

Abstract
Background
Electrocardiographic QRS duration, a measure of cardiac intraventricular conduction, varies ~2-fold in individuals without cardiac disease. Slow conduction may promote reentrant arrhythmias.
Methods and Results
We performed a genome-wide association study (GWAS) to identify genomic markers of QRS duration in 5,272 individuals without cardiac disease selected from electronic medical record (EMR) algorithms at five sites in the Electronic Medical Records and Genomics (eMERGE) network. The most significant loci were evaluated within the CHARGE consortium QRS GWAS meta-analysis. Twenty-three single nucleotide polymorphisms in 5 loci, previously described by CHARGE, were replicated in the eMERGE samples; 18 SNPs were in the chromosome 3 SCN5A and SCN10A loci, where the most significant SNPs were rs1805126 in SCN5A with p=1.2×10−8 (eMERGE) and p=2.5×10−20 (CHARGE) and rs6795970 in SCN10A with p=6×10−6 (eMERGE) and p=5×10−27 (CHARGE). The other loci were in NFIA, near CDKN1A, and near C6orf204. We then performed phenome-wide association studies (PheWAS) on variants in these five loci in 13,859 European Americans to search for diagnoses associated with these markers. PheWAS identified atrial fibrillation and cardiac arrhythmias as the most common associated diagnoses with SCN10A and SCN5A variants. SCN10A variants were also associated with subsequent development of atrial fibrillation and arrhythmia in the original 5,272 “heart-healthy” study population.
Conclusions
We conclude that DNA biobanks coupled to EMRs provide a platform not only for GWAS but may also allow broad interrogation of the longitudinal incidence of disease associated with genetic variants. The PheWAS approach implicated sodium channel variants modulating QRS duration in subjects without cardiac disease as predictors of subsequent arrhythmias.
Keywords: cardiac conduction, QRS duration, atrial fibrillation, genome-wide association study, phenome-wide association study, electronic medical records
Introduction
Electrocardiographic (ECG) parameters of cardiac conduction and repolarization, PR, QRS, and QT intervals, are widely used in clinical medicine and display substantial variability when measured across large populations. QRS duration represents activation time in the cardiac ventricle, and prolongation of this interval – representing global or regional slow conduction – has been associated with adverse outcomes such as sudden cardiac death.1 This variability reflects modulators such as abnormal electrolytes, underlying heart disease, or concomitant drug therapy, as well as heritable components; published estimates suggest that up to 40% of variability in the QRS interval is heritable.2–4 Using automated methods described further below, we have shown that 99% of QRS durations in over 30,000 normal subjects not receiving confounding medications fall between 65 to 108 msec.5
DNA repositories linked to Electronic Medical Record (EMR) systems have been proposed as one potential source of subjects for analyzing the relationship between genetic variation and a range of human traits.6–10 The advantages of this approach may include rapid generation of patient sets for study (since electronic data are already in place), and the ability to study large numbers of subjects accrued without bias with respect to factors such as disease or age. The National Human Genome Research Institute’s electronic MEdical Records and GEnomics (eMERGE) Network11 has, as one of its primary goals, the evaluation of the utility of EMR systems coupled to DNA repositories as a tool for genome science. Initial studies from eMERGE sites support the potential utility of EMR systems for discovery and validation of genotype-phenotype associations.12–16
We report here a GWAS of QRS duration in European-descent subjects whose first ECG in an EMR system was normal, and who at the time of the ECG lacked evidence of heart disease, potentially confounding medications, and electrolyte abnormalities. We then used a phenome-wide association study (PheWAS)16,17 to demonstrate that polymorphisms associated with QRS variability in subjects without cardiac disease were also associated with subsequent diagnoses of cardiac arrhythmias. This coupling of GWAS and PheWAS further validates the relationship between abnormal conduction and arrhythmias, and points to the development of genome-based predictors of arrhythmia susceptibility.
Methods
Normal QRS algorithm
We developed and deployed an algorithm to identify individuals with normal ECGs and without any cardiac disease, abnormal electrolyte values, or QRS-active medications – across the five eMERGE-1 sites identified 5,272 Caucasian patients (2,488 males and 2,784 females; Table 1). The algorithm was developed and validated in the Synthetic Derivative (SD), a de-identified image of the Vanderbilt EMR that currently contains over 120 million documents on about 2 million patients.7 The SD is refreshed regularly to add new clinical information from the EMR as it is accrued.
Table 1.
eMERGE sites contributing samples. All patients were of European ancestry.
| Site | Primary site phenotype |
Mean Age (SD) |
Genotyped samples meeting normal QRS algorithm definition |
||
|---|---|---|---|---|---|
| Males | Females | Total | |||
| Group Health | Dementia | 72.58 (7.23) | 187 | 351 | 538 |
| Marshfield | Cataract | 56.73 (9.73) | 69 | 149 | 218 |
| Mayo Clinic | Peripheral artery disease |
58.81 (8.52) | 1056 | 730 | 1786 |
| Northwestern | Type 2 diabetes |
52.49 (10.54) | 99 | 118 | 217 |
| Vanderbilt | Normal QRS | 49.95 (14.6) | 1077 | 1436 | 2513 |
| Total | 2488 | 2784 | 5272 | ||
The study population consisted of subjects with a normal ECG without evidence of cardiac disease any time before or within one month following the ECG, concurrent use of medications that interfere with ventricular conduction, and who did not have abnormal potassium, calcium, or magnesium lab values at the time of the ECG. The algorithm has been described in detail previously.13 The algorithm used natural language processing (NLP)18,19 to analyze narrative text, billing code queries, and lab queries to exclude any subjects with evidence of arrhythmia, heart failure, cardiomyopathy, myocardial ischemia/infarct, or cardiac conduction defect. The algorithm considered all physician-generated clinical documentation, including clinical notes and cardiologist-generated ECG impressions. Patients with family histories of cardiac disease were allowed by the NLP algorithm. In addition, ECGs had normal Bazett’s corrected QT intervals (<450ms), heart rates (between 50-100 bpm), and QRS (60-120 ms). The algorithm was reviewed by two physicians not involved in algorithm development, and achieved a PPV of 97% to identify patients with normal ECGs who did not have known exclusions on a random selection of 100 subjects.13 Analysis of clinical covariates in ~30,000 records with algorithm-defined normal ECGs identified gender and ancestry as modulators of QRS duration.5 Complete details of the algorithm are available from PheKB (http://phekb.org/).
The final algorithm was applied in BioVU, the Vanderbilt DNA databank that links DNA extracted from discarded blood samples to the SD.7 Patients at Vanderbilt were genotyped specifically for the purpose of studying normal QRS duration. The algorithm was then deployed across the DNA repositories at the other four eMERGE-I sites (Marshfield Clinic, Northwestern University, Mayo Clinic, Group Health Research Institute) to identify subjects with extant eMERGE-based genotyping data (based on other phenotypes; as shown in Table 1) who met algorithm-defined criteria for normal QRS. The eMERGE cohorts are described in more detail in McCarty et al. 201111 and at the Phenotype Knowledge Base (PheKB.org). Thus, all eMERGE individuals used in the analysis underwent the same algorithm to select those with normal ECGs and without prior heart disease, interfering medications, and abnormal electrolytes. To assess the performance of the algorithm when applied within external EMR systems, trained chart abstracters at Northwestern and Marshfield reviewed randomly-selected subsets of 100 subjects at Marshfield and 45 subjects at Northwestern to determine the algorithm’s accuracy at external sites. Northwestern’s evaluation also included an independent review by a board-certified internal medicine physician, with discrepancies resolved by consensus. This study included only subjects designated as “non-Hispanic white” European American in the EMR from each site. We have previously shown the EMR ancestry performs similar to self-report.20
This study was approved by each site’s Institutional Review Board. Because BioVU is de-identified and accrues individuals through left-over blood remaining after routine clinical testing, it operates as non-human subjects research according to the provisions of 45 CFR 46, as described previously.7 Individuals at other eMERGE sites were consented as part of each site’s DNA biobank.11
Genotyping and data analysis
Genotyping was performed at the Center for Genotyping and Analysis at the Broad Institute and the Center for Inherited Disease Research (CIDR) at Johns Hopkins University. Samples of European ancestry or unknown ancestry were analyzed using the Illumina Human660W-Quadv1_A genotyping platform, consisting of 561,490 SNPs and 95,876 intensity-only probes. Data were cleaned using the quality control (QC) pipeline developed by the eMERGE Genomics Working Group.21 This process includes evaluation of sample and marker call rate, gender mismatch and anomalies, duplicate and HapMap concordance, batch effects, Hardy-Weinberg equilibrium (HWE), sample relatedness, and population stratification. After QC, 528,508 SNPs were used for analysis based on the following QC criteria: SNP call rate >99%, sample call rate >99%, minor allele frequency > 0.0001, unrelated samples only (removing all parent-offspring, full and half siblings), and individuals of European-descent only (based on STRUCTURE22 analysis of >90% probability of being in the CEU cluster).
Each eMERGE site used the QC pipeline to clean their initial datasets prior to merging all the samples. QC procedures were then performed on the merged eMERGE dataset in which data from all five sites were combined, and no significant differences across sites or genotyping center were identified. As well, all sites had comparable QC results including similar SNP and sample call rates, HWE p-values overall, and minor allele frequencies. The detailed QC report on the merged dataset will be deposited in dbGaP along with the merged dataset.
Single-locus tests of association were performed using linear regression assuming an additive genetic model for all 528,508 SNPs in a total of 5,272 individuals with a normal QRS duration. Our studies of ECG intervals in 32,949 normal individuals identified sex as a major modulator of normal QRS duration, with minor effects of age and ancestry.5 All analyses were performed unadjusted and then adjusted for age, sex, BMI, and the first principal component from Eigenstrat23 to adjust for potential population stratification, without significantly changing the key results. Since only sex is significantly associated with QRS duration via the literature, we report that here. Analyses were also performed adjusting for height and/or BMI, but these did not change the results.
Associations with the lowest P values (p<10−4) were then submitted to the recent QRS Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) meta-analysis group24 and a table of P-values from that analysis was generated. The CHARGE meta-analysis of QRS duration has been described in detail previously24; briefly, it involved 40,407 individuals selected from 15 sites restricted to those of European ancestry. Individuals with prior myocardial infarction, heart failure, arrhythmias, pacemakers, antiarrhythmic medication use, or whose QRS durations >120 ms were excluded.
To calculate the variance explained by all SNPs in the dataset, we analyzed the data using GCTA.25 Only those SNPs with minor allele frequency > 0.01, genotyping efficiency > 99.9 and HWE > 0.001 were included in the analysis (n=505,502 SNPs). The genetic relationship matrix (GRM) was computed for all 5272 subjects and all SNPs using GCTA. In order to eliminate possible cryptic relationships, subjects with GRM>0.25 were pruned from the analysis, which removed 310 subjects. The proportion of variance explained by either all SNPs or all SNPs excluding the subset of 23 SNPs significant in the CHARGE GWAS was computed on the remaining subjects for QRS duration. We compared this to a linear regression analysis using the five SNPs in Table 2 to estimate the proportion of variance explained by these loci. All analyses were adjusted for age, gender and the first principal component (previously computed).
Table 2.
Replicated SNPs in the QRS GWAS analysis. Reported betas and p-values for eMERGE analysis are adjusted for sex, and all betas for CHARGE and eMERGE analyses are for the coded allele. Each SNP represents the strongest association in the region, and was the target of a PheWAS (as shown in Table 3).
| eMERGE QRS | CHARGE QRS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CHR | SNP | Location | Coded Allele |
Coded Allele Frequency |
BETA (msec) |
p-value | Coded Allele |
Coded Allele Frequency |
BETA (msec) |
p-value | Nearest Gene |
| 3 | rs1805126 | 38567410 | T* | 0.665 | −1.002 | 1.45E-08 | A | 0.655 | −0.6568 | 2.52E-20 | SCN5A |
| 3 | rs6795970 | 38741679 | A | 0.401 | 0.765 | 6.60E-06 | A | 0.396 | 0.7476 | 5.08E-27 | SCN10A |
| 6 | rs6906287 | 119069433 | C | 0.454 | 0.717 | 2.26E-05 | C | 0.451 | 0.5383 | 5.56E-16 | C6orf204 |
| 6 | rs1321313 | 36726799 | G* | 0.760 | −0.793 | 6.13E-05 | C | 0.742 | −0.8129 | 4.60E-25 | CDKN1A |
| 1 | rs2207790 | 61670555 | T* | 0.482 | −0.622 | 2.07E-04 | A | 0.461 | −0.5956 | 6.31E-18 | NFIA |
This SNP was coded on the opposite strand from that in the CHARGE study. Therefore, while the allele is not identical, the direction of effect is the same
Phenome-wide association study of QRS-associated SNPs
We selected the most significant SNP associations for analysis by PheWAS.16,17 For this analysis, we combined the entire eMERGE cohort of European American individuals (n=13,859) identified across the five eMERGE sites. These individuals represent a superset of the 5,272 individuals with normal ECGs and without heart disease used for the GWAS. To define diseases, we queried all International Classification of Disease (ICD), 9th edition, codes from the respective EMRs of the five eMERGE sites.
The PheWAS software uses occurrences of ICD codes to classify each person as having one or more of 778 possible clinical phenotypes (typically diseases). For each disease, the PheWAS algorithm constructs a control population by selecting all patients that do not have the case disease or closely related diseases (e.g., a patient with a bundle branch block cannot serve as a control for complete heart block). The PheWAS methodology has previously been validated through rediscovery of known associations.16,17 Analysis of each phenotype then proceeds using a pairwise analysis of all case and control groups for each tested SNP (n=23). We have observed that positive predictive values increase when individual codes are present more than once in the EMR, and here we required each case to have at least four instances of the same ICD code in a PheWAS case group. In addition, we did not analyze phenotypes occurring in less than 50 patients (a prevalence of 0.36% in the dataset). Association analyses were performed with PLINK using logistic regression adjusted for age, gender, and the first three principal component analyses as calculated by Eigenstrat, since on this larger population, the third principal component was statistically significant.23 Analysis adjusted with and without principal components did not substantively change the results. After identification of PheWAS case and control groups using the PheWAS software, the association analyses were performed using PLINK.26
Survival analysis of QRS population
Following PheWAS analysis, we analyzed the original set of 5272 patients that met our algorithm definition for normal cardiac conduction/normal heart for subsequent development of atrial fibrillation and cardiac arrhythmias with the SCN5A rs1805126 and SCN10A rs6795970 SNPs. Phenotype definitions were drawn from the PheWAS analysis using billing codes. Kaplan-Meier analysis and Cox proportional hazard models were calculated, using the starting time as the initial normal ECG with a time-to-event analysis. Cox proportional hazard models were adjusted for age, sex, principal components as calculated above, and QRS duration.
Results
Population identification
We identified 5,272 Caucasian patients (2,488 males and 2,784 females; Table 1) across the five eMERGE-I sites. The positive predictive value (PPV) of the automated phenotype algorithm to find cases with normal ECGs and without exclusions at the development site, Vanderbilt, to identify study subjects was 97% (95% confidence interval [CI] 91-99%).13 The PPV at Northwestern University and Marshfield Clinic were 97% (95% CI 83%-100%) and 100% (95% CI 96%-100%), respectively. Combining all reviewed samples across the three sites, the PPV would be 98% (95% CI 96%-100%). The mean QRS duration was 87.9 msec (standard deviation 9.5 msec; median 88.0 msec; Figure 1A).
Figure 1.

Panel A. Distribution of QRS durations in 5,272 normal ECGs. Panel B. Genome-wide association analysis of QRS duration, using sex-adjusted linear regression. The red line indicates genome-wide significance (p=5×10−8). The points in green are those SNPs that were also identified at genome-wide significance in the CHARGE consortium QRS meta-analysis as described in the text.
GWAS results
A total of 528,508 SNPs passed quality control of eMERGE-supported Illumina 660Quad genotyping data in these subjects. Figure 1B shows the genome-wide association analysis for QRS duration adjusted for sex; the findings were near-identical for the unadjusted analysis. There was a single association between QRS duration and a SNP (rs1805126) in SCN5A, encoding the cardiac sodium channel gene, that survived Bonferroni correction (beta=1.002 msec per copy of the T allele, p=1.45 × 10−8).
The set taken forward to the CHARGE QRS meta-analysis consortium included 108 SNPs with P-values <10−4. The retrieved P-values for this set divided into two distinct groups: 23 SNPs with P-values in the CHARGE set from 10−8 to 10−27, and 85 with P-values > 0.003. These 23 associations (Supplementary Table 1) are located in the five loci with the lowest P values reported by the CHARGE consortium: 18/23 are in the chromosome 3 locus that includes SCN5A and SCN10A, as well as other genes (e.g. EXOG and XYLB1). The other three loci are near SLC35F1 and C6orf204 (chromosome 6), near CDKN1A (chromosome 6) and in NFIA (chromosome 1). The most significant SNP for each locus is presented in Table 2. The locus zoom plot (Supplementary Figure 1) shows little linkage disequilibrium (LD) in the chromosome 3 region in HapMap Phase III (CEU), consistent with the suggestion that the SCN5A-10A finding may actually indicate multiple independent associations.24 Specifically, the most significant variants in SCN5A (rs1805126) and SCN10A (rs6795970) are not in LD (r2<0.20).
Using the GTCA25 approach, we estimated heritability for QRS at 31.1% (standard error [SE] 6.9%, p=5.7 × 10−7) using all SNPs in the dataset. Conducting the analysis without the 23 SNPs significant in CHARGE decreased the estimated heritability to 30.3%, a decrease of 0.8%. This was somewhat conservative compared to a linear regression model, which estimated an adjusted r-square value of 1.6% for the five loci in Table 2.
PheWAS analysis
The PheWAS dataset consisted of 13,859 European-American subjects in the entire genotyped eMERGE cohort. The analysis focused on the most significant SNPs in each of the five loci associated with QRS (Table 3). While no associations survived a strict Bonferroni correction for significance (p=0.05/778/5=1.3×10−5), the most significant associations were particularly relevant to cardiac disease and demonstrated significantly different patterns of associations for the five QRS-associated loci. The strongest associations for the SNPs in both SCN5A and SCN10A were with the diagnoses of cardiac arrhythmias (p=7.21×10−4 for SCN10A and p=1.1×10−3 for SCN5A) and, for SCN10A, atrial fibrillation (p=8.5×10−4, Figure 2). Table 3 lists associations for the most significant SCN5A (rs1805126) and SCN10A (rs6795970) SNPs, as well as those at the other QRS-associated loci, chromosome 1 (rs2207790) and the two chromosome 6 loci (rs6906287 and rs1321313), also graphed in Supplementary Figures 2-4. The CDKN1 and C6orf204 loci were not associated with cardiac arrhythmias (p>0.3, with 80% power to detect and OR>1.12 at p=0.05), and NFIA was weakly associated with cardiac arrhythmias (OR=0.91, p=0.02). Supplementary Table 2 presents PheWAS association data for all 23 SNPs significant in CHARGE. While most SNPs in a given gene displayed similar patterns of PheWAS associations, rs11129801, the strongest SCN10A SNP in our adjusted analysis but a lesser association in CHARGE, had a very different PheWAS pattern, with the strongest associations being epilepsy, uterine cancer, and migraines; atrial fibrillation was not associated (OR=1.07, p=0.18). In agreement with these data, rs11129801 was only in weak linkage disequilibrium to the other SCN10A SNPs, such as rs6800541 (r2=0.20). Likewise, the EXOG locus had a very different PheWAS pattern of associations (prostatic hyperplasia, sexual and gender identity disorders, liver disease, kidney disease, cerebral degenerations, diarrhea) despite being nearby the SCN5A-10A region.
Table 3.
Most significant associations between SNPs predicting normal QRS duration and diagnostic codes. For each SNP displayed, all PheWAS associations p<0.01 are displayed.
| Associated Phenotype | Case count | Odds ratio | P |
|---|---|---|---|
| SCN5A (Chr 3, rs1805126) | |||
| Cardiac arrhythmias | 3075 | 0.877 | 1.10E-03 |
| Other diseases of upper respiratory tract | 509 | 0.8131 | 3.83E-03 |
| Benign prostatic hypertrophy | 1615 | 0.8527 | 5.54E-03 |
| Chronic kidney disease | 1212 | 0.8772 | 6.02E-03 |
| Melanoma | 151 | 0.7033 | 8.18E-03 |
| Dermatophytosis | 1229 | 0.8791 | 8.77E-03 |
|
| |||
| SCN10A (Chr 3, rs6795970) | |||
|
| |||
| Cardiac arrhythmias | 3075 | 0.8781 | 7.21E-04 |
| Atrial fibrillation and flutter | 1758 | 0.8519 | 8.45E-04 |
| Arterial embolism and thrombosis | 150 | 0.6928 | 3.20E-03 |
| Cholecystitis | 121 | 0.6627 | 3.44E-03 |
| Convulsions | 311 | 1.249 | 7.01E-03 |
| Aphasia | 69 | 0.5999 | 7.25E-03 |
|
| |||
| C6orf204 (Chr 6, rs6906287) | |||
|
| |||
| Colorectal cancer | 316 | 0.7592 | 9.90E-04 |
| Benign prostatic hypertrophy | 1615 | 0.8576 | 4.19E-03 |
| Penicillin allergy | 65 | 1.68 | 4.01E-03 |
| Melanoma | 151 | 1.383 | 5.68E-03 |
| Disorders of pancreatic secretion | 84 | 0.6421 | 6.22E-03 |
| Nasal polyps | 127 | 1.413 | 7.15E-03 |
| Prostatitis | 150 | 0.7166 | 8.07E-03 |
|
| |||
| NFIA (Chr 1, rs2207790) | |||
|
| |||
| Postinflammatory pulmonary fibrosis | 173 | 0.7048 | 1.76E-03 |
| Multiple myeloma | 54 | 0.5425 | 2.80E-03 |
| Neutropenia and leukopenia | 238 | 0.7545 | 2.81E-03 |
| Facial nerve disorders | 67 | 1.677 | 3.70E-03 |
| Cardiomyopathy | 420 | 0.8033 | 4.40E-03 |
| Pneumothorax | 80 | 0.6391 | 6.59E-03 |
| Disorders of function of stomach | 355 | 1.23 | 7.73E-03 |
| Prostatitis | 150 | 0.7239 | 8.85E-03 |
| Toxic diffuse goiter | 76 | 1.541 | 9.47E-03 |
|
| |||
| CDKN1A (Chr 6, rs1321313) | |||
|
| |||
| Anorexia | 53 | 0.3649 | 1.57E-03 |
| Bacteremia | 118 | 0.5684 | 2.01E-03 |
| Anal fissure and fistula | 108 | 1.552 | 3.04E-03 |
| Abnormal loss of weight and underweight | 398 | 0.7642 | 3.39E-03 |
| Dementias | 841 | 0.833 | 5.69E-03 |
| Overweight, obesity and other | |||
| hyperalimentation | 2501 | 1.105 | 7.39E-03 |
| Spondylosis and allied disorders | 1244 | 1.156 | 9.39E-03 |
Figure 2.
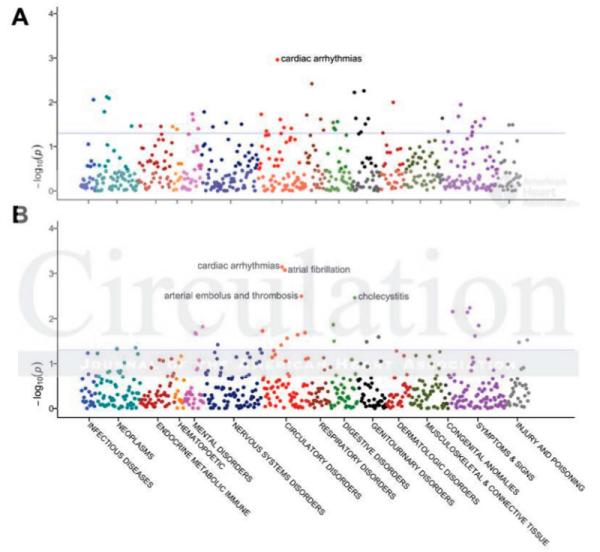
Phenome-wide association study plots of most significant SNPs associated with QRS duration. Panel A. SCN5A (rs1805126). Panel B. SCN10A (rs6795970). The blue lines indicate p<0.05.
Analysis of QRS population for arrhythmias
After PheWAS analysis, we analyzed the original set of 5,272 patients that met our algorithm definition for normal cardiac conduction/normal heart for subsequent development of atrial fibrillation and cardiac arrhythmias with the SCN5A rs1805126 and SCN10A rs6795970. In this population, 173 (3%) developed atrial fibrillation or atrial flutter at some point at least one month following the normal ECG, and 605 (11%) were coded as having any arrhythmia. QRS duration itself was associated with future development of atrial fibrillation (p=0.015) by logistic regression. As with the eMERGE PheWAS, SCN10A rs6795970 was associated with both arrhythmias (hazard ratio [HR]=0.81 per copy of the A allele, p=0.002) and atrial fibrillation/flutter (HR=0.67 per copy of the A allele, p=0.001). Moreover, this association was essentially unchanged when QRS was also included in the model (HR=0.68), indicating that the association between rs6795970 and atrial fibrillation is independent of the association between QRS and rs6795970. Similarly, the association between rs6795970 and cardiac arrhythmias was independent of QRS (HR=0.80 without QRS). Our analysis did not demonstrate an association between SCN5A rs1805126 and either atrial fibrillation (HR=1.2, p=0.14) or cardiac arrhythmias (1.03, p=0.66) in the normal QRS population. Figure 3 presents a Kaplan-Meier plot for the relationship between rs6795970 and development of atrial fibrillation.
Figure 3.
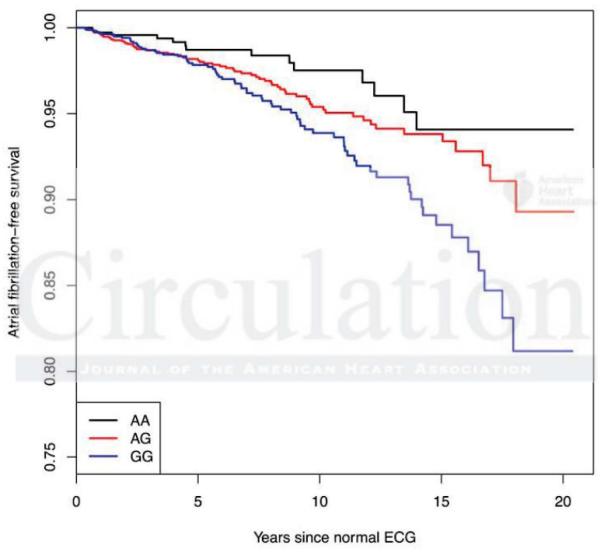
Kaplan-Meier estimate of development Atrial Fibrillation per SCN10A rs6795970 genotype. All patients were judged free of cardiac disease (including arrhythmias) at the time of the initial normal ECG.
Discussion
The current study demonstrates common variants in the SCN5A-SCN10A locus are associated with QRS duration in subjects without clinical evidence of prior heart disease. These patients were derived from clinical practice settings, adding to the growing body of evidence of supporting the utility of EMR-based genomic analysis.12–16,27 The data replicate findings from a large meta-analysis24 drawn from multiple community populations, where information on potential QRS modulators such as heart disease status and medications was not as precisely controlled or excluded from all included studies. The major new finding here is that using the PheWAS study paradigm, we were able to examine the longitudinal associations of these genomic variants on disease in a hypothesis-free manner. This analysis revealed that SNPs in SCN10A (rs6795970) and SCN5A (rs1805126) strongly associated with QRS duration, are also associated with subsequent cardiac arrhythmias. SCN10A specifically is associated with atrial fibrillation. These associations between rs6795970 and atrial fibrillation and cardiac arrhythmias were also seen specifically in the original “heart healthy” study population, and were independent of the SNP’s association with QRS duration. The latter finding suggests that while variants at the SCN5A-SCN10A locus determine QRS and subsequent arrhythmia susceptibility, they may do so by divergent (pleiotropic) pathways, or that conduction slowing occurs not only in the ventricle but also in the atrium where it contributes to susceptibility to atrial fibrillation. Importantly, the selection logic for the case selection algorithm in our GWAS required the absence of cardiovascular disease at the time of the ECG. Therefore, these associations represent subsequent development of cardiac arrhythmias in subjects with these variants. This result highlights the potential of the EMR, with multiple diagnoses and longitudinal follow-up, to identify not only variants associated with disease susceptibility or trait variability, but also subsequent outcomes associated with these variants.
Drugs that block SCN5A-encoded sodium channels slow ventricular conduction, prolong QRS duration,28 and increased mortality following myocardial infarction in the Cardiac Arrhythmia Suppression Trial (CAST).29,30 Available evidence supports the view that slow conduction, particularly in the setting of scarred or ischemic myocardium, promotes reentrant excitation that leads to fatal arrhythmias,31–33 and in CAST, longer QRS durations also predicted increased mortality among patients treated with placebo.1 In addition, a genetic disease caused by SCN5A loss-of-function mutations (Brugada Syndrome) is characterized by slowed ventricular conduction and an increased risk for fatal arrhythmias.34 Interestingly, previous analyses of variable PR duration have also identified strong associations with variants in SCN10A,13,35–37 and multiple mechanisms are currently being examined to explain this effect: expression of SCN10A-encoded channels in cardiomyocytes and/or cardiac neurons, or regulation of SCN5A-10A expression.38–40
Our PheWAS analysis suggests that SCN10A, SCN5A, and EXOG variants are associated with a cardiac arrhythmia billing codes (entered either by physicians or professional coders); this includes atrial fibrillation and flutter, supraventricular and ventricular tachycardia, cardiac arrest, and other unspecified arrhythmias. SCN10A rs6795970 was specifically associated with atrial fibrillation and flutter, which was noted by Pfeufer et al.36 but not Holm et al.35 Chambers et al.37 previously demonstrated associations between SCN10A rs6795970 and heart block and ventricular fibrillation; we also noted an association with first-degree atrioventricular block (p=0.009, Supplementary Table 2). Mouse studies have demonstrated expression of Nav1.8 in vagal and spinal afferents in gastrointestinal mucosa and myenteric plexes,41 and interestingly SCN10A was also associated with cholecystitis in the PheWAS. Variants in CDKN1A and C6orf204, however, were not associated with cardiac arrhythmias, although we cannot exclude that weak associations may exist. In contrast, the C6orf204 locus seems most associated with neoplastic disorders (colorectal cancer, prostatic hypertrophy, and melanoma); its strongest cardiovascular disease association was atherosclerosis (odds ratio 1.094, p=0.03). Thus, PheWAS suggests that although all these regions may be associated with QRS interval, only those SNPs in the SCN5A-10A region seem significantly associated with subsequent development of arrhythmias.
Recent growth in large GWAS meta-analysis has shown the power of large numbers to find genomic influences of given traits and disease. In this study, the development of the phenotype algorithm at one site was followed by its use at four other sites with different EMR systems. Algorithm performance was similar to find patients meeting inclusion and exclusion criteria at the three sites who evaluated the algorithm, providing further validation of the transportability of EMR phenotype algorithms.16,42 Estimates of heritability of QRS using all SNPs are consistent with other reports; the heritability associated with the very limited subset of 23 SNPs implicated here appears to explain a surprisingly large proportion of this heritability estimate. This analysis also indicates that, as with other traits, there is extensive “missing heritability” when QRS duration is analyzed as a function of common genomic variation.
This report highlights both limitations and real and potential advantages of EMR-based genomic research. The present study involved analysis of subjects accrued in the initial stages of the eMERGE network, and as such a major limitation was the relatively small size of the study set. Large consortia such as CHARGE have used meta-analysis to aggregate individual datasets across many sites and have demonstrated the power of the large numbers to generate highly significant results by this approach. One of the key lessons in the eMERGE experience to date has been that algorithms to identify cases and controls for genomic or other study can be successfully deployed across multiple EMR systems.16,43,44 Thus, as the number of subjects with dense genomic information across multiple EMR systems grows, this and other EMR-based studies highlight the potential for accrual of increasingly large sample sets to identify genomic predictors of variability in phenotypes such as physiologic traits or disease susceptibility.
Further, EMRs hold the promise, as suggested here, of examining longitudinal healthcare outcomes, such as disease complications or response to drug therapies. Identifying cases for such studies requires especially large resources, since subsets of subsets (e.g. drug response X in disease Y) are required. Current efforts demonstrate the feasibility of accrual of DNA collections coupled to EMRs large enough to support statistically valid analyses of rare variants and/or rare clinical events. In the current eMERGE network, there are 9 sites with EMR-based biobanks that include >250,000 subjects, and >50,000 have been genotyped on a genome-wide platform. Other resources that should expand the reach of EMR-based genomic research include the Kaiser Northern California biobank (>100,000 individuals),45 the Million Veteran’s Project (currently >100,000 individuals),46 and biobanks that will be coupled to national healthcare systems (e.g. the UK Biobank47).
Studies in the EMR environment enable PheWAS analyses: a PheWAS experiment cannot be executed in the absence of diverse diagnoses across many diagnostic classes in study subjects. The PheWAS-based associations reported here did not achieve significance using a strict Bonferroni correction; however, SCN10A rs6795970 was strongly associated with both atrial fibrillation and cardiac arrhythmias in the normal QRS population as well. The PheWAS approach is still in development and it is likely that the strategy of using only standard diagnostic codes is a limitation. The refinement of disease classifications and abstraction methods will enable more granular phenotypic subsetting, particularly when applied to increasingly large datasets described above.
A fundamental limitation of the PheWAS technique is that every diagnosis is not explicitly included or excluded in each record. Another potential limitation of EMR-based phenotyping is the accuracy of the information contained in the record. Extracting phenotypes from the EMR can result in errors if the data in the source EMR are incorrect (e.g., the EMR specifies a disease the patient does not have). Gender and ancestry testing suggest these demographic features are rarely incorrect in an EMR.7,20 Similarly, the electronic phenotyping experience in eMERGE indicates that recurrent mentions of specific diagnoses or combinations of diverse data types (such as medications plus free text plus diagnostic codes) greatly improve diagnostic accuracy of electronic phenotyping algorithms when assessed as positive predictive value using hand curation as the gold standard.48 EMRs contain data on subjects exposed to a healthcare system and as such EMR-based studies may not be generalizable to a broad population. The conduct of EMR-based studies across a variety of geographical locations and practice settings (as in eMERGE) potentially mitigates this issue.
In summary, a genome-wide association study conducted across multiple EMR systems replicated known associations for a readily available index of cardiac conduction, the QRS duration. The algorithm deployed allowed us to analyze subjects with normal electrocardiograms, and no evidence of heart disease or confounding drugs or electrolyte abnormalities, and the phenome-wide association established genomic variants predicting slower conduction in this population also associated with subsequent development of arrhythmias. Thus, the present findings are consonant with a view that individual susceptibility to serious arrhythmias is determined in part by genetically-determined variability in cardiac electrophysiologic behaviors. Furthermore, this study highlights the advantages of a genotyped EMR population to explore the subsequent emergence of clinically-important phenotypes not ascertained in the original study design.
Supplementary Material
Supplemental Figure 1: LocusZoom plot of the region in chromosome 3 in which most of the replicated associations (18/23) were identified. All variants with p<10−4 in the eMERGE data set were replicated at p<5×10−8 in the CHARGE data set.
Supplemental Figure 2: Phenome-wide association study plot for CDKN1A (rs1321313).
Supplemental Figure 3: Phenome-wide association study plot for C6orf204 (rs6906287).
Supplemental Figure 4: Phenome-wide association study plot for NFIA (rs2207790).
Supplemental Table 1: Replicated SNPs in the QRS GWAS analysis. Reported betas and pvalues for eMERGE analysis are adjusted for sex, and all betas for CHARGE and eMERGE analyses are for the coded allele.
Supplemental Table 2: PheWAS associations for replicated SNPs (N=23). Data for all associations with P<0.01 are shown. ICD9 codes for given associations can be downloaded from http://knowledgemap.mc.vanderbilt.edu/research/content/phewas.
Clinical Perspective.
QRS duration, a measure of cardiac intraventricular conduction, varies ~2-fold in individuals without cardiac disease. Slow conduction may promote reentrant arrhythmias. We identified 5,272 individuals with normal ECGs and no evidence of cardiac disease from electronic medical records at five institutions and performed genome-wide association analysis. We found variants in 5 loci associated with QRS duration in normal individuals, including SCN5A, SCN10A, NFIA, near CDKN1A, and near SLC35F1 that were replicated in the CHARGE consortium QRS meta-analysis. Subsequently, we performed phenome-wide association studies on associated SNPs. These analyses demonstrated with SCN5A-10A variants were associated with future development of atrial fibrillation and cardiac arrhythmias. We conclude that DNA biobanks coupled to EMRs provide a platform not only for GWAS but also for broadly interrogating the longitudinal incidence of disease associated with genetic variants.
Acknowledgments
Funding Sources: This work was supported by the electronic MEdical Records and GEnomics (eMERGE) Network, initiated and funded by the National Human Genome Research Institute, with additional funding from the National Institute of General Medical Sciences, through the following grants: U01-HG-004610 (Group Health Cooperative); U01-HG-004608 (Marshfield Clinic); U01-HG-04599 (Mayo Clinic); U01-HG-004609 (Northwestern University); U01-HG-04603 (Vanderbilt University, also serving as the Administrative Coordinating Center). BioVU also receives support through the National Center for Research Resources UL1 RR024975, which is now at the National Center for Advancing Translational Sciences, 2 UL1 TR000445. Dr. Sotoodehnia was supported by R01-HL088456. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest Disclosures: None.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Capone RJ, Pawitan Y, el-Sherif N, Geraci TS, Handshaw K, Morganroth J, Schlant RC, Waldo AL. Events in the cardiac arrhythmia suppression trial: baseline predictors of mortality in placebo-treated patients. J. Am. Coll. Cardiol. 1991;18:1434–1438. doi: 10.1016/0735-1097(91)90671-u. [DOI] [PubMed] [Google Scholar]
- 2.Hanson B, Tuna N, Bouchard T, Heston L, Eckert E, Lykken D, Segal N, Rich S. Genetic factors in the electrocardiogram and heart rate of twins reared apart and together. Am. J. Cardiol. 1989;63:606–609. doi: 10.1016/0002-9149(89)90907-7. [DOI] [PubMed] [Google Scholar]
- 3.Busjahn A, Knoblauch H, Faulhaber HD, Boeckel T, Rosenthal M, Uhlmann R, Hoehe M, Schuster H, Luft FC. QT interval is linked to 2 long-QT syndrome loci in normal subjects. Circulation. 1999;99:3161–3164. doi: 10.1161/01.cir.99.24.3161. [DOI] [PubMed] [Google Scholar]
- 4.Russell MW, Law I, Sholinsky P, Fabsitz RR. Heritability of ECG measurements in adult male twins. J Electrocardiol. 1998;30(Suppl):64–68. doi: 10.1016/s0022-0736(98)80034-4. [DOI] [PubMed] [Google Scholar]
- 5.Ramirez AH, Schildcrout JS, Blakemore DL, Masys DR, Pulley JM, Basford MA, Roden DM, Denny JC. Modulators of normal electrocardiographic intervals identified in a large electronic medical record. Heart Rhythm. 2011;8:271–277. doi: 10.1016/j.hrthm.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarty CA, Wilke RA, Giampietro PF, Wesbrook SD, Caldwell MD. Marshfield Clinic Personalized Medicine Research Project (PMRP): design, methods and recruitment for a large population-based biobank. Personalized Medicine. 2005;2:49–79. doi: 10.1517/17410541.2.1.49. [DOI] [PubMed] [Google Scholar]
- 7.Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, Masys DR. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 2008;84:362–369. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarty CA, Chapman-Stone D, Derfus T, Giampietro PF, Fost N. Community consultation and communication for a population-based DNA biobank: the Marshfield clinic personalized medicine research project. Am. J. Med. Genet. A. 2008;146A:3026–3033. doi: 10.1002/ajmg.a.32559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginsburg GS, Burke TW, Febbo P. Centralized biorepositories for genetic and genomic research. JAMA. 2008;299:1359–1361. doi: 10.1001/jama.299.11.1359. [DOI] [PubMed] [Google Scholar]
- 10.Lemke AA, Wolf WA, Hebert-Beirne J, Smith ME. Public and biobank participant attitudes toward genetic research participation and data sharing. Public Health Genomics. 2010;13:368–377. doi: 10.1159/000276767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCarty CA, Chisholm RL, Chute CG, Kullo IJ, Jarvik GP, Larson EB, Li R, Masys DR, Ritchie MD, Roden DM, Struewing JP, Wolf WA. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics. 2011;4:13. doi: 10.1186/1755-8794-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kullo IJ, Ding K, Shameer K, McCarty CA, Jarvik GP, Denny JC, Ritchie MD, Ye Z, Crosslin DR, Chisholm RL, Manolio TA, Chute CG. Complement receptor 1 gene variants are associated with erythrocyte sedimentation rate. Am. J. Hum. Genet. 2011;89:131–138. doi: 10.1016/j.ajhg.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denny JC, Ritchie MD, Crawford DC, Schildcrout JS, Ramirez AH, Pulley JM, Basford MA, Masys DR, Haines JL, Roden DM. Identification of genomic predictors of atrioventricular conduction: using electronic medical records as a tool for genome science. Circulation. 2010;122:2016–2021. doi: 10.1161/CIRCULATIONAHA.110.948828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kullo IJ, Ding K, Jouni H, Smith CY, Chute CG. [cited 2010 Oct 29];A genome-wide association study of red blood cell traits using the electronic medical record. PLoS ONE. 2010 :5. doi: 10.1371/journal.pone.0013011. [Internet] Available from: http://www.ncbi.nlm.nih.gov/pubmed/20927387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ritchie MD, Denny JC, Crawford DC, Ramirez AH, Weiner JB, Pulley JM, Basford MA, Brown-Gentry K, Balser JR, Masys DR, Haines JL, Roden DM. Robust replication of genotype-phenotype associations across multiple diseases in an electronic medical record. Am. J. Hum. Genet. 2010;86:560–572. doi: 10.1016/j.ajhg.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, Chai HS, Bastarache L, Zuvich R, Peissig P, Carrell D, Ramirez AH, Pathak J, Wilke RA, Rasmussen L, Wang X, Pacheco JA, Kho AN, Hayes MG, Weston N, Matsumoto M, Kopp PA, Newton KM, Jarvik GP, Li R, Manolio TA, Kullo IJ, Chute CG, Chisholm RL, Larson EB, McCarty CA, Masys DR, Roden DM, De Andrade M. Variants Near FOXE1 Are Associated with Hypothyroidism and Other Thyroid Conditions: Using Electronic Medical Records for Genome- and Phenome-wide Studies. Am. J. Hum. Genet. 2011;89:529–542. doi: 10.1016/j.ajhg.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM, Crawford DC. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denny JC, Spickard A, Johnson KB, Peterson NB, Peterson JF, Miller RA. Evaluation of a method to identify and categorize section headers in clinical documents. J Am Med Inform Assoc. 2009;16:806–815. doi: 10.1197/jamia.M3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denny JC, Miller RA, Waitman LR, Arrieta MA, Peterson JF. Identifying QT prolongation from ECG impressions using a general-purpose Natural Language Processor. Int J Med Inform. 2009;78 Suppl 1:S34–42. doi: 10.1016/j.ijmedinf.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dumitrescu L, Ritchie MD, Brown-Gentry K, Pulley JM, Basford M, Denny JC, Oksenberg JR, Roden DM, Haines JL, Crawford DC. Assessing the accuracy of observer-reported ancestry in a biorepository linked to electronic medical records. Genet Med. 2010;12:648–650. doi: 10.1097/GIM.0b013e3181efe2df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turner S, Armstrong LL, Bradford Y, Carlson CS, Crawford DC, Crenshaw AT, De Andrade M, Doheny KF, Haines JL, Hayes G, Jarvik G, Jiang L, Kullo IJ, Li R, Ling H, Manolio TA, Matsumoto M, McCarty CA, McDavid AN, Mirel DB, Paschall JE, Pugh EW, Rasmussen LV, Wilke RA, Zuvich RL, Ritchie MD. Quality control procedures for genome-wide association studies. Curr Protoc Hum Genet. 2011 doi: 10.1002/0471142905.hg0119s68. Chapter 1:Unit1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 24.Sotoodehnia N, Isaacs A, De Bakker PIW, Dörr M, Newton-Cheh C, Nolte IM, Van der Harst P, Müller M, Eijgelsheim M, Alonso A, Hicks AA, Padmanabhan S, Hayward C, Smith AV, Polasek O, Giovannone S, Fu J, Magnani JW, Marciante KD, Pfeufer A, Gharib SA, Teumer A, Li M, Bis JC, Rivadeneira F, Aspelund T, Köttgen A, Johnson T, Rice K, Sie MPS, Wang YA, Klopp N, Fuchsberger C, Wild SH, Mateo Leach I, Estrada K, Völker U, Wright AF, Asselbergs FW, Qu J, Chakravarti A, Sinner MF, Kors JA, Petersmann A, Harris TB, Soliman EZ, Munroe PB, Psaty BM, Oostra BA, Cupples LA, Perz S, De Boer RA, Uitterlinden AG, Völzke H, Spector TD, Liu F-Y, Boerwinkle E, Dominiczak AF, Rotter JI, Van Herpen G, Levy D, Wichmann H-E, Van Gilst WH, Witteman JCM, Kroemer HK, Kao WHL, Heckbert SR, Meitinger T, Hofman A, Campbell H, Folsom AR, Van Veldhuisen DJ, Schwienbacher C, O’Donnell CJ, Volpato CB, Caulfield MJ, Connell JM, Launer L, Lu X, Franke L, Fehrmann RSN, Te Meerman G, Groen HJM, Weersma RK, Van den Berg LH, Wijmenga C, Ophoff RA, Navis G, Rudan I, Snieder H, Wilson JF, Pramstaller PP, Siscovick DS, Wang TJ, Gudnason V, Van Duijn CM, Felix SB, Fishman GI, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, De Bakker PIW, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurreeman F, Liao K, Chibnik L, Hickey B, Stahl E, Gainer V, Li G, Bry L, Mahan S, Ardlie K, Thomson B, Szolovits P, Churchill S, Murphy SN, Cai T, Raychaudhuri S, Kohane I, Karlson E, Plenge RM. Genetic basis of autoantibody positive and negative rheumatoid arthritis risk in a multi-ethnic cohort derived from electronic health records. Am J Hum Genet. 2011;88:57–69. doi: 10.1016/j.ajhg.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson EA, McKinnon MG. The differential effect of quinidine and pyrilamine on the myocardial action potential at various rates of stimulation. J Pharmacol Exp Ther. 1957;120:460–468. [PubMed] [Google Scholar]
- 29.Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N Engl J Med. 1989;321:406–412. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 30.Epstein AE, Hallstrom AP, Rogers WJ, Liebson PR, Seals AA, Anderson JL, Cohen JD, Capone RJ, Wyse DG. Mortality following ventricular arrhythmia suppression by encainide, flecainide, and moricizine after myocardial infarction. The original design concept of the Cardiac Arrhythmia Suppression Trial (CAST) JAMA. 1993;270:2451–2455. [PubMed] [Google Scholar]
- 31.Akiyama T, Pawitan Y, Greenberg H, Kuo CS, Reynolds-Haertle RA. Increased risk of death and cardiac arrest from encainide and flecainide in patients after non-Q-wave acute myocardial infarction in the Cardiac Arrhythmia Suppression Trial. CAST Investigators. Am J Cardiol. 1991;68:1551–1555. doi: 10.1016/0002-9149(91)90308-8. [DOI] [PubMed] [Google Scholar]
- 32.Coromilas J, Saltman AE, Waldecker B, Dillon SM, Wit AL. Electrophysiological effects of flecainide on anisotropic conduction and reentry in infarcted canine hearts. Circulation. 1995;91:2245–2263. doi: 10.1161/01.cir.91.8.2245. [DOI] [PubMed] [Google Scholar]
- 33.Greenberg HM, Dwyer EM, Jr, Hochman JS, Steinberg JS, Echt DS, Peters RW. Interaction of ischaemia and encainide/flecainide treatment: a proposed mechanism for the increased mortality in CAST I. Br Heart J. 1995;74:631–635. doi: 10.1136/hrt.74.6.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 35.Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA, Jonasdottir A, Mathiesen EB, Njølstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Løchen M-L, Kong A, Thorsteinsdottir U, Stefansson K. Several common variants modulate heart rate, PR interval and QRS duration. Nat. Genet. 2010;42:117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 36.Pfeufer A, Van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Müller M, Sotoodehnia N, Sinner MF, Verwoert GC, Li M, Kao WHL, Köttgen A, Coresh J, Bis JC, Psaty BM, Rice K, Rotter JI, Rivadeneira F, Hofman A, Kors JA, Stricker BHC, Uitterlinden AG, Van Duijn CM, Beckmann BM, Sauter W, Gieger C, Lubitz SA, Newton-Cheh C, Wang TJ, Magnani JW, Schnabel RB, Chung MK, Barnard J, Smith JD, Van Wagoner DR, Vasan RS, Aspelund T, Eiriksdottir G, Harris TB, Launer LJ, Najjar SS, Lakatta E, Schlessinger D, Uda M, Abecasis GR, Müller-Myhsok B, Ehret GB, Boerwinkle E, Chakravarti A, Soliman EZ, Lunetta KL, Perz S, Wichmann H-E, Meitinger T, Levy D, Gudnason V, Ellinor PT, Sanna S, Kääb S, Witteman JCM, Alonso A, Benjamin EJ, Heckbert SR. Genome-wide association study of PR interval. Nat Genet. 2010;42:153–159. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chambers JC, Zhao J, Terracciano CMN, Bezzina CR, Zhang W, Kaba R, Navaratnarajah M, Lotlikar A, Sehmi JS, Kooner MK, Deng G, Siedlecka U, Parasramka S, El-Hamamsy I, Wass MN, Dekker LRC, De Jong JSSG, Sternberg MJE, McKenna W, Severs NJ, De Silva R, Wilde AAM, Anand P, Yacoub M, Scott J, Elliott P, Wood JN, Kooner JS. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152. doi: 10.1038/ng.516. [DOI] [PubMed] [Google Scholar]
- 38.London B. Whither Art Thou, SCN10A, and What Art Thou Doing? Circ Res. 2012;111:268–270. doi: 10.1161/CIRCRESAHA.112.275032. [DOI] [PubMed] [Google Scholar]
- 39.Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111:322–332. doi: 10.1161/CIRCRESAHA.112.265173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verkerk AO, Remme CA, Schumacher CA, Scicluna BP, Wolswinkel R, De Jonge B, Bezzina CR, Veldkamp MW. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res. 2012;111:333–343. doi: 10.1161/CIRCRESAHA.112.274035. [DOI] [PubMed] [Google Scholar]
- 41.Gautron L, Sakata I, Udit S, Zigman JM, Wood JN, Elmquist JK. Genetic tracing of Nav1.8-expressing vagal afferents in the mouse. J Comp Neurol. 2011;519:3085–3101. doi: 10.1002/cne.22667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carroll RJ, Thompson WK, Eyler AE, Mandelin AM, Cai T, Zink RM, Pacheco JA, Boomershine CS, Lasko TA, Xu H, Karlson EW, Perez RG, Gainer VS, Murphy SN, Ruderman EM, Pope RM, Plenge RM, Kho AN, Liao KP, Denny JC. Portability of an algorithm to identify rheumatoid arthritis in electronic health records. JAMIA. 2012;19:e162–e169. doi: 10.1136/amiajnl-2011-000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kho AN, Hayes MG, Rasmussen-Torvik L, Pacheco JA, Thompson WK, Armstrong LL, Denny JC, Peissig PL, Miller AW, Wei W-Q, Bielinski SJ, Chute CG, Leibson CL, Jarvik GP, Crosslin DR, Carlson CS, Newton KM, Wolf WA, Chisholm RL, Lowe WL. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome-wide association study. J Am Med Inform Assoc. 2012;19:212–218. doi: 10.1136/amiajnl-2011-000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carroll RJ, Thompson WK, Eyler AE, Mandelin AM, Cai T, Zink RM, Pacheco JA, Boomershine CS, Lasko TA, Xu H, Karlson EW, Perez RG, Gainer VS, Murphy SN, Ruderman EM, Pope RM, Plenge RM, Kho AN, Liao KP, Denny JC. [cited 2012 Mar 13];Portability of an algorithm to identify rheumatoid arthritis in electronic health records. JAMIA. 2012 doi: 10.1136/amiajnl-2011-000583. [Internet] Available from: http://www.ncbi.nlm.nih.gov/pubmed/22374935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. [cited 2011 Sep 13];Kaiser Permanente, UCSF Scientists Complete NIH-Funded Genomics Project Involving 100,000 People [Internet] Available from: http://www.dor.kaiser.org/external/news/press_releases/Kaiser_Permanente,_UCSF_Scientists_ Complete_NIH-Funded_Genomics_Project_Involving_100,000_People/
- 46. [cited 2012 Jun 20];Million Veteran Program (MVP) [Internet] Available from: http://www.research.va.gov/mvp/
- 47.Collins R. What makes UK Biobank special? Lancet. 2012;379:1173–1174. doi: 10.1016/S0140-6736(12)60404-8. [DOI] [PubMed] [Google Scholar]
- 48.Kho AN, Pacheco JA, Peissig PL, Rasmussen L, Newton KM, Weston N, Crane PK, Pathak J, Chute CG, Bielinski SJ, Kullo IJ, Li R, Manolio TA, Chisholm RL, Denny JC. Electronic Medical Records for Genetic Research: Results of the eMERGE Consortium. Sci Transl Med. 2011;3:79re1. doi: 10.1126/scitranslmed.3001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: LocusZoom plot of the region in chromosome 3 in which most of the replicated associations (18/23) were identified. All variants with p<10−4 in the eMERGE data set were replicated at p<5×10−8 in the CHARGE data set.
Supplemental Figure 2: Phenome-wide association study plot for CDKN1A (rs1321313).
Supplemental Figure 3: Phenome-wide association study plot for C6orf204 (rs6906287).
Supplemental Figure 4: Phenome-wide association study plot for NFIA (rs2207790).
Supplemental Table 1: Replicated SNPs in the QRS GWAS analysis. Reported betas and pvalues for eMERGE analysis are adjusted for sex, and all betas for CHARGE and eMERGE analyses are for the coded allele.
Supplemental Table 2: PheWAS associations for replicated SNPs (N=23). Data for all associations with P<0.01 are shown. ICD9 codes for given associations can be downloaded from http://knowledgemap.mc.vanderbilt.edu/research/content/phewas.