

Abstract
Accurate computer simulation of blood function can inform drug target selection, patient-specific dosing, clinical trial design, biomedical device design, as well as the scoring of patient-specific disease risk and severity. These large-scale simulations rely on hundreds of independently measured physical parameters and kinetic rate constants. However, the models can be validated against large scale, patient-specific laboratory measurements. By validation with high dimensional data, modelling becomes a powerful tool to predict clinically complex scenarios. Currently, it is possible to accurately predict the clotting rate of plasma or blood in a tube as it is activated with a dose of tissue factor, even as numerous coagulation factors are altered by exogenous attenuation or potentiation. Similarly, the dynamics of platelet activation, as indicated by calcium mobilisation or inside-out signalling, can now be numerically simulated with accuracy in cases where platelets are exposed to combinations of agonists. Multiscale models have emerged to combine platelet function and coagulation kinetics into complete physics-based descriptions of thrombosis under flow. Blood flow controls platelet fluxes, delivery and removal of coagulation factors, adhesive bonding, and von Willebrand factor conformation. The field of Blood Systems Biology has now reached a stage that anticipates the inclusion of contact, complement, and fibrinolytic pathways along with models of neutrophil and endothelial activation. Along with “-omics” data sets, such advanced models seek to predict the multifactorial range of healthy responses and diverse bleeding and clotting scenarios, ultimately to understand and improve patient outcomes.
INTRODUCTION
Perhaps no other aspect of medical biology is as well defined, from a kinetic and mechanistic perspective, as blood function during haemostasis, thrombosis, and bleeding. The majority of molecular species that control coagulation, platelet activation, platelet adhesion, fibrin polymerisation, fibrinolysis, and complement activation are well characterised. Each individual reaction has been studied in isolation to some extent. This foundational knowledge is available because no other living tissue is as readily available for clinical research as human blood. Despite these advantages, blood function can be difficult to predict due to nonlinearity, sensitivity to initial conditions, network complexity, feedback regulation, and biorheological/transport influences. In the face of these challenges, computer modelling seeks to improve prediction of the dynamics of blood function.
Bottom-up Systems Biology is the definition of distinct molecular entities, their specific molecular properties, and quantified interactions (stoichiometry, kinetics, binding, inhibition, diffusion, etc.). The resulting models then predict the regulated behaviour of biochemical pathways, cells, and tissues, either during homeostasis or during perturbation (i.e. haemostasis, thrombosis, drug regimen).
Biochemical reactions are quantified in terms of kinetic rate constants. Importantly, every rate constant requires the deployment of a mathematical rate model (e.g. r=k[A], r=k[A][B], r=kcat [E][S]/(Km+[S]), etc.) that is a conceptualisation of a putative reaction mechanism. In blood reactions, numerous species interact in solution or on surfaces, resulting in complex reaction networks. The static linkages of protein-protein interaction (PPI) networks obtained by proteomic or yeast 2-hybrid approaches simply define nodes and linkages, but lack the directionality that is highly relevant to irreversible protease reactions or platelet activation. In contrast to PPI networks, kinetic models of blood function require substantially more definition of each reaction to be useful:
reaction network
kinetic rate model (i.e. reaction mechanism)
kinetic parameters
initial condition
“Reaction topology” refers to (i) and (ii). “Model parameterisation” refers to (iii) and (iv). Disease states or therapeutic intervention can occur due to altered kinetics (e.g. inactive factor IX, drug inhibition of thrombin) or altered initial condition (e.g. low factor VIII level, elevation with recombinant VIIa). Changes in reaction topology are less common. While multicomponent kinetic models will always be approximate descriptions of reality and subject to continued improvement, their ability to describe large data sets offers confidence in their use and predictive capability.
Models that define concentrations with respect to a single volume (plasma) are homogeneous. Models that define species concentrations and kinetic rate constants for the plasma (moles/L) and on the platelet surface (mole/cm2) are heterogeneous. Models that ignore the fluid-particle nature of blood and define concentrations based only on the blood or platelet rich plasma (PRP) volume are termed pseudo-homogeneous.
In modelling, the availability of knowledge and data dictates choices: a single reaction like prothrombin activation by prothrombinase may be described with an overall rate constant or many parameters to describe multi-step activation peptide release. The availability of testable data and the anticipated application of the model helps guide which approach is optimal. In isotropic systems, there is little computational penalty for reaction complexity because solving 104 ordinary differential equations (ODEs) is relatively fast. However such large models typically lack full parameterisation. Once a kinetic reaction network is formalised, reactions can also be deployed under haemodynamic conditions that include transport/biorheological effects. Such transport equations are written for the i-th diffusive and reactive soluble species in the plasma phase ci(x,y,z,t) for i = 1 to N species and take a typical form:
The above equation is a statement of conservation of mass and is a full accounting of how each species concentration changes in space and time. At each position in a domain, a species may diffuse or convect from that location, be created or destroyed by reaction, or leave the liquid (plasma) by net binding to a surface (platelet). In the above equation, the i-j reaction Rij occurs between cj and ci and requires rate constants. If a concentration is spatially uniform (isotropic) there will be no gradients and thus no net diffusive or convective mass transfer. For isotropic systems, the partial differential equations (PDEs) above will reduce to an ODE, which captures only kinetics by reaction or adsorption. Experiments in test tubes (with or without vortexing), cone-and-plate viscometers, and aggregometers tend to be isotropic (albeit highly dynamic). Thrombosis on a wall is anisotropic. In haemodynamic systems with a velocity field ν (x,y,z,t) and spatial gradients (the convection and diffusion terms above), solving 102 PDEs could take hours to weeks of computer time depending on spatial resolution.
For a system volume where a given concentration can be counted and is generally <100, significant random fluctuations are expected. Such systems, termed “stochastic” are typically solved by Monte Carlo simulation for examples that include: (i) single bond kinetics between two adhering platelets or a platelet with a surface; (ii) sub-pM levels of tissue factor (TF)/VIIa in a small volume; (iii) calcium ions in a single platelet; or (iv) <100 platelets binding at a site of laser injury. Stochastic simulations predict both the mean behaviour of a repeated experiment and the standard deviation. System volumes that contain molecules at nM concentrations or above behave in a highly repeatable and deterministic manner, lacking the variability expected of systems with stochastic random fluctuations. Classic enzyme kinetic measurements are typically conducted in a deterministic regime.
This review focuses primarily at modelling efforts that quantify protease cascades (Section 1) with some emphasis on quantifying platelet signalling (Section 2) as well as the dynamical assembly of a thrombus under flow conditions (Section 3). For clarity, individual models are named by the convention of a first author-last author descriptor.
1. MODELS OF COAGULATION AND FIBRINOLYSIS
TF-triggered coagulation of plasma
Nesheim et al. [1] developed a hypothetical thin “interface shell” at the phospholipid vesicle surface to quantify prothrombinase assembly and function. This 2-compartment model (bulk and vesicle) called “Clotspeed” required 12 parameters for kinetic rates, Kd's, lipid-binding capacity, and initial conditions. The algebraic model solved for the initial rate of thrombin generation for various initial levels of prothrombin, Xa, and phospholipid vesicle, successfully predicting that excessively high vesicle levels can dilute reactants and reduce the rate. Even for this highly purified and idealised system, several independent literature values were required, but still allowed for reasonably quantitative predictions.
The Mann laboratory further advanced modelling of the extrinsic pathway by assuming a fully activated and excess platelet surface at t=0 and deploying pseudo-homogenous rates. The Hockin-Mann model [2] used 34 ODEs and species, 42 rate constants with 10 non-zero initial concentrations for TF, VII, VIIa, X, IX, II, VIII, V, tissue factor pathway inhibitor, and antithrombin. This model predicts the reduction in initiation time and increase in peak thrombin as [TF] is increased from 1 to 25 pM. This model accurately describes the functionality of recombinant proteins and phospholipid vesicles (i.e. reconstituted plasma) when activated with lipidated TF. A parameter sensitivity analysis [3] indicated that parameter choice around the initial interactions of VIIa and VII with TF had the greatest impact on model output. Since no contact pathway was included in this model, it cannot predict blood clotting in the absence of TF.
The discourse about the differences between reconstituted plasma, platelet free plasma (PFP), PRP, and whole blood [4, 5] only highlights opportunities for improvement of both modelling and experimental design. However, such discourse might be a tempest in a test tube when compared to the even larger qualitative and quantitative difference encountered with blood clotting under haemodynamic conditions. During clotting under flow, platelet levels in a depositing thrombus are 50-200-fold greater than that of PRP. In contrast to the closed system of a test tube, thrombosis is an open system where reactive species are rapidly diluted away under flow conditions. The effects of flow are discussed in Section 3.
The Hockin-Mann reaction network was also solved stochastically by Monte Carlo simulation [6] to reveal that small reaction volumes (~20 pL) can display highly stochastic outcomes even at high levels of 5 pM TF. This is relevant to laser injury models that can display significant variability where focal reactions occur in 30 to 50 micron diameter arterioles.
The Chatterjee-Diamond model [7] extended the Hockin-Mann reaction network to include a coarse-grained description of thrombin-mediated feedback activation of the initially resting platelet in the presence of TF and/or XIIa generation (Fig. 1). This modelling effort (76 ODEs and species, 57 reactions, 105 kinetic parameters) also included the generation of factor XIIa in the presence of corn trypsin inhibitor (CTI). The model predicted the clotting of resting and convulxin-activated human blood as well as predicted the initiation time of human blood under 50 different initial conditions that titrated increasing levels of TF, Xa, Va, XIa, IXa, and VIIa (Fig 1B-D). While resting blood will clot in this model at extremely low levels of TF (1 molecule per 100 platelets), the authors concluded that CTI-treated whole blood clots in the lab due to the lack of activity of CTI against αXIIa. The XII activation was assumed to follow first-order dependence on XII concentration. Estimating a first-order rate constant (5×10-4 s-1) for XIIa generation resolves the disparity between the Hockin-Mann model prediction and the experimentally observed control with no added TF (Fig. 1B).
Fig. 1. Systems Biology model of thrombin production in the presence of thrombin-dependent activation of platelets.

The Hockin-Mann topology (unshaded, [ref. 2]) was extended (shaded blue) to include contact activation, platelet activation which reduces protein dissociation rates from complexes, thrombin-mediated cleavage of fibrinogen and fluorogenic detector, and other reactions (A) (from [7]). The Platelet-Plasma model (dotted lines) and Hockin-Mann model (solid lines) were compared to diverse conditions where the initiation time was measured by fluorogenic assay in blood treated with increasing concentrations of TF (B), prothrombinase components (Xa and Va) (C), and intrinsic pathway components (IXa, XIa) or high doses of recombinant VIIa (D).
Distinct from the pseudo-homogeneous models that do not account explicitly for lipid binding, the Bungay-Gentry model [8] treats an isotropic reaction network (73 ODEs, 31 reactions, 74 rate constants, and 17 reversible lipid adsorption reactions, 14 non-zero initial concentrations) with special emphasis on TF/VIIa, VIIIa/IXa, Xa/Va, thrombomodulin/thrombin assembly and function on cell membranes. This system has 22 fluid phase reactions, 19 lipid bound factors, and 25 lipid bound complexes. Using an average of 100 lipid head groups per protein for all factors, they predict dynamic thrombin concentration as a function of vesicle concentration for TF(t=0) = 5 pM. They identify a low, threshold level of 25 nM lipid required for thrombin production with 30-200 nM lipid range being optimal. The simulation does not have molecular scale resolution to predict the effect of vesicle composition (% phosphatidylserine) on reaction rates. Other heterogeneous models that account for platelet activation and surface binding reactions in the presence of blood flow are also discussed in Section 3.
Contact pathway kinetics
The impaired thrombosis in a factor XII knockout mouse has renewed interested in contact activation as a pharmacological target to control thrombosis with minimal effect on haemostasis. Expanded kinetic models of contact activation would help score the therapeutic potential and risks of contact pathway inhibitors. Kinetic descriptions of XIIa activation as a function of different triggers (dextran sulphate, kaolin, polyphosphate, misfolded protein, DNA, RNA, etc.) are not yet available. The kinetics of contact activation reactions as a function of high molecular weight kininogen (HMWK) or prekallikrein are also not well established, either on artificial surfaces or on platelet membranes. Finally, the exact molecular mechanism(s) by which a surface-adsorbed XII zymogen cleaves XII to XIIa is not well defined.
Guo et al. [9] fit the plasma clotting time as a function of added XIIa and added trigger (glass). Titrating XIIa into plasma or into XIIa-depleted plasma showed that 1-50 pM of added XIIa had little effect on clotting time. Thus, they concluded that contaminating trace XIIa was not the most proximal trigger of clotting, rather generation of kinetically significant XIIa involves biomaterial-autoactivation of XII and/or kallikrein amplification as the rate controlling events. In this case, the combination of cogent experimental design and reasonably applied modelling allowed for hypothesis discrimination.
Fibrin polymerisation and fibrinolysis
Partially coupled to coagulation are the processes of fibrin polymerisation and fibrinolysis. For example, thrombin may get incorporated into fibrin fibres, an important event with respect to clotting kinetics as well as rethrombosis following thrombolytic therapy. Also the production of fibrin has a large effect on clot strength during thrombosis under flow conditions [10]. The modelling of fibrin polymerisation seeks to simulate the dynamics of fibrinopeptide release as well as prediction of gelation kinetics, fibre diameter, and branching statistics. Under flow conditions, the effect of convection will suppress fibrin formation as well as flow-align the fibrin.
Weisel and Nagaswami [11] formulated ODE's for fibrin monomer production from fibrinogen, dimer formation, protofibril growth by dimer incorporation, fibre initiation by protofibril association, and fibre growth by protofibril addition. Comparing predictions to measurement demonstrated that prediction of a time lag in turbidity required a minimum protofibril length (10 to 20-mers) prior to aggregation into fibre bundles. The authors conclude that kinetic rates dictate the extent of lateral aggregation (fibre thickness) under a wide variety of conditions.
Guy and Fogelson [12] modelled the kinetics of fibrin formation on a surface exposed to flow. Isotropic aggregation/fragmentation kinetics are routinely modelled using the Smoluchowski coagulation equation, which can be solved for continuous or discrete distributions of k-mers and can be solved deterministically or stochastically. The Guy-Fogelson model includes a source of monomer (via thrombin) and sink of kmers (via convection). This PDE model follows prothrombin, thrombin, fibrinogen in space and time. Based on available kinetics for fibrinogen activation, fibrin monomer binding, and fibrin polymerisation, the overall model predicts a fibrin gel thickness that decreases dramatically from ~50 μm to <5 μm as wall shear rates increase from 100 to ~500 s-1. Such thicknesses are typical of fibrin observed in microfluidic experiments for whole blood flow over TF/collagen surfaces [10].
Anand and Diamond [13] developed a 12-species PDE model of fibrinolysis triggered by single chain urokinase or tissue plasminogen activator (tPA) which accounted for the conversion of plasminogen to plasmin, fibrinolysis, plasmin inhibition by α2-antiplasmin and macroglobulin, and inhibition of urokinase plasminogen activator and tPA by plasminogen activator inhibitor type 1. This model solved for the pressure-driven permeation and diffusion of reactive species as they bind the dissolving fibrin. A time-evolving inlet condition of species allowed for dynamic plasma levels due to an intravenous lytic regimen. This model predicted tPA-driven lysis front movement across fibrin in the presence of constant permeation velocity. Pressure driven permeation is the dominant mode of transport allowing clinically relevant arterial thrombolysis. The biophysics of thrombolysis are reviewed in [14] and have been extended with more recent modelling efforts such as the work of Wooton et al. [15] and Bannish et al. [16].
2. MODELS OF PLATELET SIGNALING
Activated platelets are required for assembly of coagulation complexes. However, the modelling of platelet activation and function remains in early-stage development. The simplest models are coarse grained descriptions that consider the platelet as either fully resting or fully activated depending on a prevailing threshold “activator” concentration, typically with the activator representing a lumping of several important species together. This lumping of soluble platelet activators facilitates computational approaches, but prevents a quantitative evaluation of pharmacological inhibitors that uniquely target a specific clotting factor or platelet pathway.
A detailed bottom-up description of ADP-mediated signalling of P2Y1 was carried out by Purvis et al. [17] using 77 reactions and 70 species. In the Purvis-Diamond model, a total of 132 fixed kinetic parameters were obtained for reactions involving: P2Y1 G-coupled protein receptor activation, phospholipase-Cβ activation and down regulation, protein kinase C translocation and activation, phosphoinositol metabolism, IP3-receptor regulation by IP3 and Ca2+, and sarco-endoplasmic reticulum Ca2+/ATPase pump function. The model accurately predicted resting Ca2+ levels and ADP dose-responses, phosphoinositide metabolism as well as the volume of the dense tubular system. Stochastic simulation of the kinetic model demonstrated that the asynchronous Ca2+ spiking observed in single platelets was the result of stochastic fluctuations expected in cells as small as platelets. In large scale modelling of the platelet metabolism, it is important that the initial condition of the model also be a valid steady state. In other words, a resting platelet stays resting until activated by a stimulus. In this case, the steady states of smaller modules within these large metabolic ODE models can be used to efficiently predict global steady states by a principle component analysis (PCA) to reduce the search space [18].
In a similar ODE approach, the Lenoci-Hamm model [19] defines kinetics for agonist stimulation of protease activated receptor (PAR) 1, PAR1 activation of Gq and G12/13, activation of phospholipase-C, generation of IP3, followed by calcium mobilisation. The model also defines intermediate signalling through protein kinase C, protein kinase B, phospholipase D to activate Rap1, CalDAG-GEF, and RIAM, which in turn activates αIIbβ3 integrin and dense granule release. With over 80 reactions and kinetic constants and 23 non-zero initial conditions, this is the first bottom-up model to mechanistically link outside-in signalling with inside-out signalling, a critical step for embedding discrete platelets into aggregation models [20, 21] or deposition models [22] of thrombus formation.
As model development of signalling through individual receptor pathways such as P2Y1 or PAR1 progresses, the bottom-up signalling approaches have yet to address signalling through combinatorial and time-dependent activators. The first platelets at a clotting site encounter collagen and thrombin to form a core region and then the subsequent platelets in the growing thrombus shell are regulated by ADP and thromboxane signalling [23]. A top-down Systems Biology model of platelet signalling can be powerful if sufficiently powered with sufficient data. Top-down refers to the relating of inputs to outputs without necessarily defining every molecular linkage. To understand how human platelets integrate numerous signals, Chatterjee et al. [24] used a high throughput assay to measure intracellular Ca2+ in response to all pairwise combinations of 6 major agonists: ADP (P2Y1, P2Y12 and P2X1 activator), convulxin (glycoprotein (GP) VI activator), U46619 (thromboxane A2 receptor agonist), SFLLRN (PAR1 agonist), AYPGKF (PAR4 agonist) and prostaglandin E2 (IP and EP receptor agonist). The Ca2+responses to 18 single agonist stimulations (each agonist at 0.1, 1, 10 × EC50) and to 135 pairwise combinations of the 6 agonists at the 3 doses produced 34,000 data points for training a neural network (NN) model, for prediction of the entire 6-dimensional platelet response space. Once trained, the NN model then successfully predicted responses to sequential additions of agonists and ternary stimulation. With 4077 NN simulations fully spanning the 6 dimensional space, 45 combinations of 4, 5, and 6 agonists (predicted to range from strong synergism to strong antagonism) were selected and confirmed experimentally, revealing a high dimensional risk at high U46619/ prostaglandin E2 ratio, consistent with the thrombotic risk of cyclooxygenase-2 therapy.
With patient-specific NN models of platelet activation, larger scale simulations of thrombosis under flow become possible. The Flamm-Diamond model [22] takes advantage of the speed of NN calculations of individual platelet Ca2+ to conduct large scale and donor-specific simulations with 103 to 105 activating platelets under conditions of flow (Fig. 2A-B). Lattice kinetic Monte Carlo simulations of platelet motion in convective and dispersive flow fields (with red blood cell-driven drift toward the wall) allowed each platelet to activate separately in response to multiple, local and dynamic agonists including collagen, ADP, and thromboxane. Additionally, pharmacological modulators can be used in the NN training data.
Fig. 2. Multiscale models of thrombosis and inner clot dynamics.
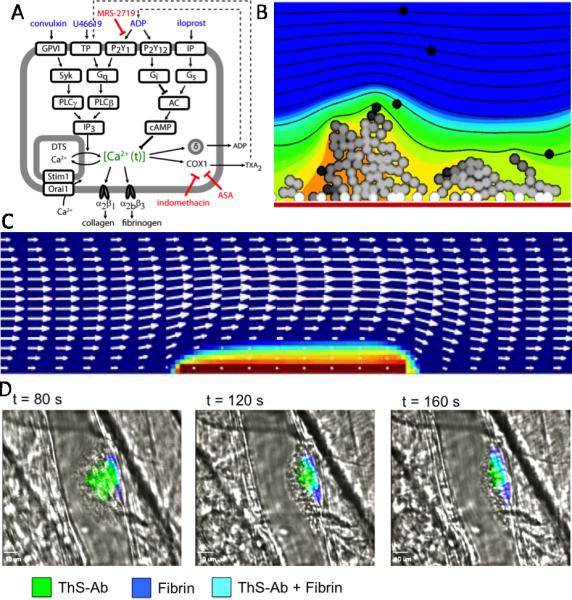
(A) Combinatorial measurements of intracellular calcium in platelets exposed to pairs of activators of GPVI, TP, P2Y1, P2Y12, and IP receptors, allowed training of a platelet activation multiscale model (B) for platelets arriving on collagen (red bar), mobilizing intracellular calcium (platelet greyscale), and releasing ADP (orange to blue colour scale) (from [22]). (C) Platelet mass and velocity field (arrows) after 30 sec perfusion at wall shear rate of 1500 s-1 over 15 fmol/cm2 of TF (from [27] used with permission). (D) Platelet accumulation at a site of laser injury with concomitant production of thrombin and fibrin (from [29]) where thrombin was detected with a thrombin sensitive fluorogenic peptide-antibody construct (ThS-Ab) that binds platelets.
For example, iloprost to activate IP receptors was part of the NN training data and allowed prediction of platelet deposition in the presence of IP activation.
3. MODELS OF BLOOD FUNCTION WITH FLOW
Thrombosis under flow
A major advance in modelling blood clotting on a TF surface under flow was the Kuharsky-Fogelson model [25], which assumes a well-mixed boundary layer near the surface. In the model of the TF surface exposed to blood flow, platelets can deposit and Xa can be generated by both extrinsic and intrinsic Xase. With 59 ODEs, the model predicted that platelet deposition is a major mechanism by which the extrinsic pathway is shut down via coverage of surface TF. The model made compelling predictions about factor deficiencies such as haemophilia A and B. The Kuharsky-Fogelson model predicted a sharp threshold concentration of surface TF necessary for the triggering of thrombin generation under flow, consistent with experimental measurements [26] that found thresholding between 2 and 10 molecules TF/μm2.
Recently, Leiderman and Fogelson [27] analysed assembly of the coagulation pathway on activating platelets that deposit on a TF surface under flow conditions. The changing velocity profile is obtained by solution of the Navier-Stokes equation with Brinkman flow through the platelet deposit. Platelets are activated by released ADP and thrombin, undergo convection-dispersion, and can adhere to the subendothelium or to other bound platelets. The kinetic model contains: 8 subendothelium reaction ODEs, 23 platelet bound reaction ODEs, 18 fluid phase PDEs, and >100 physical, kinetic, initial condition parameters. A TF density between 0 and 10 fmol/cm2 was determined to be the critical threshold level between little and maximal thrombin production (>100 nM thrombin). At a wall shear rate of 1500 s-1 and surface TF concentration of 15 fmol/cm2, ADP mediated activation of platelets was pronounced on the TF-surface at 10 to 60 sec and on the leading edge of the clot at 480-600 sec (Fig. 2C). In the simulation, thrombin-mediated activation became prominent on the outer surface of the ~30-μm thick clot between 240 and 600 sec. The addition of thrombin-mediated feedback activation of XI to XIa had relatively minor effects on total clot thrombin production indicating that this pathway might actually be difficult to observe experimentally [28]. A thrombin biosensor [29] was recently developed to detect thrombin within clots forming in microfluidic devices and in the mouse laser injury model. In these experiments, high levels of thrombin were detected in a thin ~10-50 μm layer immediately adjacent to collagen/TF in microfluidic devices or adjacent to the wall in the laser injury model (Fig. 2D).
A number of multiscale approaches are now becoming computationally feasible for the simulation of reactive platelets flowing in complex geometries, sometimes in the presence of coarse grained and approximate descriptions of coagulation proteases, platelet signalling, or fibrin polymerisation. These efforts include the use of dissipative particle dynamics [30], stochastic Cellular Potts models [31], and Lattice Boltzmann method [32].
In general, the mathematical description and numerical solution of the physical events (convection, diffusion and dispersion, cellular deformation, adhesion, embolisation) are highly complex phenomena but require only a handful of physical parameters. In contrast, the description of coagulation relies on an abundance of simple processes (e.g. E+S<->ES->E+P) that unfortunately require a large number of rate constants, many of which are not well measured for platelet surfaces or in a blood milieu. The deployment of single bond kinetics into large-scale models of thrombosis is very computationally intensive due to the near molecular length scale required to define bond formation and rupture under haemodynamic loading. The single bond models and parameterisation for von Willebrand factor/GPIb or fibrinogen/αIIbβ3 bonding remain a subject of active investigation.
Diffusional transport processes
Under static conditions, blood or plasma can be placed in contact with a surface that triggers a reaction-diffusion wave across the activating fluid. The velocity of a reaction front can be visualised and is dictated by the rate-limiting reactions and least mobile species [33]. Such experiments have revealed the role of TF in a cell monolayer controlling the initiation time and the intrinsic Xase controlling the propagation velocity [34].
When diffusion is the only transport mechanism, interesting phenomenon can be observed in plasma in contact with patterned TF. The interplay of characteristic diffusion distances and diffusion times with characteristic reaction times allows diffusion to act like an apparent “container volume”. If reactive species escape rapidly from a site by diffusion, local prevailing concentrations remain low and thus plasma clotting is extinguished. If species can build up rapidly enough relative to their diffusive dissipation, then clotting can amplify autocatalytically. Similar plasma reaction-diffusion interplays can be created with compartment volumes in microfluidic devices. Using microfluidic devices and patterned TF surfaces, Kastrup et al. [35] observed a role for a minimum TF-patch size of ~100 microns needed to trigger plasma clotting, consistent with a reaction-diffusion PDE model. It remains unclear if such a patch threshold exists in flowing blood where platelets can provide ample surface for reaction assembly and flow can alleviate diffusion limitations. For example, the patch size of a single, laser-damaged endothelial cell (~10 microns) is fully sufficient to trigger a clotting event under flow.
CONCLUSIONS
Unified blood models are emerging for combined platelet activation and coagulation. The incorporation of robust descriptions of fibrin polymerisation, fibrinolysis, and complement activation are well underway. Such models require ~10 to 100 parameters that are physics-based and account for fluid flow, membrane mechanics, and molecular/particle transport. More challenging is the validation of a robust kinetic description of platelet signalling and coagulation reactions which requires ~100 to 1000 reactions and rate parameters, either for individual reactions or from top-down data-driven approaches. Such kinetic models must at least describe the clotting rates of plasma, PRP, and whole blood when activated by TF and/or contact activators in the presence of various modulators. Prediction of known genotype/phenotype linkages is also a particularly important form of model validation.
It is a fallacy, based on curve fitting in two dimensions, to think that any model with more than 5 parameters can fit any data set. With multicomponent reaction systems where numerous pathways can be attenuated or potentiated simultaneously, an experiment on a 96- or 384-well plate or on microfluidic chips could easily follow thrombin production or platelet activation under 101 to 103 different initial conditions. Such high dimensional and time-evolving data sets are actually quite difficult and computationally challenging to fit, even with ten or a hundred parameters to adjust. When a unique set of parameters cannot be found, network topology may need revision. It is also possible to have an ensemble of different parameter sets that equally predict the data. The structure of this ensemble may indicate which parameters are highly rate controlling and which parameters have relatively minor effect on system outcome.
Even when biochemical pathways have been measured, a wide variety of values may exist for a given parameter. For example, many Km's have been published for prothrombin conversion [5] due to variations in measurement technique, experimental conditions, reagent quality, or analytical approach. Computer simulation may help: (i) reject measured values as being physiologically unbelievable, (ii) constrain acceptable bounds for a parameter to be consistent with other rates that are well known and consistently measured, (iii) demonstrate that the system output is insensitive to a particular parameter value because the species is not rate-controlling and in excess, or (iv) highlight extreme sensitivity to a parameter that requires renewed investigation [3], perhaps because it quantifies a valued drug target or the bounds of a safe pharmaceutical intervention.
Why model blood when models will always be imperfect? Blood function in the presence of flow is highly nonlinear and models help extract kinetic information from real data. When a computer model fails to predict measured data, it typically is the result of imperfect topology and not parameterisation. This means that new insights are required to explain experimental data, thus focusing hypothesis testing and experimental design. With validated models, blood responses to therapy or to disease processes are better predicted. Such Systems Biology tools, when coupled with high throughput analysis of patient-specific blood samples, would be particularly useful in characterising undefined platelet or coagulation defects as well as provide strategies for the design of patient-specific pharmaceutical therapies. As computer models of neutrophil and endothelial function emerge, the role of inflammation and vascular function will complete the description of blood as a living tissue.
ACKNOWLEDGMENTS
The author wishes to thank his many mentors, collaborators, colleagues, and students who have contributed across disciplines to the understanding of blood function. This work was funded by National Institutes of Health R01 HL-103419 to S.L.D.
Footnotes
Disclosure of Conflicts of Interest: The author states that he has no conflict of interest.
REFERENCES
- 1.Nesheim ME, Tracy RP, Mann KG. “Clotspeed,” a mathematical simulation of the functional properties of prothrombinase. J Biol Chem. 1984;259(3):1447–1453. [PubMed] [Google Scholar]
- 2.Hockin MF, Jones KC, Everse SJ, Mann KG. A model for the stoichiometric regulation of blood coagulation. J Biol Chem. 2002;277(21):18322–18333. doi: 10.1074/jbc.M201173200. [DOI] [PubMed] [Google Scholar]
- 3.Danforth CM, Orfeo T, Mann KG, Brummel-Ziedins KE, Everse SJ. The impact of uncertainty in a blood coagulation model. Math Med Biol. 2009;26(4):323–336. doi: 10.1093/imammb/dqp011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mann KG. Is there value in kinetic modeling of thrombin generation? Yes. J Thromb Haemost. 2012;10(8):1463–1469. doi: 10.1111/j.1538-7836.2012.04799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemker HC, Kerdelo S, Kremers RM. Is there value in kinetic modeling of thrombin generation? No (unless...). J Thromb Haemost. 2012;10(8):1470–1477. doi: 10.1111/j.1538-7836.2012.04802.x. [DOI] [PubMed] [Google Scholar]
- 6.Lo K, Denney WS, Diamond SL. Stochastic modeling of blood coagulation initiation. Pathophysiol Haemost Thromb. 2005;34(2-3):80–90. doi: 10.1159/000089929. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee MS, Denney WS, Jing H, Diamond SL. Systems biology of coagulation initiation: kinetics of thrombin generation in resting and activated human blood. PLoS computational biology. 2010;6(9):e1000950. doi: 10.1371/journal.pcbi.1000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bungay SD, Gentry PA, Gentry RD. A mathematical model of lipid-mediated thrombin generation. Math Med Biol. 2003;20(1):105–129. doi: 10.1093/imammb/20.1.105. [DOI] [PubMed] [Google Scholar]
- 9.Guo Z, Bussard KM, Chatterjee K, Miller R, Vogler EA, Siedlecki CA. Mathematical modeling of material-induced blood plasma coagulation. Biomaterials. 2006;27(5):796–806. doi: 10.1016/j.biomaterials.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 10.Colace TV, Muthard RW, Diamond SL. Thrombus growth and embolism on tissue factor-bearing collagen surfaces under flow: role of thrombin with and without fibrin. Arterioscler Thromb Vasc Biol. 2012;32(6):1466–1476. doi: 10.1161/ATVBAHA.112.249789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisel JW, Nagaswami C. Computer modeling of fibrin polymerization kinetics correlated with electron microscope and turbidity observations: clot structure and assembly are kinetically controlled. Biophys J. 1992;63(1):111–128. doi: 10.1016/S0006-3495(92)81594-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guy RD, Fogelson AL, Keener JP. Fibrin gel formation in a shear flow. Math Med Biol. 2007;24(1):111–130. doi: 10.1093/imammb/dql022. [DOI] [PubMed] [Google Scholar]
- 13.Anand S, Diamond SL. Computer simulation of systemic circulation and clot lysis dynamics during thrombolytic therapy that accounts for inner clot transport and reaction. Circulation. 1996;94(4):763–774. doi: 10.1161/01.cir.94.4.763. [DOI] [PubMed] [Google Scholar]
- 14.Diamond SL. Engineering design of optimal strategies for blood clot dissolution. Annual review of biomedical engineering. 1999;1:427–462. doi: 10.1146/annurev.bioeng.1.1.427. [DOI] [PubMed] [Google Scholar]
- 15.Wootton DM, Popel AS, Alevriadou BR. An experimental and theoretical study on the dissolution of mural fibrin clots by tissue-type plasminogen activator. Biotechnol Bioeng. 2002;77(4):405–419. [PubMed] [Google Scholar]
- 16.Bannish BE, Keener JP, Fogelson AL. Modelling fibrinolysis: a 3D stochastic multiscale model. Math Med Biol. 2012:4. doi: 10.1093/imammb/dqs029. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purvis JE, Chatterjee MS, Brass LF, Diamond SL. A molecular signaling model of platelet phosphoinositide and calcium regulation during homeostasis and P2Y1 activation. Blood. 2008;112(10):4069–4079. doi: 10.1182/blood-2008-05-157883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purvis JE, Radhakrishnan R, Diamond SL. Steady-State Kinetic Modeling Constrains Cellular Resting States and Dynamic Behavior. PLoS computational biology. 2009;5(3):e1000298. doi: 10.1371/journal.pcbi.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lenoci L, Duvernay M, Satchell S, Dibenedetto E, Hamm HE. Mathematical model of PAR1-mediated activation of human platelets. Mol Biosyst. 2011;7(4):1129–1137. doi: 10.1039/c0mb00250j. [DOI] [PubMed] [Google Scholar]
- 20.Laurenzi IJ, Diamond SL. Monte Carlo simulation of the heterotypic aggregation kinetics of platelets and neutrophils. Biophys J. 1999;77(3):1733–1746. doi: 10.1016/S0006-3495(99)77019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flamm MH, Sinno T, Diamond SL. Simulation of aggregating particles in complex flows by the lattice kinetic Monte Carlo method. J Chem Phys. 2011;134(3):034905. doi: 10.1063/1.3521395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flamm MH, Colace TV, Chatterjee MS, Jing H, Zhou S, Jaeger D, Brass LF, Sinno T, Diamond SL. Multiscale prediction of patient-specific platelet function under flow. Blood. 2012;120(1):190–198. doi: 10.1182/blood-2011-10-388140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R, Diamond SL, Brass LF. Hierarchical organization in the hemostatic response and its relationship to the platelet signaling network. Blood. 2013:9. doi: 10.1182/blood-2012-09-457739. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee MS, Purvis JE, Brass LF, Diamond SL. Pairwise agonist scanning predicts cellular signaling responses to combinatorial stimuli. Nat Biotechnol. 2010;28(7):727–732. doi: 10.1038/nbt.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuharsky AL, Fogelson AL. Surface-Mediated Control of Blood Coagulation: The Role of Binding Site Densities and Platelet Deposition. Biophys. J. 2001;80(3):1050–1074. doi: 10.1016/S0006-3495(01)76085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okorie UM, Denney WS, Chatterjee MS, Neeves KB, Diamond SL. Determination of surface tissue factor thresholds that trigger coagulation at venous and arterial shear rates: amplification of 100 fM circulating tissue factor requires flow. Blood. 2008;111(7):3507–3513. doi: 10.1182/blood-2007-08-106229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leiderman K, Fogelson AL. Grow with the flow: a spatial-temporal model of platelet deposition and blood coagulation under flow. Math Med Biol. 2011;28(1):47–84. doi: 10.1093/imammb/dqq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fogelson AL, Hussain YH, Leiderman K. Blood clot formation under flow: the importance of factor XI depends strongly on platelet count. Biophys J. 2012;102(1):10–18. doi: 10.1016/j.bpj.2011.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welsh JD, Colace TV, Muthard RW, Stalker TJ, Brass LF, Diamond SL. Platelet-targeting sensor reveals thrombin gradients within blood clots forming in microfluidic assays and in mouse. J Thromb Haemost. 2012;10(11):2344–2353. doi: 10.1111/j.1538-7836.2012.04928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filipovic N, Kojic M, Tsuda A. Modelling thrombosis using dissipative particle dynamics method. Philos Transact A Math Phys Eng Sci. 2008;366(1879):3265–3279. doi: 10.1098/rsta.2008.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Z, Chen N, Kamocka MM, Rosen ED, Alber M. A multiscale model of thrombus development. J R Soc Interface. 2008;5(24):705–722. doi: 10.1098/rsif.2007.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamagawa M, Kaneda H, Hiramoto M, Nagahama S. Simulation of thrombus formation in shear flows using Lattice Boltzmann Method. Artif Organs. 2009;33(8):604–610. doi: 10.1111/j.1525-1594.2009.00782.x. [DOI] [PubMed] [Google Scholar]
- 33.Dashkevich NM, Ovanesov MV, Balandina AN, Karamzin SS, Shestakov PI, Soshitova NP, Tokarev AA, Panteleev MA, Ataullakhanov FI. Thrombin activity propagates in space during blood coagulation as an excitation wave. Biophys J. 2012;103(10):2233–2240. doi: 10.1016/j.bpj.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ovanesov MV, Ananyeva NM, Panteleev MA, Ataullakhanov FI, Saenko EL. Initiation and propagation of coagulation from tissue factor-bearing cell monolayers to plasma: initiator cells do not regulate spatial growth rate. J Thromb Haemost. 2005;3(2):321–331. doi: 10.1111/j.1538-7836.2005.01128.x. [DOI] [PubMed] [Google Scholar]
- 35.Kastrup CJ, Runyon MK, Shen F, Ismagilov RF. Modular chemical mechanism predicts spatiotemporal dynamics of initiation in the complex network of hemostasis. Proc Natl Acad Sci U S A. 2006;103(43):15747–15752. doi: 10.1073/pnas.0605560103. [DOI] [PMC free article] [PubMed] [Google Scholar]