

Abstract
Susceptibility and progression of brain injury in the newborn is closely associated with an exacerbated innate immune response, but the underlying mechanisms are often unclear. Toll-like receptors (TLRs) are important innate immune sensors that may influence the vulnerability of the developing brain. In the current study, we provide novel data to show that activation of the viral innate immune receptor TLR-3 sensitizes the neonatal brain to subsequent hypoxic–ischemic (HI) damage. Poly inosinic:poly cytidylic acid (Poly I:C), a synthetic ligand for TLR-3, was administered to neonatal mice 14 h before cerebral HI. Activation of TLR-3 before HI increased infarct volume from 3.0 ± 0.5 to 15.4 ± 2.1 mm3 and augmented loss of myelin basic protein from 13.4 ± 6.0 to 70.6 ± 5.3%. The sensitizing effect of Poly I:C was specific for the TLR-3 pathway because mice deficient in the TLR-3 adaptor protein Toll/IL-1R domain-containing adaptor molecule-1 (TRIF) did not develop larger brain damage. The increased vulnerability was associated with a TRIF-dependent heightened inflammatory response, including proinflammatory cytokines, chemokines, and the apoptosis-associated mediator Fas, whereas there was a decrease in reparative M2-like CD11b+ microglia and phosphorylation of Akt. Because TLR-3 is activated via double-stranded RNA during most viral infections, the present study provides evidence that viral infections during pregnancy or in the neonate could have great impact on subsequent HI brain injury.
Introduction
Inflammation is an important contributing factor to CNS injury in neonates (Dammann and Leviton, 1997; Volpe, 2008). Toll-like receptors (TLRs) are instrumental in innate immune responses by recognizing pathogen-associated molecular patterns. TLRs also play a role in non-infectious situations, including cerebral ischemia. TLR-3 recognizes double-stranded RNA (dsRNA) released during viral infections, triggering the production of type 1 interferon and inflammatory cytokines/chemokines via the Toll/IL-1R domain-containing adaptor molecule-1 (TRIF). Although adult mice that lack TLR-3 or TRIF are not protected from cerebral ischemia (Hua et al., 2009; Hyakkoku et al., 2010; Famakin et al., 2011), TLR-3 activation exacerbates chronic neurodegeneration (Field et al., 2010) and triggers nigrostriatal dopaminergic degeneration (Deleidi et al., 2010). However, contradictory reports suggest that stimulation of the TRIF pathway may be neuroprotective by reprogramming the cerebral response to stroke (Marsh et al., 2009).
In the developing brain, TLR-3 activation impairs neural progenitor cell proliferation (Lathia et al., 2008), and maternal immune activation with poly inosinic:poly cytidylic acid (Poly I:C), a synthetic TLR-3 agonist, alters fetal brain development in an IL-6-dependent manner (Smith et al., 2007). Furthermore, maternal exposure to Poly I:C results in long-lasting effects in the offspring, including deficiencies in memory, learning tasks, and attention (Ratnayake et al., 2012; Vuillermot et al., 2012). In neonatal animals, exposure to Poly I:C increases seizure susceptibility later during adulthood (Galic et al., 2009), suggesting that TLR-3 activation during brain development has detrimental effects that may affect vulnerability to later insults. In support of a possible sensitizing effect of TLR stimulation, previous studies have shown that lipopolysaccharide (LPS), a TLR-4 ligand, can enhance the response to hypoxia–ischemia (HI) in neonatal animals (Eklind et al., 2001; Lehnardt et al., 2003).
In the present study, we hypothesized that activation of TLR-3 increases the susceptibility of the neonatal brain to HI. We used postnatal day 8 (P8) mice, which are at a brain developmental stage at which myelination has only just started, equivalent to the near-term human infant (Craig et al., 2003). We demonstrate that activation of TLR-3 markedly increased the vulnerability of the neonatal brain to HI injury in a TRIF-dependent manner, which was associated with increased induction of proinflammatory mediators, a decrease in reparative M2-like microglia phenotype and reduced phosphorylation of Akt. Because TLR-3 is activated via dsRNA during most viral infections, the present study provides evidence that viral infections during pregnancy or in the neonate could have great impact on subsequent HI brain injury.
Materials and Methods
Animals.
C57BL/6J mice lacking the gene for TRIF (C57BL/6J–Ticam1Lps2/J, TRIF KO; The Jackson Laboratory) and wild-type C57BL/6J mice (WT; Charles River) were housed and bred in a 12 h light/dark cycle at Experimental Biomedicine (University of Gothenburg, Gothenburg, Sweden). Mixed litters were obtained from WT and TRIF KO mice; approximately five generations were bred before the start of experiments. Pups of either sex were studied. Mice were provided with a standard laboratory chow diet (Brüel and Kjær) and drinking water ad libitum. All animal experiments were approved by the Ethical Committee of Gothenburg (Protocol 374-09) and performed according to the Guidelines for the Care and Use of Laboratory Animals.
Genotyping.
The genotype of TRIF KO mice was determined by RT-PCR of genomic DNA obtained from mouse tails. The tail was digested with 400 μl of lysis buffer (50 mmol/L Tris-HCl, pH 8.0, 100 mmol/L EDTA, 100 mmol/L NaCl, and 1% SDS) containing 1 mg/ml proteinase K (Roche). After incubation at 60°C overnight, 200 μl of 5 mol/L potassium acetate was added to the lysate, which was then thoroughly mixed and centrifuged at 10,000 × g for 20 min. The supernatant was transferred into a clean tube, and 800 μl of 100% ethanol was added and mixed. After incubation at −20°C for 30 min, the DNA was pelleted by centrifugation at 10,000 × g for 25 min at 4°C. The pellet was washed once with 500 μl of 75% ethanol. After removing all of the liquid, the pellet was left to dry at room temperature. The pellet of DNA was dissolved with 100 μl of sterile water, and DNA concentration was determined.
Genotypes were detected through melt-curve-based genotyping. Primers used for the amplification step were as follows: forward, 5′-CCAATCCTTTCCATCAGCCT-3′; and reverse, 5′-CACTCTGGAGTCTAAGAAG-3′ (Tib Molbiol). For melt-curve analysis, the following probes were used: 5′-CACATGTGGGGCCACACAGGGG-FL; and 5′-LC640-CCAGTCATCTGATGACAAGACTGAG-PH (Tib Molbiol). The amplification protocol comprised an initial 5 min denaturation at 95°C, followed by 40 cycles of denaturation for 30 s at 95°C and annealing/extension for 30 s at 55°C, followed by 1 min at 72°C, and thereafter, a melt-curve analysis consisted of 1 min denaturation at 95°C, followed by 3 min at 45°C and an increase of temperature to 95°C with a rate of 0.11°C/s on a LightCycler 480 (Roche). WT mice were identified with a melting temperature of 65–66°C, TRIF KO mice were identified with a melting temperature of 59–60°C, and heterozygotes were identified by the presence of both melting temperatures.
Poly I:C pretreatment and HI.
To determine the sequence of maximum TLR-3 activation, IFN-β gene expression was investigated (for details, see below) at different time points and after different doses of Poly I:C administration. P8 mice were intraperitoneally given 5 or 10 mg/kg Poly I:C [Poly(I:C)–LMW; InvivoGen], a specific synthetic TLR-3 ligand, or LPS free saline (S8776; Sigma-Aldrich) and killed at 6, 14, and 24 h after injection. IFN-β gene expression was greatest at 14 h after a Poly I:C dose of 10 mg/kg, which was used for additional studies (data not shown).
To investigate the effect of TLR-3 activation on neonatal HI, Poly I:C (10 mg/kg) or LPS free saline (S8776; Sigma-Aldrich) was administered intraperitoneally to P8 WT and TRIF KO mice of either sex (Fig. 1) 14 h before the induction of HI. At P9, mice were anesthetized with isoflurane (3.0% for induction and 1.0–1.5% for maintenance) in a mixture of nitrous oxide and oxygen (1:1). The left common carotid artery was ligated with prolene sutures (the whole procedure was <5 min). Mice were returned to the cage, allowed to recover for 1 h, and then placed in an incubator circulated with a humidified gas mixture (10.00 ± 0.01% oxygen in nitrogen) at 36°C for 50 min. After hypoxia, the pups were returned to their dam until they were killed. In some experiments, mice were subjected to sham operation 14 h after saline or Poly I:C injection. In these cases, the animals were anesthetized as above and the left common carotid artery was exposed, but not ligated and the mice were not subjected to hypoxia.
Figure 1.
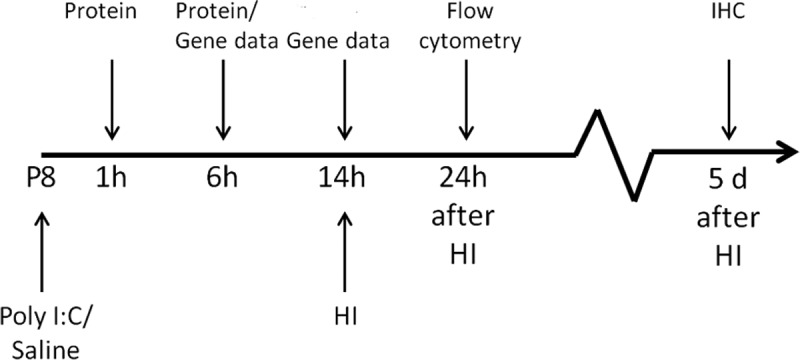
Experimental design. Poly I:C or saline was injected intraperitoneally to P8 mice. Animals were killed at 1 and 6 h for protein analysis and at 6 and 14 h for gene expression analysis. In separate animals, HI was induced at 14 h after Poly I:C/saline injection. Animals were killed 24 h after HI for flow cytometry analysis and at 5 d after HI for immunohistochemistry (IHC).
Immunohistochemistry.
Animals were anesthetized and intracardially perfused with saline and 5% buffered formaldehyde (Histofix; Histolab) 5 d after Poly I:C/HI. Brains were rapidly removed and immersion fixed in 5% formaldehyde for 24 h. After dehydration with graded series of ethanol and X-tra solv (Medite), brains were paraffin embedded and cut into 10 μm frontal sections, from ∼1 to −2.0 mm of bregma, for immunohistochemical staining. Antigen recovery was performed by boiling the sections in 10 mm sodium citrate buffer, pH 6.0, for 10 min. Nonspecific binding was blocked for 30 min in blocking solution (1% horse serum, 3% bovine serum albumin, 0.1% NaN3 in PBS). Sections were incubated in primary antibody against microtubule-associated protein-2 (MAP-2; clone HM-2, 1:1000; Sigma-Aldrich), mouse monoclonal antibody MBP (1:10,000, SMI-94R; Covance), or ionized calcium binding-adapter molecule 1 (Iba-1; 1:2000; Wako Chemicals) at 4°C overnight, followed by corresponding biotinylated secondary antibody (1:250; Vector Laboratories) for 60 min at room temperature. Visualization was performed using Vectastain ABC Elite (Vector Laboratories) with 0.5 mg/ml 3,3-diaminobenzidine enhanced with 15 mg/ml ammonium nickel sulfate, 2 mg/ml β-d-glucose, 0.4 mg/ml ammonium chloride, and 0.01 mg/ml β-glucose oxidase (all from Sigma-Aldrich).
Brain injury evaluation.
Brain gray and white matter injury and Iba-1-positive microglia response were analyzed under a Nikon Optiphot-2 microscope equipped with an AVT dolphin F145B camera (Allied Vision Technologies) as shown previously (Svedin et al., 2007). An investigator blinded to the identity of the experimental groups performed all analyses.
For quantitative evaluation of gray matter injury, every 50th section throughout the brains was stained for MAP-2. Images were captured and processed using Micro Image version 4.0 (Olympus). Infarct area was assessed as the MAP-2-negative area in the ipsilateral (injured) hemisphere. Tissue loss was calculated by subtracting the MAP-2-positive volume of the ipsilateral hemisphere from the contralateral hemisphere, and brain injury was expressed as percentage tissue loss of the non-injured hemisphere (Svedin et al., 2007). Total infarct and tissue loss volumes were calculated according to the Cavalieri principle using the following formula: V = ΣA × P × T, where V is total volume, ΣA is the sum of areas measured, P is the inverse of the sections sampling fraction, and T is the section thickness (Roda et al., 1995; Svedin et al., 2007). Atrophy was calculated by subtracting the MAP-2 positive volume and infarct volume of the ipsilateral hemisphere from the total volume of the contralateral hemisphere.
In each sham-operated animal, the MAP-2-positive area of the left and right hemisphere was measured in two stained sections at the hippocampal level.
White matter injury was analyzed by measuring the subcortical white matter area of positive staining for MBP (Micro Image version 4.0; Olympus) in both hemispheres at striatum and hippocampal levels. One section for each level was analyzed per animal. The MBP area in the ipsilateral hemisphere was compared with the contralateral hemisphere to calculate the proportion (percentage) of white matter damage.
In sham-operated animals, images of MBP-stained brain sections at two hippocampal levels were analyzed from each animal using NIH ImageJ (version 1.47f7). A region of interest (ROI; 4 mm2), including the subcortical white matter, was defined and applied to all sections. All images were converted to red–green–blue stack type, and a threshold for MBP staining was determined by the software. Within the ROI, the MBP-stained area above the threshold was measured.
Quantitative reverse transcription-PCR.
WT and TRIF KO mice were injected with Poly I:C (10 mg/kg) or LPS free saline at P8 and were killed at 6 and 14 h after injection (Fig. 1). Animals were anesthetized and intracardially perfused with saline, and brains were rapidly dissected out, snap frozen, and stored at −80°C until analysis. Brain tissue was homogenized with Qiasol lysis reagent homogenizer (Qiagen), and total RNA was extracted using RNeasy Lipid Tissue Mini Kit (Qiagen) according to the instructions of the manufacturer. RNA was measured in a spectrophotometer at 260 nm absorbance. QuantiTect Reverse Transcription Kit (Qiagen) was used to synthesize first-strand cDNA according to the instructions of the manufacturer. Each PCR (20 μl) contained 2 μl of cDNA diluted 1:4, 10 μl of Quanti Fast SYBR Green PCR Master Mix (Qiagen), 2 μl of PCR primer, and 6 μl of H2O. The following primers were used: IFN-β (QT00249662), IL-6 (QT00098875), IL-1β (QT01048355), monocyte chemoattractant protein-1 (MCP-1) (QT00167832), interferon-induced protein-10 (IP-10) (QT00093436), TNF-α (QT00104006), IL-10 (QT00106169), Fas (QT00095333), GAPDH (QT01658692), and tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide (YWHAZ; QT00105350) (all from Qiagen).
The amplification protocol comprised an initial 5 min denaturation at 95°C, followed by 40 cycles of denaturation for 10 s at 95°C and annealing/extension for 30 s at 60°C on a LightCycler 480 (Roche). Melting-curve analysis was performed to ensure that only one PCR product was obtained. For quantification and for estimating amplification efficiency, a standard curve was generated using increasing concentrations of cDNA. The amplification transcripts were quantified with the relative standard curve and normalized against the geometric mean of the reference genes GAPDH and YWHAZ.
Phospho-protein analyses.
WT mice were injected with Poly I:C (10 mg/kg) or LPS free saline at P8 and were killed at 1 and 6 h after injection (Fig. 1). Animals were anesthetized and intracardially perfused with saline, and brains were rapidly dissected out, snap frozen, and stored at −80°C until analysis. Samples were prepared for phospho-protein analysis with Bio-Plex Cell Lysis Kit (catalog #171-304011; Bio-Rad) according to the protocol from the manufacturer. Protein concentration was measured with BCA protein assay (Thermo Fisher Scientific) and adjusted to 450 μg/ml. For detection of phosphorylation of proteins, Bio-Plex Phospho 5-Plex Panel (X7000001RD; Bio-Rad) was used according to the instructions of the manufacturer and analyzed on a Bio-Plex 200 system (Bio-Rad) with the Bio-Plex Manager software 6.0 (Bio-Rad). The changes in phosphorylation were calculated using a ratio between mean fluorescence of phosphorylated protein and total protein.
Post-injury flow cytometry analysis of microglia.
Animals were deeply anesthetized and intracardially perfused with saline at 24 h after saline/HI and Poly I:C/HI or saline/sham and Poly I:C/sham (Fig. 1). Brains were dissected out and placed in Hibernate A (A12475-01; Invitrogen). Cells from the injured and uninjured hemispheres were homogenized in CellWASH (catalog #349524; BD Biosciences) using a glass homogenizer. After filtration through a 100 μm cell strainer, samples were washed three times (340 × g, 5 min, 4°C) and resuspended in CellWASH. Cells were counted with a TC10 automated cell counter (Bio-Rad), and 1 × 106 cells from each sample were used for immunostaining.
After incubation with anti-CD16/32 Fc-receptor block (1 μg/106 cells; catalog #553142; BD Biosciences Pharmingen), samples were stained with antibodies for microglia marker CD11b (anti-mouse CD11b Alexa Fluor 488; eBioscience), M1 marker CD86 (adenomatous polyposis coli anti-mouse CD86; Biolegend), and M2 marker CD206 (phycoerythrin anti-mouse CD206; Biolegend), all at a concentration of 0.5 μg/106 cells, for 30 min. Cells were washed and resuspended in CellWASH. 7-Aminoactinomycin D (7AAD; catalog #51-68981E; BD Biosciences Pharmingen) was added to each sample to distinguish between alive and dead cells. Samples were kept on ice through the whole procedure. Flow cytometric data were collected on an FACSCantoAflow cytometer (BD Biosciences) and analyzed using FACSDiva Software version 6.1.3 (BD Biosciences). For analysis, cells of interest were identified (P1 gate) in forward and side scatter. From the P1 gate, 7AAD was used to define dead cells, and the CD11b+ cells were then gated on live cells. For gating strategy, see Figure 7A. Approximately 2 × 105 live cells were analyzed from each brain hemisphere. The same gates were used on all samples run simultaneously.
Figure 7.

Poly I:C reduces reparative M2-like microglia after hypoxic–ischemic injury. A, Gating strategy for analysis of CD11b+ microglia cells. A P1 gate was defined in forward and side scatter. 7AAD was used to distinguish between live and dead cells, and microglia marker CD11b was then gated from the live cells. B, The overall population of CD11b+ microglia increased in the ipsilateral hemisphere compared with the contralateral hemisphere in Poly I:C/HI animals but not in saline/HI mice. Within the CD11b+ population, two different populations were distinguished: a CD11b+high-expressing and a CD11b+low-expressing population, which were both affected by HI. The CD11b+high population increased in the injured hemisphere in both saline/HI-and Poly I:C/HI-treated animals. In contrast, the CD11b+low population was decreased in the injured hemisphere in both saline/HI- and Poly I:C/HI-treated animals. The CD11b+high population in the injured hemisphere was lower in Poly I:C/HI compared with saline/HI animals, whereas the CD11b+low population was higher in the injured hemisphere after Poly I:C/HI compared with saline/HI. C, There were no significant changes in M1-like (proinflammatory) CD11b+CD86+ cells in the ipsilateral compared with the contralateral hemisphere after either Poly I:C/HI or saline/HI. In contrast, there was a decrease in M2-like (reparative) CD11b+CD206+cells in the injured hemisphere in both saline/HI- and Poly I:C/HI-treated animals. In the contralateral hemisphere, the percentage of CD11b+CD206+cells was lower in Poly I:C/HI-treated animals compared with saline/HI-treated animals. Data are presented as mean ± SEM; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; n = 8–9 per group.
Statistics.
Brain injuries were analyzed with two-way ANOVA with genotype and treatment or treatment and time point as factors, followed by Bonferroni's post hoc test with a 95% confidence interval. Brain injury in WT sham-operated animals with or without Poly I:C injection was compared by Student's t test. Gene expression data were analyzed with one-way ANOVA, followed by Bonferroni's post hoc test with a 95% confidence interval. WT and TRIF KO data were analyzed separately. Phospho-proteins were analyzed with Student's t test between Poly I:C and saline for each time point separately. Flow cytometry data were analyzed with a one-way ANOVA, followed by Newman–Keuls multiple comparison test with a 95% confidence interval. Data are presented as mean ± SEM, and significance was set at p < 0.05. All statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software).
Results
Poly I:C increases the vulnerability of the neonatal brain to HI
To investigate the Poly I:C effect alone on neonatal gray and white matter development, saline or Poly I:C was administered 14 h before sham surgery. No significant difference in the area of MAP-2 or MBP staining was demonstrated in Poly I:C-injected animals compared with saline-injected animals 5 d after sham surgery (data not shown). To examine the role of TLR-3 activation on neonatal HI brain injury, Poly I:C or saline was administered 14 h before HI (Poly I:C/HI and saline/HI groups, respectively) in neonatal mice, and neuropathological analyses of gray and white matter injury were performed 5 d after HI. In WT mice, Poly I:C pretreatment increased HI-induced infarct volume (Poly I:C/HI, 15.40 ± 2.06 mm3, n = 20 vs saline/HI, 3.01 ± 0.49 mm3, n = 21, p < 0.001; Fig. 2A,B), tissue loss (Poly IC/HI, 48.9 ± 2.62%, n = 20 vs saline/HI: 25.62 ± 2.07%, n = 21, p < 0.001; Fig. 2C,D), and atrophy (Poly I:C/HI, 28.83 ± 1.83 mm3, n = 20 vs saline/HI, 20.57 ± 1.44 mm3, n = 21, p < 0.01). Immunohistochemistry staining for neuronal marker MAP-2 in brain sections revealed that injury was confined to the ipsilateral hemisphere and mainly distributed in the hippocampus, cortex, striatum, and thalamus (Fig. 2E).
Figure 2.

TLR-3 activation sensitize the brain to HI. Neuropathological analysis at 5 d after HI showed a significant increase in both infarct volume (A) and tissue loss (C) in Poly I:C-treated WT mice compared with saline/HI mice. In WT mice, the infarct area at 6 levels of the brain (B) and tissue loss (D) were significantly increased throughout the brain in Poly I:C/HI compared with saline/HI animals. In TRIF KO mice, no differences were found between poly I:C/HI and saline/HI animals. E, Representative images of MAP-2-stained sections in WT and TRIF KO mice 5 d after saline/HI and Poly I:C/HI. Anteroposterior levels, from ∼1 to −2 mm of bregma, are indicated by numbers in B and D, where level 1 indicates the most posterior level. Data are presented as mean ± SEM. **p ≤ 0.01, ***p ≤ 0.001; n = 20–21 per group. Scale bar: E, 1 mm.
Compared with the saline/HI group, Poly I:C/HI mice showed more severe white matter injury as indicated by significantly increased loss of immunostaining for MBP in the subcortical white matter at both the hippocampal level (Poly I:C/HI, 70.60 ± 5.33% loss, n = 20 vs saline/HI, 13.38 ± 6.00% loss, n = 21, p < 0.001; Fig. 3A,C) and striatum level (Poly I:C/HI, 67.85 ± 3.67% loss, n = 20 vs saline/HI, 27.71 ± 2.62% loss, n = 21, p < 0.001; Fig. 3B) at 5 d after HI.
Figure 3.

Effects of TLR-3 activation on white matter injury. In WT mice, white matter injury was increased in Poly I:C/HI WT mice compared with saline/HI at both the hippocampal (A) and striatum (B) levels. No differences were seen between treatment groups in the TRIF KO mice. C, Representative images of MBP-stained sections in WT and TRIF KO mice after Poly I:C/HI or saline/HI at the hippocampal level. Data are shown as mean ±SEM percentage loss of MBP stain. ***p ≤ 0.001; n = 20–21 per group. Insets represent the area that is depicted at higher magnification. Scale bars: top, 20 μm; bottom, 5 μm.
There was no difference in either gray or white matter injury between WT and TRIF KO mice after saline/HI (p > 0.05; Figs. 2, 3). The sensitizing effect of Poly I:C to HI was blocked in TRIF KO mice, and there was no significant difference in any of the brain injury parameters between saline- and Poly I:C-treated TRIF KO animals (p > 0.05; Figs. 2, 3).
The inflammatory response as indicated by Iba-1 immunostaining showed a typical pattern of amoeboid-like cells in injured brain regions (Fig. 4). The degree and distribution of Iba-1 expression correlated with the degree of brain damage in all experimental groups. The morphology of the Iba-1-positive cells in injured areas did not differ between Poly I:C/HI- or saline/HI-treated animals or between genotypes (Fig. 4).
Figure 4.

Inflammatory response after HI. The inflammatory response, as indicated by Iba-1 immunostaining, showed a pattern of amoeboid-like cells in injured brain regions. The degree and distribution of Iba-1 expression appeared to correlate to the degree of brain damage in all experimental groups. The morphology of the Iba-1-positive cells in injured areas did not differ between Poly I:C/HI-treated (B, D) or saline/HI-treated (A, C) animals or between genotypes. Insets represent the area that is depicted at higher magnification. Scale bars: left, 1 mm; right, 5 μm.
Poly I:C induces inflammatory genes in the brain in a TRIF-dependent manner
To elucidate Poly I:C-mediated factors that may contribute to the increased vulnerability to HI, we analyzed genes associated with apoptosis (Fas), proinflammatory and anti-inflammatory immune responses (IL-6, IL-1β, TNF-α, IFN-β, and IL-10), and chemotaxis (IP-10 and MCP-1) in the brain after Poly I:C administration. The expression of IL-10 was below detection level in both WT and TRIF KO mice at both time points (data not shown).
In WT mice, Poly I:C upregulated levels of IL-6 mRNA at 6 h (n = 10, p < 0.001; Fig. 5A), IL-1β mRNA expression at 14 h (n = 10, p < 0.001; Fig. 5B), TNF-α at 6 h (n = 10, p < 0.05) and 14 h (n = 10, p < 0.001; Fig. 5C), and IFN- β at 6 h (n = 4, p < 0.05) and 14 h (n = 5, p < 0.001; Fig. 5D). Poly I:C also modulated the chemokine response shown by upregulation of IP-10 at 6 h (n = 10, p < 0.001; Fig. 5E) and MCP-1 at 6 h (n = 10, p < 0.001) and 14 h (n = 10, p < 0.001; Fig. 5F). Fas mRNA expression was upregulated at 6 h (n = 10, p < 0.001; Fig. 5G). The Poly I:C-induced inflammation for all the analyzed genes was blocked in TRIF KO mice (Fig. 5A–G).
Figure 5.

Gene expression analysis after Poly I:C administration. In WT mice, Poly I:C (10 mg/kg) increased mRNA expression of IL-6 at 6 h (A), IL-1β at 14 h (B), TNF-α at both 6 and 14 h (C), IFN-β at both 6 and 14 h (D), IP-10 at 6 h (E), MCP-1 at both 6 and 14 h (F), and Fas at 6 h (G) compared with controls (saline). In TRIF KO animals, there were no differences in expression of any of the genes between Poly I:C- and saline-injected mice. Data are presented as mean ± SEM; *p ≤ 0.05, ***p ≤ 0.001, n = 7–10 per group.
Poly I:C induces phosphorylation of nuclear factor κB inhibitor and reduces pAKT phosphorylation
Poly I:C-induced cytokine production may be regulated through mitogen-activated protein kinases (MAPKs) (Kim et al., 2008; Liu et al., 2008). To further investigate the Poly I:C sensitizing mechanism downstream of TLR-3/TRIF, we analyzed the phosphorylation of ERK, JNK, and p38 and also the pro-survival serine/threonine-specific protein kinase Akt and the general inflammatory conductor nuclear factor κB inhibitor (IκB). Poly I:C administration downregulated the phosphorylation of Akt at 1 h (n = 8, p < 0.05; Fig. 6A), whereas phosphorylation of IκB increased at 1 h (n = 8, p < 0.01; Fig. 6B). There were no differences in phosphorylation of ERK, JNK, or p38 between Poly I:C- and saline-treated animals at any of the analyzed time points (Fig. 6D,E).
Figure 6.

Poly I:C effects on MAPK and IκB phosphorylation. In WT mice, Poly I:C (10 mg/kg) reduced the phosphorylation of Akt at 1 h (A), whereas phosphorylation of IκB was increased at 1 h (B). There were no changes in the phosphorylation of ERK, JNK, or p38 (C–E). Data are presented as mean ± SEM; *p ≤ 0.05, **p ≤ 0.01; n = 8 per group.
Poly I:C reduces reparative “M2-like” microglia after HI
To evaluate the inflammatory response after Poly I:C/HI, the microglia response was analyzed by flow cytometry. We investigated the general population of microglia by examining CD11b+ cells. These cells were further classified as either proinflammatory (M1) or anti-inflammatory/reparative (M2) phenotypes (Colton, 2009), using markers CD86 and CD206, respectively. The overall population of CD11b+ microglia increased in the ipsilateral hemisphere compared with the contralateral hemisphere in Poly I:C/HI animals (Poly I:C/HI ipsilateral hemisphere, 1.06 ± 0.09% vs Poly I:C/HI contralateral hemisphere, 0.60 ± 0.13%, n = 9 per group, p < 0.05; Fig. 7B) but not in saline/HI mice (p > 0.05; Fig. 7B). Within the CD11b+ population, two different populations were distinguished: (1) a CD11b+high-expressing and (2) a CD11b+low-expressing population, which were both affected by HI but not by Poly I:C alone (saline CD11b+high, 3.70 ± 0.71% vs Poly I:C CD11b+high, 2.67 ± 0.38%; saline CD11b+low, 94.24 ± 1.03% vs Poly I:C CD11b+low, 96.14 ± 0.52%, n = 7 per group). The CD11b+high population increased in the injured hemisphere in both saline/HI-treated (ipsilateral hemisphere, 42.00 ± 3.88% vs contralateral hemisphere, 5.07 ± 1.39%, n = 8 per group, p < 0.001) and Poly I:C/HI-treated (ipsilateral hemisphere, 24.54 ± 4.86% vs contralateral hemisphere, 6.26 ± 1.64%, n = 9 per group, p < 0.001) animals (Fig. 7B). In contrast, the CD11b+low population was decreased in the injured hemisphere in both saline/HI-treated (ipsilateral hemisphere, 54.98 ± 3.88% vs contralateral hemisphere, 93.63 ± 1.79%, n = 8 per group, p < 0.001) and Poly I:C/HI-treated (ipsilateral hemisphere, 70.39 ± 5.58% vs contralateral hemisphere, 91.77 ± 1.81%, n = 9 per group, p < 0.001; Fig. 7B) animals. The CD11b+high population in the injured hemisphere was significantly lower in Poly I:C/HI compared with saline/HI animals (p < 0.001), whereas the CD11b+low population was significantly higher in the injured hemisphere after Poly I:C/HI compared with saline/HI (p < 0.01). Representative dot plots of high and low populations of CD11b+ cells are illustrated in Figure 7B.
Additional analysis of the CD11b+ microglia cells into M1-like (proinflammatory) and M2-like (reparative) phenotypes were performed by using CD86 as an M1 marker and CD206 as an M2 marker. There were no significant changes in M1-like CD11b+CD86+ cells in the ipsilateral compared with the contralateral hemisphere after either Poly I:C/HI or saline/HI (p > 0.05; Fig. 7C). In contrast, there was a decrease in M2-like CD11b+CD206+cells in the injured hemisphere in both saline/HI (ipsilateral hemisphere, 8.3 ± 1.45% vs contralateral hemisphere, 29.98 ± 4.66%, n = 8 per group, p < 0.001) and Poly I:C/HI (ipsilateral hemisphere, 3.49 ± 0.67% vs contralateral hemisphere, 14.57 ± 1.73%, n = 9 per group, p < 0.01; Fig. 7C) animals. In the contralateral hemisphere, the percentage of CD11b+CD206+cells was lower in Poly I:C/HI animals compared with saline/HI mice (p < 0.001). Similarly, the population of CD11b+CD206+cells was reduced in sham-operated animals pretreated with Poly I:C (11.14 ± 0.76%, n = 7) compared with saline pretreated pups (24.96 ± 3.99%, n = 7, p < 0.01).
Discussion
Infections by themselves or in combination with other insults are major risk factors for perinatal brain injury, but the precise etiology is often unclear. In the current study, we provide novel data to show that activation of the viral innate immune receptor TLR-3 sensitizes the neonatal brain to subsequent hypoxic–ischemic damage in a TRIF-dependent manner. The increased vulnerability to HI was associated with a TRIF-dependent heightened inflammatory response, including proinflammatory cytokine and chemokine expression and altered characteristics of CD11b+ microglia in the brain. Cell death/survival pathways were also affected with an increase in expression of the apoptosis-associated mediator Fas, whereas pro-survival pathways, such as phosphorylation of Akt, were reduced.
In adult animals, neither TLR-3 or TRIF deficiency provide protection against cerebral ischemia (Hua et al., 2009; Hyakkoku et al., 2010; Famakin et al., 2011). The present results, showing no difference in brain injury in WT or TRIF KO neonatal animals after saline/HI, coincide well with previous findings in adult ischemia models. In the adult brain, poly I:C exacerbates the inflammatory response and neurodegeneration (Field et al., 2010) and increases the susceptibility of midbrain dopaminergic neurons to subsequent neurotoxicity (Deleidi et al., 2010). These data imply that activation of TLR-3 increases the vulnerability of the brain to additional insults. However, previous stimulation of the TRIF pathway, via activation of TLR-4, confers protection against subsequent ischemic injury by reprogramming the response of the adult brain to stroke via a TRIF/interferon regulatory factor-3 (IRF-3)-induced IFN-β pathway (Marsh et al., 2009). In later studies, it was also shown that Poly I:C acts as a preconditioning factor before cerebral ischemia (Packard et al., 2012). Here, neonatal HI is produced at the time point of maximum induction of IFN-β by Poly I:C (14 h). However, despite the increased level of IFN-β expression in the brain at the time of HI, we were unable to detect any TLR3/TRIF-mediated neuroprotection. The discrepancy between our results and those in adult animals may be attributable to differences in the time interval between Poly I:C and the cerebral ischemic insult. We showed previously that activation of TLR-4, by LPS, 14 h before HI increases the vulnerability to neonatal brain injury (Wang et al., 2009) and that the time interval between immune stimulation and HI was critical for neuropathological outcome (Eklind et al., 2005). Conversely, the developmental stage at the time of TLR-3/TRIF activation may also affect outcome. The developing brain appears to be particularly sensitive to TLR-3 stimulation (Meyer et al., 2006; Ratnayake et al., 2012). Furthermore, we have evidence that TLR-3 expression in glia and neurons is altered in response to white matter injury in preterm human infants (Vontell et al., 2013), suggesting that TLR-3 may serve important functions during brain development. These studies are in agreement with the marked increased vulnerability to HI observed in the current study after previous Poly I:C administration. Together, these findings strongly suggest that activation of TLR-3/TRIF is detrimental to the developing brain, especially when followed by another insult to the brain.
Signaling through TLR-3 is mediated through the adaptor protein TRIF. Both the C- and N-terminal domains of TRIF can associate with several intracellular proteins, including receptor-interacting protein-1, TNF receptor-associated factor-6, and serine/threonine protein kinase-1, mediating activation of both nuclear factor-κB (NF-κB) and IRF-3 regulated genes as well as apoptotic pathways through FAS-associated death domain and caspase-8 (Sato et al., 2003; McWhirter et al., 2004; Meylan et al., 2004; Cusson-Hermance et al., 2005; Sasai et al., 2005). To evaluate inflammatory responses in the neonatal brain after Poly I:C, we found that IκB phosphorylation, an indicator of NF-κB transcription, was increased. Furthermore, gene expression of both type 1 interferons and other cytokines/chemokines were up regulated in the brain in a TRIF-dependent matter. These findings in WT mice correspond well with previous studies demonstrating an increased expression of IL-6, IL-1β, TNF-α, and MCP-1 in the adult mouse and guinea pig brain after Poly I:C administration (Cunningham et al., 2007; Konat et al., 2009; Field et al., 2010). Both MCP-1 and IL-1β exacerbate neonatal excitotoxic lesions (Galasso et al., 2000; Favrais et al., 2007), and deletion of the entire TNF gene cluster abolishes LPS-mediated increase in neonatal HI injury (Kendall et al., 2011), suggesting that these cytokines may play a role in inflammatory-induced neonatal brain damage. The direct influence of IL-6 on the development of brain injury is more controversial. Although studies have consistently demonstrated elevated IL-6 levels in asphyxiated infants (Sävman et al., 1998) and mice overexpressing IL-6 develop severe neurologic syndromes (Campbell et al., 1997), others find that mice deficient in IL-6 develop more severe brain injuries (Penkowa and Hidalgo, 2000). Interestingly, maternal immune activation with Poly I:C alters fetal brain development in an IL-6-dependent manner (Smith et al., 2007). However, in our experiments, the findings are correlative, and additional studies, using for example relevant knock-out animals or cytokine stimulation in TRIF KO mice, are needed to determine the specific roles of these inflammatory mediators in TLR-3-induced brain injury.
The inflammatory response was further investigated by flow cytometry analysis after Poly I:C/HI. CD11b+ microglia were markedly increased in the injured hemisphere after the combination of Poly I:C/HI, which was not seen after saline/HI. Additionally, we discovered that CD11b+ microglia were characterized by two distinct populations of cells: (1) CD11b+low-expressing and (2) CD11b+high-expressing cells. The increased population of CD11b+high -expressing microglia are not likely to represent proinflammatory CD86+ cells as we further demonstrated that M1-like CD11b+CD86+ cells were not significantly increased in the neonatal brain after saline/HI or Poly I:C/HI. This finding contrasts with reports in P11 mice having increased CD11b+CD86+ cells by 24 h after HI alone (Winerdal et al., 2012). Conversely, we found that a population with anti-inflammatory phenotype, M2-like CD11b+CD206+ cells, was significantly decreased in the injured hemisphere. These changes were apparent in both saline/HI and Poly I:C/HI brains. The Poly I:C-induced reduction in M2-like CD11b+CD206+ cells was also evident in the non-injured hemisphere, suggesting that Poly I:C by itself, without HI, has an effect on this cell population. In support, we found that Poly I:C-treated sham-operated animals also demonstrate a decrease in CD11b+CD206+ cells. CD206 is a C-type lectin carbohydrate binding protein expressed by alternatively activated macrophages and associated with recovery and restoration of function (Bhatia et al., 2011). Therefore, our data indicate that poly I:C has a general detrimental effect on “protective” microglia and suggest that, rather than affecting the proinflammatory response, deleterious Poly I:C-mediated effects may instead predominantly downregulate the reparative response. At present, it is unclear whether the CD11b+ cells that we studied represent mainly resident microglia or whether there may also be recruitment of peripheral immune cells into the brain.
In the ischemic brain, Akt signaling pathway is neuroprotective by phosphorylating its substrates and preventing inflammation and apoptosis (Mullonkal and Toledo-Pereyra, 2007). Neonatal HI decreases Akt phosphorylation (Brywe et al., 2005) and in turn increases axonal injury (Xiong et al., 2012). Akt phosphorylation plays an important role in the TRIF signaling pathway and promotes cell survival in response to innate immune stimulation (Park et al., 2006; Joung et al., 2011). In the current neonatal model, Poly I:C transiently decreased Akt phosphorylation and upregulated mRNA for Fas ligand. This later finding is in agreement with the reported Poly I:C-exacerbated neurodegeneration linked to Fas ligand upregulation (Field et al., 2010). Together, our data indicate that dephosphorylation of Akt and release of pro-apoptotic Fas may contribute to the vulnerability of the immature brain against additional injuries such as HI, although additional studies need to confirm this using, for example, specific inhibitors.
To conclude, we demonstrate that activation of the TLR-3/TRIF pathway before HI markedly increases both white and gray matter injury in a TRIF-dependent manner in neonatal mice. We observed a TRIF-dependent increase in expression of both type I interferons (IFN-β), proinflammatory cytokines (IL-6, IL-1β, and TNF-α) and chemotaxic mediators (IP-10 and MCP-1) after Poly I:C administration, which suggest that both pathways involving NF-κB and IRF-3 transcription factors are involved in the cerebral response to TLR-3/TRIF stimulation. The inflammatory response to Poly I:C/HI was characterized by a decrease in number of M2-like microglia, suggesting that anti-inflammatory/restorative mechanisms in the brain are diminished after Poly I:C. Poly I:C is a synthetic mimic of dsRNA viral products. Therefore, the Poly I:C-induced increase in vulnerability to HI observed in the present study may indicate that viral infections in the neonate, acquired prenatally or perinatally, could have great impact on hypoxic–ischemic brain injury in the newborn.
Footnotes
This research was supported by Swedish Research Council Grants VR2009-2630 and 2012-2992, Sahlgrenska University Hospital Government Grant to a Researcher in Public Health Service ALFGBG-142881, European Union Grant FP7 (Neurobid, HEALTHF2-2009-241778), Leducq Foundation Grant DSRR_P34404, the Åhlén Foundation, the Wilhelm and Martina Lundgren Foundation, the Olle Engkvist Foundation, and National Institutes of Health Grant R01HD070996. X.W. is supported by Swedish Research Council Grant VR K2009-54X-21119-01-4 and Swedish Governmental Agency for Innovation Systems Grant 2011-03458.
The authors declare no competing financial interests.
References
- Bhatia S, Fei M, Yarlagadda M, Qi Z, Akira S, Saijo S, Iwakura Y, van Rooijen N, Gibson GA, St Croix CM, Ray A, Ray P. Rapid host defense against Aspergillus fumigatus involves alveolar macrophages with a predominance of alternatively activated phenotype. PLoS One. 2011;6:e15943. doi: 10.1371/journal.pone.0015943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brywe KG, Mallard C, Gustavsson M, Hedtjärn M, Leverin AL, Wang X, Blomgren K, Isgaard J, Hagberg H. IGF-I neuroprotection in the immature brain after hypoxia–ischemia, involvement of Akt and GSK3beta? Eur J Neurosci. 2005;21:1489–1502. doi: 10.1111/j.1460-9568.2005.03982.x. [DOI] [PubMed] [Google Scholar]
- Campbell IL, Stalder AK, Chiang CS, Bellinger R, Heyser CJ, Steffensen S, Masliah E, Powell HC, Gold LH, Henriksen SJ, Siggins GR. Transgenic models to assess the pathogenic actions of cytokines in the central nervous system. Mol Psychiatry. 1997;2:125–129. doi: 10.1038/sj.mp.4000225. [DOI] [PubMed] [Google Scholar]
- Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig A, Ling Luo N, Beardsley DJ, Wingate-Pearse N, Walker DW, Hohimer AR, Back SA. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp Neurol. 2003;181:231–240. doi: 10.1016/S0014-4886(03)00032-3. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Teeling J, Felton L, Perry VH. The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C) Brain Behav Immun. 2007;21:490–502. doi: 10.1016/j.bbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA. Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-kB activation but does not contribute to interferon regulatory factor 3 activation. J Biol Chem. 2005;280:36560–36566. doi: 10.1074/jbc.M506831200. [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- Deleidi M, Hallett PJ, Koprich JB, Chung CY, Isacson O. The Toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. J Neurosci. 2010;30:16091–16101. doi: 10.1523/JNEUROSCI.2400-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklind S, Mallard C, Leverin AL, Gilland E, Blomgren K, Mattsby-Baltzer I, Hagberg H. Bacterial endotoxin sensitizes the immature brain to hypoxic–ischaemic injury. Eur J Neurosci. 2001;13:1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- Eklind S, Mallard C, Arvidsson P, Hagberg H. Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain. Pediatr Res. 2005;58:112–116. doi: 10.1203/01.PDR.0000163513.03619.8D. [DOI] [PubMed] [Google Scholar]
- Famakin BM, Mou Y, Ruetzler CA, Bembry J, Maric D, Hallenbeck JM. Disruption of downstream MyD88 or TRIF Toll-like receptor signaling does not protect against cerebral ischemia. Brain Res. 2011;1388:148–156. doi: 10.1016/j.brainres.2011.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favrais G, Schwendimann L, Gressens P, Lelièvre V. Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiol Dis. 2007;25:496–505. doi: 10.1016/j.nbd.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Field R, Campion S, Warren C, Murray C, Cunningham C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun. 2010;24:996–1007. doi: 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galasso JM, Liu Y, Szaflarski J, Warren JS, Silverstein FS. Monocyte chemoattractant protein-1 is a mediator of acute excitotoxic injury in neonatal rat brain. Neuroscience. 2000;101:737–744. doi: 10.1016/S0306-4522(00)00399-7. [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Henderson AK, Tsutsui S, Pittman QJ. Viral-like brain inflammation during development causes increased seizure susceptibility in adult rats. Neurobiol Dis. 2009;36:343–351. doi: 10.1016/j.nbd.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, Wang J, Sayeed I, Ishrat T, Atif F, Stein DG. The TRIF-dependent signaling pathway is not required for acute cerebral ischemia/reperfusion injury in mice. Biochem Biophys Res Commun. 2009;390:678–683. doi: 10.1016/j.bbrc.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, Akira S, Inagaki N, Nagai H, Hara H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171:258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- Joung SM, Park ZY, Rani S, Takeuchi O, Akira S, Lee JY. Akt contributes to activation of the TRIF-dependent signaling pathways of TLRs by interacting with TANK-binding kinase 1. J Immunol. 2011;186:499–507. doi: 10.4049/jimmunol.0903534. [DOI] [PubMed] [Google Scholar]
- Kendall GS, Hristova M, Horn S, Dafou D, Acosta-Saltos A, Almolda B, Zbarsky V, Rumajogee P, Heuer H, Castellano B, Pfeffer K, Nedospasov SA, Peebles DM, Raivich G. TNF gene cluster deletion abolishes lipopolysaccharide-mediated sensitization of the neonatal brain to hypoxic ischemic insult. Lab Invest. 2011;91:328–341. doi: 10.1038/labinvest.2010.192. [DOI] [PubMed] [Google Scholar]
- Kim H, Yang E, Lee J, Kim SH, Shin JS, Park JY, Choi SJ, Kim SJ, Choi IH. Double-stranded RNA mediates interferon regulatory factor 3 activation and interleukin-6 production by engaging Toll-like receptor 3 in human brain astrocytes. Immunology. 2008;124:480–488. doi: 10.1111/j.1365-2567.2007.02799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konat GW, Borysiewicz E, Fil D, James I. Peripheral challenge with double-stranded RNA elicits global up-regulation of cytokine gene expression in the brain. J Neurosci Res. 2009;87:1381–1388. doi: 10.1002/jnr.21958. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Okun E, Tang SC, Griffioen K, Cheng A, Mughal MR, Laryea G, Selvaraj PK, ffrench-Constant C, Magnus T, Arumugam TV, Mattson MP. Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. J Neurosci. 2008;28:13978–13984. doi: 10.1523/JNEUROSCI.2140-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kimura K, Yanai R, Chikama T, Nishida T. Cytokine, chemokine, and adhesion molecule expression mediated by MAPKs in human corneal fibroblasts exposed to poly(I:C) Invest Ophthalmol Vis Sci. 2008;49:3336–3344. doi: 10.1167/iovs.07-0972. [DOI] [PubMed] [Google Scholar]
- Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29:9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2004;101:233–238. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- Mullonkal CJ, Toledo-Pereyra LH. Akt in ischemia and reperfusion. J Invest Surg. 2007;20:195–203. doi: 10.1080/08941930701366471. [DOI] [PubMed] [Google Scholar]
- Packard AE, Hedges JC, Bahjat FR, Stevens SL, Conlin MJ, Salazar AM, Stenzel-Poore MP. Poly-IC preconditioning protects against cerebral and renal ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2012;32:242–247. doi: 10.1038/jcbfm.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Lapteva N, Seethammagari M, Slawin KM, Spencer DM. An essential role for Akt1 in dendritic cell function and tumor immunotherapy. Nat Biotechnol. 2006;24:1581–1590. doi: 10.1038/nbt1262. [DOI] [PubMed] [Google Scholar]
- Penkowa M, Hidalgo J. IL-6 deficiency leads to reduced metallothionein-I+II expression and increased oxidative stress in the brain stem after 6-aminonicotinamide treatment. Exp Neurol. 2000;163:72–84. doi: 10.1006/exnr.2000.7383. [DOI] [PubMed] [Google Scholar]
- Ratnayake U, Quinn TA, Castillo-Melendez M, Dickinson H, Walker DW. Behaviour and hippocampus-specific changes in spiny mouse neonates after treatment of the mother with the viral-mimetic Poly I:C at mid-pregnancy. Brain Behav Immun. 2012;26:1288–1299. doi: 10.1016/j.bbi.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Roda JM, Carceller F, Díez-Tejedor E, Avendaño C. Reduction of infarct size by intra-arterial nimodipine administered at reperfusion in a rat model of partially reversible brain focal ischemia. Stroke. 1995;26:1888–1892. doi: 10.1161/01.STR.26.10.1888. [DOI] [PubMed] [Google Scholar]
- Sasai M, Oshiumi H, Matsumoto M, Inoue N, Fujita F, Nakanishi M, Seya T. Cutting Edge: NF-kappaB-activating kinase-associated protein 1 participates in TLR3/Toll-IL-1 homology domain-containing adapter molecule-1-mediated IFN regulatory factor 3 activation. J Immunol. 2005;174:27–30. doi: 10.4049/jimmunol.174.1.27. [DOI] [PubMed] [Google Scholar]
- Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, Akira S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol. 2003;171:4304–4310. doi: 10.4049/jimmunol.171.8.4304. [DOI] [PubMed] [Google Scholar]
- Sävman K, Blennow M, Gustafson K, Tarkowski E, Hagberg H. Cytokine response in cerebrospinal fluid after birth asphyxia. Pediatr Res. 1998;43:746–751. doi: 10.1203/00006450-199806000-00006. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedin P, Hagberg H, Sävman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia–ischemia. J Neurosci. 2007;27:1511–1518. doi: 10.1523/JNEUROSCI.4391-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Neonatal encephalitis and white matter injury: more than just inflammation? Ann Neurol. 2008;64:232–236. doi: 10.1002/ana.21466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vontell R, Supramaniam V, Thornton C, Wyatt-Ashmead J, Mallard C, Gressens P, Rutherford M, Hagberg H. Toll-like receptor 3 expression in glia and neurons alters in response to white matter injury in preterm infants. Dev Neurosci. 2013 doi: 10.1159/000346158. doi: 10.1159/000346158. Advance online publication. Retrieved June 18, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S, Joodmardi E, Perlmann T, Ögren SO, Feldon J, Meyer U. Prenatal immune activation interacts with genetic Nurr1 deficiency in the development of attentional impairments. J Neurosci. 2012;32:436–451. doi: 10.1523/JNEUROSCI.4831-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, Mallard C. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol. 2009;183:7471–7477. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- Winerdal M, Winerdal ME, Kinn J, Urmaliya V, Winqvist O, Adén U. Long lasting local and systemic inflammation after cerebral hypoxic ischemia in newborn mice. PLoS One. 2012;7:e36422. doi: 10.1371/journal.pone.0036422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong T, Tang J, Zhao J, Chen H, Zhao F, Li J, Qu Y, Ferriero D, Mu D. Involvement of the Akt/GSK-3beta/CRMP-2 pathway in axonal injury after hypoxic-ischemic brain damage in neonatal rat. Neuroscience. 2012;216:123–132. doi: 10.1016/j.neuroscience.2012.04.052. [DOI] [PubMed] [Google Scholar]