

Abstract
Amygdala excitatory synaptic strengthening is thought to contribute to both conditioned fear and anxiety. Thus, one basis for behavioral flexibility could allow these pathways to be weakened and corresponding emotion to be attenuated. However, synaptic depression within the context of amygdala-dependent behavior remains poorly understood. Previous work identified lateral amygdala (LA) calcium-permeable AMPA receptors (CP-AMPARs) as a key target for synaptic removal in long-term depression (LTD) and persistent fear attenuation. Here we demonstrate that LA neurons express two equally potent forms of LTD with contrasting requirements for protein kinase and phosphatase activity and differential impact on CP-AMPAR trafficking. Selective removal of CP-AMPARs from synapses is contingent on group 1 metabotropic glutamate receptor (mGluR1) and PKC signaling, in contrast to an alternate LTD pathway that nonselectively removes AMPARs and requires calcineurin (PP2b). Intriguingly, the balance between these forms of LTD is shifted by posttraining activation of β-adrenergic receptors in fear conditioned mice, resulting in selective augmentation of mGluR-dependent depression. These results highlight the complexity of core mechanisms in LTD and suggest that norepinephrine exposure mediates a form of synaptic metaplasticity that recalibrates fear memory processing.
Introduction
The lateral amygdala (LA) figures prominently in appetitive (Murray, 2007) and aversive (Johansen et al., 2011) pavlovian conditioning. In particular, auditory fear conditioning involves strengthening of thalamic pathways to the LA (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997; Rumpel et al., 2005; Kwon and Choi, 2009; Clem and Huganir, 2010), a process that has been modeled by induction of long-term potentiation (LTP) at thalamic input synapses (Rogan and LeDoux, 1995; Huang et al., 2000; Humeau et al., 2007; Tully et al., 2007; Asrar et al., 2009). Conversely, fear responses are extinguished by repeated exposure to the auditory cue, which sometimes but not always leads to thalamo-amygdala synaptic weakening and occlusion of long-term depression (LTD) (Mao et al., 2006; Kim et al., 2007; Clem and Huganir, 2010). Consequently, the mechanisms governing LTD may in part determine the behavioral outcome of extinction training or its clinical equivalent, exposure-based therapy.
We demonstrated previously that both LTD and extinction-dependent fear erasure involve synaptic removal of GluA2-lacking calcium-permeable AMPA receptors (CP-AMPARs) in the LA (Clem and Huganir, 2010). Here we report that CP-AMPAR trafficking is stimulus protocol specific. Furthermore, although LA neurons express both CP-AMPAR-dependent and -independent forms of LTD, these LTD mechanisms rely differentially on phosphatase and kinase signaling. After auditory fear conditioning, group 1 metabotropic glutamate receptor (mGluR1)- and CP-AMPAR-dependent LTD is selectively enhanced as a result of β-adrenergic receptor (β-AR) signaling during memory consolidation. Our results indicate that a discrete LTD pathway is augmented in parallel with fear-related synaptic strengthening and suggest that strategies for fear attenuation can selectively target CP-AMPARs for removal without globally saturating LTD processes.
Materials and Methods
Subjects.
All subjects in wild-type experiments were adolescent, male C57BL/6N mice (Charles River) aged 4–6 postnatal weeks. Subjects were housed in groups of three on a 12 h light/dark cycle and given food and water ad libitum. Protein interacting with C-kinase 1 (PICK1) knock-outs were generated as reported previously (Gardner et al., 2005) and backcrossed at least 11 times to C57BL/6 before heterozygous mating to generate wild-type and knock-out mice.
Slice electrophysiology.
Brains were dissected into 2°C buffer containing the following (in mm): 210.3 sucrose, 11 glucose, 2.5 KCl, 1 NaH2PO4, 26.2 NaHCO3, 0.5 ascorbate, 0.5 CaCl2, and 4 MgCl2. Acute coronal slices were obtained at 350 μm and transferred to normal ACSF composed of the following (in mm): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 26.2 NaHCO3, 11 glucose, 2 CaCl2, and 2 MgCl2. After recovery at 35°C for 40 min, slices were maintained at 22–25°C. Whole-cell recordings were performed using borosilicate glass electrodes (3–5 MΩ) on slices containing LA as well as a sharply defined bundle of thalamic axons traversing the amygdalo-striatal transition area. All recordings were targeted to principal excitatory neurons in the dorsolateral amygdala based on pyramidal morphology. Current-clamp recordings with a potassium methanesulfonate internal solution confirmed that these neurons had a regular firing phenotype. For voltage-clamp recording, electrode internal solution contained the following (in mm): 130 cesium-methanesulfonate, 10 HEPES, 0.5 EGTA, 8 NaCl, 4 Mg-ATP, 1 QX-314, 10 Na-phosphocreatine, and 0.4 Na-GTP. Monosynaptic responses (3–4 ms latency) were evoked by field depolarization (50 μs duration) of thalamic axons using a matrix microelectrode. All experiments were performed in picrotoxin (100 μm) to block fast GABAergic transmission. To prevent spontaneous bursting under these conditions, extracellular Ca2+ and Mg2+ were increased to 4 mm each to reduce neuronal membrane excitability. Data were filtered at 3 kHz and acquired at 10 kHz using Multiclamp 700B and pClamp 10 (Molecular Devices). EPSCs were monitored by stimulation at 0.1 Hz. Series resistance was continuously monitored, and recordings were discarded when this measurement changed by >20%. Two protocols were used for LTD induction. The first was adapted from a pairing protocol used in hippocampal CA1 neurons and consisted of 1 Hz stimulation for 10 min while the neuron was clamped at −40 mV. In the second protocol, stimulation for 3 min at 3 Hz was performed with paired pulses (50 ms interpulse interval) while the neuron was clamped at −50 mV. Blockade of CP-AMPARs was performed with 1-naphthylacetyl spermine (NASPM; 50 μm; Sigma-Aldrich). 1-Amino-phosphovaleric acid (APV; Ascent), LY367385 [(S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid] (Tocris Bioscience), and 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP; Tocris Bioscience) stocks were made in aqueous solution and applied to the recording bath for the duration of LTD experiments. Okadaic acid (OA) and FK506 [(3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy-3-[(1E)-2-[(1R,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propen-1-yl)-15,19-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)tetrone] (Sigma-Aldrich) were applied in 0.1% DMSO. Protein kinase C (PKC) inhibitor PKI[19–31] (EMD Millipore) and GluA2-3A and GluA2-3Y peptides (AnaSpec) were added to the recording internal solution.
Fear conditioning.
Unpaired and paired groups received training that was divided into two sessions, separated by 30 min. Unpaired animals were presented with six unpaired tones [conditioned stimuli (CS)] in session 1 and six unsignaled footshocks [unconditioned stimuli (US)] in session 2. Paired animals were presented with no stimuli in session 1 and six paired CS and US during session 2, in which CS and US were coterminating. During each session, a period of acclimation lasting 200 s preceded the presentation of cues. The CS consisted of an 80 dB, 2 kHz tone lasting 20 s. The US consisted of scrambled 1.0 mA footshock. Cue presentations were separated by 100 s. After conditioning, mice were returned to their home cages until preparation of brain slices. For β-AR modulation, mice were given intraperitoneal injections of propranolol (20 mg/kg) or PBS vehicle within 5 min after training.
Statistical analyses.
Significance of observations was established by unpaired Student's t test in the case of independent groups, paired t test for within-cell comparison with baseline measures, or one-way ANOVA followed by Tukey's post hoc test for more than two independent groups, with resulting p values given in text. All graphs represent group means ± SE.
Results
To examine the contribution of CP-AMPARs to thalamo-amygdala transmission, we targeted LA principal excitatory neurons for whole-cell voltage-clamp recordings in acute brain slices from naive mice. Bath application of the CP-AMPAR antagonist NASPM during stimulation of thalamic inputs reduced AMPAR–EPSCs by 17.9 ± 2.3% (n = 6; Fig. 1B), consistent with previous reports that CP-AMPARs contribute to synaptic transmission in LA principal neurons (Mahanty and Sah, 1998; Clem and Huganir, 2010). Importantly, NASPM reduces the apparent rectification of AMPAR–EPSCs in LA neurons (Clem and Huganir, 2010), further demonstrating that functional CP-AMPARs exist at thalamo-amygdala synapses.
Figure 1.

Induction protocol-dependent removal of CP-AMPARs during LTD. A, Position of stimulating and recording electrodes for brain slice electrophysiology. BA, Basal amygdala. B, Application of NASPM (50 μm) to acute slices from LA in the continuous presence of APV (100 μm). Example traces are sampled from representative neurons in the shaded regions. Calibration: 100 pA, 20 ms. C, LTD induction by LFS pairing (1 Hz, 10 min, −40 mV) in normal ACSF followed by NASPM application (50 μm). Calibration: 200 pA, 20 ms. D, LTD induction by ppLFS pairing (3 Hz, 50 ms interpulse interval, 3 min, −50 mV) in normal ACSF followed by NASPM application. Calibration: 200 pA, 20 ms. E, F, Mean LTD, as determined by comparison of the blue and green shaded regions (E) and NASPM block, as determined by comparison of the green and red shaded regions (F) from experiments in A–C. Example traces in C and D are sampled from the shaded regions. All data points represent group means ± SE.
To determine the impact of synaptic activity on CP-AMPARs, we examined two stimulus conditions for LTD. In the first, neurons were depolarized to −40 mV during delivery of low-frequency afferent stimulation (LFS pairing). In the second, milder depolarization was used (−50 mV) together with low-frequency paired-pulse stimulation (ppLFS pairing). Although both conditions yield reliable depression, the use of paired versus single pulses can affect the pharmacological requirements of resulting LTD (Huber et al., 2000; Kemp et al., 2000), which may be attributable the capacity for paired pulses to more effectively activate extrasynaptic glutamate receptors. After either form of stimulation, NASPM was applied to slices to reveal the residual contribution of CP-AMPARs.
Surprisingly, although stimulus protocol did not affect the magnitude of depression (Fig. 1C–E) [EPSC % baseline: LFS = 68.8 ± 4.6 (n = 7), ppLFS = 73.3 ± 2.6 (n = 7), p > 0.1], only ppLFS triggered CP-AMPAR removal. The inhibitory effect of NASPM was unaltered after LFS pairing (Fig. 1F) [EPSC % inhibition = 20.0 ± 2.6 (n = 7), p > 0.1 vs naive], indicating that this form of LTD is not AMPAR subtype selective. Conversely, ppLFS pairing completely and selectively abolished CP-AMPAR transmission [EPSC % inhibition = 1.07 ± 3.6 (n = 7), p < 0.001 vs naive]. Given the longer duration of LFS pairing, it is unlikely that differences in CP-AMPAR trafficking could be explained by a threshold effect.
Synaptic plasticity depends on intracellular signaling pathways linked to glutamate receptor activation. In particular, LTD has been reported to require NMDA-type glutamate receptors (NMDARs) and/or mGluR1/5 (Malenka and Bear, 2004; Lüscher and Huber, 2010). We therefore asked whether CP-AMPAR trafficking exhibits a differential dependence on these receptor classes. Both LFS and ppLFS LTD were blocked by the NMDAR antagonist APV (Fig. 2A–D) [EPSC % baseline: LFS control = 63.4 ± 3.7 (n = 6), LFS APV = 97.7 ± 2.4 (n = 6), p < 0.0001 vs control; ppLFS control = 78.2 ± 5.4 (n = 6), ppLFS APV = 97.9 ± 3.3 (n = 7), p < 0.05 vs control]. In contrast, an mGluR1 antagonist completely abolished ppLFS LTD (Fig. 2E,F) [EPSC % baseline: 96.6 ± 3.8 (n = 7), p < 0.05 vs control] but did not block LFS LTD [EPSC % baseline: 66.6 ± 3.7 (n = 6), p = 0.98 vs control]. Neither form of LTD required activation of mGluR5 (Fig. 2E,F) [EPSC % baseline: LFS MPEP = 57.0 ± 3.6 (n = 6), p = 0.76 vs control; ppLFS MPEP = 71.1 ± 6.6 (n = 7), p = 0.82 vs control].
Figure 2.

Specific requirement for mGluR1 and PKC in ppLFS LTD. A, B, Induction of LFS and ppLFS LTD in normal ACSF. Calibration: 100 pA, 20 ms. C, D, Induction of LFS (C) and ppLFS LTD (D) in the presence of APV (100 μm). Calibration: C, 80 pA, 20 ms; D, 100 pA, 20 ms. E, F, Induction of LFS (E) and ppLFS LTD (F) in the presence of MPEP (10 μm) or LY367385 (100 μm). Calibration: E, LY367385, 100 pA, 20 ms; MPEP, 150 pA, 20 ms; F, LY367385, 80 pA, 20 ms; MPEP, 60 pA, 20 ms. G, H, Induction of LFS (G) and ppLFS LTD (H) with the PKC inhibitor peptide PKI[19–31] (10 μm) in the recording pipette internal solution. Calibration: 200 pA, 20 ms. Example traces are sampled from the shaded regions. All data points represent group means ± SE.
Because of the role of mGluR1 exclusively in ppLFS LTD, we considered the involvement of mGluR1 downstream effectors. Acting through phospholipase C, mGluR1 can stimulate PKC, which can also be activated by NMDARs under some conditions (Sun and June Liu, 2007; Matta et al., 2011). Intracellular application of a highly selective peptide inhibitor of PKC (PKI[19–31]) (House and Kemp, 1987; Smith et al., 1990) completely abolished ppLFS LTD (Fig. 2H) [EPSC % baseline: 97.6 ± 3.2 (n = 6), p < 0.05 vs control]. This was unlikely to have resulted from nonspecific effects of the PKI[19–31], because ppLFS LTD was unaffected by an unrelated peptide (see Fig. 4B), and PKI[19–31] did not occlude LFS LTD (Fig. 2G) [EPSC % baseline: 63.1 ± 5.8 (n = 6), p = 1.00 vs control]. Thus, PKC likely mediates downstream effects of mGluR1 stimulation.
Figure 4.
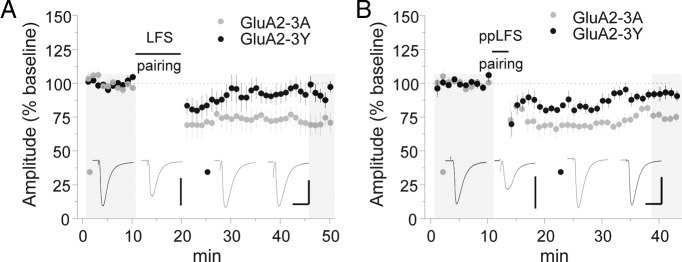
Peptide inhibitor of GluA2 trafficking blocks both LFS and ppLFS LTD. A, B, Induction of LFS (A) and ppLFS LTD (B) with GluA2-3A or GluA2-3Y in the recording pipette internal solution (100 μg/ml). Calibration: A, GluA2-3A, 250 pA, 20 ms; GluA2-3Y, 150 pA, 20 ms; B, GluA2-3A, 200 pA, 20 ms; GluA2-3Y, 200 pA, 20 ms. Example traces are sampled from the shaded regions. All data points represent group means ± SE.
Pharmacological studies of NMDAR-dependent LTD have implicated postsynaptic protein phosphatases (Malenka and Bear, 2004). In particular, both PP1/PP2a and calcineurin (PP2b) inhibitors interfere with LTD in the hippocampal CA1 area (Mulkey et al., 1993, 1994). Because we previously demonstrated that CP-AMPAR synaptic accumulation is driven by phosphorylation of the GluA1 subunit residue serine 845 (Clem and Huganir, 2010), we hypothesized that PP2b-mediated dephosphorylation of this site might drive CP-AMPAR removal (Kameyama et al., 1998; Banke et al., 2000; Tavalin et al., 2002; He et al., 2009). However, neither PP1/2a nor PP2b inhibitors blocked ppLFS LTD (Fig. 3B) [EPSC % baseline: control = 69.1 ± 7.4 (n = 6), OA = 65.1 ± 4.8 (n = 6), p = 0.87 vs control; FK506 = 71.6 ± 4.3 (n = 6), p = 0.95 vs control]. Conversely, LFS LTD was inhibited by PP2b but not PP1/2a antagonism (Fig. 3A) [EPSC % baseline: control = 70.8 ± 3.5 (n = 6), FK506 = 88.2 ± 3.9 (n = 6), p < 0.05 vs control; OA = 65.9 ± 6.3 (n = 6), p = 0.76 vs control].
Figure 3.
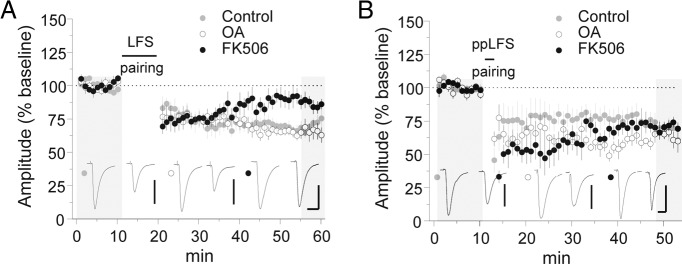
Calcineurin activity is required for LFS but not ppLFS LTD. A, B, Induction of LFS (A) and ppLFS LTD (B) in the presence of 0.1% DMSO vehicle, OA (1 μm), or FK506 (1 μm). Calibration: A, control, 100 pA, 20 ms; OA, 100 pA, 20 ms; FK506, 80 pA, 20 ms; B, control, 100 pA, 20 ms; OA, 150 pA, 20 ms; FK506, 100 pA, 20 ms. Example traces are sampled from the shaded regions. All data points represent group means ± SE.
Given these significant discrepancies in signal transduction, we considered the role of synaptic substrates in LTD. An eventual effector for synaptic weakening in several brain areas is trafficking of GluA2-containing AMPARs, for which regulatory sequences have been identified within the GluA2 C terminus. In particular, phosphorylation of GluA2 tyrosine residues has been shown to regulate AMPAR endocytosis, an effect that is blocked by intracellular application of a synthetic peptide containing these sites (GluA2-3Y) (Brebner et al., 2005; Migues et al., 2010; Li et al., 2011). Intriguingly, GluA2-3Y inhibited both LFS and ppLFS LTD in the LA (Fig. 4A,B) [EPSC % baseline: LFS GluA2-3Y = 93.5 ± 6.1 (n = 6), p < 0.05 vs GluA2-3A; ppLFS GluA2-3Y = 92.7 ± 3.5 (n = 6), p < 0.01 vs control GluA2-3A], although CP-AMPARs removed by ppLFS pairing are considered devoid of GluA2. This was not attributable to nonspecific peptide effects, because LTD was unaffected by a control peptide in which alanine substitutions occur at each tyrosine residue (Fig. 4A,B) [EPSC % baseline: LFS GluA2-3A = 72.3 ± 5.9 (n = 5), p < 0.01 vs baseline by paired t test; ppLFS GluA2-3A = 75.1 ± 1.2 (n = 5), p < 0.01vs baseline by paired t test].
Because GluA2-3Y did not effectively discriminate between ppLFS and LFS LTD, we next considered a region in the extreme C terminus of GluA2 that acts as a PDZ ligand for PICK1 as well as glutamate receptor interacting protein 1/2 (GRIP1/2) (Xia et al., 1999; Chung et al., 2000; Osten et al., 2000). Disruption of GRIP1/2 and facilitation of PICK1 binding to this region are triggered by GluA2 serine 880 phosphorylation (Chung et al., 2000), a mechanism that has been implicated in GluA2 synaptic removal (Kim et al., 2001; Seidenman et al., 2003; Steinberg et al., 2006; Lin and Huganir, 2007; Citri et al., 2010; Clem et al., 2010). Therefore, we examined the effect of disrupting PICK1–GluA2 interaction with the specific interfering peptide pep2-EVKI, but unfortunately this reagent induced rundown of baseline responses and was thus not suitable for application during LTD. However, to gain more insight into these baseline effects, we applied pep2-EVKI or a control peptide that lacks PICK1 specificity, pep2-SGKA. Significant rundown of AMPAR–EPSCs was observed after pep2-EVKI but not pep2-SGKA application in wild-type mice (Fig. 5A,B) [EPSC % baseline: pep2-EVKI = 79.0 ± 5.4 (n = 7); pep2-SGKA = 112.0 ± 6.2 (n = 6), p < 0.01]. After pep2-EVKI-induced rundown, rectification of AMPAR–EPSCs was reduced, consistent with CP-AMPAR synaptic removal (Fig. 5C). To examine the role of PICK1 in this process, we analyzed basal synaptic transmission and ppLFS in PICK1 knock-out mice. Interestingly, PICK1 mice had reduced rectification under all conditions and abolished ppLFS LTD (Fig. 5D) [EPSC % baseline: +/+ mice = 69.2 ± 4.4 (n = 10); −/− mice = 117.8 ± 16.6 (n = 8), p < 0.005], indicating that failure to accumulate CP-AMPARs in PICK1 knock-outs eliminates a principal substrate for ppLFS LTD. Moreover, the pep2-EVKI peptide had no effect on baseline transmission in slices from PICK1 knock-out mice [EPSC % baseline: pep2-EVKI = 102.6 ± 6.6 (n = 5); pep2-SGKA = 100.6 ± 4.2 (n = 4), p = 0.81].
Figure 5.

Release of GluA2 from PICK1 leads to displacement of synaptic CP-AMPARs. A, B, Intracellular application of pep2-EVKI or pep2-SGKA in wild-type (A) or PICK1 knock-out (B) mice. Calibration: A, pep2-EVKI, 150 pA, 20 ms; pep2-SGKA, 100 pA, 20 ms; B, pep2-EVKI, 75 pA, 20 ms; pep2-SGKA, 75 pA, 20 ms. C, Current–voltage plots for AMPAR–EPSCs after intracellular peptide application. *p < 0.001. D, Comparison of ppLFS LTD in wild-type and PICK1 knock-out mice. Example traces are sampled from the shaded regions. All data points represent group means ± SE.
Because plasticity can be influenced by previous neuronal activity, coexistence of discrete forms of LTD in LA neurons raises the question of which LTD pathways predominate in emotionally experienced mice. Therefore, we obtained brain slice recordings from auditory fear conditioned mice or unpaired controls to determine the impact of fear memory storage on LTD expression. Twenty-four hours after training, no difference in LFS LTD was observed in mice receiving paired versus unpaired training (Fig. 6B) [EPSC % baseline: unpaired = 65.0 ± 4.1 (n = 7); paired = 71.0 ± 10.6 (n = 6), p = 0.59]. However, ppLFS LTD was significantly enhanced by paired fear conditioning (Fig. 6C) [EPSC % baseline: unpaired = 84.0 ± 6.1 (n = 8), paired = 58.0 ± 2.9 (n = 9), p < 0.005]. In addition, direct pharmacological stimulation of mGluR1 induced LTD after paired training but not after unpaired training (Fig. 6D) [EPSC % baseline: unpaired = 91.7 ± 3.2 (n = 5), paired = 73.2 ± 4.9 (n = 5), p < 0.01], indicating that LA synapses acquire sensitivity to mGluR–LTD during fear memory storage.
Figure 6.

Selective enhancement of mGluR-dependent LTD after fear conditioning. A, Mice received six pairings of tone (CS) and footshock (US) 24 h before slice preparation. In the control condition, CS and US were explicitly unpaired. B–D, Comparison of LFS LTD (B), ppLFS LTD (C), and (RS)-3,5-dihydroxyphenylglycine LTD (D; 50 μm bath application) in mice receiving unpaired versus paired fear conditioning. Calibration: B, unpaired, 120 pA, 20 ms; paired, 80 pA, 20 ms; C, unpaired, 80 pA, 20 ms; paired, 150 pA, 20 ms; D, unpaired, 200 pA, 20 ms; paired, 150 pA, 20 ms. Example traces are sampled from the shaded regions. All data points represent group means ± SE.
To gain additional insight into metaplasticity of mGluR–LTD, we considered the impact of fear conditioning on synaptic AMPAR composition. Previously, we demonstrated that fear conditioning leads to the accumulation of CP-AMPARs at thalamic synapses, an effect that was not observed in mice containing alanine mutations of GluA1 serine 845. Because β-AR agonists can induce serine 845 phosphorylation (Hu et al., 2007; Makino et al., 2011; Huang et al., 2012), we examined whether β-ARs were involved in AMPAR trafficking after fear conditioning. Surprisingly, posttraining injections of the β-AR antagonist propranolol did not affect synaptic strengthening (Fig. 7B,C) [AMPA/NMDAR: vehicle = 3.70 ± 0.20 (n = 7), propranolol = 4.68 ± 0.50 (n = 7), p = 0.15]. However, propranolol inhibited CP-AMPAR synaptic accumulation, as revealed by reduced rectification of AMPAR–EPSCs (Fig. 7D–F) [rectification index: vehicle = 2.17 ± 0.18 (n = 6), propranolol = 1.27 ± 0.04 (n = 10), p < 0.00001]. Importantly, fear conditioning did not affect the decay time of NMDAR–EPSCs [weighted tau decay: unpaired = 166.1 ± 13.9 ms (n = 11), paired = 168.8 ± 12.5 ms (n = 12), p = 0.89], indicating that experience does not alter the subunit composition of NMDARs. Coincident with this effect, ppLFS LTD was strongly reduced in propranolol-treated mice (Fig. 7G) [EPSC % baseline: vehicle = 56.1 ± 7.55 (n = 5), propranolol = 79.0 ± 4.64 (n = 5), p < 0.01]. Thus, although β-ARs are not required to consolidate fear-related synaptic strengthening, posttraining β-AR stimulation mediates metaplasticity at thalamic synapses.
Figure 7.

Norepinephrine-dependent AMPAR redistribution underlies metaplasticity. A, Schematic of β-AR treatments after fear conditioning (FC). B, Representative examples of compound AMPAR/NMDAR–EPSCs at the following membrane holding potentials: −70, 0, and +40 mV. C, Mean AMPAR/NMDAR ratio. p < 0.001. D, Representative examples of AMPAR–EPSCs at the following membrane holding potentials: −70, −60, −40, −20, 0, +20, +40, and +50 mV. E, Current–voltage plot of AMPAR–EPSCs. *p < 0.01. F, Mean AMPAR–EPSC rectification index. *p < 0.01. G, Induction of ppLFS LTD 24 h after paired fear conditioning, combined with vehicle or propranolol treatment. Calibration: vehicle, 80 pA, 20 ms; propranolol, 100 pA, 20 ms. Example traces are sampled from the shaded regions. All data points represent group means ± SE. prop, Propranolol.
Discussion
The trafficking of CP-AMPARs in behavioral contexts has piqued broad interest in this unique receptor population. Likely comprising homomeric assemblies of GluA1 (Wenthold et al., 1996; Gao and Wolf, 2007; He et al., 2009; Billa et al., 2010; Clem et al., 2010; Rozov et al., 2012), these receptors accumulate at synapses after associative fear conditioning in a manner dependent on GluA1 phosphorylation at serine 845 (Clem and Huganir, 2010), a target of PKA (Roche et al., 1996) and cyclic GMP-dependent kinase II (Serulle et al., 2007). Because neural activity abolishes CP-AMPAR currents, we initially hypothesized that CP-AMPARs may be unstably associated with the postsynaptic apparatus. However, we demonstrate here that CP-AMPAR trafficking specifically characterizes a mechanistically distinct form of LTD at thalamo-amygdala synapses. The existence of this discrete form of synaptic weakening suggests that changes in AMPAR subunit composition can lead to molecularly defined adjustments to LTD, a prediction that was confirmed by specific enhancement of mGluR1-dependent LTD in fear conditioned mice.
By comparing two induction protocols, we show that LTD of similar magnitude can result from highly distinct signal transduction. Although ppLFS pairing selectively reduced the NASPM-sensitive fraction of receptors at LA synapses, LFS pairing equally affected NASPM-sensitive and -insensitive currents. Therefore, CP-AMPARs are not a generally unstable receptor population but are instead more highly sensitive to high-frequency activity (in this case, 20 Hz). Such rates are typically exceeded by spike bursts of nonlemniscal medial geniculate neurons that convey auditory input to the LA (He and Hu, 2002) and may be a key factor in extinction-dependent synaptic weakening. The imposition of pyramidal cell hyperpolarization, for example, by feedforward inhibitory circuits (Bissiere et al., 2003), might squelch this process under most conditions. Dynamic regulation of GluA2 receptor content may function together with these local network states to confine synaptic depression to specific behavioral contexts and maturational stages of memory.
A major difference from LFS LTD was the dependence of ppLFS LTD on mGluR1 and PKC. These results agree closely with studies in principal neurons of the nucleus accumbens (McCutcheon et al., 2011) and ventral tegmentum (Bellone and Lüscher, 2005), as well as stellate cells of the cerebellum (Kelly et al., 2009), in which mGluR1 has been associated with CP-AMPAR removal. Interestingly, mGluR1 activation did not supersede a role for NMDARs in ppLFS LTD. This additional requirement for NMDARs might reflect the activity of an extrasynaptic NMDAR population, which has been linked to PKC-dependent AMPAR trafficking (Sun and June Liu, 2007). However, whether mGluR1 and NMDARs play a redundant role in CP-AMPAR dynamics remains to be established. In contrast to ppLFS, our results indicate that PKC is not required for LFS LTD, which depends instead on the phosphatase calcineurin. Thus, a balance of kinase and phosphatase activity, which can arise from the recruitment of perisynaptic versus synaptic glutamate receptor activity, may determine whether CP-AMPARs are targeted for removal.
A component of fear attenuation by extinction involves reversal of fear-conditioned plasticity (Kim et al., 2007; Clem and Huganir, 2010; Lai et al., 2012), in which some CS pathways may undergo LTD after fear memory retrieval. In particular, the timing of extinction to coincide with memory reconsolidation, a technique referred to as reconsolidation update, more effectively attenuates fear and weakens amygdala AMPAR transmission. Although amygdala GluA1-containing CP-AMPARs mediate fear erasure by reconsolidation update (Clem and Huganir, 2010), a recent report concluded that this form of extinction requires hippocampal GluA2 internalization for inhibiting contextual fear because these effects could be blocked by hippocampal GluA2-3Y application (Rao-Ruiz et al., 2011). However, we would caution that effects of GluA2-3Y be carefully interpreted, because this reagent may not discriminate between underlying plasticity mechanisms. Indeed, we found that GluA2-3Y blocked both mGluR1-dependent and -independent forms of LTD, despite their differential engagement of GluA2-lacking AMPAR trafficking. A possible explanation for these effects may be the requirement for GluA2 trafficking in both forms of LTD, wherein GluA2-3Y disrupts the forward trafficking of GluA2 and its subsequent displacement of synaptic CP-AMPARs. Consistent with this interpretation, inhibition of PICK1–GluA2 binding leads to EPSC rundown, implying that relief of PICK1-mediated extrasynaptic retention of GluA2 (Lin and Huganir, 2007) may facilitate its displacement of CP-AMPARs. Thus, detailed examination of compartmental trafficking may be required to delineate the precise role of GluA2 in mGluR1-dependent and -independent LTD.
Although both forms of LTD that we observed have the potential to reverse fear-associated synaptic potentiation, engaging CP-AMPAR LTD may enable more selective targeting of recent or reactivated fear associations due to the transient increase in CP-AMPAR content at LA thalamic inputs 24 h after fear learning (Clem and Huganir, 2010) or after recent reactivation of fear memories (our unpublished results). The increased level of CP-AMPARs during this transient window may provide a substrate for selective fear attenuation by mGluR1-dependent LTD. The role of recent experience in forming this window is mediated by norepinephrine, the release of which governs multiple aspects of fear-related behavior. In particular, acquisition of auditory fear conditioning requires amygdalar β-ARs at the time of CS–US pairing (Bush et al., 2010). Although our synaptic physiology confirms previous studies suggesting that consolidation of amygdala synaptic strengthening does not require β-ARs (Debiec and Ledoux, 2004; Bush et al., 2010), we demonstrate that posttraining β-AR activity regulates amygdala synaptic plasticity and thus may affect the subsequent updating of emotional responses. Curiously, β-AR activation by the experience of innate fear has been linked to the delayed recomposition of synaptic AMPARs in cerebellar stellate cells but instead mediates a switch from GluA2-lacking to GluA2-containing subtypes in these inhibitory interneurons (Liu et al., 2010). Therefore, fear experience may be sufficient to trigger cell-type-specific adaptations throughout the brain via the actions of norepinephrine.
In conclusion, our results indicate that the mechanism by which thalamo-amygdala LTD is expressed depends on the configuration of synaptic stimuli. An induction protocol that incorporates paired pulses, ostensibly similar to the high-frequency firing of thalamic neurons, selectively displaces CP-AMPARs from synapses. This distinct form of LTD, which uniquely relies on mGluR1 and PKC signaling, coexists at LA synapses with a more conventional LTD pathway defined by NMDAR–calcineurin signaling. During fear memory storage, β-ARs mediate a selective enhancement of synaptic CP-AMPARs and mGluR1-dependent LTD. These data suggest how extinction training might drive excitatory synaptic weakening and corresponding fear erasure (Clem and Huganir, 2010) and designate molecular strategies for facilitating these effects based on CP-AMPAR trafficking.
Footnotes
This work was supported by grants from the National Institutes of Health and the Howard Hughes Medical Institute (R.L.H.). R.L.C. was supported by National Research Service Award MH087037 from the National Institute of Mental Health. We thank Yilin Yu and Monica Coulter for expert technical assistance.
Under a licensing agreement between Millipore Corporation and Johns Hopkins University, R.L.H. is entitled to a share of royalties received by the university on sales of products described in this article. R.L.H. is a paid consultant to Millipore Corporation. The terms of this arrangement are being managed by Johns Hopkins University in accordance with its conflict-of-interest policies.
References
- Asrar S, Zhou Z, Ren W, Jia Z. Ca(2+) permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II. PLoS One. 2009;4:e4339. doi: 10.1371/journal.pone.0004339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. Eur J Neurosci. 2005;21:1280–1288. doi: 10.1111/j.1460-9568.2005.03979.x. [DOI] [PubMed] [Google Scholar]
- Billa SK, Liu J, Bjorklund NL, Sinha N, Fu Y, Shinnick-Gallagher P, Morón JA. Increased insertion of GluR2-lacking AMPA receptors at hippocampal synapses upon repeated morphine administration. Mol Pharmacol. 2010;77:874–883. doi: 10.1124/mol.109.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissière S, Humeau Y, Lüthi A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci. 2003;6:587–592. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- Bush DE, Caparosa EM, Gekker A, Ledoux J. Beta-adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front Behav Neurosci. 2010;4:154. doi: 10.3389/fnbeh.2010.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Bhattacharyya S, Ma C, Morishita W, Fang S, Rizo J, Malenka RC. Calcium binding to PICK1 is essential for the intracellular retention of AMPA receptors underlying long-term depression. J Neurosci. 2010;30:16437–16452. doi: 10.1523/JNEUROSCI.4478-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Anggono V, Huganir RL. PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J Neurosci. 2010;30:6360–6366. doi: 10.1523/JNEUROSCI.6276-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiec J, Ledoux JE. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience. 2004;129:267–272. doi: 10.1016/j.neuroscience.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Gao C, Wolf ME. Dopamine alters AMPA receptor synaptic expression and subunit composition in dopamine neurons of the ventral tegmental area cultured with prefrontal cortex neurons. J Neurosci. 2007;27:14275–14285. doi: 10.1523/JNEUROSCI.2925-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, Huganir RL. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron. 2005;45:903–915. doi: 10.1016/j.neuron.2005.02.026. [DOI] [PubMed] [Google Scholar]
- He J, Hu B. Differential distribution of burst and single-spike responses in auditory thalamus. J Neurophysiol. 2002;88:2152–2156. doi: 10.1152/jn.2002.88.4.2152. [DOI] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee HK. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1–S845 phosphorylation. Proc Natl Acad Sci U S A. 2009;106:20033–20038. doi: 10.1073/pnas.0910338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Huang S, Treviño M, He K, Ardiles A, Pasquale Rd, Guo Y, Palacios A, Huganir R, Kirkwood A. Pull-push neuromodulation of LTP and LTD enables bidirectional experience-induced synaptic scaling in visual cortex. Neuron. 2012;73:497–510. doi: 10.1016/j.neuron.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Martin KC, Kandel ER. Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci. 2000;20:6317–6325. doi: 10.1523/JNEUROSCI.20-17-06317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Reisel D, Johnson AW, Borchardt T, Jensen V, Gebhardt C, Bosch V, Gass P, Bannerman DM, Good MA, Hvalby Ø, Sprengel R, Lüthi A. A pathway-specific function for different AMPA receptor subunits in amygdala long-term potentiation and fear conditioning. J Neurosci. 2007;27:10947–10956. doi: 10.1523/JNEUROSCI.2603-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/S0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- Kelly L, Farrant M, Cull-Candy SG. Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nat Neurosci. 2009;12:593–601. doi: 10.1038/nn.2309. [DOI] [PubMed] [Google Scholar]
- Kemp N, McQueen J, Faulkes S, Bashir ZI. Different forms of LTD in the CA1 region of the hippocampus: role of age and stimulus protocol. Eur J Neurosci. 2000;12:360–366. doi: 10.1046/j.1460-9568.2000.00903.x. [DOI] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci U S A. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, Kim H, Lee C, Sun W, Kim K, Shin KS, Choi S. Amygdala depotentiation and fear extinction. Proc Natl Acad Sci U S A. 2007;104:20955–20960. doi: 10.1073/pnas.0710548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JT, Choi JS. Cornering the fear engram: long-term synaptic changes in the lateral nucleus of the amygdala after fear conditioning. J Neurosci. 2009;29:9700–9703. doi: 10.1523/JNEUROSCI.5928-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CS, Franke TF, Gan WB. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- Li YQ, Xue YX, He YY, Li FQ, Xue LF, Xu CM, Sacktor TC, Shaham Y, Lu L. Inhibition of PKMzeta in nucleus accumbens core abolishes long-term drug reward memory. J Neurosci. 2011;31:5436–5446. doi: 10.1523/JNEUROSCI.5884-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Formisano L, Savtchouk I, Takayasu Y, Szabó G, Zukin RS, Liu SJ. A single fear-inducing stimulus induces a transcription-dependent switch in synaptic AMPAR phenotype. Nat Neurosci. 2010;13:223–231. doi: 10.1038/nn.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty NK, Sah P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature. 1998;394:683–687. doi: 10.1038/29312. [DOI] [PubMed] [Google Scholar]
- Makino Y, Johnson RC, Yu Y, Takamiya K, Huganir RL. Enhanced synaptic plasticity in mice with phosphomimetic mutation of the GluA1 AMPA receptor. Proc Natl Acad Sci U S A. 2011;108:8450–8455. doi: 10.1073/pnas.1105261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mao SC, Hsiao YH, Gean PW. Extinction training in conjunction with a partial agonist of the glycine site on the NMDA receptor erases memory trace. J Neurosci. 2006;26:8892–8899. doi: 10.1523/JNEUROSCI.0365-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta JA, Ashby MC, Sanz-Clemente A, Roche KW, Isaac JT. mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron. 2011;70:339–351. doi: 10.1016/j.neuron.2011.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JE, Loweth JA, Ford KA, Marinelli M, Wolf ME, Tseng KY. Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J Neurosci. 2011;31:14536–14541. doi: 10.1523/JNEUROSCI.3625-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Murray EA. The amygdala, reward and emotion. Trends Cogn Sci. 2007;11:489–497. doi: 10.1016/j.tics.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Osten P, Khatri L, Perez JL, Köhr G, Giese G, Daly C, Schulz TW, Wensky A, Lee LM, Ziff EB. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27:313–325. doi: 10.1016/S0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Rao-Ruiz P, Rotaru DC, van der Loo RJ, Mansvelder HD, Stiedl O, Smit AB, Spijker S. Retrieval-specific endocytosis of GluA2-AMPARs underlies adaptive reconsolidation of contextual fear. Nat Neurosci. 2011;14:1302–1308. doi: 10.1038/nn.2907. [DOI] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/S0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15:127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Rozov A, Sprengel R, Seeburg PH. GluA2-lacking AMPA receptors in hippocampal CA1 cell synapses: evidence from gene-targeted mice. Front Mol Neurosci. 2012;5:22. doi: 10.3389/fnmol.2012.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23:9220–9228. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MK, Colbran RJ, Soderling TR. Specificities of autoinhibitory domain peptides for four protein kinases. Implications for intact cell studies of protein kinase function. J Biol Chem. 1990;265:1837–1840. [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Sun L, June Liu S. Activation of extrasynaptic NMDA receptors induces a PKC-dependent switch in AMPA receptor subtypes in mouse cerebellar stellate cells. J Physiol. 2007;583:537–553. doi: 10.1113/jphysiol.2007.136788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, Bolshakov VY. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc Natl Acad Sci U S A. 2007;104:14146–14150. doi: 10.1073/pnas.0704621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/S0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]