

Abstract
In the central nervous system (CNS), mastocytes and glial cells (microglia, astrocytes and oligodendrocytes) function as sensors of neuroinflammatory conditions, responding to stress triggers or becoming sensitized to subsequent proinflammatory challenges. The corticotropin-releasing hormone and glucocorticoids are critical players in stress-induced mastocyte degranulation and potentiation of glial inflammatory responses, respectively. Mastocytes and glial cells express different toll-like receptor (TLR) family members, and their activation via proinflammatory molecules can increase the expression of connexin hemichannels and pannexin channels in glial cells. These membrane pores are oligohexamers of the corresponding protein subunits located in the cell surface. They allow ATP release and Ca2+ influx, which are two important elements of inflammation. Consequently, activated microglia and astrocytes release ATP and glutamate, affecting myelinization, neuronal development, and survival. Binding of ligands to TLRs induces a cascade of intracellular events leading to activation of several transcription factors that regulate the expression of many genes involved in inflammation. During pregnancy, the previous responses promoted by viral infections and other proinflammatory conditions are common and might predispose the offspring to develop psychiatric disorders and neurological diseases. Such disorders could eventually be potentiated by stress and might be part of the etiopathogenesis of CNS dysfunctions including autism spectrum disorders and schizophrenia.
1. Introduction
Signaling between nervous and immune systems is in part due to the fact that these two systems share ligands and receptors. The cellular components involved in these interactions within the central nervous system (CNS) are mainly mastocytes, also called mast cells, and glia. In human brain, mastocytes are very scarce and are preferentially located in perivascular territories. By contrast, glial cells comprise about 90% of the total cell content in the CNS and are classified as microglia and macroglia (astrocytes, oligodendrocytes, and ependymal cells) [1]. Representative of the immune system in the CNS are mastocytes and microglia, two cell types derived from hematopoietic cells of the bone marrow that migrate to the brain before closure of the blood brain barrier (BBB) [2, 3].
The CNS challenged by different aggressions frequently elicits immune and inflammatory responses [4, 5]. Mastocytes and microglia are efficient sensors of adverse endogenous or exogenous conditions of the CNS [2, 6]. Moreover, stress conditions induce rapid mastocyte degranulation via the hypothalamic peptide corticotropin-releasing hormone (CRH) [7] and exogenous danger molecules like polyinosinic-polycytidylic acid (poly (I:C)), bacterial lipopolysaccharide (LPS), and peptidoglycan (PGN), which are detected by mastocytes and microglia via toll-like receptors (TLRs) [8, 9]. Also, glucocorticoids (GCs) play a relevant role in stress-induced potentiation of neuroinflammatory responses by sensitizing microglia to proinflammatory stimuli [10]. As part of these responses, glial TLRs, connexin hemichannels (Cx HCs), pannexin (Panx) channels might be key players in acute and chronic neurodegenerative diseases characterized by open BBB, demyelinization, and neuronal degeneration [11].
The causes of various chronic diseases that affect the CNS, such as Alzheimer's disease (AD), Parkinson's disease (PD), and multiple sclerosis (MS), are complex and can be related to multiple factors. Notably, the innate host defense has been demonstrated to play an active role in promoting neurodegeneration [12, 13]. However, the possible role of these cellular and molecular elements during brain ontogenesis and the consequences in the adult CNS remain unknown. This review presents possible implications of glial toll-like receptors (TLRs) and Cx HC and Panx channels activation after potentiation by stress in CNS dysfunctions.
During pregnancy, viral infections are common and emerge to predispose the offspring to develop psychiatric diseases [14, 15]. Viral mimic polyinosinic:polycytidylic acid [poli (I:C)] resembles the structure of double-stranded RNA (dsRNA) generated in host cells during viral replication, and it is recognized by TLR3 that activates the innate immune response [16]. The administration of poly (I:C) is a way to trigger the innate immune response, which mimics the early phase of viral infections [17], and avoids the use of infectious agents, and treatments can be standardized and experiments may be easily compared [18]. All together, they represent an interesting area because perinatal infections, particularly those of viral etiology, are frequent and have been associated with diverse alterations of adult CNS, including schizophrenia and autism [19, 20].
2. Toll-Like Receptors (TLRs): Their Expression and Functions in Brain Cells
TLRs are highly conserved germ line-encoded pattern-recognition receptors that initiate innate immune responses via recognition of pathogen-associated molecular patterns (PAMPs) as well by recognition of danger-associated molecular patterns (DAMPS) that correspond to endogenous ligands released after tissue injury or cellular stress, such ATP, histones, heat-shock proteins, mRNA, high-mobility group box-1 protein (HMGB1), surfactant proteins A and D, and mitochondrial proteins [21]. Activation of TLRs triggers a cascade of intracellular events leading to activation of several transcription factors, including NF-κB, activator protein-1 (AP-1), and IFN-regulatory factor-3 (IRF-3) and -7 that regulate the expression of various cytokines and chemokines, responses that are performed in the CNS mainly by mastocytes and microglia. In addition, activation of innate immune responses via TLRs is a prerequisite for the generation of adaptive immune responses [22] that become relevant in autoimmune diseases such as experimental autoimmune encephalomyelitis (EAE).
The number of molecular members that comprise the TLR family is ten in humans (TLRs 1–10) and twelve in mice (TLRs 1–9; TLRs 11–13) [22]. Some TLRs can be expressed on the cell surface (TLRs 1, 2, 4, 5, 6, and 10) or in intracellular compartments (TLRs 3, 7/8, and 9), but others can be found in both the cell membrane and intracellular compartments (TLR3 and TLR7; endosomes and endoplasmic reticulum) [21]. Each TLR detects distinct PAMPs derived from viruses, bacteria, mycobacteria, fungi, or parasites. For example, TLR3 and TLR7/8 detect ds and single-stranded (ss) RNAs from virus, respectively; TLR4 responds to LPS from Gram-negative bacteria; and TLR9 senses bacterial DNA that contains unmethylated cytosine-guanosine dinucleotides (CpG) [22–25].
In the adult brain, mastocytes are mainly found in leptomeninges [2] and thalamus close to the BBB [26, 27], but they are also present early in brain ontogeny [28, 29]. Mastocytes can be activated by antigens that induce crosslinking of IgE bound to mast cells, CD47 recognition, calcium ionophore, ATP, compound 48/80, and also by recognition of DAMPS or PAMPS [26, 27]. If these activators bind to mastocytes for a short period of time (from seconds to a few minutes), they lead to rapid degranulation and release bioamines, proteoglycans, proteases, ATP, TNF-α and chemokines stored in preformed granules, whereas activations of longer durations lead to the release of newly formed cytokine (TNF-α, IL1β, and granulocyte macrophage colony-stimulating factor (GM-CSF)), and chemokine (C–C motif) ligand 3 (CCL3), enzymes (tryptase, chymase, carboxypeptidase), lipid mediators (prostaglandins, leukotrienes, thromboxanes, and platelet-activating factor), and nitric oxide (NO), mediating the recruitment of effector cells, fluid extravasation, and tissue inflammation [30, 31].
Murine mastocytes express the mRNA of TLRs 1–4 and 6–9 but not TLR5 [32–36]. Moreover, human mastocytes express the mRNA of TLRs 1–10 with the exception of TLR8 [9, 37–39]. In mastocytes, TLR ligands, such as poly (I:C), LPS, R-848, and CpG oligodeoxynucleotide, promote IL-6 and TNF-α secretion as well as regulated upon activation, normal T cell expressed and secreted (RANTES) and macrophage inflammatory protein (MIP) without significant degranulation [35, 38, 40, 41]. More specifically, in rodent mastocytes, binding of LPS to TLR4 induces the release of de novo expressed (without degranulation) and secreted TNF-α, IL-5, IL-10, and IL-13 but not GM-CSF, IL-1, or leukotriene C4 (LTC4), while binding of PGN to TLR2 induces degranulation that includes histamine release [9, 34, 37].
In three different mouse models, where TLR3, TLR4, and TLR7 were specifically deleted in mastocytes, the recruitment of effector CD8+ T cells, neutrophils, and dendritic cells, respectively, was totally avoided after agonist stimulation [33, 42, 43]. This implies that mastocytes recognize, respond, and coordinate immune responses, features that are suppressed by TRLs 3, 4, and 7.
Not only ligands, but also immunological host environments are decisive for mastocyte activity. In human mastocytes, prolonged lymphotoxin-alpha (LTA) and PGN exposure downregulate FcεRI, decreasing degranulation products after an antigen crosslinking reaction [39]. Poly (I:C) treatment also decreases degranulation in an in vitro allergic model, affecting mastocyte adhesion to fibronectin and vitronectin through conformational inactivation of CD29, the receptor of fibronectin [44]. Moreover, LPS and PGN induce mastocytes migration in vitro after brief treatment with IL-6 and CCL5/RANTES, respectively [45].
The activation and migration of mastocytes occur in several neurologic disorders including MS [46, 47], PD [48], amyotrophic lateral sclerosis (ALS) [49, 50], AD [51], traumatic injury [52], ischemic and hemorrhagic stroke [53, 54], and viral infections [55]. Mastocytes activation and migration are critical for the increased BBB permeability and progression of neuroinflammation. Mastocytes also degranulate upon recognition of myelin basic protein and purinergic P2 receptors [56]. Additionally, proteases released during mastocyte degranulation can also degrade myelin components [57], contributing to myelin damage in the CNS and peripheral nervous system.
Microglia can rapidly respond to pathogens through their TLRs but do not sense apoptotic cells through the same mechanism [58, 59]. They express mRNAs encoding for TLRs 1 to 9. Moreover, levels of TLRs expressed by microglia vary depending on the stages of development or pathological conditions [8]. TLR activation induces a cascade of intracellular events leading to the activation of several transcription factors, including NF-κB, AP-1, IRF-3, and IRF-7 that regulate the expression of many molecular elements of inflammatory responses [60].
In human microglia, activation of TLR3 by agonists such as poly (I:C) induces a strong proinflammatory response that allows microglia to mediate the development of T-helper 1 (Th1) cells [61]. Moreover, infection with the West Nile virus (a retrovirus that produces dsRNA) in mice lacking TLR3 shows reduced microglial activation and more resistance to lethal infection with reduced viral load and inflammatory responses in the brain compared to wild-type mice [62].
Mastocytes release several cytokines in response to TLR2 activation including TNF-α, IL-4, IL-5, IL-6, and IL-13. Meanwhile, the activation of TLR4 causes release of TNF-α, IL-6, IL-13, IL-5, IL-10, and eotaxin [34, 63–65]. Also, numerous chemokines including CCL5/RANTES, can also induce a proinflammatory profile in microglia [37, 38, 59, 66]. IL-33 derived from microglia modulates the activation of P2 receptors on mastocytes inducing secretion of IL-6, IL-13, and monocyte chemoattractant protein-1 (MIP-1), which in turn can modulate the microglia activity [67]. Tryptase is the main protease secreted by human mastocytes. It is elevated in the CSF of patients with MS [68]. It induces microglia to secrete TNF-α, IL-6 [69], and ROS and activate in microglia proteinase-activated receptor-2 (PAR-2), a G protein-coupled receptors widely expressed in neurons, astrocytes, and microglia that are implicated in the pathogenesis of ischemia and neurodegeneration [70], because it induces widespread inflammation [71–73]. The activation of microglial PAR-2 also upregulates P2X4 receptors and promotes release of brain-derived neurotrophic factor, TNF-α, and IL-6 that upregulate the expression mastocyte of PAR-2, which results in activation and release of TNF-α [67].
It is interesting to note that mastocytes but not microglia have been described to be the first responder in CNS injuries, such as perinatal hypoxia-ischemia. Many cells produce TNF-α in response to several stimuli, but mastocytes store TNF-α in granules, and thus they can release it before other cells including microglia and endothelial cells. Additionally, the recruitment and activation of mastocytes occur previous to responses elicited by neurons, glia, and endothelial cells. Therefore, mastocytes initiate acute inflammations in response to a stimulus, and when inhibited, the brain damage decreases, as observed when the early mastocyte response is inhibited with cromolyn (a mastocyte stabilizer), and then significant neuroprotection is observed [74].
A strong link between LPS, the TLR4 agonist, and brain injury both in fetal and newborn animals has been demonstrated [75]. LPS injected into developing mouse and rat brains has been shown to induce injury in white matter [76]. Moreover, systemic LPS administration to preterm fetal sheep induces cerebellar white matter injury [77], and in vitro assays demonstrate that TLR4 gene deletion prevents LPS-induced oligodendrocyte death [78].
In astrocytes, TLRs mediate the first step of innate immune cell activation. The expression of TLRs is limited in astrocytes, probably because of the neuroectodermal origin of astroglia [79]. These cells express TLR2, which increases in response to proinflammatory stimuli [22, 80]. They also express TLR3 that responds to poly (I:C), hence producing among other cytokines IL-6 that contributes to inflammation in humans and mice [80–82]. The gene profile of astrocytes activated via TLR3 shows neuroprotective mediators and cell growth factors, that is, differentiation and migration molecules comprising a neuroprotective response rather than a proinflammatory phenotype [83, 84].
TLR4 has been shown to participate in stroke-caused brain damage [85–87] and in AD [88, 89]. Likewise, TLR4 could play a pivotal role in demyelinating diseases, such as MS [90]. TLR activation in astrocytes also induces the release of several cytokines and chemokines [91]. Both TLR agonists and cytokines induce the expression of chemokines CCL2, CCL3, CCL5, intercellular cell adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1). Moreover, LPS and poly (I:C) induce the production of IL-6, TNF-α, IFN-α4, IFN-β, and iNOS [80]. In addition, poly (I:C) activation induces CXCL-10 production [92]. LPS and dsRNA in parallel induce astrocyte activation, which leads to IL-1α, IL-1β, IL-6, TNF-α, GM-CSF, LTβ, and TGF-β3 secretion, although macrophage migration inhibitory factor (MIF) secretion is inhibited. However, no effect has been found on anti-inflammatory cytokines such as IL-2, IL-3, IL-4, IL-5, IL-10, TGF-β1, TGF-β2, and TNF-β [11, 93].
Recently, in addition to TLR2, TLR3, and TLR4, TLR1, TLR5, TLR6, and TLR7/8 have been found in astrocytes, but their functional roles remain unknown [22, 84]. Therefore, the understanding of the detailed mechanisms of TLR signaling in astrocyte activation in CNS inflammatory conditions still needs further investigation.
The expression and function of TLRs in oligodendrocytes, unlike other glial cells, have been poorly studied. Only TLR2, -3, and -4 have been evaluated [94], being these receptors related to the regulation of inflammatory processes, gliosis, and remyelination after injury [95, 96]. Knockout mice for TLR2 and TLR4 exposed to spinal cord injuries show a lower remyelination capacity, and thus it is believed that these receptors would have a key role in the formation of myelin [84].
Astrocyte dysfunction triggers primary microglial activation, which induces demyelination [78, 97]. Furthermore, injection of LPS in the bone marrow induces a rapid oligodendrocyte loss, followed by an increase in oligodendrocyte number [98]. After acute demyelination induced by LPS, a more widespread distribution of oligodendrocyte precursor cells is triggered by the activation of microglia/macrophages, which is an event that accelerates remyelination [99, 100].
Rats treated with zymosan, a TLR2 agonist, show oligodendrocyte and axonal loss without regeneration [98]. In addition, rats treated with LPS, that is, a TLR4 agonist, show oligodendrocyte death and demyelination [76, 101]. Also, LPS-induced spinal cord damage shows significant demyelinization associated with an important reduction in the amount of oligodendrocytes [102]. Other researchers have shown that TNF-α and TNFR1 play a relevant role in oligodendrocyte death induced by TLR activation [103–105]. However, Bsibsi et al. [100] showed that zymosan and LPS reduce survival, differentiation, and myelin-like membrane formation, while poly (I:C) triggers apoptosis in rat oligodendrocyte cultures. These findings suggest that TLRs play a pivotal role in oligodendrocyte differentiation and myelination, both in physiological and pathological conditions. Compared to other cell types, TLRs play direct roles in regulating various aspects of oligodendrocyte's behavior. However, the apparent contradiction between the effects of LPS and zymosan on oligodendrocytes in different models has not been clarified. Future research could help to determine the functionality of TLR receptors in oligodendrocytes under physiological and pathological conditions.
With regard to the neuroendocrine modulation of the activity of TLRs, this can take local, regional, and systemic routes [106]. Local components include neuropeptides such as substance P, CRH, calcitonin gene-related peptide (CGRP), and endogenous opioids [106] released by peripheral nervous system. Among the regional components, the sympathetic and parasympathetic innervations release neurotransmitters (adrenaline and acetyl choline), and neuropeptides (neuropeptide Y or vasoactive intestinal peptide (VIP)) play a relevant role. Also at a regional level, a neuronal component regulates immunity through the innervation of immune organs and release of noradrenaline, and also a hormonal component regulates immunity systemically by means of adrenaline released from the medulla of the adrenal glands [106], whereas the systemic factors include the neuroendocrine system through the hypothalamic-pituitary-adrenal (HPA) axis and the anti-inflammatory effects of GCs. Furthermore, neuropeptides including cholecystokinin (CCK), somatostatin, melanocyte-stimulating hormone (MSH), VIP, and gastrin also reduce the inflammatory response [107].
Additionally, IL-1β participates in several aspects of the immune response to infections such as regulation of inflammation and modulation of adaptive immune responses against viral infections [108, 109]. The inflammasome is a multiprotein complex that activates a platform for caspase-1 and caspase-1-dependent proteolytic maturation and secretion of interleukin-1β (IL-1β). Several inflammasomes have been described being the NLRP3 inflammasome the most extensively studied [110]. It requires two signals. The signal 1 corresponds to TLR ligands or TNF-α, and the signal 2 includes ATP, amyloid-β (Aβ), K+ efflux, pore-forming toxins, and silicic and uric acid crystals [111–113]. After TLR2 and TLR4 activation, secretion and maturation of cytokines IL-1β and IL-18 depend on caspase-1 cleavage of their premature forms. In both cases, inflammasome complex proteins mediate caspase-1 activation in the presence of high concentrations of extracellular ATP through activation of P2X7 receptors [114, 115]. Activation of P2X7 receptor leads to a large membrane pore formation identified as Panx1 channels [116, 117], which recently has been found critical for caspase-1 activation [116, 118]. Not only in immune cells but also in neurons and astrocytes, Panx1 recruitment mediates caspase-1 activation [119], suggesting that during infections, overall TLRs and Panx1 channels could enhance inflammatory responses.
3. Cx HCs and Panx1 Channels in Glial Cell and Mastocytes
One HC corresponds to one-half of a gap junction channel and is located at unapposed cell surfaces serving as communication pathway between the intra- and extracellular compartments. Two types of HCs are formed in most cells, and they are generally coexpressed [120]. One of them is formed by connexins (Cxs, 21 in humans) and the other by Panxs 1–3. HCs provide a membrane pathway for releasing signaling molecules (e.g., ATP, glutamate, PGE2, and NAD+) and thus are recognized as paracrine/autocrine communication pathways under normal and pathological conditions [121, 122]. Inflammation is a key condition in neurodegeneration that occurs in postischemic brain, diabetes, MS, PD, AD, and possibly in various other neurodegenerative diseases [123, 124]. In neuroinflammatory conditions, the successive activation of different glial cells via HCs has been partially demonstrated [125, 126], and mastocytes are likely to be involved in early steps of different pathological conditions (Figure 1).
Figure 1.
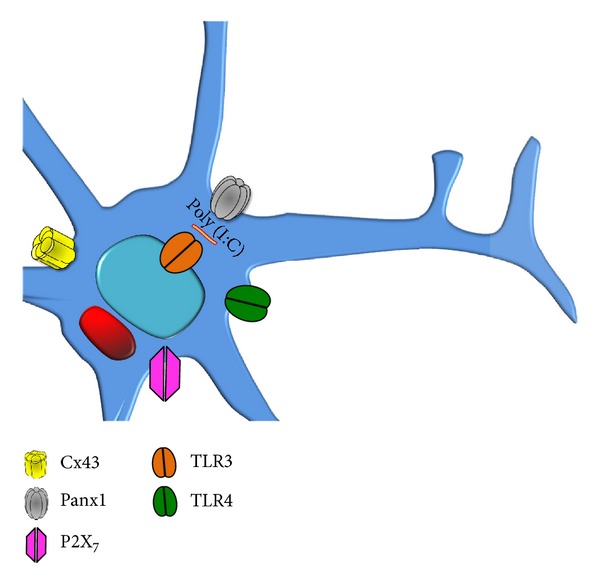
Expression of toll-like receptors in microglia and their relationship with pannexin channels and P2X7 receptors. Microglia express TLRs 1 to 9, and in this figure only, TLRs 3 and 4 are shown responding to pathogen-associated molecular patterns of virus (TLR3) and Gram negative bacteria (TLR4). TLR3 is expressed in endosomal membranes (and also in cell surface) and recognizes nucleic acids of virus (dsRNA) and poly (I:C). We propose that microglia under activation with TLR ligands increase the expression of Panx1 channels and connexin HCs and the activity of P2X7 receptors.
As mentioned previously, the degranulation response of mastocytes is an early and rapid response and might require precise coordination where HCs could be essential. Mastocytes express Cxs 32 and 43 [127], but to our knowledge, it remains unknown whether they form functional HCs. In addition, no clear evidence of Panx1 expression in mastocytes has been published, but activation of P2X7 receptors leads to the formation of membrane pores permeable to molecules up to about 900 kDa with single currents, similar to what has been described for Panx1 channels, along with histamine release [117, 128]. Since the degranulation process depends on influx of extracellular Ca2+ [129], it is possible that Panx1 channels participate in ATP release, and then ATP activates P2X7 receptors, which are Ca2+ permeable, allowing the influx of Ca2+ required for the mastocyte degranulation response. Then, glial cells become involved and microglial cells respond before astrocytes (within several minutes to few hours).
In the normal CNS, microglial cells are in a resting state and are sparsely distributed. They express the macrophage marker CD11b, low levels of CD45, and practically undetectable levels of major histocompatibility complex (MHC) class II molecules, CD40, and CD86. In vitro, the microglia activation process is characterized by an upregulation of CD45, MHC class II, and the costimulatory molecules CD40 and CD86 [130, 131]. The expression of MHC II antigens is a characteristic feature of antigen-presenting cells, and their coexpression with costimulatory molecules is a hallmark of microglial cells' ability to interact with other cells, such as T cells.
Activated, microglia proliferate and migrate to the injury site where they form cell aggregates and secrete pro- and anti-inflammatory cytokines and chemokines, NO, and growth factors [132]. The activation of microglia can be acute or chronic, and this would depend not only on the duration of an external stimulus but also on the quality of the stimulus (stress, infection, inflammation, and signals from damaged neurons) [133]. In fact, they show differences when activation is induced by stress or inflammation. For instance, acute stress induces morphological activation of microglia and increased c-Fos expression in the periaqueductal gray matter but not in the surrounding midbrain. If activation is chronic, it can lead to microglial overactivation followed by microglial degeneration [134]. Therefore, activated microglia secrete TNF-α and IL-1β, which in astrocytes induce opening of Cx43 HCs leading to the release of ATP and glutamate by astrocytes, which can kill neurons through the activation of Panx1 channels, P2X7 receptors, and NMDA receptors in neurons [135].
Another way of cell-cell interaction used by activated microglia can be found in Cx- and Panx-based channels. Microglia express low to undetectable levels of Cx32, Cx36, Cx43, and Cx45 [136–139]. They also express Panx1, and treatment with Aβ 25–35 has been shown to increase its surface levels [126]. Similarly, the expression of Cx43 is upregulated in cultured rat/mouse microglia treated with LPS or TNF-α plus IFN-γ [136], calcium ionophore plus phorbol 12-myristate 13-acetate [140], or PGN derived from Staphylococcus aureus [139]. However, the possible functional role of Cx-based HCs expressed by activated microglia remains to be elucidated.
Under normal conditions, astrocytes are highly coupled with each other, forming intercellular networks [141], through which Ca2+ waves propagate [142]. Extracellular ATP acts as a paracrine messenger in these waves, since it activates purinergic receptors (P2X and P2Y) in astrocytes of surrounding cells, thus resulting in an increase of [Ca2+]i [143]. The mechanisms for ATP release from astrocytes may include vesicle-mediated exocytosis [144] and diffusion through Cx43 HCs [125, 145, 146] and/or channels formed by Panx1 [147]. Astrocytes also release several transmitters called “gliotransmitters,” including glutamate [148], GABA [149], ATP [150], and adenosine [151]. Increases in [Ca2+]i can induce the release of gliotransmitters that promote increases in [Ca2+]i in neighboring neurons, for example, through ATP- and glutamate receptor-dependent pathways [148]. The increased [Ca2+]i occurs in local astroglia as well as in astrocytes located more distantly. Gliotransmitters might affect diverse neuronal functions including arborization and neuronal plasticity [142] as well as more complex functions such as fear memory [152]. Thus, astrocytic Cx HCs and Panx1 channels might be molecular targets to prevent undesired effects induced by stress.
Most astrocytes also express Cx30 and Cx43 [153], and at least Cx43 forms HCs that are activated by proinflammatory cytokines, hypoxia-reoxygenation, and high glucose [135]. For instance, LPS does not induce cell permeabilization to fluorescent dyes in primary cultures highly enriched with astrocytes of newborn brains, but astrocytes cocultured with microglia respond to LPS with a large increase in Cx43 HC activity [154]. Moreover, the effect of LPS is mimicked by exogenous applied TNF-α and IL-β, indicating that astrocytes do not respond to LPS in the absence of microglia. Moreover, astrocytes previously exposed for 24 h to medium conditioned by Aβ-treated microglia (CM-Aβ) are permeabilized via Cx43 HCs [126]. As part of the mechanism, TNF-α and IL-1β have been shown to mimic the effect of CM-Aβ, and neutralizing TNF-α with soluble receptors and IL-1β antagonists abrogated this effect [125]. Recent in vivo studies have demonstrated that Cx43 HCs are critical mediators of postischemic white and gray matter dysfunction and injury [155]. Moreover, upregulation of astroglial Panx1 channels and Cx43 HCs has been found using an experimental model of brain abscess [156], suggesting that both channel types could play an orchestrated function in some inflammatory responses. Cx43 HCs of reactive astrocytes favor the release of excitotoxic compounds, ATP, and glutamate, which activate neuronal P2X7 receptors, NMDA receptors, and Panx1 channels, hence promoting neurodegeneration [125]. Activation of neuronal Panx1 channels by ATP and glutamate released through Cx43 HCs from astrocytes exposed to CM-Aβ was shown to induce neuronal death [126]. Therefore, it has been proposed that blockade of astroglia and/or neuronal Cx HCs and Panx1 channels of the inflamed nervous system may represent a strategy to reduce neuronal loss in various pathological states [157–159]. Additionally, the effect of the maternal environment on the developing CNS in the offspring has been analyzed in fetal nonhuman primates. To this end, mothers were subject to a high-fat diet (HFD), and the CNS of the fetuses showed increased levels of IL-1β and IL-1 type 1 receptor, as well as a rise in microglia activation markers, suggesting the activation of the local inflammatory response [160]. Under the previous conditions, it is possible that microglia and astrocytes also present upregulation of HC activity, but this needs experimental demonstration.
Oligodendrocytes might respond within the same time frame as astrocytes, since they can communicate via gap junctions as previously described herein. These cells are responsible for producing and maintaining myelin from the earliest stages of embryonic development to adulthood [161]. Like other cells of the CNS, oligodendrocytes have low renewal capacity [162]. However, oligodendrocyte precursor cells induce remyelination, following the loss of myelin as a consequence of an injury [163]. Many of their functions are accomplished by the expression of a variety of interactions between Cx- and pannexin-based channels. Oligodendrocytes form gap junction channels with cell bodies of adjacent oligodendrocytes and between layers of myelin, called reflective gap junctions [164]; oligodendrocytes form gap junctions with astrocytes as well [165]. Collectively, this gap junction communicated network helps to absorb and remove extracellular K+ and glutamate released during neuronal activity, thus generating a spatial buffer where ions and molecules are diluted among cell communicated via gap junction channels [165–167].
The study of demyelinating diseases, consisting of loss or destruction of myelin, has revealed Panx1 channels, Cx HCs, and gap junction channels as key factors in oligodendrocyte survival, as well as neuroprotection and myelin maintenance [168]. Oligodendrocytes express three different connexins: Cx29, Cx32, and Cx47 [169]. Cx32, but not Cx29 or Cx47, is known to form functional HCs. Moreover, by means of the qPCR technique, the mRNA of Panxs 1 and 2 was detected in primary cultures of oligodendrocytes obtained from optic nerves of 12-day-old rats. Both were located in somas as well as in the layers of the myelin sheath [170]. Extracellular ATP mediates the ischemic damage to oligodendrocytes and is partially explained by the activation of Panx1 channels [170].
Both genetic and/or inflammatory diseases triggered by viral or toxic sources may affect myelin formation (hypomyelinating diseases) or its maintenance (demyelinating diseases) as it has been found in human diseases associated with HCs formed by mutated Cxs [161]. The first event in pathological manifestations of demyelinating disease of the CNS is the disruption of the BBB that leads to access of demyelinating antibodies [161, 171–174]. Also, activated T cells entering the CNS mediate the release of inflammatory cells, which together with activated microglia release proinflammatory cytokines that promote oligodendrocyte death in vitro [175–178]. TNF-α binding to its receptor can induce oligodendrocyte apoptosis directly [179]. Indirectly, TNF-α and IFN-γ can activate microglia and/or macrophage that destroy oligodendrocytes by oxidative stress [180, 181].
Myelin repair occurs after acute inflammatory lesions, such as MS. This repair is called remyelination, and its process, mediated by oligodendrocyte progenitor cells, is associated with functional recovery [163]. It has been shown that chemokine- (CXCL-) 2 and proinflammatory cytokines, such as IL-1β and IL-6, promote oligodendrocyte progenitor cell proliferation, differentiation, and remyelination [163]. Under inflammatory conditions, oligodendrocytes show upregulation of MHC I molecules, which are constitutively expressed, as well as Fas, IFN-γ, and TNF-α receptors (TNFRI-II), transforming them into targets for CD8+ cells [175, 176, 182–185]. Under control conditions there is no expression of MHC II molecules in these cells [186, 187]. However, cultured oligodendrocytes treated with IFN-γ in the presence of the synthetic GC (dexamethasone) express MHC II molecules [188], suggesting that under stress they could interact with CD4 T lymphocytes and either activate immune reactions or become the targets of T-cell-mediated cytotoxic attack.
An excess of extracellular ATP is an activator of both innate and acquired immunities, acting as a DAMP that is chemotactic factor for neutrophils, and a strong regulator of activation, death, and survival of microglial cells [189–191]. Pathway for ATP release is highly variable and includes connexin HCs, Panx1 channels, volume-regulated anion channel (VRAC), purinergic P2X7 receptor, and/or vesicular exocytosis [192–195]. Moreover, mastocytes represent an abundant source of ATP stored in granules that are released under activation conditions [196–198] such as specific (e.g., IgE + antigen) and nonspecific stimulation (e.g., stress, mechanic stimulation, and osmotic swelling). With regard to the participation of mastocytes in CNS alterations, ATP can be released by trauma-induced degranulation and thus stimulates adjacent neurites via P2X and P2Y receptors. Additionally, the neuropeptide SP released from nerve terminals upon bradykinin stimulation participates in nerve mastocyte communication [199]. This enables interactions between nerve and mast cells and initiates and represents the development of neuroimmunological synapses. Also, glial cells are involved in neuroimmune cross-communication, and ATP induces glial cells to release IL-1β, TNF-α, and IL-33. Therefore, ATP released from mastocytes is an important autocrine/paracrine/exocrine factor that mediates cross-communication between different cell types [200]. Moreover, human LAD2 mast cells stimulated with IgE, anti-IgE, or substance P (SP) secrete mitochondrial particles, mitochondrial DNA (mtDNA), and ATP in absence of cell death. Furthermore, mitochondria added to mast cells trigger degranulation and release of histamine, PGD2, IL-8, TNF-α, and IL-1β, and this response is partially inhibited by DNAse and ATP receptor antagonists [201].
4. Activation of Glial Cells and Mastocytes during Stress and Infection
Only 30 min of immobilization stress can stimulate the HPA axis and cause degranulation in ~70% of rat dura mastocytes [202]. This response could be triggered by neurotensin (NT) and CRH acting on mastocytes increasing the permeability of the BBB [203–206]. As mentioned previously, activated mastocytes release proinflammatory cytokines and ATP among other bioactive compounds that promote microglia, and astrocyte activation and both reactive glia promote neuronal damage [123, 124]. Related to this, acute or chronic stress through GCs sensitizes microglia to a subsequent proinflammatory challenge [10], suggesting that stress should worsen the outcome of neuroinflammation. To our knowledge, it remains unknown if signal transduction of proinflammatory agents via TLRs and activity of HCs is enhanced by GCs or stress.
Related to the issue presented previously, various neurodegenerative disorders present activation of microglia in different brain regions [124] and restraint combined with water immersion induces massive microglial activation in the hippocampus, hypothalamus, thalamus, and periaqueductal gray matter [207, 208]. Although the precise mechanism of microglia activation induced by stress remains unknown, it is likely that bioactive molecules released by activated mastocytes (see what is mentioned previously) lead to the activation of microglia and, therefore, induce progression of neurodegenerative changes. In an ex vivo approach, rats were first pretreated in vivo with RU486 (GC receptor antagonist) and then exposed to an acute stressor (inescapable tail shock; IS), and 24 h later, hippocampal microglia were isolated and stimulated with LPS. Microglia obtained from rats not treated with a GC receptor antagonist showed an increase in gene expression of proinflammatory cytokines (IL-1β and IL-6). However, in rats pretreated with RU486, the sensitization of microglial to proinflammatory stimuli did not occur [10]. Astrocytic signaling is potentiated by GCs (i.e., methylprednisolone and dexamethasone) via long-range calcium waves, and an increase is observed in resting cytosolic Ca2+ levels, as well as the extent and amplitude of calcium wave propagation (twofold) compared to control conditions [209]. Furthermore, it is known that stress affects microglial function and viability during adulthood and early postnatal life [210]. Experiments both in vitro and in vivo have shown that stress hormones can affect the function and viability of microglia. However, little is known if stress during pregnancy affects microglia of the offspring. In a recent report, prenatal stress effects on microglia of the offspring were studied. In this model, prenatal stress during embryonic days 10–20 consisted of 20 min of forced swimming. In the offspring, a reduction in the number of immature microglia in the two main brain reservoirs of amoeboid microglia, corpus callosum, and internal capsule was observed. Moreover, accelerated microglial differentiation into ramified forms in the internal capsule and brain regions, such as the entorhinal cortex, parietal lobe neocortex, thalamus, and septum, was seen in the neonates in relation to an increase in plasma corticosterone in the pregnant dam [211].
The stimulation of microglial TLR3 with its ligand leads to the release of IL-6, IL-12, TNF-α, and IFN-γ among others (Figure 1). In connection to this, the importance of TLR in various CNS diseases (i.e., infection, trauma, stroke, neurodegeneration, and autoimmunity) has been described [212]. This is how viral infections have been implicated in the onset of MS by stimulation of TLR3 [105]. Additionally, in an animal model of schizophrenia, the stimulation of pregnant mothers with poly (I:C) results in reduced neuronal arborization of the offspring, which is correlated with a status of higher activation [213]. Interestingly, Cx HCs participate in neurite outgrowth [214] and release of ATP and glutamate [125], which also affect neuronal arborization [214, 215].
It is interesting to note that sensitivity to drug abuse behavior, as well the neuroinflammatory response to a subsequent proinflammatory challenge (as noted previously), is associated with stress and stress-induced release of GCs. Neuroinflammatory mediators derived from glia have an important role in the development of drug abuse [216]. This is how neuroinflammatory mediators, such as proinflammatory cytokines, are induced by opioids, psychostimulants, and alcohol, all of which modulate many effects including drug reward, dependence, tolerance, and analgesic properties. An interesting aspect is that drugs of abuse may directly activate microglial and astroglial cells via TLRs, which mediate the innate immune response to pathogens [216]. A key aspect is the timing of stress exposure relative to inflammatory challenge, and if a proinflammatory stimulus (e.g., LPS) is added immediately before stress exposure, stress induces an anti-inflammatory effect, which is reflected in the inhibition of the increase in brain IL-1β levels [217].
The importance of stress associated with infections is given by the fact that the acute or chronic stress sensitizes the inflammatory responses of the CNS to immunological challenges. Microglia show an increase in expression of MHC II, TLR4, and the F4/80 antigens. Therefore, stress changes the microenvironment of the CNS to a phenotype with inflammatory characteristics. One explanation to this phenomenon is that GCs sensitize microglia to infections [10, 218]. In peripheral blood monocytes from individuals under chronic stress, an increase in the expression of genes with promoter response elements for NF-κB is observed as well as allows expression of genes that have promoter elements for GC receptors [219]. Otherwise, in older stressed or chronically depressed adults, an increase in inflammatory response occurs when they are challenged with antigens, showing depressive characteristics and elevated levels of IL-6 after immunization with influenza vaccines. Further evidence that supports this notion comes from observations in older caregivers of patients with dementia, who also presented an elevation of IL-6 for over four weeks after vaccination with influenza vaccines, whereas this elevation was not observed in non stressed individuals [220].
Furthermore, stress worsens immunity and brain inflammation, which is important in MS and neuropsychiatric disorders [221–226]. Under stress, the neuropeptides CRH and NT are secreted and thus can activate microglia and mast cells, which in turn release molecules with proinflammatory properties. This results in maturation and activation of Th17 autoimmune cells and disruption of the BBB that leads to T cells entry into the CNS enhancing the brain inflammation, which might support the pathogenesis of MS. NT also stimulates secretion of vascular endothelial growth factor (VEGF) and induces expression of CRH receptor-1 in mast cells [20, 206, 227]. Several lines of evidence associate microglia with the pathogenesis of MS because activation of microglia is prominent and precedes T-lymphocyte infiltration and demyelination [228]. Activated microglia release glutamate and NO causing neuronal death and BBB disruption [228, 229]. With regard to the participation of mastocytes in the pathogenesis of MS, patients with this disease show elevated levels of tryptase (that activate microglia) and histamine in cerebrospinal fluid (CSF) [68, 230]. Therefore, several lines of evidence suggest an important role of mastocytes and microglia in neuroinflammatory diseases [67]. Therefore, both cell types represent therapeutic targets to be considered for treatment of MS and other neuroinflammatory diseases.
Among the factors relevant to the development of autism spectrum disorders (ASD), stress during pregnancy and the first 6 months of postnatal life has been associated with increased risk of ASD [231]. Stress induces the secretion of CRH from the hypothalamus and activates the HPA axis [232]. As mentioned previously, CRH also activates mast cells, resulting in the release of several proinflammatory cytokines [233] including IL-6, which in turns may increase the BBB permeability [222, 234, 235].
Recently, a decrease in the mitochondrial function in approximately 60% of patients with autism has been demonstrated [236–238]. The brain of these patients shows lines of evidence of neuroinflammation [239–242], with high levels of mitochondrial DNA [243]. Additionally, elevated levels of NT that could activate mast cells have been detected in children with autism [244]. The involvement of mast cells and brain inflammation is related to mitochondrial fission and translocation to the cell surface during degranulation [245], which leads to release of ATP and mitochondrial DNA [243]. The importance of ATP is that it can maintain inflammation by activating mast cells [225, 246].
5. Concluding Remarks
Stress potentiates neuroinflammatory responses by sensitizing microglia to proinflammatory stimuli [10]. This is how prenatal stress modifies the phenotype, distribution, and activation statuses of microglia in the offspring [211]. Different stressors, together with the activation of the inflammatory immune response, enhance the effects of proinflammatory molecules or conditions, showing synergistic effects [247]. Viral infections are the most common causes of infection during prenatal life, and maternal respiratory infection can also increase the risk of the offspring to develop certain mental disorders. The most direct evidence for this comes from a prospective study of pregnant women with medically documented respiratory infections, where the risk for schizophrenia in the offspring is increased 3-fold by infection in the second trimester [248]. Evidence that supports this phenomenon comes from models of cocultures between astroglia and microglia treated with dexamethasone. In these experiments, functional membrane properties of astrocytes in cocultures are differentially regulated, which might reflect steroid effects in adjacent glial components in vivo. In cocultures with 30% microglia, dexamethasone-treated cocultures show significant increased gap junctional intercellular communication [249], which could facilitate the propagation of inflammatory signal along astrocytic networks. Therefore, if a stressor is sufficiently sustained, this may reflect neurochemical processes that can make the organism more vulnerable to pathological stimuli producing behavioral and neurochemical responses [250, 251]. This can be reflected in an increased susceptibility to diseases of the nervous system, such as the progression of depressive disorders and anxiety, and can even affect the course of neurological diseases [250, 251]. Furthermore, activated microglia affect the expression of Cx HCs in astrocytes, which in turn increases the astrocytic ATP and glutamate release with deleterious consequences on neurons [125]. Therefore, these lines of evidence represent an aspect to be addressed in a model of stress in pregnant animals, in which one can analyze the effects of stress on microglia of the offspring in terms of activation and its effect on astrocytes, which could promote neuronal damage, with Cx HCs and Panx1 channels being possible therapeutic targets. Additionally, the synergistic effect of stress and stimulation with viral infection (for which RNA viral mimics poly (I:C)) has not been studied in offspring of pregnant females subjected to stress, which is also a novel approach and can be correlated with a possible susceptibility of offspring to diseases of the nervous system.
An important aspect is that when microglia are strongly activated, they remain in a preactivate state for years, which means that microglia are excessively responsive to even slight stimuli. This fact also has been linked to the activation of microglia by viral infections early in life and that can be later reactivated more rapidly compared to microglia in normal state [252, 253]. Therefore, the possibility of having microglia (using minocycline) and mastocytes activation (with GRH-R antagonists) as therapeutic targets opens the possibility of their modulation as treatment for various neuropsychiatric disorders, viral infections, and other neuroinflammatory pathologies of the CNS.
In summary, parental stress is proposed to induce potentiation of neuroinflammatory responses by first: activating directly mast cells through CRH recognition. Second: mast cells proinflammatory mediators prime microglia, astrocytes and olygodendrocytes, modifying their phenotype, distribution, and activation statuses in the offspring, but mainly promoting HC expression. Third: sensitized microglia exposed to inflammatory stimuli (i.e., TLR3 ligands) (Figure 1) are activated and secrete cytokines (TNF-α, IL-1β). They also show increased functional expression of Panx1 channels and Cx HCs through which ATP and glutamate are released to the extracellular milieu. Astrocyte and oligodendrocyte become activated and release ATP and glutamate in an HC depending way, and thus they promote neurodegeneration (Figure 2). Therefore, HCs represent a novel target with clinical applications in neuroinflammatory diseases.
Figure 2.
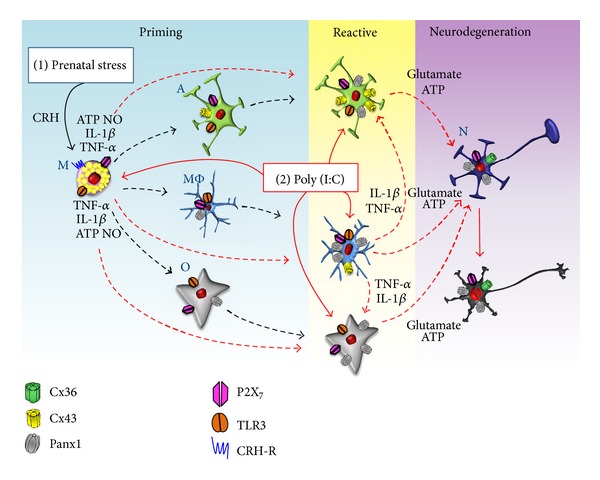
Model of the involvement of mastocytes and microglia in neuroinflammatory responses and potentiation of their responses by stress. Stress increases the levels of CRH and glucocorticoids which are critical players in stress-induced mastocytes (M) degranulation and potentiation of glial inflammatory responses (sensibilization). Furthermore, perinatal infections, particularly those of viral etiology (poly (I:C)), are frequent and have been associated with diverse alterations of CNS. Mastocytes and microglia (MΦ) express toll-like receptor 3 (TLR3). Activated microglia and mastocytes increase the hemichannel activity in reactive astrocytes (A) and oligodendrocytes (O). Both activated microglia and astrocytes release ATP and glutamate that induce neurodegeneration through the activation of P2X7 receptors and Panx1 channels in neurons (N) (neurodegeneration) (modified from Orellana et al., 2011) [125].
Acknowledgments
This work was partially funded by FONDECYT grants FONDECYT postdoctoral fellowship 3130632 (to A. Aguirre), CONICYT Ph.D. student fellowship 21100401 (to C. J. Maturana) and 24121474 (to P. A. Harcha), and 1111033, Anillo ACT-71, FONDEF DO7I1086, and Chilean Science Millennium Institute P09-022 (to J. C. Sáez).
References
- 1.Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunological Reviews. 2006;213(1):48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theoharides TC. Mast cells: the immune gate to the brain. Life Sciences. 1990;46(9):607–617. doi: 10.1016/0024-3205(90)90129-f. [DOI] [PubMed] [Google Scholar]
- 3.Turrin NP, Rivest S. Molecular and cellular immune mediators of neuroprotection. Molecular Neurobiology. 2006;34(3):221–242. doi: 10.1385/MN:34:3:221. [DOI] [PubMed] [Google Scholar]
- 4.Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Molecular Neurodegeneration. 2009;4(1, article 47) doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rivest S. Regulation of innate immune responses in the brain. Nature Reviews Immunology. 2009;9(6):429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 6.Block ML, Zecca L, Hong J-S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews Neuroscience. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 7.Theoharides TC, Donelan J, Kandere-Grzybowska K, Konstantinidou A. The role of mast cells in migraine pathophysiology. Brain Research Reviews. 2005;49(1):65–76. doi: 10.1016/j.brainresrev.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Mallard C, Wang X, Hagberg H. The role of Toll-like receptors in perinatal brain injury. Clinics in Perinatology. 2009;36(4):763–772. doi: 10.1016/j.clp.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 9.McCurdy JD, Olynych TJ, Maher LH, Marshall JS. Cutting edge: distinct toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. Journal of Immunology. 2003;170(4):1625–1629. doi: 10.4049/jimmunol.170.4.1625. [DOI] [PubMed] [Google Scholar]
- 10.Frank MG, Thompson BM, Watkins LR, Maier SF. Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain, Behavior, and Immunity. 2012;26(2):337–345. doi: 10.1016/j.bbi.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okun E, Griffioen KJ, Lathia JD, Tang S-C, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Research Reviews. 2009;59(2):278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iribarren P, Zhou Y, Hu J, Le Y, Wang JM. Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunologic Research. 2005;31(3):165–176. doi: 10.1385/IR:31:3:165. [DOI] [PubMed] [Google Scholar]
- 13.Arroyo DS, Soria JA, Gaviglio EA, Rodriguez-Galan MC, Iribarren P. Toll-like receptors are key players in neurodegeneration. International Immunopharmacology. 2011;11(10):1415–1421. doi: 10.1016/j.intimp.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown AS, Begg MD, Gravenstein S, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Archives of General Psychiatry. 2004;61(8):774–780. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- 15.Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophrenia Bulletin. 2006;32(2):200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer U, Nyffeler M, Engler A, et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. Journal of Neuroscience. 2006;26(18):4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer U, Feldon J. To poly(I:C) or not to poly(I:C): advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology. 2012;62(3):1308–1321. doi: 10.1016/j.neuropharm.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Arrode-Brusés G, Brusés JL. Maternal immune activation by poly I:C induces expression of cytokines IL-1β and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. Journal of Neuroinflammation. 2012;9:p. 83. doi: 10.1186/1742-2094-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown AS. Exposure to prenatal infection and risk of schizophrenia. Frontiers in Psychiatry. 2011;2:p. 63. doi: 10.3389/fpsyt.2011.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asadi S, Theoharides TC. Corticotropin-releasing hormone and extracellular mitochondria augment IgE-stimulated human mast-cell vascular endothelial growth factor release, which is inhibited by luteolin. Journal of Neuroinflammation. 2012:p. 85. doi: 10.1186/1742-2094-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Liu T, Gao Y-J, Ji R-R. Emerging role of Toll-like receptors in the control of pain and itch. Neuroscience Bulletin. 2012:1–14. doi: 10.1007/s12264-012-1219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via till-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 24.Yarovinsky F, Zhang D, Andersen JF, et al. Immunology: TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308(5728):1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 25.Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. Microglia recognize double-stranded RNA via TLR3. Journal of Immunology. 2006;176(6):3804–3812. doi: 10.4049/jimmunol.176.6.3804. [DOI] [PubMed] [Google Scholar]
- 26.Dimitriadou V, Lambracht-Hall M, Reichler J, Theoharides TC. Histochemical and ultrastructural characteristics of rat brain perivascular mast cells stimulated with compound 48/80 and carbachol. Neuroscience. 1990;39(1):209–224. doi: 10.1016/0306-4522(90)90234-u. [DOI] [PubMed] [Google Scholar]
- 27.Manning KA, Pienkowski TP, Uhlrich DJ. Histaminergic and non-histamine-immunoreactive mast cells within the cat lateral geniculate complex examined with light and electron microscopy. Neuroscience. 1994;63(1):191–206. doi: 10.1016/0306-4522(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 28.Michaloudi H, Batzios C, Chiotelli M, Papadopoulos GC. Developmental changes of mast cell populations in the cerebral meninges of the rat. Journal of Anatomy. 2007;211(4):556–566. doi: 10.1111/j.1469-7580.2007.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khalil M, Ronda J, Weintraub M, Jain K, Silver R, Silverman A-J. Brain mast cell relationship to neurovasculature during development. Brain Research. 2007;1171(1):18–29. doi: 10.1016/j.brainres.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marshall JS. Mast-cell responses to pathogens. Nature Reviews Immunology. 2004;4(10):787–799. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- 31.Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nature Immunology. 2005;6(2):135–142. doi: 10.1038/ni1158. [DOI] [PubMed] [Google Scholar]
- 32.McCurdy JD, Lin T-J, Marshall JS. Toll-like receptor 4-mediated activation of murine mast cells. Journal of Leukocyte Biology. 2001;70(6):977–984. [PubMed] [Google Scholar]
- 33.Supajatura V, Ushio H, Nakao A, Okumura K, Ra C, Ogawa H. Protective roles of mast cells against enterobacterial infection are mediated by Toll-like receptor 4. Journal of Immunology. 2001;167(4):2250–2256. doi: 10.4049/jimmunol.167.4.2250. [DOI] [PubMed] [Google Scholar]
- 34.Masuda A, Yoshikai Y, Aiba K, Matsuguchi T. Th2 cytokine production from mast cells is directly induced by lipopolysaccharide and distinctly regulated by c-Jun N-terminal kinase and p38 pathways. Journal of Immunology. 2002;169(7):3801–3810. doi: 10.4049/jimmunol.169.7.3801. [DOI] [PubMed] [Google Scholar]
- 35.Matsushima H, Yamada N, Matsue H, Shimada S. TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. Journal of Immunology. 2004;173(1):531–541. doi: 10.4049/jimmunol.173.1.531. [DOI] [PubMed] [Google Scholar]
- 36.Mrabet-Dahbi S, Metz M, Dudeck A, Zuberbier T, Maurer M. Murine mast cells secrete a unique profile of cytokines and prostaglandins in response to distinct TLR2 ligands. Experimental Dermatology. 2009;18(5):437–444. doi: 10.1111/j.1600-0625.2009.00878.x. [DOI] [PubMed] [Google Scholar]
- 37.Varadaradjalou S, Féger F, Thieblemont N, et al. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human mast cells. European Journal of Immunology. 2003;33(4):899–906. doi: 10.1002/eji.200323830. [DOI] [PubMed] [Google Scholar]
- 38.Kulka M, Alexopoulou L, Flavell RA, Metcalfe DD. Activation of mast cells by double-stranded RNA: evidence for activation through Toll-like receptor 3. Journal of Allergy and Clinical Immunology. 2004;114(1):174–182. doi: 10.1016/j.jaci.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 39.Yoshioka M, Fukuishi N, Iriguchi S, et al. Lipoteichoic acid downregulates FcεRI expression on human mast cells through Toll-like receptor 2. Journal of Allergy and Clinical Immunology. 2007;120(2):452–461. doi: 10.1016/j.jaci.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 40.Leal-Berumen I, Conlon P, Marshall JS. IL-6 production by rat peritoneal mast cells is not necessarily preceded by histamine release and can be induced by bacterial lipopolysaccharide. Journal of Immunology. 1994;152(11):5468–5476. [PubMed] [Google Scholar]
- 41.Zhu F-G, Marshall JS. CpG-containing oligodeoxynucleotides induce TNF-α and IL-6 production but not degranulation from murine bone marrow-derived mast cells. Journal of Leukocyte Biology. 2001;69(2):253–262. [PubMed] [Google Scholar]
- 42.Orinska Z, Bulanova E, Budagian V, Metz M, Maurer M, Bulfone-Paus S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood. 2005;106(3):978–987. doi: 10.1182/blood-2004-07-2656. [DOI] [PubMed] [Google Scholar]
- 43.Heib V, Becker M, Warger T, et al. Mast cells are crucial for early inflammation, migration of Langerhans cells, and CTL responses following topical application of TLR7 ligand in mice. Blood. 2007;110(3):946–953. doi: 10.1182/blood-2006-07-036889. [DOI] [PubMed] [Google Scholar]
- 44.Kulka M, Metcalfe DD. TLR3 activation inhibits human mast cell attachment to fibronectin and vitronectin. Molecular Immunology. 2006;43(10):1579–1586. doi: 10.1016/j.molimm.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 45.Wierzbicki M, Brzezińska-Blłaszczyk E. Diverse effects of bacterial cell wall components on mast cell degranulation, cysteinyl leukotriene generation and migration. Microbiology and Immunology. 2009;53(12):694–703. doi: 10.1111/j.1348-0421.2009.00174.x. [DOI] [PubMed] [Google Scholar]
- 46.Secor VH, Secor WE, Gutekunst C-A, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. Journal of Experimental Medicine. 2000;191(5):813–821. doi: 10.1084/jem.191.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sayed BA, Walker ME, Brown MA. Cutting edge: mast cells regulate disease severity in a relapsing-remitting model of multiple sclerosis. Journal of Immunology. 2011;186(6):3294–3298. doi: 10.4049/jimmunol.1003574. [DOI] [PubMed] [Google Scholar]
- 48.Tunçel N, Şener E, Cerit C, et al. Brain mast cells and therapeutic potential of vasoactive intestinal peptide in a Parkinson’s disease model in rats: brain microdialysis, behavior, and microscopy. Peptides. 2005;26(5):827–836. doi: 10.1016/j.peptides.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 49.Graves MC, Fiala M, Dinglasan LAV, et al. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and t cells. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2004;5(4):213–219. doi: 10.1080/14660820410020286. [DOI] [PubMed] [Google Scholar]
- 50.Fiala M, Chattopadhay M, La Cava A, et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. Journal of Neuroinflammation. 2010;7, article 76 doi: 10.1186/1742-2094-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kvetnoi IM, Kvetnaya TV, Ryadnova IY, Fursov BB, Ernandes-Jago J, Blesa JR. Expression of β-amyloid and tau-protein in mastocytes in Alzheimer’s disease. Arkhiv Patologii. 2003;65(1):36–39. [PubMed] [Google Scholar]
- 52.Lozada A, Maegele M, Stark H, Neugebauert EMA, Panula P. Traumatic brain injury results in mast cell increase and changes in regulation of central histamine receptors. Neuropathology and Applied Neurobiology. 2005;31(2):150–162. doi: 10.1111/j.1365-2990.2004.00622.x. [DOI] [PubMed] [Google Scholar]
- 53.Strbian D, Karjalainen-Lindsberg M-L, Tatlisumak T, Lindsberg PJ. Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. Journal of Cerebral Blood Flow and Metabolism. 2006;26(5):605–612. doi: 10.1038/sj.jcbfm.9600228. [DOI] [PubMed] [Google Scholar]
- 54.Strbian D, Tatlisumak T, Ramadan UA, Lindsberg PJ. Mast cell blocking reduces brain edema and hematoma volume and improves outcome after experimental intracerebral hemorrhage. Journal of Cerebral Blood Flow and Metabolism. 2007;27(4):795–802. doi: 10.1038/sj.jcbfm.9600387. [DOI] [PubMed] [Google Scholar]
- 55.Mokhtarian F, Griffin DE. The role of mast cells in virus-induced inflammation in the murine central nervous system. Cellular Immunology. 1984;86(2):491–500. doi: 10.1016/0008-8749(84)90404-0. [DOI] [PubMed] [Google Scholar]
- 56.Brenner T, Soffer D, Shalit M, Levi-Schaffer F. Mast cells in experimental allergic encephalomyelitis: characterization, distribution in the CNS and in vitro activation by myelin basic protein and neuropeptides. Journal of the Neurological Sciences. 1994;122(2):210–213. doi: 10.1016/0022-510x(94)90300-x. [DOI] [PubMed] [Google Scholar]
- 57.Johnson D, Seeldrayers PA, Weiner HL. The role of mast cells in demyelination. 1. Myelin proteins are degraded by mast cell proteases and myelin basic protein and P2 can stimulate mast cell degranulation. Brain Research. 1988;444(1):195–198. doi: 10.1016/0006-8993(88)90929-8. [DOI] [PubMed] [Google Scholar]
- 58.Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158(3):1030–1038. doi: 10.1016/j.neuroscience.2008.06.046. [DOI] [PubMed] [Google Scholar]
- 59.Ribes S, Adam N, Ebert S, et al. The viral TLR3 agonist poly(I:C) stimulates phagocytosis and intracellular killing of Escherichia coli by microglial cells. Neuroscience Letters. 2010;482(1):17–20. doi: 10.1016/j.neulet.2010.06.078. [DOI] [PubMed] [Google Scholar]
- 60.Venero JL, Burguillos MA, Brundin P, Joseph B. The executioners sing a new song: killer caspases activate microglia. Cell Death and Differentiation. 2011;18(11):1679–1691. doi: 10.1038/cdd.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kees T, Lohr J, Noack J, et al. Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro-Oncology. 2012;14(1):64–78. doi: 10.1093/neuonc/nor182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nature Medicine. 2004;10(12):1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 63.Supajatura V, Ushio H, Nakao A, et al. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. Journal of Clinical Investigation. 2002;109(10):1351–1359. doi: 10.1172/JCI14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kubo Y, Fukuishi N, Yoshioka M, et al. Bacterial components regulate the expression of Toll-like receptor 4 on human mast cells. Inflammation Research. 2007;56(2):70–75. doi: 10.1007/s00011-006-6064-4. [DOI] [PubMed] [Google Scholar]
- 65.Nigo YI, Yamashita M, Hirahara K, et al. Regulation of allergic airway inflammation through Toll-like receptor 4-mediated modification of mast cell function. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2286–2291. doi: 10.1073/pnas.0510685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feuser K, Thon K-P, Bischoff SC, Lorentz A. Human intestinal mast cells are a potent source of multiple chemokines. Cytokine. 2012;58(2):178–185. doi: 10.1016/j.cyto.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 67.Skaper SD, Giusti P, Facci L. Microglia and mast cells: two tracks on the road to neuroinflammation. The FASEB Journal. 2012;26:3103–3117. doi: 10.1096/fj.11-197194. [DOI] [PubMed] [Google Scholar]
- 68.Rozniecki JJ, Hauser SL, Stein M, Lincoln R, Theoharides TC. Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Annals of Neurology. 1995;37(1):63–66. doi: 10.1002/ana.410370112. [DOI] [PubMed] [Google Scholar]
- 69.Malamud V, Vaaknin A, Abramsky O, et al. Tryptase activates peripheral blood mononuclear cells causing the synthesis and release of TNF-α, IL-6 and IL-1β: possible relevance to multiple sclerosis. Journal of Neuroimmunology. 2003;138(1-2):115–122. doi: 10.1016/s0165-5728(03)00090-0. [DOI] [PubMed] [Google Scholar]
- 70.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiological Reviews. 2004;84(2):579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 71.Molino M, Barnathan ES, Numerof R, et al. Interactions of mast cell tryptase with thrombin receptors and PAR-2. Journal of Biological Chemistry. 1997;272(7):4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- 72.Olejár T, Matěj R, Zadinová M, Poučková P. Proteinase-activated receptor-2 expression on cerebral neurones after radiation damage: immunohistochemical observation in Wistar rats. International Journal of Tissue Reactions. 2002;24(3):81–88. [PubMed] [Google Scholar]
- 73.Rohatgi T, Henrich-Noack P, Sedehizade F, et al. Transient focal ischemia in rat brain differentially regulates mRNA expression of protease-activated receptors 1 to 4. Journal of Neuroscience Research. 2004;75(2):273–279. doi: 10.1002/jnr.10847. [DOI] [PubMed] [Google Scholar]
- 74.Jin Y, Silverman AJ, Vannucci SJ. Mast cells are early responders after hypoxia-ischemia in immature rat brain. Stroke. 2009;40(9):3107–3112. doi: 10.1161/STROKEAHA.109.549691. [DOI] [PubMed] [Google Scholar]
- 75.Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-Ischemic, and excitotoxic insults. Mental Retardation and Developmental Disabilities Research Reviews. 2002;8(1):30–38. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- 76.Pang Y, Cai Z, Rhodes PG. Disturbance of oligodendrocyte development, hypomyelination and white matter injury in the neonatal rat brain after intracerebral injection of lipopolysaccharide. Developmental Brain Research. 2003;140(2):205–214. doi: 10.1016/s0165-3806(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 77.Dean JM, Wang X, Kaindl AM, et al. Microglial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microglia in vitro. Brain, Behavior, and Immunity. 2010;24:776–783. doi: 10.1016/j.bbi.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 78.Lehnard S, Lachance C, Patrizi S, et al. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. Journal of Neuroscience. 2002;22(7):2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends in Immunology. 2007;28(3):138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 80.Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49(3):360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- 81.Scumpia PO, Kelly KM, Reeves WH, Stevens BR. Double-stranded RNA signals antiviral and inflammatory programs and dysfunctional glutamate transport in TLR3-expressing astrocytes. Glia. 2005;52(2):153–162. doi: 10.1002/glia.20234. [DOI] [PubMed] [Google Scholar]
- 82.Kim H, Yang E, Lee J, et al. Double-stranded RNA mediates interferon regulatory factor 3 activation and interleukin-6 production by engaging Toll-like receptor 3 in human brain astrocytes. Immunology. 2008;124(4):480–488. doi: 10.1111/j.1365-2567.2007.02799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bsibsi M, Persoon-Deen C, Verwer RWH, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53(7):688–695. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- 84.Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clinical Science. 2011;121(9):367–387. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115(12):1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 86.Tang S-C, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiology of Disease. 2008;31(1):33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 88.Cashman JR, Ghirmai S, Abel KJ, Fiala M. Immune defects in Alzheimer’s disease: new medications development. BMC Neuroscience. 2008;9(2, article S13) doi: 10.1186/1471-2202-9-S2-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tang S-C, Lathia JD, Selvaraj PK, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid β-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Experimental Neurology. 2008;213(1):114–121. doi: 10.1016/j.expneurol.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marta M. Toll-like receptors in multiple sclerosis mouse experimental models. Annals of the New York Academy of Sciences. 2009;1173:458–462. doi: 10.1111/j.1749-6632.2009.04849.x. [DOI] [PubMed] [Google Scholar]
- 91.Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. GLIA. 2011;59(2):242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- 92.Jack CS, Arbour N, Manusow J, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. Journal of Immunology. 2005;175(7):4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 93.Krasowska-Zoladek A, Banaszewska M, Kraszpulski M, Konat GW. Kinetics of inflammatory response of astrocytes induced by TLR3 and TLR4 ligation. Journal of Neuroscience Research. 2007;85(1):205–212. doi: 10.1002/jnr.21088. [DOI] [PubMed] [Google Scholar]
- 94.Bsibsi M, Ravid R, Gveric D, Van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. Journal of Neuropathology and Experimental Neurology. 2002;61(11):1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 95.Setzu A, Lathia JD, Zhao C, et al. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia. 2006;54(4):297–303. doi: 10.1002/glia.20371. [DOI] [PubMed] [Google Scholar]
- 96.Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. Journal of Neurochemistry. 2007;102(1):37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- 97.Sharma R, Fischer M-T, Bauer J, et al. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathologica. 2010;120(2):223–236. doi: 10.1007/s00401-010-0704-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schonberg DL, Popovich PG, McTigue DM. Oligodendrocyte generation is differentially influenced by toll-like receptor (TLR) 2 and TLR4-mediated intraspinal macrophage activation. Journal of Neuropathology and Experimental Neurology. 2007;66(12):1124–1135. doi: 10.1097/nen.0b013e31815c2530. [DOI] [PubMed] [Google Scholar]
- 99.Glezer I, Lapointe A, Rivest S. Innate immunity triggers oligodendrocyte progenitor reactivity and confines damages to brain injuries. The FASEB Journal. 2006;20(6):750–752. doi: 10.1096/fj.05-5234fje. [DOI] [PubMed] [Google Scholar]
- 100.Bsibsi M, Nomden A, van Noort JM, Baron W. Toll-like receptors 2 and 3 agonists differentially affect oligodendrocyte survival, differentiation, and myelin membrane formation. Journal of Neuroscience Research. 2012;90(2):388–398. doi: 10.1002/jnr.22767. [DOI] [PubMed] [Google Scholar]
- 101.Lehnardt S, Massillon L, Follett P, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Felts PA, Woolston A-M, Fernando HB, et al. Inflammation and primary demyelination induced by the intraspinal injection of lipopolysaccharide. Brain. 2005;128(7):1649–1666. doi: 10.1093/brain/awh516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li J, Ramenaden ER, Peng J, Koito H, Volpe JJ, Rosenberg PA. Tumor necrosis factor α mediates lipopolysaccharide-induced microglial toxicity to developing oligodendrocytes when astrocytes are present. Journal of Neuroscience. 2008;28(20):5321–5330. doi: 10.1523/JNEUROSCI.3995-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim S, Steelman AJ, Koito H, Li J. Astrocytes promote TNF-mediated toxicity to oligodendrocyte precursors. Journal of Neurochemistry. 2011;116(1):53–66. doi: 10.1111/j.1471-4159.2010.07084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Steelman AJ, Li J. Poly(I:C) promotes TNFα/TNFR1-dependent oligodendrocyte death in mixed glial cultures. Journal of Neuroinflammation. 2011;8, article 89 doi: 10.1186/1742-2094-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nature Reviews Immunology. 2006;6(4):318–328. doi: 10.1038/nri1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gomariz RP, Gutiérrez-Cañas I, Arranz A, et al. Peptides targeting toll-like receptor signalling pathways for novel immune therapeutics. Current Pharmaceutical Design. 2010;16(9):1063–1080. doi: 10.2174/138161210790963841. [DOI] [PubMed] [Google Scholar]
- 108.Ben-Sasson SZ, Caucheteux S, Crank M, Hu-Li J, Paul WE. IL-1 acts on T cells to enhance the magnitude of in vivo immune responses. Cytokine. 2011;56(1):122–125. doi: 10.1016/j.cyto.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kanneganti T-D. Central roles of NLRs and inflammasomes in viral infection. Nature Reviews Immunology. 2010;10(10):688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunological Reviews. 2011;243(1):136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 111.Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death and Differentiation. 2007;14(9):1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 112.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Craven RR, Gao X, Allen IC, et al. Staphylococcus aureus α-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE. 2009;4(10) doi: 10.1371/journal.pone.0007446.e7446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 115.Sutterwala FS, Ogura Y, Szczepanik M, et al. Critical role for NALP3/CIAS1/cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24(3):317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 116.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. The EMBO Journal. 2006;25(21):5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Locovei S, Wang J, Dahl G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Letters. 2006;580(1):239–244. doi: 10.1016/j.febslet.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 118.Kanneganti T-D, Lamkanfi M, Kim Y-G, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26(4):433–443. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 119.Silverman WR, de Rivero Vaccari JP, Locovei S, et al. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. Journal of Biological Chemistry. 2009;284(27):18143–18151. doi: 10.1074/jbc.M109.004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sáez JC, Schalper KA, Retamal MA, Orellana JA, Shoji KF, Bennett MVL. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Experimental Cell Research. 2010;316(15):2377–2389. doi: 10.1016/j.yexcr.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 121.Bennett MVL, Contreras JE, Bukauskas FF, Sáez JC. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends in Neurosciences. 2003;26(11):610–617. doi: 10.1016/j.tins.2003.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sáez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MVL. Connexin-based gap junction hemichannels: gating mechanisms. Biochimica et Biophysica Acta. 2005;1711(2):215–224. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Thornton P, Pinteaux E, Gibson RM, Allan SM, Rothwell NJ. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. Journal of Neurochemistry. 2006;98(1):258–266. doi: 10.1111/j.1471-4159.2006.03872.x. [DOI] [PubMed] [Google Scholar]
- 124.Orellana JA, Sáez PJ, Shoji KF, et al. Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxidants and Redox Signaling. 2009;11(2):369–399. doi: 10.1089/ars.2008.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Orellana JA, Froger N, Ezan P, et al. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. Journal of Neurochemistry. 2011;118(5):826–840. doi: 10.1111/j.1471-4159.2011.07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Orellana JA, Shoji KF, Abudara V, et al. Amyloid β-induced death in neurons involves glial and neuronal hemichannels. Journal of Neuroscience. 2011;31(13):4962–4977. doi: 10.1523/JNEUROSCI.6417-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vliagoftis H, Hutson AM, Mahmudi-Azer S, et al. Mast cells express connexins on their cytoplasmic membrane. Journal of Allergy and Clinical Immunology. 1999;103(4):656–662. doi: 10.1016/s0091-6749(99)70239-3. [DOI] [PubMed] [Google Scholar]
- 128.Tatham PER, Lindau M. ATP-induced pore formation in the plasma membrane of rat peritoneal mast cells. Journal of General Physiology. 1990;95(3):459–476. doi: 10.1085/jgp.95.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parekh AB, Putney JW., Jr. Store-operated calcium channels. Physiological Reviews. 2005;85(2):757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 130.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. Journal of Neuroscience Research. 2005;81(3):374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 131.Ponomarev ED, Shriver LP, Dittel BN. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. Journal of Immunology. 2006;176(3):1402–1410. doi: 10.4049/jimmunol.176.3.1402. [DOI] [PubMed] [Google Scholar]
- 132.Chen K, Huang J, Gong W, Zhang L, Yu P, Wang JM. CD40/CD40L dyad in the inflammatory and immune responses in the central nervous system. Cellular & Molecular Immunology. 2006;3(3):163–169. [PubMed] [Google Scholar]
- 133.Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathologica. 2010;119(1):89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- 134.Polazzi E, Contestabile A. Overactivation of LPS-stimulated microglial cells by co-cultured neurons or neuron-conditioned medium. Journal of Neuroimmunology. 2006;172(1-2):104–111. doi: 10.1016/j.jneuroim.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 135.Bennett MV, Garre JM, Orellana JA, Bukauskas FF, Nedergaard M, Saez JC. Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Research. 2012;1487:3–15. doi: 10.1016/j.brainres.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Eugenín EA, Eckardt D, Theis M, Willecke K, Bennett MVL, Sáez JC. Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-γ and tumor necrosis factor-α . Proceedings of the National Academy of Sciences of the United States of America. 2001;98(7):4190–4195. doi: 10.1073/pnas.051634298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Parenti R, Campisi A, Vanella A, Cicirata F. Immunocytochemical and RT-PCR analysis of connexin36 in cultures of mammalian glial cells. Archives Italiennes de Biologie. 2002;140(2):101–108. [PubMed] [Google Scholar]
- 138.Dobrenis K, Chang H-Y, Pina-Benabou MH, et al. Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. Journal of Neuroscience Research. 2005;82(3):306–315. doi: 10.1002/jnr.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garg S, Syed MM, Kielian T. Staphylococcus aureus-derived peptidoglycan induces Cx43 expression and functional gap junction intercellular communication in microglia. Journal of Neurochemistry. 2005;95(2):475–483. doi: 10.1111/j.1471-4159.2005.03384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Martinez AD, Hayrapetyan V, Moreno AP, Beyer EC. Connexin43 and connexin45 form heteromeric gap junction channels in which individual components determine permeability and regulation. Circulation Research. 2002;90(10):1100–1107. doi: 10.1161/01.res.0000019580.64013.31. [DOI] [PubMed] [Google Scholar]
- 141.Giaume C, McCarthy KD. Control of gap-junctional communication in astrocytic networks. Trends in Neurosciences. 1996;19(8):319–325. doi: 10.1016/0166-2236(96)10046-1. [DOI] [PubMed] [Google Scholar]
- 142.Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54(7):716–725. doi: 10.1002/glia.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cotrina ML, Lin JH-C, López-García JC, Naus CCG, Nedergaard M. ATP-mediated glia signaling. Journal of Neuroscience. 2000;20(8):2835–2844. doi: 10.1523/JNEUROSCI.20-08-02835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Calegari F, Coco S, Taverna E, et al. A regulated secretory pathway in cultured hippocampal astrocytes. Journal of Biological Chemistry. 1999;274(32):22539–22547. doi: 10.1074/jbc.274.32.22539. [DOI] [PubMed] [Google Scholar]
- 145.Stout C, Charles A. Modulation of intercellular calcium signaling in astrocytes by extracellular calcium and magnesium. Glia. 2003;43(3):265–273. doi: 10.1002/glia.10257. [DOI] [PubMed] [Google Scholar]
- 146.Kang J, Kang N, Lovatt D, et al. Connexin 43 hemichannels are permeable to ATP. Journal of Neuroscience. 2008;28(18):4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E. Pannexin 1: the molecular substrate of astrocyte “hemichannels”. Journal of Neuroscience. 2009;29(21):7092–7097. doi: 10.1523/JNEUROSCI.6062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369(6483):744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 149.Albrecht J, Rafalowska U. Enhanced potassium-stimulated γ-aminobutyric acid release by astrocytes derived from rats with early hepatogenic encephalopathy. Journal of Neurochemistry. 1987;49(1):9–11. doi: 10.1111/j.1471-4159.1987.tb03385.x. [DOI] [PubMed] [Google Scholar]
- 150.Queiroz G, Gebicke-Haerter PJ, Schobert A, Starke K, Von Kügelgen I. Release of ATP from cultured rat astrocytes elicited by glutamate receptor activation. Neuroscience. 1997;78(4):1203–1208. doi: 10.1016/s0306-4522(96)00637-9. [DOI] [PubMed] [Google Scholar]
- 151.Albrecht J, Simmons M, Dutton GR, Norenberg MD. Aluminium chloride stimulates the release of endogenous glutamate, taurine and adenosine from cultured rat cortical astrocytes. Neuroscience Letters. 1991;127(1):105–107. doi: 10.1016/0304-3940(91)90905-9. [DOI] [PubMed] [Google Scholar]
- 152.Stehberg J, Moraga-Amaro R, Salazar C, et al. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. The FASEB Journal. 2012;26:3649–3657. doi: 10.1096/fj.11-198416. [DOI] [PubMed] [Google Scholar]
- 153.Nagy JI, Ionescu A-V, Lynn BD, Rash JE. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: implications from normal and connexin32 knockout mice. Glia. 2003;44(3):205–218. doi: 10.1002/glia.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Retamal MA, Froger N, Palacios-Prado N, et al. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. Journal of Neuroscience. 2007;27(50):13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Davidson JO, Green CR, B. Nicholson LF, et al. Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Annals of Neurology. 2012;71(1):121–132. doi: 10.1002/ana.22654. [DOI] [PubMed] [Google Scholar]
- 156.Karpuk N, Burkovetskaya M, Fritz T, Angle A, Kielian T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. Journal of Neuroscience. 2011;31(2):414–425. doi: 10.1523/JNEUROSCI.5247-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Eugenin EA, Basilio D, Sáez JC, et al. The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. Journal of Neuroimmune Pharmacology. 2012;7(3):499–518. doi: 10.1007/s11481-012-9352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Orellana JA, Hernández DE, Ezan P, et al. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia. 2010;58(3):329–343. doi: 10.1002/glia.20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rovegno M, Soto PA, Sáez JC, von Bernhardi R. Biological mechanisms involved in the spread of traumatic brain damage. Medicina Intensiva. 2012;36(1):37–44. doi: 10.1016/j.medin.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 160.Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology. 2010;151(4):1622–1632. doi: 10.1210/en.2009-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiological Reviews. 2001;81(2):871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- 162.Dougherty JD, Fomchenko EI, Akuffo AA, et al. Candidate pathways for promoting differentiation or quiescence of oligodendrocyte progenitor-like cells in glioma. Cancer Research. 2012;72:4856–4868. doi: 10.1158/0008-5472.CAN-11-2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Miron VE, Kuhlmann T, Antel Jack P. JP. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochimica et Biophysica Acta. 2011;1812(2):184–193. doi: 10.1016/j.bbadis.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 164.Magnotti LM, Goodenough DA, Paul DL. Deletion of oligodendrocyte Cx32 and astrocyte Cx43 causes white matter vacuolation, astrocyte loss and early mortality. Glia. 2011;59(7):1064–1074. doi: 10.1002/glia.21179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nualart-Marti A, Solsona C, Fields RD. Gap junction communication in myelinating glia. Biochimica et Biophysica Acta. 2013;1828(1):69–78. doi: 10.1016/j.bbamem.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schousboe A, Westergaard N, Sonnewald U, et al. Glutamate and glutamine metabolism and compartmentation in astrocytes. Developmental Neuroscience. 1993;15(3–5):359–366. doi: 10.1159/000111356. [DOI] [PubMed] [Google Scholar]
- 167.Kamasawa N, Sik A, Morita M, et al. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: implications for ionic homeostasis and potassium siphoning. Neuroscience. 2005;136(1):65–86. doi: 10.1016/j.neuroscience.2005.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bruce CC, Zhao C, Franklin RJM. Remyelination—an effective means of neuroprotection. Hormones and Behavior. 2010;57(1):56–62. doi: 10.1016/j.yhbeh.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 169.Ahn M, Lee J, Gustafsson A, et al. Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. Journal of Neuroscience Research. 2008;86(5):992–1006. doi: 10.1002/jnr.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Domercq M, Perez-Samartin A, Aparicio D, Alberdi E, Pampliega O, Matute C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia. 2010;58(6):730–740. doi: 10.1002/glia.20958. [DOI] [PubMed] [Google Scholar]
- 171.Brosnan CF, Raine CS. Mechanisms of immune injury in multiple sclerosis. Brain Pathology. 1996;6(3):243–257. doi: 10.1111/j.1750-3639.1996.tb00853.x. [DOI] [PubMed] [Google Scholar]
- 172.Genain CP, Abel K, Belmar N, et al. Late complications of immune deviation therapy in a nonhuman primate. Science. 1996;274(5295):2054–2057. doi: 10.1126/science.274.5295.2054. [DOI] [PubMed] [Google Scholar]
- 173.Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nature Medicine. 1999;5(2):170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- 174.Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. American Journal of Pathology. 1988;130(3):443–454. [PMC free article] [PubMed] [Google Scholar]
- 175.Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Annals of Neurology. 1988;23(4):339–346. doi: 10.1002/ana.410230405. [DOI] [PubMed] [Google Scholar]
- 176.Vartanian T, Li You LY, Zhao Meijuan ZM, Stefansson K. Interferon-gamma-induced oligodendrocyte cell death: implications for the pathogenesis of multiple sclerosis. Molecular Medicine. 1995;1(7):732–743. [PMC free article] [PubMed] [Google Scholar]
- 177.Curatolo L, Valsasina B, Caccia C, Raimondi GL, Orsini G, Bianchetti A. Recombinant human IL-2 is cytotoxic to oligodendrocytes after in vitro self aggregation. Cytokine. 1997;9(10):734–739. doi: 10.1006/cyto.1997.0228. [DOI] [PubMed] [Google Scholar]
- 178.Hisahara S, Shoji S, Okano H, Miura M. ICE/CED-3 family executes oligodendrocyte apoptosis by tumor necrosis factor. Journal of Neurochemistry. 1997;69(1):10–20. doi: 10.1046/j.1471-4159.1997.69010010.x. [DOI] [PubMed] [Google Scholar]
- 179.Jurewicz A, Matysiak M, Tybor K, Kilianek L, Raine CS, Selmaj K. Tumour necrosis factor-induced death of adult human oligodendrocytes is mediated by apoptosis inducing factor. Brain. 2005;128(11):2675–2688. doi: 10.1093/brain/awh627. [DOI] [PubMed] [Google Scholar]
- 180.Agresti C, Meomartini ME, Amadio S, et al. ATP regulates oligodendrocyte progenitor migration, proliferation, and differentiation: involvement of metabotropic P2 receptors. Brain Research Reviews. 2005;48(2):157–165. doi: 10.1016/j.brainresrev.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 181.Merrill JE, Scolding NJ. Mechanisms of damage to myelin and oligodendrocytes and their relevance to disease. Neuropathology and Applied Neurobiology. 1999;25(6):435–458. doi: 10.1046/j.1365-2990.1999.00200.x. [DOI] [PubMed] [Google Scholar]
- 182.Dowling P, Shang G, Raval S, Menonna J, Cook S, Husar W. Involvement of the CD95 (APO-1/Fas) receptor/ligand system in multiple sclerosis brain. Journal of Experimental Medicine. 1996;184(4):1513–1518. doi: 10.1084/jem.184.4.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pouly S, Becher B, Blain M, Antel JP. Interferon-γ modulates human oligodendrocyte susceptibility to Fas-mediated apoptosis. Journal of Neuropathology and Experimental Neurology. 2000;59(4):280–286. doi: 10.1093/jnen/59.4.280. [DOI] [PubMed] [Google Scholar]
- 184.Höftberger R, Aboul-Enein F, Brueck W, et al. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathology. 2004;14(1):43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Patel J, Balabanov R. Molecular mechanisms of oligodendrocyte injury in multiple sclerosis and experimental autoimmune encephalomyelitis. International Journal of Molecular Sciences. 2012;13:10647–10659. doi: 10.3390/ijms130810647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bö L, Mörk S, Kong PA, Nyland H, Pardo CA, Trapp BD. Detection of MHC class II-antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis lesions. Journal of Neuroimmunology. 1994;51(2):135–146. doi: 10.1016/0165-5728(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 187.Redwine JM, Buchmeier MJ, Evans CF. In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. American Journal of Pathology. 2001;159(4):1219–1224. doi: 10.1016/S0002-9440(10)62507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bergsteinsdottir K, Brennan A, Jessen KR, Mirsky R. In the presence of dexamethasone, γ interferon induces rat oligodendrocytes to express major histocompatibility complex class II molecules. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(19):9054–9058. doi: 10.1073/pnas.89.19.9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Harada K, Hide I, Seki T, Tanaka S, Nakata Y, Sakai N. Extracellular ATP differentially modulates Toll-like receptor 4-mediated cell survival and death of microglia. Journal of Neurochemistry. 2011;116(6):1138–1147. doi: 10.1111/j.1471-4159.2011.07170.x. [DOI] [PubMed] [Google Scholar]
- 191.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nature Reviews Immunology. 2011;11(3):201–212. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bal-Price A, Moneer Z, Brown GC. Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia. 2002;40(3):312–323. doi: 10.1002/glia.10124. [DOI] [PubMed] [Google Scholar]
- 193.Parpura V, Scemes E, Spray DC. Mechanisms of glutamate release from astrocytes: gap junction “hemichannels”, purinergic receptors and exocytotic release. Neurochemistry International. 2004;45(2-3):259–264. doi: 10.1016/j.neuint.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 194.Schenk U, Westendorf AM, Radaelli E, et al. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Science Signaling. 2008;1(39):p. ra6. doi: 10.1126/scisignal.1160583. [DOI] [PubMed] [Google Scholar]
- 195.Tokunaga A, Tsukimoto M, Harada H, Moriyama Y, Kojima S. Involvement of SLC17A9-dependent vesicular exocytosis in the mechanism of ATP release during T cell activation. Journal of Biological Chemistry. 2010;285(23):17406–17416. doi: 10.1074/jbc.M110.112417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dahlquist R, Diamant B. Interaction of ATP and calcium on the rat mast cell: effect on histamine release. Acta Pharmacologica et Toxicologica. 1974;34(5):368–384. doi: 10.1111/j.1600-0773.1974.tb03533.x. [DOI] [PubMed] [Google Scholar]
- 197.McCloskey MA, Fan Y, Luther S. Chemotaxis of rat mast cells toward adenine nucleotides. Journal of Immunology. 1999;163(2):970–977. [PubMed] [Google Scholar]
- 198.Bulanova E, Budagian V, Orinska Z, et al. Extracellular ATP induces cytokine expression and apoptosis through P2X7 receptor in murine mast cells. Journal of Immunology. 2005;174(7):3880–3890. doi: 10.4049/jimmunol.174.7.3880. [DOI] [PubMed] [Google Scholar]
- 199.Suzuki R, Furuno T, McKay DM, et al. Direct neurite-mast cell communication in vitro occurs via the neuropeptide substance P. Journal of Immunology. 1999;163(5):2410–2415. [PubMed] [Google Scholar]
- 200.Hudson CA, Christophi GP, Gruber RC, Wilmore JR, Lawrence DA, Massa PT. Induction of IL-33 expression and activity in central nervous system glia. Journal of Leukocyte Biology. 2008;84(3):631–643. doi: 10.1189/jlb.1207830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhang B, Asadi S, Weng Z, Sismanopoulos N, Theoharides TC. Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0049767.e49767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Theoharides TC, Spanos C, Pang X, et al. Stress-induced intracranial mast cell degranulation: a corticotropin-releasing hormone-mediated effect. Endocrinology. 1995;136(12):5745–5750. doi: 10.1210/endo.136.12.7588332. [DOI] [PubMed] [Google Scholar]
- 203.Theoharides TC, Singh LK, Boucher W, et al. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology. 1998;139:403–413. doi: 10.1210/endo.139.1.5660. [DOI] [PubMed] [Google Scholar]
- 204.Cao J, Papadopoulou N, Kempuraj D, et al. Human mast cells express corticotropin-releasing hormone (CRH) receptors and CRH leads to selective secretion of vascular endothelial growth factor. Journal of Immunology. 2005;174(12):7665–7675. doi: 10.4049/jimmunol.174.12.7665. [DOI] [PubMed] [Google Scholar]
- 205.Esposito P, Chandler N, Kandere K, et al. Corticotropin-releasing hormone and brain mast cells regulate blood-brain-barrier permeability induced by acute stress. Journal of Pharmacology and Experimental Therapeutics. 2002;303(3):1061–1066. doi: 10.1124/jpet.102.038497. [DOI] [PubMed] [Google Scholar]
- 206.Alysandratos KD, Asadi S, Angelidou A, et al. Neurotensin and CRH interactions augment human mast cell activation. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0048934.e48934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sugama S. Stress-induced microglial activation may facilitate the progression of neurodegenerative disorders. Medical Hypotheses. 2009;73(6):1031–1034. doi: 10.1016/j.mehy.2009.02.047. [DOI] [PubMed] [Google Scholar]
- 208.Sugama S, Takenouchi T, Fujita M, Conti B, Hashimoto M. Differential microglial activation between acute stress and lipopolysaccharide treatment. Journal of Neuroimmunology. 2009;207(1-2):24–31. doi: 10.1016/j.jneuroim.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 209.Simard M, Couldwell WT, Zhang W, et al. Glucocorticoids-potent modulators of astrocytic calcium signaling. Glia. 1999;28:1–12. doi: 10.1002/(sici)1098-1136(199910)28:1<1::aid-glia1>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 210.Mirescu C, Peters JD, Gould E. Early life experience alters response of adult neurogenesis to stress. Nature Neuroscience. 2004;7(8):841–846. doi: 10.1038/nn1290. [DOI] [PubMed] [Google Scholar]
- 211.Gómez-González B, Escobar A. Prenatal stress alters microglial development and distribution in postnatal rat brain. Acta Neuropathologica. 2010;119(3):303–315. doi: 10.1007/s00401-009-0590-4. [DOI] [PubMed] [Google Scholar]
- 212.Helmut K, Hanisch U-K, Noda M, Verkhratsky A. Physiology of microglia. Physiological Reviews. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 213.Juckel G, Manitz MP, Brüne M, Friebe A, Heneka MT, Wolf RJ. Microglial activation in a neuroinflammational animal model of schizophrenia—a pilot study. Schizophrenia Research. 2011;131(1–3):96–100. doi: 10.1016/j.schres.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 214.Belliveau DJ, Bani-Yaghoub M, McGirr B, Naus CCG, Rushlow WJ. Enhanced neurite outgrowth in PC12 cells mediated by connexin hemichannels and ATP. Journal of Biological Chemistry. 2006;281(30):20920–20931. doi: 10.1074/jbc.M600026200. [DOI] [PubMed] [Google Scholar]
- 215.Joseph DJ, Williams DJ, MacDermott AB. Modulation of neurite outgrowth by activation of calcium-permeable kainate receptors expressed by rat nociceptive-like dorsal root ganglion neurons. Developmental Neurobiology. 2011;71(10):818–835. doi: 10.1002/dneu.20906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Frank MG, Watkins LR, Maier SF. Stress- and glucocorticoid-induced priming of neuroinflammatory responses: potential mechanisms of stress-induced vulnerability to drugs of abuse. Brain, Behavior, and Immunity. 2011;25(1):S21–S28. doi: 10.1016/j.bbi.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Goujon E, Parnet P, Laye S, Combe C, Kelley KW, Dantzer R. Stress downregulates lipopolysaccharide-induced expression of proinflammatory cytokines in the spleen, pituitary, and brain of mice. Brain, Behavior, and Immunity. 1995;9(4):292–303. doi: 10.1006/brbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- 218.Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. Journal of Psychiatry and Neuroscience. 2009;34(1):4–20. [PMC free article] [PubMed] [Google Scholar]
- 219.Pace TWW, Miller AH. Cytokines and glucocorticoid receptor signaling: relevance to major depression. Annals of the New York Academy of Sciences. 2009;1179:86–105. doi: 10.1111/j.1749-6632.2009.04984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gouin J-P, Hantsoo L, Kiecolt-Glaser JK. Immune dysregulation and chronic stress among older adults: a review. NeuroImmunoModulation. 2008;15(4-6):251–259. doi: 10.1159/000156468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Nakata A. Psychosocial job stress and immunity: a systematic review. Methods in Molecular Biology. 2012;934:39–75. doi: 10.1007/978-1-62703-071-7_3. [DOI] [PubMed] [Google Scholar]
- 222.Angelidou A, Asadi S, Alysandratos KD, Karagkouni A, Kourembanas S, Theoharides TC. Perinatal stress, brain inflammation and risk of autism-review and proposal. BMC Pediatrics. 2012;12:p. 89. doi: 10.1186/1471-2431-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Garate I, Garcia-Bueno B, Madrigal JL, et al. Stress-induced neuroinflammation: role of the Toll-like receptor-4 pathway. Biological Psychiatry. 2013;73:32–43. doi: 10.1016/j.biopsych.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 224.Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(5):1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Theoharides TC, Zhang B, Conti P. Decreased mitochondrial function and increased brain inflammation in bipolar disorder and other neuropsychiatric diseases. Journal of Clinical Psychopharmacology. 2011;31(6):685–687. doi: 10.1097/JCP.0b013e318239c190. [DOI] [PubMed] [Google Scholar]
- 226.Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Annals of Neurology. 2012;71(4):444–457. doi: 10.1002/ana.22620. [DOI] [PubMed] [Google Scholar]
- 227.Vasiadi M, Therianou A, Sideri K, et al. Increased serum CRH levels with decreased skin CRHR-1 gene expression in psoriasis and atopic dermatitis. Journal of Allergy and Clinical Immunology. 2012;129(5):1410–1413. doi: 10.1016/j.jaci.2012.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Sanders P, De Keyser J. Janus faces of microglia in multiple sclerosis. Brain Research Reviews. 2007;54(2):274–285. doi: 10.1016/j.brainresrev.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 229.de Vries HE, Kuiper J, De Boer AG, Van Berkel TJC, Breimer DD. The blood-brain barrier in neuroinflammatory diseases. Pharmacological Reviews. 1997;49(2):143–155. [PubMed] [Google Scholar]
- 230.Tuomisto L, Kilpeläinen H, Riekkinen P. Histamine and histamine-N-methyltransferase in the CSF of patients with multiple sclerosis. Agents and Actions. 1983;13(2-3):255–257. doi: 10.1007/BF01967346. [DOI] [PubMed] [Google Scholar]
- 231.Kinney DK, Munir KM, Crowley DJ, Miller AM. Prenatal stress and risk for autism. Neuroscience and Biobehavioral Reviews. 2008;32(8):1519–1532. doi: 10.1016/j.neubiorev.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Chrousos GP, Epstein F, Flier J, Reichlin S, Pavlou S. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. The New England Journal of Medicine. 1995;332(20):1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- 233.Theoharides TC, Donelan JM, Papadopoulou N, Cao J, Kempuraj D, Conti P. Mast cells as targets of corticotropin-releasing factor and related peptides. Trends in Pharmacological Sciences. 2004;25(11):563–568. doi: 10.1016/j.tips.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 234.Theoharides TC, Konstantinidou AD. Corticotropin-releaslng hormone and the blood-brain-barrier. Frontiers in Bioscience. 2007;12(5):1615–1628. doi: 10.2741/2174. [DOI] [PubMed] [Google Scholar]
- 235.Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cellular and Molecular Neurobiology. 2000;20(2):131–147. doi: 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Giulivi C, Zhang Y-F, Omanska-Klusek A, et al. Mitochondrial dysfunction in autism. Journal of the American Medical Association. 2010;304(21):2389–2396. doi: 10.1001/jama.2010.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Palmieri L, Persico AM. Mitochondrial dysfunction in autism spectrum disorders: cause or effect? Biochimica et Biophysica Acta. 2010;1797(6-7):1130–1137. doi: 10.1016/j.bbabio.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 238.Frye RE, Rossignol DA. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatric Research. 2011;69(5) doi: 10.1203/PDR.0b013e318212f16b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Enstrom AM, Van De WAJ, Ashwood P. Autoimmunity in autism. Current Opinion in Investigational Drugs. 2009;10(5):463–473. [PMC free article] [PubMed] [Google Scholar]
- 240.Theoharides TC, Kempuraj D, Redwood L. Autism: an emerging “neuroimmune disorder” in search of therapy. Expert Opinion on Pharmacotherapy. 2009;10(13):2127–2143. doi: 10.1517/14656560903107789. [DOI] [PubMed] [Google Scholar]
- 241.Goines P, Van De Water J. The immune system’s role in the biology of autism. Current Opinion in Neurology. 2010;23(2):111–117. doi: 10.1097/WCO.0b013e3283373514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wills S, Cabanlit M, Bennett J, Ashwood P, Amaral DG, Van de Water J. Detection of autoantibodies to neural cells of the cerebellum in the plasma of subjects with autism spectrum disorders. Brain, Behavior, and Immunity. 2009;23(1):64–74. doi: 10.1016/j.bbi.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Zhang B, Angelidou A, Alysandratos K-D, et al. Mitochondrial DNA and anti-mitochondrial antibodies in serum of autistic children. Journal of Neuroinflammation. 2010;7, article 80 doi: 10.1186/1742-2094-7-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Angelidou A, Francis K, Vasiadi M, et al. Neurotensin is increased in serum of young children with autistic disorder. Journal of Neuroinflammation. 2010;7, article 48 doi: 10.1186/1742-2094-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Zhang B, Alysandratos K-D, Angelidou A, et al. Human mast cell degranulation and preformed TNF secretion require mitochondrial translocation to exocytosis sites: relevance to atopic dermatitis. Journal of Allergy and Clinical Immunology. 2011;127(6):1522–e8. doi: 10.1016/j.jaci.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.von zur Muhlen F, Eckstein F, Penner R. Guanosine 5′-[β-thio]triphosphate selectively activates calcium signaling in mast cells. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(3):926–930. doi: 10.1073/pnas.88.3.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gibb J, Hayley S, Gandhi R, Poulter MO, Anisman H. Synergistic and additive actions of a psychosocial stressor and endotoxin challenge: circulating and brain cytokines, plasma corticosterone and behavioral changes in mice. Brain, Behavior, and Immunity. 2008;22(4):573–589. doi: 10.1016/j.bbi.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 248.Shi L, Smith SEP, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain, Behavior, and Immunity. 2009;23(1):116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hinkerohe D, Wolfkühler D, Haghikia A, Meier C, Faustmann PM, Schlegel U. Dexamethasone differentially regulates functional membrane properties in glioma cell lines and primary astrocytes in vitro. Journal of Neuro-Oncology. 2011;103(3):479–489. doi: 10.1007/s11060-010-0456-6. [DOI] [PubMed] [Google Scholar]
- 250.Gibb J, Hayley S, Poulter MO, Anisman H. Effects of stressors and immune activating agents on peripheral and central cytokines in mouse strains that differ in stressor responsivity. Brain, Behavior, and Immunity. 2011;25(3):468–482. doi: 10.1016/j.bbi.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 251.Hayley S, Mangano E, Strickland M, Anisman H. Lipopolysaccharide and a social stressor influence behaviour, corticosterone and cytokine levels: divergent actions in cyclooxygenase-2 deficient mice and wild type controls. Journal of Neuroimmunology. 2008;197(1):29–36. doi: 10.1016/j.jneuroim.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 252.Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Frontiers in Behavioral Neuroscience. 2009;3:p. 14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kato TA, Kanba S. Are microglia minding us? Digging up the unconscious mind-brain relationship from a neuropsychoanalytic approach. Frontiers in Human Neuroscience. 2013;7:p. 13. doi: 10.3389/fnhum.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]