

Abstract
Stressful life events are important contributors to relapse in recovering cocaine addicts, but the mechanisms by which they influence motivational systems are poorly understood. Studies suggest that stress may “set the stage” for relapse by increasing the sensitivity of brain reward circuits to drug-associated stimuli. We examined the effects of stress and corticosterone on behavioral and neurochemical responses of rats to a cocaine prime after cocaine self-administration and extinction. Exposure of rats to acute electric footshock stress did not by itself reinstate drug-seeking behavior but potentiated reinstatement in response to a subthreshold dose of cocaine. This effect of stress was not observed in adrenalectomized animals, and was reproduced in nonstressed animals by administration of corticosterone at a dose that reproduced stress-induced plasma levels. Pretreatment with the glucocorticoid receptor antagonist RU38486 did not block the corticosterone effect. Corticosterone potentiated cocaine-induced increases in extracellular dopamine in the nucleus accumbens (NAc), and pharmacological blockade of NAc dopamine receptors blocked corticosterone-induced potentiation of reinstatement. Intra-accumbens administration of corticosterone reproduced the behavioral effects of stress and systemic corticosterone. Corticosterone treatment acutely decreased NAc dopamine clearance measured by fast-scan cyclic voltammetry, suggesting that inhibition of uptake2-mediated dopamine clearance may underlie corticosterone effects. Consistent with this hypothesis, intra-accumbens administration of the uptake2 inhibitor normetanephrine potentiated cocaine-induced reinstatement. Expression of organic cation transporter 3, a corticosterone-sensitive uptake2 transporter, was detected on NAc neurons. These findings reveal a novel mechanism by which stress hormones can rapidly regulate dopamine signaling and contribute to the impact of stress on drug intake.
Introduction
Cocaine addicts report that craving responses to drug-associated stimuli are intensified during periods of stress, resulting in heightened susceptibility to relapse of drug use (Sinha, 2001; Hyman et al., 2006). These reports are paralleled by preclinical findings that stress can potentiate the reinstatement of compulsive drug-seeking behavior by drug-associated cues in rodents (Shelton and Beardsley, 2005; Feltenstein and See, 2006; Buffalari and See, 2009). Together, these studies suggest that stress may act as a “stage-setter,” inducing state-dependent changes in the sensitivity of brain reward circuits to the reinforcing properties of drugs (Preston and Epstein, 2011) and enhancing the potency of drugs of abuse or drug-associated cues to induce relapse. Thus, interactions between pathways activated by stress and by cocaine-associated stimuli are likely to be critical determinants of relapse vulnerability. However, the potential mechanisms underlying these interactions have not been well studied.
In addition to directly engaging motivational circuitry, stressful stimuli activate the hypothalamic-pituitary-adrenocortical (HPA) axis, resulting in the release of glucocorticoid hormones, corticosterone in rodents and cortisol in humans. Clinical and preclinical studies suggest a complex relationship between glucocorticoids and drug relapse. In human addicts, stress-, cue-, and drug-induced craving for cocaine is associated with elevated cortisol (Berger et al., 1996; Reid et al., 2003; Sinha et al., 2003) and heightened HPA reactivity can predict relapse susceptibility (Sinha et al., 2006, 2011; Fatseas et al., 2011; McKee et al., 2011). In rodents, glucocorticoids can enhance behavioral and neurochemical responses to drugs of abuse (Piazza et al., 1996; Marinelli et al., 1997; Barrot et al., 2000), but eliminating increases in glucocorticoids does not prevent stress- or cocaine-induced reinstatement of compulsive drug seeking in rats (Erb et al., 1998; Mantsch et al., 2008; Graf et al., 2011) or cocaine-induced reinstatement in monkeys (Lee et al., 2003), and studies examining the ability of elevated glucocorticoids alone to reinstate cocaine seeking have yielded mixed results (Deroche et al., 1997; Lee et al., 2003; Shalev et al., 2003). Together, these studies indicate that, although glucocorticoids alone may not drive relapse, they do interact with drugs and drug-associated stimuli to influence the likelihood of drug use. In the present studies, we examined the effects of stress and glucocorticoids on the behavioral and neurochemical responses of rats to a relapse-promoting stimulus.
Materials and Methods
Animals.
Adult male Sprague Dawley rats (Harlan Laboratories), weighing 275–325 g, were housed individually in a temperature- and humidity-controlled, Association for Assessment and Accreditation of Laboratory Animal Care-accredited vivarium under a 12 h/12 h reversed light-dark cycle (lights off at 0700 h) with ad libitum access to food and water. Procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Jugular catheterization.
Rats received indwelling jugular catheters under ketamine/xylazine (100 mg/kg; 2 mg/kg, i.p.) anesthesia as previously described (Graf et al., 2011). Lines were connected to syringe pumps (Razel) via fluid swivels (Instech Laboratory) suspended above the chambers. Swivel/leash assemblies were balanced to permit unrestrained movement.
Cocaine self-administration, extinction, and reinstatement.
Self-administration was conducted using computer-interfaced operant conditioning chambers contained in sound-attenuating cubicles (Med Associates). Rats were trained to self-administer cocaine (0.5 mg/kg/200 μl, i.v.) by pressing a lever under a fixed ratio one schedule during daily 2 h sessions. Cocaine infusions were accompanied by illumination of a light followed by a 25 s timeout period during which the light was extinguished but the lever remained extended. Responding on a second, inactive lever was also recorded. Once stable responding was observed (<10% variation in daily responding over ≥3 consecutive sessions), rats underwent an additional 14 daily self-administration sessions followed by extinction training consisting of daily sessions during which the cocaine solution was replaced with saline. Once extinction criterion was met (≤10 active responses/session across 3 sessions), each rat was tested for reinstatement. Reinstatement sessions were identical to extinction, except that they were preceded by the delivery of an experimental treatment (e.g., footshock, corticosterone, or vehicle) followed by noncontingent intraperitoneal injection of cocaine (2.5 mg/kg) or saline. Reinstatement was defined as an increase in responding on the lever previously reinforced by cocaine compared with the preceding extinction session. Inactive lever pressing was recorded during reinstatement but is not reported because of a lack of significant effects.
Behavioral testing.
Behavioral experiments were conducted during the light phase. Each rat in the reinstatement experiments was tested four times in a counterbalanced sequence, such that each rat received each treatment. Between consecutive reinstatement tests, rats underwent additional extinction sessions until they again met extinction criterion.
Microdialysis.
Drug-naive rats were anesthetized under ketamine/xylazine anesthesia and implanted with bilateral guide cannulae (20 gauge; 14 mm; Plastics One) directed at the nucleus accumbens (NAc), using the following coordinates (Paxinos and Watson, 1998): +0.9 mm AP, ±2.5 mm ML to bregma, −4.4 mm from the surface of the skull, at an angle 6° from vertical, and allowed to recover for 5 d. The night before testing, rats were housed in self-administration chambers. The following day, microdialysis probes constructed as previously described (Madayag et al., 2010) were inserted into guide cannulae and dialysis buffer (5 mm glucose, 140 mm NaCl, 1.4 mm CaCl2, 1.2 mm MgCl2, and 0.15% PBS, pH 7.4) was pumped through at a rate of 1 μl/min for ≥3 h to allow neurotransmitter levels to stabilize. Samples were collected at 20 min intervals throughout the study. Dopamine concentrations were quantified by comparing peak heights from samples and external standards using HPLC coupled to electrochemical detection. The mobile phase consisted of 15% acetonitrile, 10% methanol, 150 mm NaH2PO4, 4.76 mm citric acid, 3 mm SDS, and 50 μm EDTA, pH 5.6. Dopamine was separated using a reversed-phase column (3 μm; 80 × 3.2 mm; ESA) and detected using an ESA Coulochem II detector coupled to two electrodes set a t −0.075 V and +0.25 V, respectively.
Experiment 1: effects of stress on cocaine-primed reinstatement.
After extinction training, rats (n = 7) were placed into self-administration chambers for 15 min, during which they received either no shocks (control) or a series of intermittent uncontrollable electric footshocks (0.5 mA, 0.5 s duration, mean intershock interval = 40 s, range 10–70 s) through the grid floor. Response levers were retracted during this period. After footshock, rats received an injection of cocaine (2.5 mg/kg, i.p.) or saline, and were tested for reinstatement.
Experiment 2: effects of adrenalectomy on footshock- and cocaine-primed reinstatement.
After acquiring stable self-administration, rats (n = 6) underwent bilateral adrenalectomy under ketamine/xylazine anesthesia and were implanted with subcutaneous 25% corticosterone (Sigma-Aldrich) pellets in the nape of the neck, which results in plasma corticosterone concentrations approximating those observed at the nadir of the diurnal cycle (Mantsch et al., 2008). Rats received 0.025% corticosterone in the drinking water (ethanol concentration = 0.0001%). During the active phase, when most drinking occurs, this elevates plasma corticosterone concentrations to those observed at the circadian peak (Mantsch et al., 2008). Drinking water also contained 0.9% NaCl. Upon recovery, rats underwent extinction training, followed by reinstatement testing.
Experiment 3: effects of corticosterone on basal and cocaine-primed reinstatement.
After extinction training, adrenal-intact rats (n = 6) were tested for reinstatement after an injection of corticosterone (2 mg/kg, i.p.) or vehicle (0.03% EtOH, i.p.) 40 min before administration of cocaine (2.5 mg/kg, i.p.) or saline. This dose of corticosterone reproduces electric footshock-induced plasma levels (Table 1).
Table 1.
Summary of treatment effects on plasma corticosterone concentrations
| Control | Electric footshock | Corticosterone (2.0 mg/kg) | |
|---|---|---|---|
| Vehicle | 122 ± 28 | 265 ± 57 | 384 ± 34 |
| Cocaine (2.5 mg/kg) | 129 ± 32 | 244 ± 47 | 360 ± 46 |
Experiment 4: effects of RU38486 and corticosterone on cocaine-primed reinstatement.
To test the role of glucocorticoid receptor (GR) in corticosterone-induced potentiation of reinstatement, adrenal-intact rats (n = 10) received an injection of RU38486 (12.5 mg/kg, s.c.) or vehicle (45% hydroxypropyl β-cyclodextrin) 1 h before intraperitoneal administration of corticosterone (2 mg/kg) or vehicle (0.03% ethanol). Forty minutes later, rats were tested for cocaine-induced reinstatement (2.5 mg/kg, i.p.).
Experiment 5: effects of corticosterone on cocaine-induced increases in NAc dopamine.
After collection of baseline samples, rats received an injection of corticosterone (2.0 mg/kg, i.p.; n = 13) or vehicle (0.03% EtOH; n = 12), followed 40 min later by an injection of cocaine (2.5 mg/kg, i.p.). Samples were collected for an additional 2 h. After the study, rats were anesthetized with an overdose of pentobarbital (60 mg/kg, i.p.) and transcardially perfused with 0.9% saline and 2.5% formalin. Brains were removed, postfixed, and sectioned in the coronal plane (100 μm) for verification of probe placement.
Experiment 6: effects of intra-accumbens fluphenazine on corticosterone-induced potentiation of cocaine-primed reinstatement.
At the time of jugular implantation, rats received bilateral 11 mm, 26-gauge cannula aimed 0.5 mm above the target region, and the following coordinates were used (Paxinos and Watson, 1998): core: anteroposterior (AP), +1.0; mediolateral (ML), ±2.2; dorsoventral (DV), −6.5 at 0°; shell: AP, +2.5; ML, ±0.75; DV, −6.5 at 0°. Rats received bilateral injections of the nonselective dopamine receptor antagonist, fluphenazine (30 nmol/0.3 μl/side over 1 min) or vehicle (aCSF) into the NAc core (n = 6) or shell (n = 7) 25 min after administration of corticosterone (2 mg/kg, i.p.) and 15 min before low-dose cocaine (2.5 mg/kg, i.p.) administration and reinstatement testing.
Experiment 7: effects of intra-accumbens corticosterone on cocaine-primed reinstatement.
Rats with cannulae aimed at the NAc core (n = 6) or shell (n = 6) received intra-accumbens injections of hydroxypropyl β-cyclodextrin-complexed corticosterone (0.05 μg/0.3 μl/side over 1 min) or vehicle (aCSF) 15 min before administration of low-dose cocaine (2.5 mg/kg, i.p.) or saline and reinstatement testing.
Experiment 8: effects of intra-accumbens normetanephrine on cocaine-primed reinstatement.
Rats with cannulae aimed at the NAc shell (n = 6) received intra-accumbens injections of the nonglucocorticoid OCT3 inhibitor, normetanephrine (25 ng/0.3 μl/side over 1 min) or vehicle (aCSF) 15 min before administration of low-dose cocaine (2.5 mg/kg, i.p.) or saline and reinstatement testing.
Voltammetry.
Rats (n = 5 experimental, 5 control) were anesthetized with urethane (1.5 g/kg, i.p.; Sigma-Aldrich) and placed in a stereotaxic frame (David Kopf Instruments). AP, ML, and DV positions were referenced from bregma. The dura mater was punctured, carefully removed, and a single carbon-fiber microelectrode was lowered into the NAc shell (AP, +1.7 mm; ML, +0.8 mm; DV, 6.0–8.0 mm), coordinates taken from an atlas of Paxinos and Watson (1998). The NAc shell was targeted based on our previous studies, which demonstrated that, although OCT3 is expressed in both the core and shell of the NAc, it is expressed more densely in the shell (Gasser et al., 2009). An Ag/AgCl reference electrode was placed in the contralateral cortex. Electrical stimulation was accomplished with a bipolar, stainless-steel electrode (0.2 mm in diameter, Plastics One) placed in the ventral tegmental area (AP, −5.2 mm; ML, +1.0 mm; DV, 8.0–9.0 mm). The tips of the bipolar electrode were separated by ∼1.0 mm.
Electrical stimulation.
Computer-generated stimulus trains (NL 800A, Neurolog, Medical Systems) were optically isolated from the electrochemical system. Trains consisted of 24 biphasic pulses (300 μA, 2 ms each phase) with a stimulation frequency of 60 Hz applied through the bipolar electrode. An interval of 3 min was allowed to elapse between electrical stimulations to ensure reproducible catecholamine release with each stimulation.
Voltammetric procedures.
Voltammetric procedures have been described in detail previously (Heien et al., 2005). Briefly, glass-encased, cylindrical carbon-fiber microelectrodes (T-650 carbon fibers, 5.1 μm in nominal diameter, Thornel, Amoco) with an exposed length of 75–100 μm and Ag/AgCl reference electrodes were prepared. The fiber was held at −0.4 V against Ag/AgCl between scans and then driven to +1.3 V and back in a triangular fashion at 400 V/s for each voltammetric measurement. Application of this triangle waveform causes oxidation and reduction of chemical species that are electroactive within this potential range, producing a change in current at the carbon fiber. Dopamine was identified by plotting changes in current against applied potential to produce a cyclic voltammogram. Current arising from electrode surface processes was subtracted as background. Background-subtracted cyclic voltammograms were obtained by digitally subtracting voltammograms collected during stimulation from those collected during baseline recording. Voltammetric responses were viewed as color plots with the abscissa as voltage, the ordinate as acquisition time, and the current encoded in color. Temporal responses were determined by monitoring the current at the peak oxidation potential for dopamine by principal component regression (Heien et al., 2005). Because the carbon-fiber microelectrode was used to lesion the brain and mark the precise recording site, postcalibration of the electrode was impossible. Instead, postcalibration factors/carbon fiber area (6.9 ± 0.3 pA/[μmol/L × μm2]) were used on the basis of the average response data obtained from calibrating multiple electrodes in the manner of Park et al. (2010).
Data analysis.
The Demon Voltammetry software suite was used to analyze all data in the manner of Yorgason et al. (2011). τ was extrapolated from an exponential fit of the decay curve, as the point of 63.2% decline in the signal. Full-width at half-height was defined as the time between the midpoint of decline of the decay curve extended back in time to the intersecting point of incline of the ascending curve of the dopamine signal. Km is the apparent affinity of dopamine for transporters, solved for from the Michaelis-Menten equation (Wightman et al., 1988; Yorgason et al., 2011). The decay times τ20 and τ80 were defined as the time to 20% and 80% signal reductions in the decay curve. Data are represented as mean change after treatment ± SEM.
Immunofluorescence.
For immunodetection of OCT3, an affinity-isolated antibody (rabbit anti-OCT3, catalog #OCT31A, Alpha Diagnostics International) raised against an 18-amino acid sequence in the large intracellular loop of rat OCT3 (amino acids 313–330: HLSSNYSEITVTDEEVSN) was used. This amino acid sequence is 100% conserved in mouse and rat OCT3 and has no significant sequence homology with other OCTs or with any organic cation/carnitine transporters. The specificity of this antibody was confirmed previously in immunohistochemical and immunofluorescence applications (Vialou et al., 2004; Lips et al., 2005; Gasser et al., 2006, 2009). Additional proteins were detected using primary antibodies directed against tyrosine hydroxylase (MAB 318, Millipore; diluted 1:1600), dopamine D1 receptor (MAB 5290, Millipore; diluted 1:500), and β-III tubulin (ab 7751, Abcam; diluted 1:1200). Primary antibodies were detected using AlexaFluor-594-conjugated donkey anti-rabbit and AlexaFluor-488-conjugated donkey anti-mouse IgG antibodies (1:200; Invitrogen). Photomicrographs were acquired using a Retiga 2000R digital camera (QImaging) on a Nikon 80i microscope using NIS Elements software (Nikon Instruments). Each combination of antibodies was used in at least three independent studies with similar results.
Statistical analysis.
Statistical analyses were conducted using IBM SPSS Statistics software. Sample sizes (n) were determined by expected effect sizes and the power of the analysis. Statistical significance was determined using ANOVA or Student's t tests (two-tailed) followed, when appropriate, by further analyses of main effects using ANOVA and/or post hoc testing using Bonferroni-corrected t tests. Normality testing for all data was conducted using the Shapiro-Wilk test; and in all cases, data were determined to be sufficiently normal to merit parametric analysis. For within-subjects analyses, sphericity of data was confirmed using the Mauchly's test. For between-subjects analyses, homogeneity of data was confirmed using the Levene's test.
Results
Stress and corticosterone potentiate cocaine-induced reinstatement of drug-seeking behavior
We demonstrate here that a dose of cocaine (2.5 mg/kg, i.p.), which is insufficient to reinstate extinguished cocaine seeking under baseline conditions, produces robust reinstatement when delivered after exposure to an acute stressor, uncontrollable electric footshock. Significant reinstatement was observed when rats were tested for cocaine-induced reinstatement after footshock (post hoc, p = 0.034 vs extinction) but not when rats received cocaine alone or footshock in combination with a saline injection (two-way ANOVA; significant footshock × reinstatement condition interaction: F(3,18) = 3.424; p = 0.04) (Fig. 1A).
Figure 1.

Stress potentiates cocaine-induced drug-seeking behavior by a corticosterone-dependent, GR-independent mechanism. A, B, Cocaine lever responses on the last day of extinction and during reinstatement testing of (A) adrenal-intact (n = 7) or (B) adrenalectomized (ADX; n = 6) rats. On the day of reinstatement testing, rats were subjected to 15 min of uncontrollable, intermittent electric footshock (EFS) or no shock, followed by injection of cocaine (2.5 mg/kg, i.p.) or saline, and cocaine lever responses were recorded for 2 h. C, Cocaine lever responses on the last day of extinction and during reinstatement testing. Rats (n = 6) received an injection of corticosterone (CORT; 2.0 mg/kg, i.p.) or vehicle, followed 40 min later by an injection of cocaine (2.5 mg/kg, i.p.) or saline. D, Cocaine lever responses on the last day of extinction and during reinstatement testing. Rats (n = 10) received an injection of the glucocorticoid receptor antagonist RU38486 (12.5 mg/kg, s.c.) or vehicle, followed 60 min later by injection of corticosterone (2.0 mg/kg, i.p.) or vehicle, followed 40 min later by an injection of cocaine (2.5 mg/kg, i.p.) (see also Table 1). *p < 0.05, significantly different from extinction responding. Error bars represent mean ± SEM.
To test the hypothesis that the observed potentiation of reinstatement by footshock was mediated by elevated corticosterone, we conducted two experiments. First, we tested for footshock-potentiated reinstatement in a group of adrenalectomized rats in which plasma corticosterone concentrations were maintained at basal (nonstress) concentrations. Adrenalectomy was performed after acquisition of self-administration but before reinstatement testing. Footshock failed to potentiate cocaine-induced reinstatement in adrenalectomized rats (two-way ANOVA; no significant footshock × reinstatement condition interaction: F(3,21) = 0.976; p = 0.423) (Fig. 1B), indicating that stress-induced potentiation of cocaine-seeking behavior requires elevation of corticosterone concentrations to stress levels. Second, we examined (in adrenal-intact rats) the effects of acute stress-level corticosterone treatment on cocaine-primed reinstatement in the absence of stress. In this study, administration of corticosterone at a dose (2 mg/kg, i.p.) that produced stress levels in plasma (Table 1) was sufficient to potentiate cocaine-induced reinstatement in the absence of stress (Fig. 1C). Like footshock, corticosterone alone (before a saline injection) failed to reinstate cocaine seeking but potentiated reinstatement in response to low-dose cocaine (two-way ANOVA, significant corticosterone treatment × reinstatement condition interaction: F(3,15) = 20.997; p = 0.001). When administered alone, neither corticosterone nor low-dose cocaine produced significant reinstatement. However, the combination of corticosterone pretreatment followed by low-dose cocaine produced robust reinstatement (p = 0.003 vs extinction).
Role of the glucocorticoid receptor in corticosterone-induced potentiation of drug-seeking
Although many actions of corticosteroids require activation of the GR, some acute actions appear to be mediated by GR-independent mechanisms (Di et al., 2003). To determine the contribution of GR-dependent signaling to corticosterone effects on reinstatement, we examined the effects of pretreatment with the GR antagonist RU486 on corticosterone-induced potentiation of cocaine-primed reinstatement. In reinstatement studies, pretreatment with RU486 (12.5 mg/kg, s.c.) failed to block the potentiation of low-dose cocaine-induced reinstatement by corticosterone administration (Fig. 1D). Once again, corticosterone potentiated reinstatement after low-dose cocaine administration (three-way ANOVA, significant corticosterone × reinstatement condition interaction: F(1,9) = 11.141, p = 0.009). RU-486 treatment did not disrupt this effect (significant main effects or interactions involving RU-486 were not found), indicating that corticosterone's potentiating action involves a GR-independent, nongenomic mechanism. To confirm that the dose of RU486 (12.5 mg/kg) used in this study resulted in occupation of central GR, we examined the effects of RU486 on subcellular localization of GR in hippocampal cells using immunofluorescence. Consistent with previous studies demonstrating that RU486 translocates hippocampal GR from cytosolic to nuclear fractions (Spiga et al., 2010), GR immunostaining in the hippocampi of RU486-treated rats was concentrated in cell nuclei, whereas GR in vehicle-treated rats was observed as diffuse cellular staining (data not shown), indicating that treatment with RU486 at this dose and via this route of administration resulted in occupation of central GR.
Corticosterone-dopamine interactions in the NAc underlie potentiation of reinstatement
Activation of dopamine receptors in subregions of the NAc is sufficient to reinstate drug seeking in rats (Cornish and Kalivas, 2000; Bachtell et al., 2005; Schmidt et al., 2006), and the magnitude of the cocaine-induced increase in accumbens dopamine appears to be a key determinant of the magnitude of reinstatement (Madayag et al., 2010). Previous reports have demonstrated that evoked increases in NAc dopamine are enhanced when corticosterone is elevated to stress concentrations at the time of testing (Piazza et al., 1996; Deroche et al., 1997; Marinelli et al., 1997), suggesting that the potentiating effects of corticosterone and stress on cocaine seeking may involve augmentation of the NAc dopamine response to cocaine. To test this hypothesis, we used in vivo microdialysis to examine the effects of stress-level corticosterone treatment on cocaine-induced increases in extracellular dopamine concentrations in the NAc. Figure 2A illustrates extracellular dopamine levels in the NAc before and after intraperitoneal injection of low-dose cocaine (2.5 mg/kg) in animals pretreated with stress-level corticosterone (2 mg/kg, i.p.) or vehicle. Cumulative postcocaine extracellular dopamine levels (calculated as areas under the curve using all postcocaine samples) were significantly higher in corticosterone-pretreated rats relative to vehicle controls (Fig. 2B; unpaired t test, t(23) = 1.793, p = 0.043).
Figure 2.

Corticosterone potentiates cocaine-induced increases in extracellular dopamine concentrations in the NAc. A, In vivo microdialysis was used to measure extracellular dopamine levels in the NAc in drug-naive rats. After collection of baseline samples, rats received an injection of corticosterone (2.0 mg/kg, i.p.; n = 13; ●) or vehicle (n = 12; ○) at the time indicated. Forty minutes later, all animals received an injection of cocaine (2.5 mg/kg, i.p.). Data represent mean dopamine concentrations at each time point. B, Cumulative dopamine (area under the curve) during the postcocaine period (t = 40–160 min). *p < 0.05, significantly different from the vehicle group (unpaired t test). C, Black lines indicate the locations of the microdialysis probes aimed at the NAc.
Based on these findings, we hypothesized that the observed corticosterone-potentiated increase in NAc dopaminergic neurotransmission was responsible for the potentiating effects of corticosterone on cocaine-induced reinstatement. To test this hypothesis, we examined the effects of bilateral intra-accumbens infusions of the nonselective dopamine receptor antagonist fluphenazine or vehicle on corticosterone-induced potentiation of cocaine-primed reinstatement. Intra-accumbens fluphenazine treatment prevented the increase in reinstatement that was observed when rats received both corticosterone and low-dose cocaine (Fig. 3A; two-way ANOVA; significant treatment × reinstatement condition interaction: F(1,12) = 6.007; p = 0.031). Low-dose cocaine administration after corticosterone pretreatment reinstated cocaine seeking after delivery of vehicle (p = 0.007), but not fluphenazine, into the NAc. Subgroups of rats received injections targeting the NAc core and shell. Because the effects of fluphenazine on reinstatement did not differ between these two brain regions, data from the two regions were combined for analysis.
Figure 3.

Dopamine-dependent actions in the NAc underlie corticosterone-induced potentiation of drug-seeking behavior. A, Cocaine lever responses on the last day of extinction and during reinstatement testing. Rats (n = 13) received an intraperitoneal injection of corticosterone (2 mg/kg). After 25 min, all animals received intra-accumbens injections of fluphenazine (30 nmol/0.3 μl) or vehicle followed, 15 min later, by an intraperitoneal cocaine injection (2.5 mg/kg). *p < 0.05, significantly different from extinction responding. #p < 0.05, significantly different from reinstatement responding after vehicle treatment. ns, Not significant. B, Cocaine lever responses on the last day of extinction and during reinstatement testing. On the day of reinstatement testing, rats (n = 12) received intra-accumbens injections of corticosterone (CORT, 0.05 g/0.3 μl) followed, 15 min later, by an intraperitoneal injection of saline or cocaine (2.5 mg/kg). *p < 0.05, significantly different from extinction responding). C, Locations of microinjection sites from animals used in A and B.
Corticosterone acts in the NAc to potentiate reinstatement
Because we found that (1) corticosterone acutely potentiates cocaine-induced increases in extracellular dopamine in the NAc and (2) dopaminergic neurotransmission in the NAc is necessary for corticosterone-induced potentiation of behavior, we sought to determine whether corticosterone actions within the NAc are sufficient to promote reinstatement. To accomplish this, we tested the ability of intra-accumbens corticosterone infusions to reproduce the effects of systemic corticosterone or footshock on low-dose cocaine-induced reinstatement. As was the case with the fluphenazine study, subgroups of rats received infusions of corticosterone or vehicle into the NAc shell or core; and because the effects of corticosterone administered into the shell or core did not differ, data from the two regions were combined for analysis. As in the systemic corticosterone experiments, neither low-dose cocaine (after intra-accumbens vehicle) nor intra-accumbens corticosterone (before intraperitoneal saline administration) reinstated cocaine seeking when administered alone. However, when rats received bilateral intra-accumbens corticosterone before administration of low-dose cocaine, significant reinstatement was observed (two-way ANOVA, significant treatment × reinstatement condition interaction: F(2,22) = 4.182; p = 0.029; post hoc testing p = 0.01 vs extinction) (Fig. 3B).
Effects of acute corticosterone treatment on dopamine clearance in the NAc
Together, our results demonstrate that corticosterone acutely potentiates cocaine-induced dopamine signaling in the NAc and that this potentiation promotes cocaine seeking. However, corticosterone alone has no effect on extracellular dopamine and does not induce cocaine seeking. Further, this effect appears to occur independently of GR activation, suggesting a rapid, nongenomic mechanism of action. Such a mechanism, which would enhance cocaine effects on dopaminergic neurotransmission without directly driving increases in dopamine, could involve either inhibition of dopamine metabolism or of transporter-mediated clearance. Corticosteroids have long been known to acutely block catecholamine clearance mediated by uptake2, a high-capacity monoamine transport system originally described in peripheral tissues (Iversen and Salt, 1970). Uptake2 activity has more recently been attributed to a group of broadly specific transporters, including the organic cation transporter (OCT) family (OCTs 1, 2, and 3) and the plasma membrane monoamine transporter (PMAT). All of these transporters are capable of transporting, with varying efficiencies, dopamine and other monoamines (Grundemann et al., 1998b; Shang et al., 2003; Gorboulev et al., 2005; Schomig et al., 2006; Duan and Wang, 2010) and, with the exception of OCT1, all are expressed in the brain (Engel et al., 2004; Vialou et al., 2004; Amphoux et al., 2006; Gasser et al., 2009). Although all are sensitive to inhibition by corticosterone, their sensitivities vary widely. PMAT is the least sensitive to corticosterone (Ki = 450 μm), followed by OCT1 (IC50 = 150 μm), OCT2 (IC50 = 5 μm), and OCT3 (IC50 = 0.04–0.2 μm) (Grundemann et al., 1998b; Shang et al., 2003; Gorboulev et al., 2005; Schomig et al., 2006; Duan and Wang, 2010). Recent studies have demonstrated that corticosterone inhibition of OCT-mediated transport occurs by direct binding to the transporter, through a GR-independent, nongenomic mechanism (Grundemann et al., 1999; Horvath et al., 2003; Gorboulev et al., 2005).
To test the hypothesis that the potentiation of cocaine-primed reinstatement by corticosterone involves blockade of dopamine clearance, we used fast-scan cyclic voltammetry, an electrochemical technique with the temporal resolution necessary for resolving dopamine release and uptake (Wightman et al., 1988). Dopamine release was electrically evoked in anesthetized rats by stimulating dopamine neurons in the ventral tegmental area while uptake was measured in the NAc shell under transport saturating conditions, permitting Michaelis-Menten kinetic modeling (Wightman et al., 1988; Yorgason et al., 2011). Electrically evoked dopamine release and uptake were measured in the NAc in drug naive rats to establish a baseline (Fig. 4). All rats then received an injection of GBR12909 (15 mg/kg, i.p.), an inhibitor that, unlike cocaine, selectively blocks the dopamine transporter (DAT) and reliably reduces dopamine clearance (Andersen, 1989; Park et al., 2010). We observed the GBR12909-induced reduction in DA clearance to be stable on a time scale consistent with previous reports (Budygin et al., 2000; Espana et al., 2008; Park et al., 2010), and we verified stability of the pharmacological effect of GBR12909 as a persistent increase in both amplitude and duration of the stimulated dopamine signal. After stabilization of the response, rats received an additional injection either of corticosterone (2 mg/kg), or vehicle to test the hypothesis that corticosterone treatment would further augment the dopamine signal.
Figure 4.
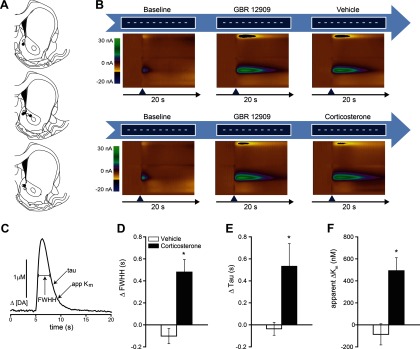
Corticosterone administration decreases dopamine uptake after DAT blockade. A, NAc recording sites for dopamine uptake measurements in vehicle-treated (○) and corticosterone-treated (●) rats. B, Experimental design and example of two-dimensional color representation of cyclic voltammetric data collected for 20 s around a single stimulation. The ordinate is the applied voltage (Eapp), and the abscissa is time (in seconds). Changes in current at the carbon-fiber electrode are indicated in color. After 10 baseline stimulations (white horizontal lines), rats were injected with GBR12909 (15 mg/kg) and stimulated until stabilization of the drug response (center color plot). Rats were then either injected with corticosterone (2 mg/kg, n = 5, right color plot, top), or vehicle (n = 5, right color plot, bottom). C, Measures of dopamine uptake. D–F, Mean ± SEM change in measures of uptake after corticosterone treatment (D, FWHH; E, τ; and F, apparent Km), compared with vehicle-injected controls. *p < 0.05, compared with vehicle (Student's unpaired t test).
Electrical stimulation of dopamine neurons in the VTA reliably induced dopamine release in the NAc (Fig. 4B). This effect was augmented by blocking the DAT with GBR12909. After stabilization of the GBR effect, injection of corticosterone at a dose that reproduces stress-induced plasma levels (Table 1) further augmented the dopamine signal (Fig. 4B–D; Table 2). Kinetics of dopamine transport were modeled with the Demon Voltammetry and Analysis software suite. Several measures of uptake were analyzed (Fig. 4; Table 2) with each measure in agreement, consistent with previous studies (Yorgason et al., 2011). Corticosterone treatment significantly increased full-width at half-height (t(8) = −4.375, p = 0.002, Fig. 4D), τ (t(8) = −2.643, p = 0.03, Fig. 4E), and apparent Km (t(8) = −3.804, p = 0.005, Fig. 4F), compared with vehicle-injected controls, indicating a reduction in dopamine clearance.
Table 2.
Summary of treatment effects on measures of dopamine clearance in the NAc
| Δ Full-width at half-height (s) | Δ Tau (s) | Δ Apparent Km (nm) | Δ Half-life (ms) | Δ t20 (ms) | Δ t80 (ms) | Δ Peak height [DA] (nm) | |
|---|---|---|---|---|---|---|---|
| Vehicle | −0.102 (0.06931) | −0.036 (0.05887) | −86.118 (96.09) | −0.02 (0.03937) | −0.018 (0.09074) | −0.094 (0.09796) | −0.68 (0.16775) |
| Corticosterone | 0.48 (0.11353) | 0.532 (0.2067) | 492.282 (117.814) | 0.368 (0.14267) | 0.284 (0.09293) | 1.122 (0.51043) | 0.69 (0.51905) |
Intra-accumbens normetanephrine potentiates cocaine-induced drug seeking
Although the results of the voltammetry studies strongly suggest that corticosterone acts at least in part by inhibiting dopamine clearance, there are additional GR-independent mechanisms through which glucocorticoids may alter neurotransmission (Di et al., 2003). To more rigorously test our hypothesis that uptake inhibition mediates the potentiating effects of stress and corticosterone on cocaine seeking, we examined the ability of intra-accumbens administration of normetanephrine, the O-methylated metabolite of norepinephrine and a potent nonglucocorticoid inhibitor of uptake2-mediated transport (Martel et al., 1999), to reproduce the effects of corticosterone and footshock on low-dose cocaine-induced reinstatement. Although normetanephrine has been reported to act as a weak agonist at α1-adrenergic receptors (Langer and Rubio, 1973), to our knowledge the sole overlapping mechanism of action between normetanephrine and glucocorticoids is blockade of uptake2-mediated transport. Based on our demonstration that corticosterone decreased dopamine clearance in the NAc shell, and on our previous studies demonstrating that, although OCT3 is expressed in both the shell and core of the NAc, it is expressed more densely in the shell (Gasser et al., 2009), our injections of normetanephrine were targeted to the shell (Fig. 5). Like intra-accumbens corticosterone, intra-accumbens normetanephrine potentiated low-dose cocaine-induced reinstatement but had no effect on cocaine seeking when administered alone (two-way ANOVA, significant treatment × reinstatement condition interaction: F(2,8) = 8.765; p = 0.01) (Fig. 5). Post hoc testing indicated that intra-accumbens normetanephrine infusions potentiated cocaine-induced reinstatement (p = 0.01 vs extinction).
Figure 5.

Normetanephrine potentiates cocaine-induced drug-seeking behavior. A, Effects of intra-accumbens normetanephrine on cocaine-primed reinstatement. Rats (n = 6) received bilateral infusions of normetanephrine (25 ng/0.3 μl) or vehicle (aCSF) followed, 15 min later, by an injection of cocaine (2.5 mg/kg, i.p.) or saline. Data represent mean ± SEM of active lever responding over the 2 h immediately after the second injection. Significant increases in active lever responding were observed only when animals had received both normetanephrine and cocaine. *p < 0.05, compared with all other groups. B, Locations of microinjection sites from animals used in B.
OCT3 is expressed on NAc neurons
The uptake2 transporters vary widely in their sensitivity to inhibition by corticosterone. Of these transporters, OCT3 is by far the most sensitive to inhibition by corticosterone and is the only transporter inhibited by corticosterone at physiological concentrations (Shang et al., 2003; Gorboulev et al., 2005; Gasser et al., 2006; Duan and Wang, 2010; Hill et al., 2011). Thus, we hypothesized that the acute corticosterone-induced inhibition of dopamine clearance in the NAc reported here is the result of inhibition of OCT3-mediated transport. Although our previous studies identified OCT3-expressing cells in the NAc (Gasser et al., 2009), the identity of these cells is unknown, and there is no information available about the distribution of OCT3 with respect to dopaminergic terminals and dopamine receptors. In the present studies, we used immunofluorescence techniques to confirm the expression of OCT3 in the NAc, to identify the phenotype of OCT3-expressing cells, and to describe the localization of OCT3 immunoreactivity in relation to dopaminergic terminals and dopamine receptors in the region. OCT3-like immunoreactivity was observed as punctae distributed throughout the NAc core and shell, and was also concentrated in small-diameter (8–10 μm) perikarya (Fig. 6). Staining was not observed in sections incubated in the absence of the OCT3 antibody (data not shown). In both regions, OCT3-ir punctae were observed in close proximity to, but not colocalized with, tyrosine hydroxylase-immunoreactive fibers (Fig. 6A,B), and were observed in close proximity to dopamine D1 receptor-immunoreactive punctae (Fig. 6C). OCT3-immunoreactive perikarya also displayed immunoreactivity for the neuron-specific cytoskeletal marker β-III tubulin (Fig. 6D).
Figure 6.

OCT3 is expressed proximal to dopamine terminals and D1 receptors in the NAc. A, B, Representative photomicrographs depicting immunostaining for OCT3 (red) and tyrosine hydroxylase (green) in adult rat NAc core (A) and shell (B). C, Immunostaining for OCT3 (red) and dopamine D1 receptor (green) in NAc core. Square in C indicates area shown at higher magnification in the inset. D, Immunostaining for OCT3 (red) and the neuronal marker B-III tubulin (green) in NAc core. E, Line drawing (adapted from Paxinos and Watson, 1998) of a coronal section containing the areas of the NAc used for immunostaining and depicted in the indicated panels. Labeled black rectangles indicate the approximate locations from which the images were acquired. Scale bars: A, B, 33.3 μm; C, D, 20 μm; D inset, 6.75 μm.
Discussion
Our findings provide new insight into the mechanisms by which stressful life events increase the likelihood of relapse in recovering addicts. The data establish that acute stress potentiates the effects of low-dose cocaine on drug-seeking behavior in rats, and identify corticosterone as a necessary mediator of this potentiating interaction. They identify the NAc as a critical site at which corticosterone acts to potentiate cocaine effects on dopaminergic signaling and reinstatement. They demonstrate that corticosterone acutely decreases dopamine clearance within the NAc, and provide evidence that corticosterone acts by inhibiting the high-capacity monoamine transporter OCT3. We conclude that corticosterone-induced inhibition of dopamine clearance increases sensitivity to the effects of cocaine on dopaminergic neurotransmission and behavior and may represent a fundamental mechanism through which stress modulates dopaminergic neurotransmission, resulting in alterations in motivated behavior.
Although acute stress can, under certain conditions, act as a primary trigger for the reinstatement of drug-seeking behavior (Erb et al., 1998; Graf et al., 2011), it has been proposed that stress more often acts as a “stage-setter,” increasing the likelihood of relapse in response to other drug-associated stimuli (Preston and Epstein, 2011). The present studies model the stage-setting actions of stress and demonstrate that, even under conditions when stress alone does not trigger reinstatement, it can potentiate reinstatement in response to another subthreshold stimulus. Furthermore, these studies demonstrate that the stage-setting actions of stress are mediated by a mechanism distinct from that underlying reinstatement triggered by stress alone. Specifically, reinstatement induced primarily by stress does not require increases in corticosterone to stress levels (Erb et al., 1998; Graf et al., 2011). In contrast, the potentiation of cocaine-induced reinstatement by stress requires elevation of corticosterone to stress levels, and treatment with stress levels of corticosterone in the absence of stress is sufficient to potentiate cocaine-induced reinstatement. Thus, although stress-induced increases in corticosterone do not by themselves drive reinstatement of drug-seeking behavior, they appear to enhance sensitivity to other relapse-promoting stimuli. This may represent an important mechanism by which stress influences the likelihood of relapse in recovering addicts. Given that a history of exposure to cocaine and other drugs of abuse can alter HPA axis function (for review, see Kreek and Koob, 1998) and determines the pattern and magnitude of drug seeking and/or self-administration (Ahmed and Koob, 1998; Mantsch et al., 2008), the present findings suggest the following: (1) altered HPA reactivity and glucocorticoid regulation of dopamine clearance may contribute to the transition to compulsive use that defines addiction; and (2) the contribution of this mechanism to drug use may vary depending on the drug and the phase of the addiction process.
Our data indicate that corticosterone enhances the behavioral effects of low-dose cocaine by potentiating cocaine-induced increases in NAc dopamine. Dopamine signaling in the NAc is an important contributor to cocaine-induced reinstatement of drug seeking (Cornish and Kalivas, 2000; Bachtell et al., 2005; Schmidt et al., 2006); and the magnitude of cocaine-primed reinstatement appears to be determined in part by the level of extracellular dopamine in the NAc (Madayag et al., 2010). Our findings are consistent with this model and strongly suggest that the potentiation of relapse by stress or corticosterone is mediated by augmentation of cocaine-induced increases in NAc dopamine. In the present studies, noncontingent injection of low-dose cocaine was insufficient either to elevate NAc dopamine or to induce reinstatement of compulsive drug-seeking unless it was preceded by elevation of corticosterone to stress levels, either by acute stress exposure or by corticosterone injection. Furthermore, elevation of corticosterone within the NAc was sufficient to potentiate subthreshold cocaine-induced reinstatement, and the ability of systemic corticosterone to potentiate cocaine-primed reinstatement was eliminated by intra-accumbens blockade of dopamine receptors. These results are consistent with previous studies demonstrating that removal of corticosterone by adrenalectomy or metyrapone treatment blunts cocaine-induced increases in NAc dopamine (Rouge-Pont et al., 1995; Barrot et al., 2000).
Our finding that stress-level corticosterone treatment potentiated cocaine-induced increases in NAc dopamine, but had no effect on its own, is consistent with the results of previous studies demonstrating that corticosterone-induced increases in striatal dopamine concentrations are state-dependent (Piazza et al., 1996). This state dependency led to the hypothesis that corticosterone does not directly stimulate dopamine release in the NAc but rather acts indirectly to enhance dopaminergic responses to other stimuli (e.g., by inhibiting catabolic enzymes or uptake mechanisms) (Piazza et al., 1996). Under this hypothesis, corticosterone effects on NAc dopamine would require a threshold level of activity of dopaminergic inputs. Below this threshold, corticosterone would have little effect on dopamine concentrations.
The results of our voltammetry studies demonstrate that corticosterone treatment rapidly decreases dopamine clearance in the NAc. This is consistent with previous ex vivo findings that glucocorticoids rapidly decrease the uptake of dopamine by crude striatal synaptosomes (Gilad et al., 1987), a preparation that contains both presynaptic and postsynaptic compartments. The rapidity with which glucocorticoids decreased uptake in our studies and in the ex vivo studies indicates that corticosterone-induced decreases in dopamine clearance involve a nongenomic mechanism. This is consistent with our finding that the GR antagonist RU38486 did not block corticosterone-induced potentiation of cocaine-primed reinstatement. The ability of corticosterone to decrease dopamine clearance in animals in which the activity of the DAT was inhibited by treatment with a saturating dose of GBR12909 indicates that corticosterone-sensitive dopamine clearance does not involve the DAT. These findings suggest that corticosterone decreases extracellular dopamine clearance either by inhibiting dopamine catabolism or by blocking a non-DAT dopamine transporter. Although recent studies have demonstrated acute glucocorticoid-induced decreases in monoamine oxidase protein and activity, these effects were only observed after 2–4 h of glucocorticoid exposure (Soliman et al., 2012), well beyond the time points for measurement of uptake and behavior in the current studies. Thus, the most likely mechanism involves inhibition of dopamine transport.
Our finding that intra-accumbens administration of normetanephrine, a nonglucocorticoid inhibitor of uptake2-mediated transport (Martel et al., 1999), mimicked the effects of corticosterone on cocaine-induced reinstatement, strongly suggests that the behavioral and physiological effects of corticosterone reported here result from corticosterone-induced inhibition of dopamine clearance mediated by an uptake2 transporter. Uptake2 is a high-capacity, corticosteroid-sensitive monoamine transport system originally described in peripheral tissues (Iversen and Salt, 1970). Recent studies have identified a small group of transporters, including OCTs 1, 2, and 3 and PMAT, as mediators of uptake2-like monoamine transport and have demonstrated their expression in the brain (Engel et al., 2004; Vialou et al., 2004; Amphoux et al., 2006). Of these transporters, OCT3 and PMAT transport dopamine with the greatest efficiency, whereas OCT1 and 2 are not thought to transport dopamine under physiological conditions (Grundemann et al., 1998a, b, 1999; Engel et al., 2004; Duan and Wang, 2010; Bacq et al., 2011). OCT3 and PMAT transport dopamine with similar efficiencies, but they differ markedly in sensitivity to inhibition by corticosterone. Whereas the IC50 for corticosterone inhibition of OCT3-mediated transport (0.04–0.2 μm) (Shang et al., 2003) is within the physiological range reported for stress-induced concentrations of corticosterone in the brain (Droste et al., 2008), PMAT-mediated transport is inhibited only at very high concentrations of corticosterone (Ki = 450 μm) (Duan and Wang, 2010). Based upon these properties, we hypothesize that OCT3 inhibition underlies the effects of stress level corticosterone treatment on dopamine clearance and cocaine-seeking behavior in the present studies. Definitive testing of this hypothesis will require the examination of stress and corticosterone effects on drug-seeking behavior and dopamine clearance in OCT3-deficient animals. Consistent with our hypothesis, OCT3-deficient mice exhibit enhanced behavioral and neurochemical responses to methamphetamine and cocaine (Vialou et al., 2008; Cui et al., 2009).
The results of our immunofluorescence studies suggest that OCT3 represents a postsynaptic dopamine clearance mechanism in the NAc. We confirmed the expression of OCT3 in the core and shell of the rat NAc, and we began to characterize the phenotype of OCT3-expressing cells and the subcellular localization of OCT3 in this area. Our observations indicate that OCT3 is expressed on the somata and processes of postsynaptic, likely medium spiny neurons in the NAc, and not on dopaminergic terminals. As in the amygdala (Hill and Gasser, 2013), OCT3 in the NAc is localized in close proximity to D1 receptors. These findings raise the interesting possibility that OCT3 represents a previously uncharacterized postsynaptic dopamine clearance mechanism, and make OCT3 a compelling mechanism through which glucocorticoids may act to potentiate cocaine effects on dopaminergic neurotransmission and behavior. Additional studies, including electron microscopic examination, are required to more clearly identify the subcellular localization of OCT3.
The effects of stress on motivated behavior are powerful, pervasive, and context-dependent, suggesting complex interactions between stress-response systems and the circuitry underlying motivated behavior. However, rather than driving specific behaviors, stress often modulates ongoing behaviors in a context-dependent manner. We have demonstrated a central role for glucocorticoid hormones in the heightened susceptibility to drug relapse during times of stress, and we have identified a mechanism by which stress-level glucocorticoids act in the NAc to decrease dopamine clearance, enhancing the effects of cocaine on dopaminergic neurotransmission and behavior. This may represent a general mechanism by which stress, by elevating glucocorticoids, modulates ongoing behavior, shaping appropriate responses to environmental stimuli.
Footnotes
This work was supported by National Institutes of Health Grants DA017328, DA15758, and DA025679.
The authors declare no competing financial interests.
References
- Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science. 1998;282:298–300. doi: 10.1126/science.282.5387.298. [DOI] [PubMed] [Google Scholar]
- Amphoux A, Vialou V, Drescher E, Brüss M, Mannoury La Cour C, Rochat C, Millan MJ, Giros B, Bönisch H, Gautron S. Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology. 2006;50:941–952. doi: 10.1016/j.neuropharm.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Andersen PH. The dopamine inhibitor GBR 12909: selectivity and molecular mechanism of action. Eur J Pharmacol. 1989;166:493–504. doi: 10.1016/0014-2999(89)90363-4. [DOI] [PubMed] [Google Scholar]
- Bachtell RK, Whisler K, Karanian D, Self DW. Effects of intra-nucleus accumbens shell administration of dopamine agonists and antagonists on cocaine-taking and cocaine-seeking behaviors in the rat. Psychopharmacology (Berl) 2005;183:41–53. doi: 10.1007/s00213-005-0133-1. [DOI] [PubMed] [Google Scholar]
- Bacq A, Balasse L, Biala G, Guiard B, Gardier AM, Schinkel A, Louis F, Vialou V, Martres MP, Chevarin C, Hamon M, Giros B, Gautron S. Organic cation transporter 2 controls brain norepinephrine and serotonin clearance and antidepressant response. Mol Psychiatry. 2012;17:926–939. doi: 10.1038/mp.2011.87. [DOI] [PubMed] [Google Scholar]
- Barrot M, Marinelli M, Abrous DN, Rougé-Pont F, Le Moal M, Piazza PV. The dopaminergic hyper-responsiveness of the shell of the nucleus accumbens is hormone-dependent. Eur J Neurosci. 2000;12:973–979. doi: 10.1046/j.1460-9568.2000.00996.x. [DOI] [PubMed] [Google Scholar]
- Berger SP, Hall S, Mickalian JD, Reid MS, Crawford CA, Delucchi K, Carr K, Hall S. Haloperidol antagonism of cue-elicited cocaine craving. Lancet. 1996;347:504–508. doi: 10.1016/S0140-6736(96)91139-3. [DOI] [PubMed] [Google Scholar]
- Budygin EA, Kilpatrick MR, Gainetdinov RR, Wightman RM. Correlation between behavior and extracellular dopamine levels in rat striatum: comparison of microdialysis and fast-scan cyclic voltammetry. Neurosci Lett. 2000;281:9–12. doi: 10.1016/S0304-3940(00)00813-2. [DOI] [PubMed] [Google Scholar]
- Buffalari DM, See RE. Footshock stress potentiates cue-induced cocaine-seeking in an animal model of relapse. Physiol Behav. 2009;98:614–617. doi: 10.1016/j.physbeh.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Aras R, Christian WV, Rappold PM, Hatwar M, Panza J, Jackson-Lewis V, Javitch JA, Ballatori N, Przedborski S, Tieu K. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc Natl Acad Sci U S A. 2009;106:8043–8048. doi: 10.1073/pnas.0900358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroche V, Marinelli M, Le Moal M, Piazza PV. Glucocorticoids and behavioral effects of psychostimulants: II. Cocaine intravenous self-administration and reinstatement depend on glucocorticoid levels. J Pharmacol Exp Ther. 1997;281:1401–1407. [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droste SK, de Groote L, Atkinson HC, Lightman SL, Reul JM, Linthorst AC. Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology. 2008;149:3244–3253. doi: 10.1210/en.2008-0103. [DOI] [PubMed] [Google Scholar]
- Duan H, Wang J. Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. J Pharmacol Exp Ther. 2010;335:743–753. doi: 10.1124/jpet.110.170142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K, Zhou M, Wang J. Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem. 2004;279:50042–50049. doi: 10.1074/jbc.M407913200. [DOI] [PubMed] [Google Scholar]
- Erb S, Shaham Y, Stewart J. The role of corticotropin-releasing factor and corticosterone in stress- and cocaine-induced relapse to cocaine seeking in rats. J Neurosci. 1998;18:5529–5536. doi: 10.1523/JNEUROSCI.18-14-05529.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- España RA, Roberts DC, Jones SR. Short-acting cocaine and long-acting GBR-12909 both elicit rapid dopamine uptake inhibition following intravenous delivery. Neuroscience. 2008;155:250–257. doi: 10.1016/j.neuroscience.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatseas M, Denis C, Massida Z, Verger M, Franques-Rénéric P, Auriacombe M. Cue-induced reactivity, cortisol response and substance use outcome in treated heroin dependent individuals. Biol Psychiatry. 2011;70:720–727. doi: 10.1016/j.biopsych.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Feltenstein MW, See RE. Potentiation of cue-induced reinstatement of cocaine-seeking in rats by the anxiogenic drug yohimbine. Behav Brain Res. 2006;174:1–8. doi: 10.1016/j.bbr.2006.06.039. [DOI] [PubMed] [Google Scholar]
- Gasser PJ, Lowry CA, Orchinik M. Corticosterone-sensitive monoamine transport in the rat dorsomedial hypothalamus: potential role for organic cation transporter 3 in stress-induced modulation of monoaminergic neurotransmission. J Neurosci. 2006;26:8758–8766. doi: 10.1523/JNEUROSCI.0570-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser PJ, Orchinik M, Raju I, Lowry CA. Distribution of organic cation transporter 3, a corticosterone-sensitive monoamine transporter, in the rat brain. J Comp Neurol. 2009;512:529–555. doi: 10.1002/cne.21921. [DOI] [PubMed] [Google Scholar]
- Gilad GM, Rabey JM, Gilad VH. Presynaptic effects of glucocorticoids on dopaminergic and cholinergic synaptosomes: implications for rapid endocrine-neural interactions in stress. Life Sci. 1987;40:2401–2408. doi: 10.1016/0024-3205(87)90754-5. [DOI] [PubMed] [Google Scholar]
- Gorboulev V, Shatskaya N, Volk C, Koepsell H. Subtype-specific affinity for corticosterone of rat organic cation transporters rOCT1 and rOCT2 depends on three amino acids within the substrate binding region. Mol Pharmacol. 2005;67:1612–1619. doi: 10.1124/mol.104.008821. [DOI] [PubMed] [Google Scholar]
- Graf EN, Hoks MA, Baumgardner J, Sierra J, Vranjkovic O, Bohr C, Baker DA, Mantsch JR. Adrenal activity during repeated long-access cocaine self-administration is required for later CRF-induced and CRF-dependent stressor-induced reinstatement in rats. Neuropsychopharmacology. 2011;36:1444–1454. doi: 10.1038/npp.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründemann D, Köster S, Kiefer N, Breidert T, Engelhardt M, Spitzenberger F, Obermüller N, Schömig E. Transport of monoamine transmitters by the organic cation transporter type 2, OCT2. J Biol Chem. 1998a;273:30915–30920. doi: 10.1074/jbc.273.47.30915. [DOI] [PubMed] [Google Scholar]
- Gründemann D, Schechinger B, Rappold GA, Schömig E. Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nat Neurosci. 1998b;1:349–351. doi: 10.1038/1557. [DOI] [PubMed] [Google Scholar]
- Gründemann D, Liebich G, Kiefer N, Köster S, Schömig E. Selective substrates for non-neuronal monoamine transporters. Mol Pharmacol. 1999;56:1–10. doi: 10.1124/mol.56.1.1. [DOI] [PubMed] [Google Scholar]
- Heien ML, Khan AS, Ariansen JL, Cheer JF, Phillips PE, Wassum KM, Wightman RM. Real-time measurement of dopamine fluctuations after cocaine in the brain of behaving rats. Proc Natl Acad Sci U S A. 2005;102:10023–10028. doi: 10.1073/pnas.0504657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JE, Gasser PJ. Organic cation transporter 3 is densely expressed in the intercalated cell groups of the amygdala: anatomical evidence for a stress hormone-sensitive dopamine clearance system. J Chem Neuroanat. 2013 doi: 10.1016/j.jchemneu.2013.04.007. doi: 10.1016/j.jchemneu.2013.04.007. Advance online publication. Retrieved May 18, 2013. [DOI] [PubMed] [Google Scholar]
- Hill JE, Makky K, Shrestha L, Hillard CJ, Gasser PJ. Natural and synthetic corticosteroids inhibit uptake 2-mediated transport in CNS neurons. Physiol Behav. 2011;104:306–311. doi: 10.1016/j.physbeh.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Horvath G, Sutto Z, Torbati A, Conner GE, Salathe M, Wanner A. Norepinephrine transport by the extraneuronal monoamine transporter in human bronchial arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:829–837. doi: 10.1152/ajplung.00054.2003. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Koob GF. Drug dependence: stress and dysregulation of brain reward pathways. Drug Alcohol Depend. 1998;51:23–47. doi: 10.1016/s0376-8716(98)00064-7. [DOI] [PubMed] [Google Scholar]
- Iversen LL, Salt PJ. Inhibition of catecholamine uptake-2 by steroids in the isolated rat heart. Br J Pharmacol. 1970;40:528–530. doi: 10.1111/j.1476-5381.1970.tb10637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer SZ, Rubio MC. Effects of the noradrenaline metabolites on the adrenergic receptors. Naunyn Schmiedebergs Arch Pharmacol. 1973;276:71–88. doi: 10.1007/BF00500780. [DOI] [PubMed] [Google Scholar]
- Lee B, Tiefenbacher S, Platt DM, Spealman RD. Role of the hypothalamic-pituitary-adrenal axis in reinstatement of cocaine-seeking behavior in squirrel monkeys. Psychopharmacology (Berl) 2003;168:177–183. doi: 10.1007/s00213-003-1391-4. [DOI] [PubMed] [Google Scholar]
- Lips KS, Volk C, Schmitt BM, Pfeil U, Arndt P, Miska D, Ermert L, Kummer W, Koepsell H. Polyspecific cation transporters mediate luminal release of acetylcholine from bronchial epithelium. Am J Respir Cell Mol Biol. 2005;33:79–88. doi: 10.1165/rcmb.2004-0363OC. [DOI] [PubMed] [Google Scholar]
- Madayag A, Kau KS, Lobner D, Mantsch JR, Wisniewski S, Baker DA. Drug-induced plasticity contributing to heightened relapse susceptibility: neurochemical changes and augmented reinstatement in high-intake rats. J Neurosci. 2010;30:210–217. doi: 10.1523/JNEUROSCI.1342-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantsch JR, Baker DA, Serge JP, Hoks MA, Francis DM, Katz ES. Surgical adrenalectomy with diurnal corticosterone replacement slows escalation and prevents the augmentation of cocaine-induced reinstatement in rats self-administering cocaine under long-access conditions. Neuropsychopharmacology. 2008;33:814–826. doi: 10.1038/sj.npp.1301464. [DOI] [PubMed] [Google Scholar]
- Marinelli M, Rougé-Pont F, De Jesus-Oliveira C, Le Moal M, Piazza PV. Acute blockade of corticosterone secretion decreases the psychomotor stimulant effects of cocaine. Neuropsychopharmacology. 1997;16:156–161. doi: 10.1016/S0893-133X(96)00169-8. [DOI] [PubMed] [Google Scholar]
- Martel F, Ribeiro L, Calhau C, Azevedo I. Comparison between uptake2 and rOCT1: effects of catecholamines, metanephrines and corticosterone. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:303–309. doi: 10.1007/PL00005356. [DOI] [PubMed] [Google Scholar]
- McKee SA, Sinha R, Weinberger AH, Sofuoglu M, Harrison EL, Lavery M, Wanzer J. Stress decreases the ability to resist smoking and potentiates smoking intensity and reward. J Psychopharmacol. 2011;25:490–502. doi: 10.1177/0269881110376694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Aragona BJ, Kile BM, Carelli RM, Wightman RM. In vivo voltammetric monitoring of catecholamine release in subterritories of the nucleus accumbens shell. Neuroscience. 2010;169:132–142. doi: 10.1016/j.neuroscience.2010.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Rougé-Pont F, Deroche V, Maccari S, Simon H, Le Moal M. Glucocorticoids have state-dependent stimulant effects on the mesencephalic dopaminergic transmission. Proc Natl Acad Sci U S A. 1996;93:8716–8720. doi: 10.1073/pnas.93.16.8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston KL, Epstein DH. Stress in the daily lives of cocaine and heroin users: relationship to mood, craving, relapse triggers, and cocaine use. Psychopharmacology (Berl) 2011;218:29–37. doi: 10.1007/s00213-011-2183-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MS, Prichep LS, Ciplet D, O'Leary S, Tom M, Howard B, Rotrosen J, John ER. Quantitative electroencephalographic studies of cue-induced cocaine craving. Clin Electroencephalogr. 2003;34:110–123. doi: 10.1177/155005940303400305. [DOI] [PubMed] [Google Scholar]
- Rougé-Pont F, Marinelli M, Le Moal M, Simon H, Piazza PV. Stress-induced sensitization and glucocorticoids: II. Sensitization of the increase in extracellular dopamine induced by cocaine depends on stress-induced corticosterone secretion. J Neurosci. 1995;15:7189–7195. doi: 10.1523/JNEUROSCI.15-11-07189.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Anderson SM, Pierce RC. Stimulation of D1-like or D2 dopamine receptors in the shell, but not the core, of the nucleus accumbens reinstates cocaine-seeking behaviour in the rat. Eur J Neurosci. 2006;23:219–228. doi: 10.1111/j.1460-9568.2005.04524.x. [DOI] [PubMed] [Google Scholar]
- Schömig E, Lazar A, Gründemann D. Extraneuronal monoamine transporter and organic cation transporters 1 and 2: a review of transport efficiency. Handb Exp Pharmacol. 2006;175:151–180. doi: 10.1007/3-540-29784-7_8. [DOI] [PubMed] [Google Scholar]
- Shalev U, Marinelli M, Baumann MH, Piazza PV, Shaham Y. The role of corticosterone in food deprivation-induced reinstatement of cocaine seeking in the rat. Psychopharmacology (Berl) 2003;168:170–176. doi: 10.1007/s00213-002-1200-5. [DOI] [PubMed] [Google Scholar]
- Shang T, Uihlein AV, Van Asten J, Kalyanaraman B, Hillard CJ. 1-Methyl-4-phenylpyridinium accumulates in cerebellar granule neurons via organic cation transporter 3. J Neurochem. 2003;85:358–367. doi: 10.1046/j.1471-4159.2003.01686.x. [DOI] [PubMed] [Google Scholar]
- Shelton KL, Beardsley PM. Interaction of extinguished cocaine-conditioned stimuli and footshock on reinstatement in rats. Int J Comp Psychol. 2005;18:154–166. [Google Scholar]
- Sinha R. How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 2001;158:343–359. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- Sinha R, Talih M, Malison R, Cooney N, Anderson GM, Kreek MJ. Hypothalamic-pituitary-adrenal axis and sympatho-adreno-medullary responses during stress-induced and drug cue-induced cocaine craving states. Psychopharmacology (Berl) 2003;170:62–72. doi: 10.1007/s00213-003-1525-8. [DOI] [PubMed] [Google Scholar]
- Sinha R, Garcia M, Paliwal P, Kreek MJ, Rounsaville BJ. Stress-induced cocaine craving and hypothalamic-pituitary-adrenal responses are predictive of cocaine relapse outcomes. Arch Gen Psychiatry. 2006;63:324–331. doi: 10.1001/archpsyc.63.3.324. [DOI] [PubMed] [Google Scholar]
- Sinha R, Fox HC, Hong KI, Hansen J, Tuit K, Kreek MJ. Effects of adrenal sensitivity, stress- and cue-induced craving, and anxiety on subsequent alcohol relapse and treatment outcomes. Arch Gen Psychiatry. 2011;68:942–952. doi: 10.1001/archgenpsychiatry.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman A, Udemgba C, Fan I, Xu X, Miler L, Rusjan P, Houle S, Wilson AA, Pruessner J, Ou XM, Meyer JH. Convergent effects of acute stress and glucocorticoid exposure upon MAO-A in humans. J Neurosci. 2012;32:17120–17127. doi: 10.1523/JNEUROSCI.2091-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiga F, Knight DM, Droste SK, Conway-Campbell B, Kershaw Y, MacSweeney CP, Thomson FJ, Craighead M, Peeters BW, Lightman SL. Differential effect of glucocorticoid receptor antagonists on glucocorticoid receptor nuclear translocation and DNA binding. J Psychopharmacol. 2011;25:211–221. doi: 10.1177/0269881109348175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Amphoux A, Zwart R, Giros B, Gautron S. Organic cation transporter 3 (Slc22a3) is implicated in salt-intake regulation. J Neurosci. 2004;24:2846–2851. doi: 10.1523/JNEUROSCI.5147-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Balasse L, Callebert J, Launay JM, Giros B, Gautron S. Altered aminergic neurotransmission in the brain of organic cation transporter 3-deficient mice. J Neurochem. 2008;106:1471–1482. doi: 10.1111/j.1471-4159.2008.05506.x. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Amatore C, Engstrom RC, Hale PD, Kristensen EW, Kuhr WG, May LJ. Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience. 1988;25:513–523. doi: 10.1016/0306-4522(88)90255-2. [DOI] [PubMed] [Google Scholar]
- Yorgason JT, España RA, Jones SR. Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods. 2011;202:158–164. doi: 10.1016/j.jneumeth.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]