

Abstract
Gastrokine 1 (GKN1) plays an important role in the gastric mucosal defense mechanism and also acts as a functional gastric tumor suppressor. In this study, we examined the effect of GKN1 on the expression of inflammatory mediators, including NF-κB, COX-2 and cytokines in GKN1-transfected AGS cells and shGKN1-transfected HFE-145 cells. Lymphocyte migration and cell viability were also analyzed after treatment with GKN1 and inflammatory cytokines in AGS cells by transwell chemotaxis and an MTT assay, respectively. In GKN1-transfected AGS cells, we observed inactivation and reduced expression of NF-κB and COX-2, whereas shGKN1-transfected HFE-145 cells showed activation and increased expression of NF-κB and COX-2. GKN1 expression induced production of inflammatory cytokines including IL-8 and -17A, but decreased expression of IL-6 and -10. We also found IL-17A expression in 9 (13.6%) out of 166 gastric cancer tissues and its expression was closely associated with GKN1 expression. GKN1 also acted as a chemoattractant for the migration of Jurkat T cells and peripheral B lymphocytes in the transwell assay. In addition, GKN1 significantly reduced cell viability in both AGS and HFE-145 cells. These data suggest that the GKN1 gene may inhibit progression of gastric epithelial cells to cancer cells by regulating NF-κB signaling pathway and cytokine expression.
Keywords: GKN1, gastric cancer, NF-κB, inflammatory mediator, cytokine
Introduction
The gastrointestinal tract is subject to constant trauma from ingested food particles and various noxious agents, in addition to the stress due to acidic environment. Gastric mucosal inflammation is generally believed to be caused by chronic Helicobacter pylori (H. pylori) infection; while atrophic gastritis, intestinal metaplasia and dysplasia represent the different stages of the gastric carcinogenesis cascade [Yuasa, 2003]. Thus, H. pylori infection likely increases the risk of carcinogenesis by differentially affecting host inflammatory responses and epithelial cell physiology [Peek et al., 2002]. For example, H. pylori infection induces production of pro-inflammatory mediators, including cytokines and pro-proliferative factors [Lindhilm et al., 1998; Holck et al., 2003; Basso et al., 1996]. More importantly, infiltrating inflammatory cells develop into cancer cells with the help of an array of cytokines, chemokines and cytotoxic mediators, which participate in the regulation of tumor growth, angiogenesis, invasion and metastasis [Zhou et al., 2007]. However, little is known about the regulation of these cytokines in the gastric mucosa or the molecular mechanisms underlying the contribution of these cytokines to gastric carcinogenesis.
Recently, gastrokine 1 (GKN1) has been isolated from gastric mucosal cells of several mammalian species, including rats [Martin et al., 2003]. Toback et al. reported that GKN1 protects the antral mucosa and promotes healing by facilitating restoration and proliferation after injury [Toback et al., 2003]. In addition, GKN1 also protects the intestinal mucosal barrier by acting on specific tight junction proteins and stabilizing peri-junctional actin [Walsh-Reitz et al., 2005]. Clinically, GKN1 is downregulated in H. pylori-infected gastric epithelial cells, and this loss of GKN1 expression is detected in gastric cancer tissues and precancerous lesions, such as intestinal metaplasia [Nardone et al., 2007; Shiozaki et al., 2001]. In accordance with these previous findings, we and other researchers also witnessed frequent loss of GKN1 expression in gastric cancer tissues and GKN1 tumor suppressor activity in a functional analysis [Yoon et al., 2011b; Rippa et al., 2011]. Moreover, GKN1 plays an important role in epithelial-mesenchymal transition and migration of gastric cancer cells by regulating reactive oxygen species (ROS) and the PI3K/Akt pathway [Yoon et al., 2011a]. Recently, it has been suggested that GKN1 induces senescence through activating p16/Rb pathway in gastric cancer cells [Xing et al., 2012]. Thus, we hypothesized that GKN1 plays an important role in the development or progression of gastric cancer by regulating pro-proliferative inflammatory mediators.
In this study, we investigated the effect of GKN1 on lymphocyte migration and on the expression levels of inflammatory mediators, including nuclear factor (NF)-κB, COX-2 and cytokines, in AGS and HFE-145 cells. Overall, we intend to demonstrate that GKN1 inhibits pro-carcinogenic inflammatory mediators, including NF-κB, COX-2 and pSTAT-3, while inducing anti-carcinogenic cytokine production and stimulating lymphocyte migration.
Materials and Methods
Ethics Statement
Total of 166 gastric cancer tissues from formalin-fixed paraffin embedded specimens were obtained from St. Mary’s Hospital, Seoul, Korea. For lymphocyte study, the peripheral blood monocyte cells (PBMCs) from healthy individuals were obtained from Hematopoietic Stem Cell Bank in Catholic University of Korea. Informed consent was provided according to the Declaration of Helsinki. Written informed consent was obtained from all subjects, and the study was approved by the Ethics Committee of the Catholic University of Korea, College of Medicine (IRB approval number; CUMC09U089).
Cell culture and transfection of GKN1
AGS gastric cancer cell lines and HFE-145 normal gastric mucosa cells were cultured at 37 °C in 5% CO2 in RPMI-1640 medium (Lonza, Basel, Switzerland) with 10% heat-inactivated fetal bovine serum. GKN1 cDNA was cloned into the pcDNA3.1 expression vector (Invitrogen, Carlsbad, CA, USA) and shGKN1 cloned into pSilencer neo vector (Invitrogen, Carlsbad, CA, USA). AGS and HFE-145 cells were transfected in 60 mm-diameter dishes with the expression plasmids and shRNA plasmids (2 μg total DNA), respectively, using Lipofectamine Plus transfection reagent (Invitrogen), according to the manufacturers recommendations. After transfection, the conditioned media were collected from mock- or GKN1-transfected AGS cells, and the concentration of GKN1 (USCN, Wuhan, China), IL-17 and TNF-α (R&D, Abingdon, U.K.) was calculated using an ELISA kit according to the manufacturer’s instructions.
Western blotting
To determine whether GKN1 is involved in the regulation of inflammatory mediators, expression of p-STAT3, Erk and NF-κB related proteins, including NF-κB p-p65, NF-κB p65, p-IKKα/β, IKKα/β, p-IκB and IκB, was examined in AGS cells 24 hrs after GKN1 transfection and in HFE-145 cells 48 hrs after shGKN1 transfection. Cell lysates were separated on a 10% polyacrylamide gel and transferred onto a Hybond PVDF membrane (Amersham Pharmacia Biotech, Piscataway, NJ, USA). After blocking, the membrane was subsequently probed with antibodies against NF-κB p65, p-IKKα/β, IκB (Santa Cruz Biotech, Santa Cruz, CA, USA), NF-κB p-p65, IKKα/β, p-IκB, cyclin D1, and Foxp3 (Cell Signaling Technology, Danvers, MA, USA). Protein bands were detected using enhanced chemiluminescence reagents (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
Immunofluorescence analysis
The effect of GKN1 on the expression of NF-κB p65 in AGS cells was determined by immunofluorescence and confocal microscopy. Cells transfected with GKN1 were grown on 2-well chamber slides and fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4), for 10 min at room temperature, rinsed with PBS, and incubated in 10% normal donkey serum for 30 min to block nonspecific binding. The cells were then incubated with mouse monoclonal antibody against NF-κB p65 (Santa Cruz Biotech, Santa Cruz, CA, USA) in PBS containing 0.5% Triton X-100 overnight. The cells were rinsed with PBS and incubated with a Cy3-conjugated donkey anti-mouse IgG (1:200 dilution; Jackson Immunoresearch, West Grove, PA, USA) for 1 hr at room temperature. Counterstaining of cell nuclei was carried out by incubating the cells with DAPI (4′,6-diamidino–2′-phenyindole; Roche; dilution 1:1000) for 10 min. Slides were viewed with a confocal microscope (LSM 510 Meta, Carl Zeiss Co., Ltd., Germany). Images were converted to TIFF format, and contrast levels were adjusted using Adobe Photoshop v. 7.0 (Adobe Systems, San Jose, CA, USA).
Real-time RT-PCR
To investigate the effects of GKN1 on cytokines and COX-2 expression in AGS and HFE-145 cells, real-time reverse transcriptase-polymerase chain reaction (RT-PCR) analysis was performed. cDNA was synthesized using the reverse transcription kit from Roche Molecular Systems (Roche, Mannheim, Germany) according to the manufacturer’s protocol. For quantitative PCR (QPCR), 50 ng cDNA was amplified using Fullvelocity SYBR Green QPCR Master Mix (Stratagene, La Jolla, CA, USA) and 20 pmol/μl of each set of forward and reverse primers on the Stratagene Mx 3000P QPCR system with techniques previously published [Yoon et al., 2011b]. The specific oligonucleotide primers for mRNA were designed using the primer3 program (available: http://frodo.wi.mit.edu/primer3/) for COX-2 and inflammatory cytokine genes, including IL-6, -8, -10, -17A and TNF-α. The primer sequences for each gene were as follow; TNFα: 5′-CGAGTGACAAGCCTGTAGC-3′ for forward and 5′-GGTGTGGGTGAGGAGCACAT-3′ for reverse, IL-6: 5′-AAATGCCAGCCTGCTGA CGAAC-3′ for forward and 5′-AACAACAATCTGAGGTGCCCATGCTAC-3′ for reverse, IL-8: 5′-AACTTCTCCACAACCCTCTG-3′ for forward and 5′-TTGGCAGCCTTCCTGATTTC-3′ for reverse, IL-10: 5′-GTGATGCCCCAAGCTGAGA-3′ for forward and 5′-CACGGCCTTGCTCTTGTTTT-3′ for reverse, IL-17A: 5′-ACCGCAATGAAGACCCTGAT-3′ for forward and 5′-TCCCTCCGCATTGACACA-3′ for reverse, COX-2: 5′-CTTGCTGTTCCCACCCATGTCAAA-3′ for forward and 5′-TGCACTGTGTTTGGAGTGGGTTTC-3′ for reverse. To ensure the fidelity of mRNA extraction and reverse transcription, all samples were subjected to PCR amplification with oligonucleotide primers specific to the constitutively expressed gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and the expression levels of the above-mentioned genes were normalized to GAPDH expression. The primer sequences of the GAPDH were as follows; 5′-AAATCAAGTGGGGCGATGCTG-3′ for forward and 5′-GCAGAGATGATGACCCTTTTG-3′ for reverse. The standard curve method was used for quantification of the relative amounts of gene expression products. This method provides unitless normalized expression values that can be used for direct comparison of the relative amount of mRNA in different samples. All samples were tested in duplicate and average values were used for quantification.
A sensitive ELISA kit (R&D, Abingdon, U.K) was used to detect IL -17 and TNF-α in AGS cells after mock and GKN1 transfection, according to the manufacturer’s recommendations. All samples were assayed in duplicate. The amount of IL-17 and TNF-α was measured as nanograms per milliliter of AGS cell culture supernatant.
Lymphocytes migration assay
Jurkat T cells and immortalized peripheral B lymphocytes with SV40 from healthy individuals were cultured at 37 °C in 5% CO2 in RPMI-1640 medium (Lonza, Basel, Switzerland) with 10% heat-inactivated fetal bovine serum.
Cell migration was assayed in 48-well microchemotaxis chambers with gelatin-coated 8 μm polyvinylpyrrolidine-free polycarbonate filters (Neuroprobe, Cabin John, MD, USA) using mock- or GKN1-transfected AGS cells as a chemoattractant. A suspension containing 6 × 104 cells was added to each upper chamber, and the cell number was determined by calculating the number of cells in the membrane in a 20x field from three independent experiments. To confirm that IL-17 produced by GKN1 plays a role in recruiting lymphocytes, cell migration assay was done after neutralization of IL-8 and IL-17A with treatment of human anti-IL-8 (1 ug/ml) and anti-IL-17A (1 μg/ml) in lower chamber containing AGS cells transfected with wild-type GKN1.
Immunohistochemistry
Since the predominant source of IL-17 remains in the CD4+ T-cell population, Th17 subset [Weaver et al., 2006], we examined via immunohistochemistry whether IL-17A production is associated with GKN1 expression in gastric cancer tissues. For immunohistochemical analysis, tissue microarray recipient blocks were constructed containing 166 gastric cancer tissues from formalin-fixed paraffin embedded specimens. To maximize the immunohistochemistry signal, two strategies were used: antigen retrieval in citrate buffer and signal amplification with biotinylated tyramide, as previously described [Yoon et al., 2011b]. 2 μm sections were incubated overnight at 4°C with IL-17A antibody (1/100; R&D, Abingdon, U.K.). Detection was carried out using biotinylated goat anti-mouse antibodies (Sigma, St. Louis, MO, USA), followed by incubation with a peroxidase-linked avidin-biotin complex. Diaminobenzidine was used as a chromogen and the slides were counterstained with Mayer’s hematoxylin. For negative controls, primary antibodies were replaced with non-immune serum. We compared IL-17A expression patterns in 166 gastric cancer tissues with our previous GKN1 expression data [Yoon et al., 2011b].
Measurement of cell viability
To investigate whether inflammatory mediators, including IL-10 and IL-17A, are involved in gastric mucosal cell viability, an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide] assay was performed in AGS cells at 24 hrs following transfection with GKN1 (0.1, 0.3, and 0.5 ug), treatment with recombinant IL-10 (10, 50, and 100 ng/ml), and IL-17A (10, 50, and 100 ng/ml) and in HFE-145 cells at 48 hrs following transfection with shGKN1 (0.5 ug). Absorbance was measured at 540 nm using a spectrophotometer to evaluate viability relative to the control.
To find out the effect of GKN1 on T lymphocyte viability, peripheral blood mononuclear cells (PBMCs) from 4 healthy individuals were isolated by density centrifugation using Ficoll-Hypaque (Pharmacia LKB, Uppsala, Sweden). Anti-CD4 microbeads were used as previously described to sort out Th0 cells [Heo et al., 2010]. In brief, PBMCs were resuspended in 100 μl MACS buffer [1% bovine serum albumin (BSA), 5 mM EDTA, and 0.01% sodium azide]. Anti-CD4 microbeads (10 μl/1 × 107 cells) were added and incubated for 15 min at 4 °C. Cells were diluted in 10 μl MACS buffer, pelleted, resuspended in 500 μl, and separated magnetically in an AutoMACS magnet fitted with a MACS MS column (Miltenyi Biotec, Bergisch Gladbach, Germany). The cell suspension was adjusted to a concentration of 1 × 106/ml in RPMI 1640 supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM L-glutamine. The isolated cells were then dispensed into 24-well plates (Nunc, Roskilde, Denmark) and incubated at 37 °C in 5% CO2. Anti-CD3 monoclonal antibody and anti-CD28 monoclonal antibody were obtained from BD Biosciences (San Diego, CA, USA). Recombinant IL-6 and IL-1β were purchased from R&D Systems, and recombinant TGF-β was purchased from Peprotech (Rocky Hill, NJ, USA). Phorbol 12-myristate 13-acetate (PMA) and ionomycin were from Sigma–Aldrich Chemical Co. (St Louis, MO, USA), and monoclonal anti-human IFN-γ and IL-4 were purchased from R&D Systems.
For the apoptotic assay, an Annexin V-binding assay was performed according to the manufacturer’s instructions. To differentiate Th cells, isolated CD4+ T cells (5 × 105) and radiated antigen-presenting cells (APC; 5 × 105) from healthy individuals were mixed and incubated under different conditions to stimulate Th0 or Th17 cells. Anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml) were used to differentiate Th0 cells. IL-1β (20 ng/ml), IL-6 (10 ng/ml), neutralizing anti INF-γ (10 μg/ml) and anti-IL-4 (10 μg/ml) were added to stimulate differentiation of Th17 cells.
Flow Cytometry Analysis
For all intracellular staining, cells were stimulated for 4 h with PMA (25 ng/ml; Sigma) and ionomycin (250ng/ml; Sigma), and were treated with GolgiPlug (1 μg per 1 × 106 cells; BD Pharmingen). In the staining procedure, cells were stained in the presence of Fcγ Blocker (Clone 24G2, BD Pharmingen, San Diego, CA) with PerCP-labeled anti-CD4 antibody and APC-labeled anti-CD25 antibody. Cells were then washed in Fix/Perm solution (eBioscience, San Diego, CA, USA). Cells were assessed for IL-17 and Foxp3 expression. Data were acquired using a Becton Dickinson FACS Calibur and analyzed with Cell Quest software (BD Bioscience).
Statistical analysis
A Student’s t-test was used to analyze the effect of GKN1 on cytokine expression and cell viability. Data are expressed as means ± S.D. from at least two or three independent experiments. The chi-square test was used to examine the relationship of expression between GKN1 and IL-17A proteins. A P-value less than 0.05 was considered to be the limit of statistical significance.
Results
GKN1 inactivates the NF-κB pathway by activating IκB
As shown in Figure 1, ectopic GKN1 expression suppressed p-IKKα/β and p-NF-κB expression and NF-κB nuclear translocation, whereas it increased IκB expression. We also found that GKN1 increased Foxp3 and decreased nuclear expression of cyclin D1 (Fig. 1). In addition, repressed GKN1 expression in HFE-145 cells transfected with shGKN1 showed increased p-IKKα/β, p-NF-κB, and NF-κB expression and decreased IκB expression (Fig. 1B). To confirm this result, AGS cells transfected with GKN1 were stained with antibodies NF-κB p65, and compared to the respective controls (mock transfection). Ectopic GKN1 expression inhibited NF-κB p65 expression and nuclear translocation (Fig. 1C).
Figure 1.

Effects of GKN1 on NF-κB inactivation in AGS and HFE-145 cells. A & B: Western blot analysis for p-IKKα/β, IKKα/β, p-IκB, IκB, p-NF-κB p65, NF-κB p65, Foxp3, p-STAT3, and Erk expression was performed on AGS cells after GKN1 transfection (A) and on HFE-145 after shGKN1 transfection (B). GKN1 expression in AGS cells induced decreased expression of p-STAT3, p-IKKα/β, and nuclear p65 and cyclin D1, and increased expression of p-Erk, IκB and Foxp3. Knock-down of GKN1 expression in HFE-145 cells induced increased expression of p-IKKα/β, IKKα/β, p-IκB, p-NF-κB p65, NF-κB p65, and decreased IκB expression. The data are representative of three separate experiments. C: Immunofluorescence and confocal microscopy showed that ectopic expression of GKN1 inhibited NF-κB p65 expression and nuclear translocation.
GKN1 regulates the expression of inflammatory cytokines
Ectopic GKN1 expression led to 2.9 and 2.6 fold changes in IL-8 and -17A mRNA expression, respectively (P < 0.05), while IL-6 and IL-10 expression was significantly decreased (Fig. 2A). However, no significant change in TNF-α mRNA expression was observed after GKN1 transfection (Fig. 2A). Additionally, knock-down of GKN1 expression in shGKN1-transfected HFE-145 cells led to −2.2, and −3.6 fold changes in IL-8 and -17A mRNA expression, respectively, whereas IL-6 and IL-10 expression was significantly increased. To find out the underlying mechanism of altered cytokine expression, we examined p-STAT3 and p-Erk expression in GKN1-transfected AGS cells. Expectedly, GKN1 expression induced decreased expression of p-STAT3 and increased expression of p-Erk protein (Fig. 1A).
Figure 2.

Effect of GKN1 on the expression of inflammatory cytokines. A: After GKN1 transfection in AGS cells and after shGKN1 transfection in HFE-145 cells, expressions of IL-6, IL-8, IL-10, IL-17A, and TNF-α mRNA were measured by real-time RT PCR. Values are represented as the fold changes presented as means ± S.D. of 2 independent experiments. B: After GKN1 transfection in AGS cells, TNF-α and IL-17A were measured in conditioned culture media by ELISA at 24 hrs. The level of IL-17A in culture media was significantly increased from 16.966 ± 3.85 to 22.668 ± 4.215 ng/ml. Values are the means ± S.D. of 2 independent experiments. C: In immunohistochemistry, IL-17 was expressed in cytoplasm and membrane of gastric mucosal epithelial (left panel) and cancer cells (right). Original magnification, 200X.
In AGS cells transfected with mock- or GKN1, IL-17A and TNF-α level were also measured in culture media by ELISA. GKN1 expression induced a significant increase in IL-17A secretion in the culture media of AGS cells compared to the media of mock-transfected AGS cells (P = 0.0143) (Fig. 2B). GKN1 tended to induce TNF-α secretion, but no statistical significance was observed (P=0.1051) (Fig. 2B). In immunohistochemistry, non-cancerous gastric mucosae and cancer cells showed a moderate-to-strong IL-17A expression in the membrane and cytoplasm of the cells (Fig. 2C). We found IL-17A expression in 9 (13.6%) out of 166 gastric cancer tissues (Fig. 2C). Of them, 7 cases were of diffuse-type gastric cancer and 2 cases were of intestinal-type. When we compared IL-17A expression with GKN1 expression in these cases, 5 cancer cases with IL-17A expression also showed immunoreactivity for GKN1 [Yoon et al., 2011b]. Statistically, there was close association between IL-17A and GKN1 expression in gastric cancer (P=0.0001).
GKN1 induces lymphocyte migration in vitro
In in vitro transwell-chemotaxis assay, Jurkat T cells and peripheral B lymphocytes significantly migrated into the lower chamber containing AGS cells transfected with GKN1 compared to the lower chamber containing mock-transfected AGS cells (Fig. 3). Interestingly, Jurkat T cell migration was significantly inhibited after neutralization of IL-8 and IL-17 with treatment of human anti-IL-8 and anti-IL-17 antibodies in lower chamber containing GKN1-transfected AGS cells (Fig. 3).
Figure 3.
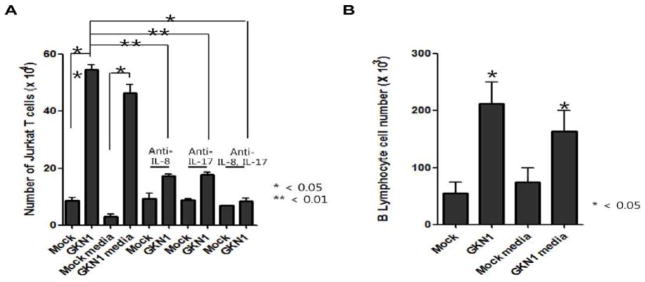
Effects of GKN1 on cell migration. A & B: Cell migration assays were performed with Jurkat T cells (A) and peripheral B lymphocytes (B) by transwell microchemotaxis. Mock or GKN1-transfected AGS cells and conditioned media collected from mock- and GKN1-transfected cells were used as chemoattractants. The results are presented as mean ± SD of a triplicate from a representative experiment. Cell migration to GKN1-overexpressed AGS cells and conditioned media containing GKN1 (10 ng/ml) was significantly increased by 93.75% and 74.12% in Jurkat T cells and peripheral B lymphocytes, respectively. However, cell migration was significantly inhibited after neutralization of IL-17 and/or IL-8 with treatment of a human anti-IL-17 and anti IL-8 antibodies in Jurkat T cells.
GKN1 suppresses COX-2 expression
Since overexpression of COX-2 in gastric cancer tissues is thought to be mediated in part by enhanced NF-κB activity [Lim et al., 2001], we examined COX-2 expression levels in non-treated and IL-1β-treated AGS cells and GKN1 knock-downed HFE-145 cells by real time RT-PCR. Treatment of IL-1β resulted in increased COX-2 expression, whereas GKN1 expression significantly decreased COX-2 mRNA expression (Fig. 4). Additionally, knock-down of GKN1 expression in HFE-145 cells significantly increased COX-2 mRNA expression (Fig. 4).
Figure 4.
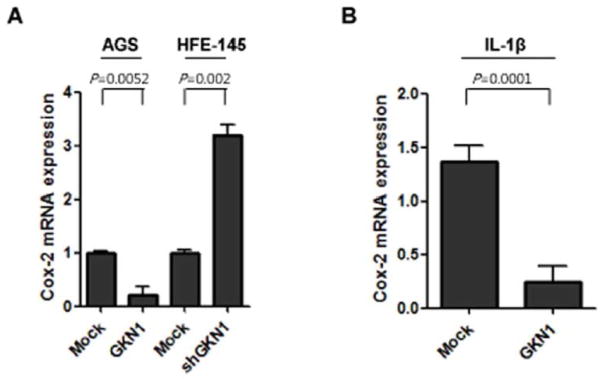
Effect of GKN1 on COX-2 mRNA expression in AGS or HFE-145 cells. A: Significantly decreased COX-2 mRNA expression was observed in GKN1-transfected AGS cells and increased COX-2 mRNA expression was found in shGKN1-transfected HFE-145 cells. B: GKN1 also decreased COX-2 mRNA expression in IL-1β-treated AGS cells. The results are presented as mean ± SD of a triplicate from a representative experiment.
GKN1 reduces cell viability in gastric epithelial cells, but not in lymphocytes
Effects of GKN1 and cytokines on AGS cell viability were determined 24 hrs after cytokine treatment. Effects of GKN1 on HFE-145 cell viability were measured 48 hrs after shGKN1 transfection by an MTT assay. A significant dose-dependent reduction in cell viability in the AGS cells transfected with GKN1 and induction in cell viability in the HFE-145 cells transfected with shGKN1 were observed; however, IL-10 or IL-17 showed no effects on cell viability in AGS cells (Fig. 5).
Figure 5.
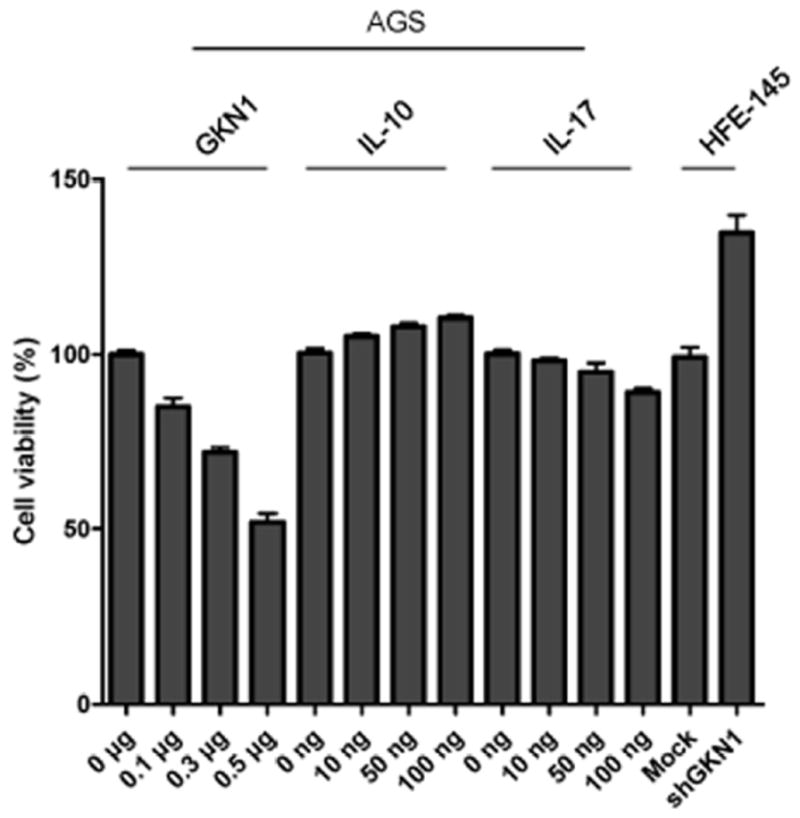
Cell viability was measured at 24 hrs in AGS cells after treatment with GKN1 or recombinant IL-10 and IL-17 and at 48 hrs in shGKN1 transfected HFE-145 cells. GKN1-transfected AGS cells showed dose-dependent inhibition of cell viability and shGKN1-transfected HFE-145 cells induced cell proliferation. Treatment of IL-10 or IL-17 in AGS cells did not show significant difference in cell viability.
In addition, we used flow cytometry to examine whether GKN1 has an influence on T-cell viability and differentiation. Under the Th17-polarizing condition, recombinant GKN1 was added at concentrations of 1, 5, 25, 50 and 100 ng/ml. Recombinant GKN1 treatment did not induce apoptosis in both Th0 and Th17 cells (Fig. 6A). In addition, no significant effect on IL-17A and Foxp3 expression was seen in these cells (Fig. 6B & 6C).
Figure 6.

Effect of GKN1 on T lymphocyte viability and differentiation. A: Cell death was measured by an annexin V-binding assay in Th0 and Th17 cells following recombinant GKN1 treatment. No significant increase in annexin V staining was seen in treated cells. B: Flow cytometry for intracellular Foxp3 and IL-17 expression in GKN1-treated (1, 5, 25, 50, and 100 ng/ml) Th0 cells. The data are representative of two independent experiments. C: The concentration of IL-17 was measured in Th0 and Th17 cells by ELISA after recombinant GKN1 treatment. Recombinant GKN1 treatment did not result in significant difference in IL-17 production. The results are presented as mean ± SD of a duplicate from a representative experiment. G1, 1 ng/ml GKN1 treatment; G5, 5 ng/ml GKN1 treatment; G25, 25 ng GKN1 treatment; G50, 50 ng/ml GKN1 treatment; G100, 100 ng/ml GKN1 treatment.
Discussion
The gastric epithelial cells and infiltrating inflammatory cells produce several kinds of cytokines, ROS and inflammatory mediators which participate in the development and progression of gastric cancer. A critical balance between the pathways of pro- and anti-inflammatory cytokines maintains a homeostatic environment in the gastric epithelium, which effectively neutralizes harmful environmental insults to prevent excessive tissue damage [Guang et al., 2010]. Thus, loss of the homeostatic cytokine balance in the gastric epithelium may lead to gastric mucosal defects, rendering the gastric mucosa vulnerable to exposure from carcinogens and resulting injuries.
The mucosal barrier function of GKN1 protects the gastric mucosa from systemic exposure to foreign antigens, bacteria and gastric acid [Toback et al., 2003; Oien et al., 2004]. In our previous studies, we reported that GKN1 function as a tumor suppressor by inhibiting cell proliferation and epithelial-mesenchymal transition via regulating cancer cell migration and invasiveness [Yoon et al., 2011a]. Furthermore, the anti-inflammatory drug, such as celecoxib, has potent anti-tumor activity [Koki and Masferrer, 2002]. Thus, we hypothesized that the GKN1 gene may also have anticancer properties by regulating expression of inflammatory mediators associated with tumorigenesis. NF-κB is a potent transcriptional factor that orchestrates many biological functions essential for inflammatory and immune processes induced by H. pylori, and its activation stimulates IL-1 and TNF-α production [Isomoto et al., 2000; Sharma et al., 1998]. Aberrant activation of NF-κB results in enhanced proliferation, evasion of apoptosis, and genomic instability in gastric cancer cells [Kang et al., 2008; Liu et al., 2004; Matsumoto et al., 2007]. Here, we found that GKN1 expression suppresses activation and nuclear translocation of NF-κB by inhibiting degradation and phosphorylation of IκB, inactivating IKKα/β, and increasing Foxp3 expression (Fig. 1). Previously, Grant et al. demonstrated that Foxp3 can block activation of key inducible proteins, such as NF-κB and cAMP-responsive element binding protein [Grant et al., 2006]. In addition, it has been reported that Foxp3 interacts directly with NF-κB and modulates its transcriptional activity [Chung et al., 2010]. As NF-κB regulates the expression of various pro-inflammatory cytokines, these results suggest that GKN1 may play an important role in the development of gastric cancer by inhibiting oncogenic NF-κB signaling.
H. pylori infection is regarded as a major cause of gastritis since it induces production of several cytokines including IL-17 [Luzza et al., 2000], which is a pro-inflammatory cytokine produced by CD4+ T lymphocytes of the new T cell lineage, Th17 [Weaver et al., 2006]. IL-17 induces secretion of IL-8 by activating the ERK 1/2 MAP kinase pathway, and the released IL-8 attracts neutrophils, resulting in inflammation and reduced bacterial load [Kabir, 2011]. IL-6, another pro-inflammatory cytokine, also plays an important role in gastric tumor progression because it promotes cell motility and invasiveness by activating the Src/Rho/ROCK signaling pathway [Lin et al., 2007]. IL-6 and IL-10 both regulate the same signaling molecule, STAT3, leading to phosphorylation, nuclear localization, and cytokine-specific gene activation pattern [Braun et al., 2012]. Clinically, a low level of IL-17 expression is associated with poor prognosis for gastric cancer patients, whereas high levels of serum IL-6 and IL-10 correlate with tumor progression and serve as independent predictors of poor survival in gastric cancer [Liao et al., 2008; Ikeguchi et al., 2009]. In this study, GKN1 overexpression induced a significant increase in mRNA IL-8 and -17A expression, while IL-6 and IL-10 expression was significantly decreased (Fig. 2A). In immunohistochemistry, IL-17A production detected in 9 (13.6%) out of 166 gastric cancer tissues was closely associated with GKN1 expression (P=0.0001). In addition, ectopic GKN1 expression induced decreased p-STAT3 expression and increased p-Erk protein expression (Fig. 1). These results suggest that GKN1 contributes to anti-tumor properties by regulating inflammatory cytokine expression.
We therefore examined whether GKN1 could modulate the recruitment of inflammatory cells. Transwell chemotaxis assays showed that Jurkat T cells and peripheral B lymphocytes significantly migrated to the lower chamber containing GKN1 (Fig. 3). Since GKN1 expression activated the production of pro-inflammatory cytokines, including IL-8 and IL-17A (Fig. 2), these results indicate that GKN1 may play a role as a chemoattractant during the infiltration of immune cells in gastric mucosa.
In addition, disruption of COX-2 blocks the upregulation of cell proliferation, suggesting that COX-2 deficiency suppresses H. pylori-induced cell proliferation [Li et al., 2006]. In this study, we found that GKN1 expression led to decreased COX-2 expression even in IL-1β treated AGS cells, and knock-down of GKN1 expression in HFE-145 cells increased COX-2 expression (Fig. 4). Since activation of COX-2 has been shown to be involved in many processes leading to tumor progression, such as angiogenesis, survival, proliferation and invasion [Wang et al., 2010], it is likely that GKN1 is protective against gastritis-induced gastric carcinoma via not only inducing apoptosis but also inhibiting cell proliferation by COX-2 down-regulation.
Next, we went on to determine whether increased production of inflammatory cytokines promotes the proliferation of gastric mucosal cells. Expectedly, GKN1 expression lowered cell viability in AGS cells in a dose dependent manner, but recombinant IL-10 and IL-17 protein treatment did not induce apoptosis or cell proliferation in AGS cells (Fig. 5). Interestingly, recombinant GKN1 treatment did not induce apoptosis or cell differentiation of T lymphocytes (Fig. 6). These results further support that increased expression of Th17-related factors may not correlate with gastric tumorigenesis [Kennedy et al., 2011]. Hence, it is likely that GKN1 may affect cell viability of gastric cancer cells, but transient exposure to these inflammatory cytokines may not affect cell viability of gastric mucosal cells. Further studies are urgently necessary to verify these initial observations.
To summarize these findings, the role of GKN1 in gastric mucosa is multi-functional. GKN1 expression induced the inactivation of the NF-kB and COX-2, which may inhibit gastric mucosal cell proliferation. In addition, GKN1 stimulated infiltration of immune cells by producing inflammatory cytokines, such as IL-8 and -17A. GKN1 also lowered viability of gastric mucosal cell, but did not lower that of inflammatory cells. All of these results suggest that GKN1 may have anti-carcinogenic effects via suppression of NF-kB and COX-2, regulation of cytokine production, and recruitment of inflammatory cells. Additional functional and translational GKN1 studies will certainly broaden our understanding of the pathogenesis of gastritis and provide us with novel preventive and therapeutic modalities for treating gastric inflammation and cancer.
Acknowledgments
This work was supported by the Happy Tech. (2010-0020764) and the Basic Science Research Program programs (2012R1A2A2A01002531) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology.
Footnotes
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: [10.1002/jcb.24524]
Conflicts of interest
The authors disclose no conflicts.
Author Contributions
Conceived and designed the experiments: JHY, MLC, WSP.
Performed the experiments: JHY, MLC, YJC, JYB, MKP, SWL, BJC, HA, DTS. Analyzed the data: JHY, WSP.
Contributed reagents/materials/analysis tools: HA, DTS, SWN, JYL
Wrote the paper: WSP, MLC.
References
- Basso D, Scrigner M, Toma A, Navaglia F, Di Mario F, Rugge M, Plebani M. Helicobacter pylori infection enhances mucosal interleukin-1β, interleukin-6, and the soluble receptor of interleukin-2. Int J Clin Lab Res. 1996;26:207–210. doi: 10.1007/BF02592984. [DOI] [PubMed] [Google Scholar]
- Braun DA, Fribourg M, Sealfon SC. Cytokine response is determined by duration of receptor and STAT3 activation. J Biol Chem. 2012 doi: 10.1074/jbc.M112.386573. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HS, Lee JH, Kim H, Lee HJ, Kim SH, Kwon HK, Im SH, Bae H. Foxp3 is a novel repressor of microglia activation. Glia. 2010;58:1247–1256. doi: 10.1002/glia.21006. [DOI] [PubMed] [Google Scholar]
- Grant C, Oh U, Fugo K, Takenouchi N, Griffith C, Yao K, Newhook TE, Ratner L, Jacobson S. Foxp3 represses retroviral transcription by targeting both NF-kappaB and CREB pathways. PLoS Pathog 2006. 2006;2:e33. doi: 10.1371/journal.ppat.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang W, Ding H, Czinn SJ, Kim KC, Blanchard TG, Lillehoj EP. Muc1 cell surface mucin attenuates epithelial inflammation in response to a common mucosal pathogen. J Biol Chem. 2010;285:20547–20557. doi: 10.1074/jbc.M110.121319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo YJ, Joo YB, Oh HJ, Park MK, Heo YM, Cho ML, Kwok SK, Ju JH, Park KS, Cho SG, Park SH, Kim HY, Min JK. IL-10 suppresses Th17 cells and promotes regulatory T cells in the CD4+ T cell population of rheumatoid arthritis patients. Immunol Lett. 2010;127:150–156. doi: 10.1016/j.imlet.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Holck S, Nørgaard A, Bennedsen M, Permin H, Norn S, Andersen LP. Gastric mucosal cytokine responses in Helicobacter pylori-infected patients with gastritis and peptic ulcers. Association with inflammatory parameters and bacteria load. FEMS Immunol Med Microbiol. 2003;36:175–180. doi: 10.1016/S0928-8244(03)00028-2. [DOI] [PubMed] [Google Scholar]
- Ikeguchi M, Hatada T, Yamamoto M, Miyake T, Matsunaga T, Miyake T, Fukumoto Y, Yamada Y, Fukuda K, Saito H, Tatebe S. Serum interleukin-6 and -10 levels in patients with gastric cancer. Gastric cancer. 2009;12:95–100. doi: 10.1007/s10120-009-0509-8. [DOI] [PubMed] [Google Scholar]
- Isomoto H, Mizuta Y, Miyazaki M, Takeshima F, Omagari K, Murae K, Nishiyama T, Inoue K, Murata I, Kohno S. Implication of NF-kappaB in Helicobacter pylori-associated gastritis. Am J Gastroenterol. 2000;95:2768–2776. doi: 10.1111/j.1572-0241.2000.02304.x. [DOI] [PubMed] [Google Scholar]
- Kabir S. The role of interleukin-17 in the Helicobacter pylori induced infection and immunity. Helicobacter. 2011;16:1–8. doi: 10.1111/j.1523-5378.2010.00812.x. [DOI] [PubMed] [Google Scholar]
- Kang MJ, Ryu BK, Lee MG, Han J, Lee JH, Ha Tk, Byun DS, Chae KS, Lee BH, Chun HS, Lee KY, Kim HJ, Chi SG. NF-kappaB activates transcription of the RNA-binding factor HuR, via PI3K-AKT signaling to promote gastric tumorigenesis. Gastroenterology. 2008;135:2030–2042. doi: 10.1053/j.gastro.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Kennedy CL, Najdovska M, Jones GW, McLeod L, Hughes NR, Allison C, Ooi CH, Tan P, Ferrero RL, Jones SA, Dev A, Sievert W, Bhathal PS, Jenkins BJ. The molecular pathogenesis of STAT3-driven gastric tumourigenesis in mice is independent of IL-17. J Pathol. 2011;225:255–264. doi: 10.1002/path.2933. [DOI] [PubMed] [Google Scholar]
- Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Control. 2002;9:28–35. doi: 10.1177/107327480200902S04. [DOI] [PubMed] [Google Scholar]
- Li GQ, Xia HH, Chen MH, Gu Q, Wang JD, Peng JZ, Chan AO, Cho CH, So HL, Lam SK, Hu PH, Liang YJ, Lin HL, Berg DE, Feng ZH, Langenbach R, Wong BC. Effects of cyclooxygenase-1 and -2 gene disruption on Helicobacter pylori-induced gastric inflammation. J Infect Dis. 2006;193:1037–1046. doi: 10.1086/500984. [DOI] [PubMed] [Google Scholar]
- Liao WC, Lin JT, Wu CY, Huang SP, Lin MT, Wu AS, Huang YJ, Wu MS. Serum interleukin-6 level but genotype predicts survival after resection in stages II and III gastric carcinoma. Clin Cancer Res. 2008;14:428–434. doi: 10.1158/1078-0432.CCR-07-1032. [DOI] [PubMed] [Google Scholar]
- Lim JW, Kim H, Kim KH. Nuclear factor-kappaB regulates cylooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab Invest. 2001;81:349–360. doi: 10.1038/labinvest.3780243. [DOI] [PubMed] [Google Scholar]
- Lin MT, Lin BR, Chang CC, Chu CY, Su HJ, Chen ST, Jeng YM, Kuo ML. IL-6 induces AGS gastric cancer cell invasion via activation of the c-Src/RhoA/ROCK signaling pathway. Int J Cancer. 2007;120:2600–2608. doi: 10.1002/ijc.22599. [DOI] [PubMed] [Google Scholar]
- Lindholm C, Quiding-Järbrink M, Lönroth H, Hamlet A, Svennerholm AM. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–5971. doi: 10.1128/iai.66.12.5964-5971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CA, Wang MJ, Chi CW, Wu CW, Chen JY. Rho/Rhotekin-mediated NF-kappaB activation confers resistance to apoptosis. Oncogene. 2004;23:8731–8742. doi: 10.1038/sj.onc.1208106. [DOI] [PubMed] [Google Scholar]
- Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- Martin TE, Powell CT, Wang Z, Bhattacharyya S, Walsh-Reitz MM, Agarwal K, Toback FG. A novel mitogenic protein that is highly expressed in cells of the gastric antrum mucosa. Am J Physiol Gastrointest Liver Physiol. 2003;285:G332–G343. doi: 10.1152/ajpgi.00453.2002. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki IM, Honjo T, Chiba T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- Nardone G, Rippa E, Martin G, Rocco A, Siciliano RA, Fiengo A, Cacace G, Malorni A, Budillon G, Arcari P. Gastrokine 1 expression in patients with and without Helicobacter pylori infection. Dig Liver Dis. 2007;39:122–129. doi: 10.1016/j.dld.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Oien KA, McGregor F, Butler S, Ferrier RK, Downie I, Bryce S, Burns S, Keith WN. Gastrokine 1 is abundantly and specifically expressed in superficial gastric epithelium, down-regulated in gastric carcinoma, and shows high evolutionary conservation. J Pathol. 2004;203:789–797. doi: 10.1002/path.1583. [DOI] [PubMed] [Google Scholar]
- Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- Rippa E, La Monica G, Allocca R, Romano MF, De Palma M, Arcari P. Overexpression of gastrokine 1 in gastric cancer cells induces Fas-mediated apoptosis. J Cell Physiol. 2011;226:2571–2578. doi: 10.1002/jcp.22601. [DOI] [PubMed] [Google Scholar]
- Sharma SA, Tummuru MK, Blaser MJ, Kerr LD. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol. 1998;160:2401–2407. [PubMed] [Google Scholar]
- Shiozaki K, Nakamori S, Tsujie M, Okami J, Yamamoto H, Nagano H, Dono K, Umeshita K, Sakon M, Furukawa H, Hiratsuka M, Kasugai T, Ishiguro S, Monden M. Human stomach-specific gene, CA11, is down-regulated in gastric cancer. Int J Oncol. 2001;19:701–707. doi: 10.3892/ijo.19.4.701. [DOI] [PubMed] [Google Scholar]
- Toback FG, Walsh-Reitz MM, Musch MW, Chang EB, Del Valle J, Ren H, Huang E, Martin TE. Peptide fragments of AMP-18, a novel secreted gastric antrum mucosal protein, are mitogenic and motogenic. Am J Physiol Gastrointest Liver Physiol. 2003;285:G344–G353. doi: 10.1152/ajpgi.00455.2002. [DOI] [PubMed] [Google Scholar]
- Walsh-Reitz MM, Huang EF, Musch MW, Chang EB, Martin TE, Kartha S, Toback FG. AMP-18 protects barrier function of colonic epithelial cells: role of tight junction proteins. Am J Physiol Gastrointest Liver Physiol. 2005;289:G163–G171. doi: 10.1152/ajpgi.00013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effect or CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Xing R, Li W, Cui J, Zhang J, Kang B, Wang Y, Wang Z, Liu S, Lu Y. Gastrokine 1 induces senescence through p16/Rb pathway activation in gastric cancer cells. Gut. 2012;61:43–52. doi: 10.1136/gut.2010.230623. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Kang YH, Choi YJ, Park IS, Nam SW, Lee JY, Lee YS, Park WS. Gastrokine 1 Functions as a Tumor Suppressor by Inhibition of Epithelial-Mesenchymal Transition in Gastric Cancers. J Cancer Res & Clin Oncol. 2011a;137:1697–1704. doi: 10.1007/s00432-011-1051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Song JH, Zhang C, Jin M, Kang YH, Nam SW, Lee JY, Park WS. Inactivation of the Gastrokine 1 Gene in Gastric Adenomas and Carcinomas. J Pathol. 2011b;223:618–625. doi: 10.1002/path.2838. [DOI] [PubMed] [Google Scholar]
- Yuasa Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat Rev Cancer. 2003;3:592–600. doi: 10.1038/nrc1141. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Toh ML, Zrioual S, Miossec P. IL-17A versus IL-17F induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in AGS gastric adenocarcinoma cells. Cytokine. 2007;38:157–164. doi: 10.1016/j.cyto.2007.06.002. [DOI] [PubMed] [Google Scholar]