzed by flow cytometry as described in materials and methods. (B) Representative flow plots gated on live cells are shown, n=3 mice per genotype. (D) Percentage of CD4+ T cells that express IL-17, from lungs tissue homogenates, analyzed through flow cytometry. Data are representative of three mice of the Ndfip1+/+, Ndfip1−/− IL-4−/−, and Ndfip1+/+ IL-4−/− genotypes, and 4 Ndfip1−/− mice.
DISCUSSION
A previously published report from our lab shows that mice deficient in Ndfip1 develop TH2-mediated inflammation.27 This phenotype can be explained by the role of Ndfip1 in the regulation of Itch-mediated ubiquitilation and degradation of JunB, a transcription factor that regulates the expression of the TH2 cytokines IL-4 and IL-5.29 The inflammation seen in these mice is characterized by high levels of IgE and IL-5 in serum and an influx of immune cells in the lung, and at other mucosal surfaces. In the lungs of Ndfip1−/− mice, infiltrating inflammatory cells, including eosinophils, were observed in the perivascular regions. Goblet cell hyperplasia is also evident.27 We have now shown that in addition to this TH2 response, Ndfip1−/− mice have elevated numbers of neutrophils and increased percentages of TH17 cells in the lungs.
The increase in lung TH17 cells in these mice led us to hypothesize that Ndfip1 might prevent TH17 differentiation, and that its absence would cause an increase in TH17 cells in vitro and in vivo. Thus, we tested the role of this adaptor protein in TH17 differentiation in vitro. Unexpectedly, our results showed that Ndfip1−/− CD4 T cells are defective in differentiating into the TH17 subset in vitro. Ndfip1−/− CD4 T cells are more likely to become TH2 cells compared to WT counterparts, in the presence of TH2 driving conditions. Our results further show that even under neutral or TH17-driving conditions, Ndfip1−/− CD4 T cells make IL-4 after activation. IL-4 has been previously shown to inhibit TH17 differentiation.9,10,38 Consistent with these results, the IL-4 produced by Ndfip1−/− CD4 T cells significantly inhibited their TH17 differentiation in vitro. This is not necessarily due to a direct inhibition of TH17 differentiation by JunB, as these results indicated that the defect in TH17 differentiation seen in Ndfip1−/− cells is dependent on signaling through the IL-4 receptor. Given the role of Ndfip1 in the negative regulation of TH2 cytokine expression, our results show that this adaptor protein acts to promote TH17 differentiation by preventing IL-4 production.
It has been reported that TGF-β plays an important role in the inhibition of TH2 cytokine expression during TH17 differentiation as well as during iTreg cell differentiation.39 We showed recently that TGF-β dampens IL-4 production by Ndfip1-dependent as well as Ndfip1-independent mechanisms.34 We now show that this is true during TH17 differentiation as well. Supporting this, in Ndfip1−/− CD4 T cells TGF-β is not sufficient to completely prevent IL-4 production and allow differentiation into the TH17 subtype. Thus, CD4 T cells have at least two mechanisms that prevent IL-4 production during TH17 differentiation, one of which depends on Ndfip1.
Our in vitro experiments show that Ndfip1−/− CD4 T cells are defective in differentiating into TH17 cells; however, in vivo, Ndfip1−/− mice show increased numbers in lung TH17 cells. Given that IL-6 plays a major role in the differentiation of TH17 cells,4,5 we analyzed the levels of IL-6 in the serum of Ndfip1−/− mice. The levels of IL-6 were elevated in the serum of Ndfip1−/− mice and ex-vivo analysis of splenocytes from these mice revealed high percentages of IL-6 producing eosinophils. Blocking IL-6 in vivo resulted in a decrease in lung TH17 cells in Ndfip1−/− mice indicating that this cytokine promotes TH17 differentiation in our system. Therefore, although Ndfip1−/− CD4 T cells are less likely to undergo TH17 differentiation in vitro, the presence of IL-6 promotes TH17 differentiation in vivo leading to an increase in lung TH17 cells and neutrophilia as compared to healthy Ndfip1+/+ mice.
The inflammatory environment within Ndfip1-deficient mice contains factors that promote and inhibit TH17 differentiation. Our results indicate that high levels of IL-6 in Ndfip1−/− mice are conducive to TH17 differentiation. On the other hand, Ndfip1−/− mice have a highly TH2 polarized cytokine environment, where IL-4 could prevent TH17 differentiation. In order to test the effect of the observed TH2 environment on TH17 differentiation in vivo, we crossed Ndfip1−/− to IL-4−/− mice. Analysis of mice lacking both Ndfip1 and IL-4 revealed a lower level of IL-6 in serum and fewer IL-6 producing eosinophils compared to mice lacking only Ndfip1, indicating that the TH2 inflammation in Ndfip1−/− mice might lead to higher IL-6 expression. Furthermore, Ndfip1−/− IL-4−/− mice contained fewer lung TH17 cells compared to an Ndfip1−/−IL-4+/+ littermate controls. These results suggest that the TH2 cytokine production in Ndfip1−/− mice leads to recruitment and activation of IL-6 producing eosinophils, which promotes TH17 differentiation. While IL-4 can directly decrease the percentage of CD4 T cells differentiating into the TH17 subtype, a TH2 response can lead to significant inflammation and promotes the production of IL-6 by eosinophils, which increases TH17 differentiation. Therefore, a balance between the direct inhibition of IL-4 and that of inflammation and IL-6 can determine the outcome of an accompanying TH17 response. Our results indicate that even in the presence of IL-4, the inflammatory environment and the increase in IL-6 expression observed in Ndfip1−/− mice can tilt the balance towards TH17 differentiation (Figure 7). Furthermore, our results help to resolve the apparent discrepancy between in vitro data showing that IL-4 can directly impair TH17 differentiation9,10,38 and ex vivo data showing elevated levels of TH17 cells in asthmatic patients despite a predominant TH2 response12–14. Based on the results presented here, we propose that the missing link connecting the TH2 and TH17 responses in vivo is eosinophil production of IL-6.
Figure 7. Model representing the role of Ndfip1 in TH17 differentiation.
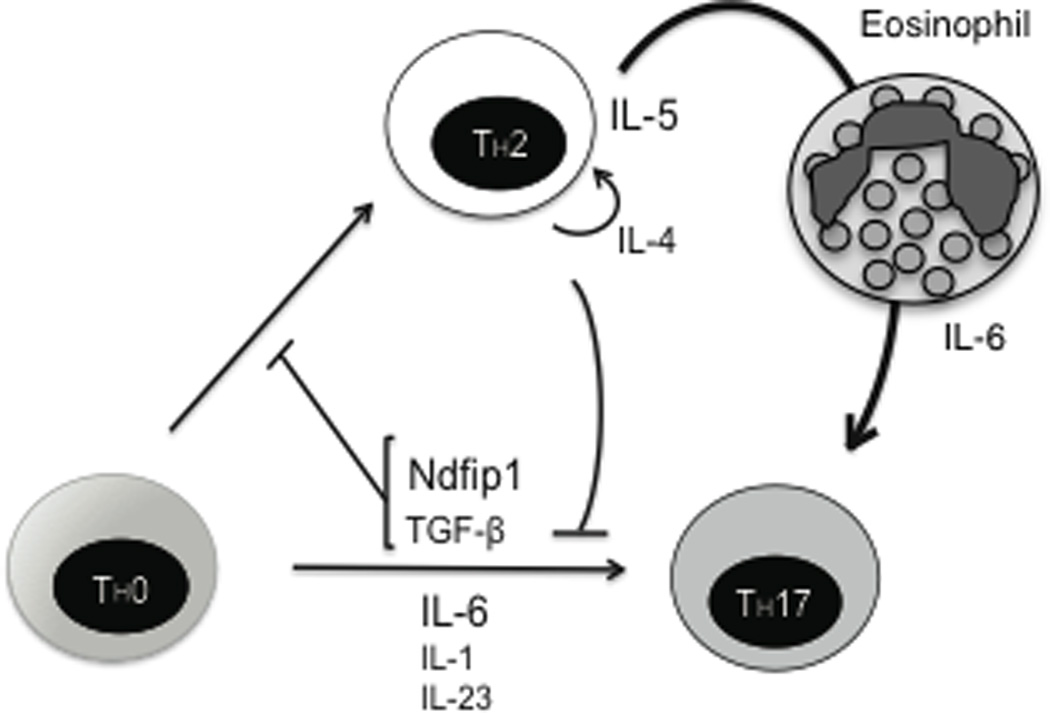
Ndfip1, along with TGF-β, promotes TH17 differentiation by inhibiting IL-4 expression. While IL-4 can directly inhibit TH17 differentiation, a TH2 response can lead to significant inflammation and IL-6 production predominantly by eosinophils to create an environment that supports the differentiation of TH17 cells.
Here we show that Ndfip1−/− mice have an ongoing TH17 response in the lung in addition to their previously described TH2-mediated inflammation. This is also seen in atopic asthma, since high levels of IL-17, as well as an increase in TH17 cells, have been observed in the lungs of asthmatic patients.12–14 Using mouse models of asthma, it has been shown that TH17 cells induce the recruitment of neutrophils into the lungs through their production of IL-17, which can induce the expression of neutrophil chemoattractants by bronchial fibroblasts.24 In addition, several reports have shown that a TH17 response can promote a TH2 response and increase lung eosinophilia,20–22 which is mediated by an increase in the expression of eotaxin.40 Here we show that a TH2 response can further promote TH17 differentiation through the production of the TH17-driving cytokine IL-6 by activated eosinophils. Based on previous data and our data, it is therefore possible that during lung inflammation, there is cross-talk between the TH2 and TH17 responses that ultimately leads to amplification of both responses and significantly elevated levels of inflammation.
IL-6 is a cytokine that is normally expressed during both acute and chronic inflammation. IL-6 is elevated in cases of TH1-mediated autoimmune disease such as rheumatoid arthritis or CD,41 but also expressed during TH2-mediated diseases such as asthma.42 Although there are cases where an immune response is directly guided towards a TH17 response, it is possible that inflammation and its consequent tissue damage generally promote TH17 differentiation, explaining why TH17 cells are found along with both TH1 or TH2 responses. The results presented here support the idea that inflammation, even if it is highly TH2-polarized, can lead to the differentiation of TH17 cells through the induction of the proinflammatory cytokine IL-6.
Supplementary Material
ACKNOWLEDGMENTS
We thank Amy LaRoche for technical assistance and we thank members of the Children’s Hospital of Philadelphia pathology core and the University of Pennsylvania flow cytometry core.
Footnotes
This manuscript was funded in part by the NIH grant T-32-AI-055428-06, R-03-AR-057144, R-01-AI-093566
References
- 1.Dubin PJ, Kolls JK. Th17 cytokines and mucosal immunity. Immunol Rev. 2008;226:160–171. doi: 10.1111/j.1600-065X.2008.00703.x. [DOI] [PubMed] [Google Scholar]
- 2.Korn T, et al. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 3.Louten J, Boniface K, de Waal Malefyt R. Development and function of TH17 cells in health and disease. J Allergy Clin Immunol. 2009;123(5):1004–1011. doi: 10.1016/j.jaci.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 5.Veldhoen M, et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung Y, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30(4):576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26(3):371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 11.Herrick CA, Bottomly K. To respond or not to respond: T cells in allergic asthma. Nat Rev Immunol. 2003;3(5):405–412. doi: 10.1038/nri1084. [DOI] [PubMed] [Google Scholar]
- 12.Bullens DM, et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pene J, et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. 2008;180(11):7423–7430. doi: 10.4049/jimmunol.180.11.7423. [DOI] [PubMed] [Google Scholar]
- 14.Matsunaga K, et al. Airway cytokine expression measured by means of protein array in exhaled breath condensate: correlation with physiologic properties in asthmatic patients. J Allergy Clin Immunol. 2006;118(1):84–90. doi: 10.1016/j.jaci.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Chakir J, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111(6):1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 16.Barczyk A, Pierzchala W, Sozanska E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir Med. 2003;97(6):726–733. doi: 10.1053/rmed.2003.1507. [DOI] [PubMed] [Google Scholar]
- 17.Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol. 2010;72:495–516. doi: 10.1146/annurev-physiol-021909-135926. [DOI] [PubMed] [Google Scholar]
- 18.Pavord ID, et al. Non-eosinophilic corticosteroid unresponsive asthma. Lancet. 1999;353(9171):2213–2214. doi: 10.1016/S0140-6736(99)01813-9. [DOI] [PubMed] [Google Scholar]
- 19.Hellings PW, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28(1):42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 20.Kim SR, et al. PTEN down-regulates IL-17 expression in a murine model of toluene diisocyanate-induced airway disease. J Immunol. 2007;179(10):6820–6829. doi: 10.4049/jimmunol.179.10.6820. [DOI] [PubMed] [Google Scholar]
- 21.Park SJ, et al. Peroxisome proliferator-activated receptor gamma agonist down-regulates IL-17 expression in a murine model of allergic airway inflammation. J Immunol. 2009;183(5):3259–3267. doi: 10.4049/jimmunol.0900231. [DOI] [PubMed] [Google Scholar]
- 22.Wilson RH, et al. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. 2009;180(8):720–730. doi: 10.1164/rccm.200904-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fossiez F, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183(6):2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molet S, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108(3):430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, et al. IL-17A and TNF-a exert synergistic effects on expression of CXCL5 by alveolar type II cells in vivo and in vitro. J Immunol. 2011;186(5):3197–3205. doi: 10.4049/jimmunol.1002016. [DOI] [PubMed] [Google Scholar]
- 26.Huang F, et al. Requirement for both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-kappaB activation by IL-17A in enhancing cytokine expression in human airway epithelial cells. J Immunol. 2007;179(10):6504–6513. doi: 10.4049/jimmunol.179.10.6504. [DOI] [PubMed] [Google Scholar]
- 27.Oliver PM, et al. Ndfip1 protein promotes the function of itch ubiquitin ligase to prevent T cell activation and T helper 2 cell-mediated inflammation. Immunity. 2006;25(6):929–940. doi: 10.1016/j.immuni.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao M, et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science. 2004;306(5694):271–275. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- 29.Li B, et al. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J. 1999;18(2):420–432. doi: 10.1093/emboj/18.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramon HE, et al. The ubiquitin ligase adaptor Ndfip1 regulates T cell-mediated gastrointestinal inflammation and inflammatory bowel disease susceptibility. Mucosal Immunol. 2010 doi: 10.1038/mi.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhn R, et al. Generation and analysis of interleukin-4 deficient mice. Science. 1991;254(5032):707–710. doi: 10.1126/science.1948049. [DOI] [PubMed] [Google Scholar]
- 32.Mei J, et al. CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection. Immunity. 2010;33(1):106–117. doi: 10.1016/j.immuni.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez GJ, et al. Regulation and function of proinflammatory TH17 cells. Ann N Y Acad Sci. 2008;1143:188–211. doi: 10.1196/annals.1443.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beal AM, et al. TGF-β induces the expression of the adaptor Ndfip1 to silence IL-4 production during iTreg cell differentiation. Nat Immunol. 2012;13:77–85. doi: 10.1038/ni.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103(21):8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zubiaga AM, et al. Regulation of interleukin 6 production in T helper cells. Int Immunol. 1990;2(11):1047–1054. doi: 10.1093/intimm/2.11.1047. [DOI] [PubMed] [Google Scholar]
- 37.Bloemen K, et al. The allergic cascade: Review of the most important molecules in the asthmatic lung. Immunol Letters. 2007;113(1):6–18. doi: 10.1016/j.imlet.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 38.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 39.Das J, et al. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med. 2009;206(11):2407–2416. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wakashin H, et al. IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008;178(10):1023–1032. doi: 10.1164/rccm.200801-086OC. [DOI] [PubMed] [Google Scholar]
- 41.Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Traves SL, Donnelly LE. Th17 cells in airway diseases. Curr Mol Med. 2008;8(5):416–426. doi: 10.2174/156652408785160998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.