

Abstract
Herein we describe in full our investigations leading to the first total syntheses of ent-dioxepandehydrothyrsiferol and armatol A. Discovery of a bromonium-initiated epoxide-opening cascade enabled novel tactics for constructing key fragments found in both natural products and have led us to revise the proposed biogeneses. Other common features found in the routes include convergent fragment coupling strategies to assemble the natural products’ backbones and the use of epoxide-opening cascades for rapid constructions of the fused polyether subunits. Through de novo synthesis of armatol A, we elucidate the absolute and relative configuration of this natural product.
Keywords: Natural products, Total synthesis, Structure elucidation, Terpenoids, Epoxidation
1. Introduction
Halogenated squalene-derived terpenoid polyethers comprise a class of structurally intriguing natural products bearing various sizes of oxygen heterocycles and stereochemical relationships by which they are connected (Figure 1).1 Dioxepandehydrothyrsiferol (1),2 along with venustatriol (2),3 thyrsiferol (3),4 and enshuol (4)5 were isolated from red algae of the genus Laurencia. A common feature found in these natural products is the presence of a secondary neopentyl bromide moiety within a bromo-oxane or bromo-oxepane ring. In 1, the bromo-oxepane ring is part of a fused tricycle with a unique trans-anti-trans ring connectivity. A direct and efficient construction of such a substructure was unknown prior to this study and hence served as a motivation to undertake the total synthesis of ent-1.6 This account will outline approaches that eventually led to the successful first total synthesis of this natural product.7
Figure 1.

Representative examples of the halogenated squalene-derived terpenoid polyethers.
In contrast, armatols A–F (5–10) are derived from a related alga of the genus Chondria and generally possess a trans-syn-trans topography on the fused tricyclic portion of the molecule (Figure 1).8 In particular, armatol A (5) is the only natural product among the oxasqualenoid polyethers that contains a 7,7,6-fused tricycle in which one of the rings is an oxepene. Our interest in this family of natural products is bolstered by a lack of absolute and complete relative stereochemical assignment, either by advanced spectroscopic techniques or synthetic studies.
In line with the ongoing program in our group to access polyether natural products through epoxide-opening cascades,9 we embarked on the syntheses of ent-1 and 5 using such an approach. While a cascade cyclization of a polyepoxide precursor has been proposed in the biosynthesis of 1,2 no biogenetic proposal has been put forth for the armatol natural products. Given the structural similarities between 1 and 5–10 (the 7,7,6-fused polyether fragments and bromo-oxepanes; either as part of a fused tricycle or by itself), we hypothesize that the two classes of natural products share unified biogenetic mechanisms.
A universal strategy utilized at the beginning of our study involved Lewis acid-initiated epoxide-opening cascades to construct the fused tricycles found in both natural products (Scheme 1). Both of these cascades would rely on Me groups to direct the desired regioselectivity in the epoxide-opening events.10 The trans-anti-trans ring connectivity in ent-1 could retrosynthetically be traced to polyepoxide 13, bearing epoxides of both R and S configurations. A bromide installation would follow this cyclization. On the other hand, the trans-syn-trans ring connectivity in 5 would come from 14, and an oxepene installation via elimination of an activated alcohol moiety would follow this cascade. The initial strategy employed an allylic alcohol as a trapping nucleophile in both cascades. This functional group is found in the backbone of ent-1 and would also allow for elaboration toward fragment coupling to eventually access 5 (vide infra).
Scheme 1.

A unified approach toward the tricyclic fragments of ent-1 and 5.
2. Results and Discussion
2.1. Synthetic studies toward ent-dioxepandehydrothyrsiferol
Our first approach toward ent-1 is delineated in Scheme 2. We planned to access the natural product from an intermediate such as 11,11 which would be obtained via a Lewis acid-initiated epoxide-opening cascade of triepoxide 13. The allylic alcohol moiety in the backbone of this intermediate could be prepared by Ni-catalyzed reductive coupling of aldehyde 15 and allene 16, as previously described by our group.12
Scheme 2.

First-generation retrosynthetic analysis of ent-1.
Toward this end, 15 and 17 were coupled in the presence of stoichiometric Ni(cod)2 and tBuMe2SiH to provide the desired allylic ether as a 1:1 mixture of diastereomers at C14 (Scheme 3).13 After desilylation, allylic alcohols 18 (as a mixture of diastereomers) were obtained for cyclization studies. We felt that these substrates were appropriate model systems to explore the feasibility of formation of the tetrahydropyran (THP) ring in the fused tricyclic portion of ent-1. However, cyclization of the desired stereoisomer of 18 under thermal, Lewis acidic, or BrØnsted acidic conditions gave predominantly the undesired tetrahydrofuran (THF) product 19 instead of the desired THP product 20 (Scheme 3). This result could be accounted for using a chair transition state model (21) to form the THP product, which has also been suggested by Forsyth.14 This produces an unfavorable 1,3-diaxial interaction between the Me group of the epoxide and alkenyl group of the allylic alcohol.
Scheme 3.

Reductive coupling to access 18 and cyclization studies. Reagents and conditions: a) Ni(cod)2, Cyp3P, tBuMe2SiH, THF, rt, 67%, 1:1 dr; b) TBAF, THF, rt.
We postulated that a way to remove the unfavorable interaction in the transition state leading to the desired six-membered ring product was to use a trapping nucleophile attached to an sp2-hybridized carbon to give a transition state such as 25 (Scheme 4). As a proof of principle, after oxidation of 18 to enone 22, exposure to Amberlyst 15 gave dihydropyran 23 as the sole product over dihydrofuran 24. Alternatively, oxidation of aldehyde 15 to carboxylic acid 26 (used crude in the cyclization reaction) also provided a substrate capable of selective δ-lactone 27 formation over γ-lactone 28 under BF3•OEt2-promotion.
Scheme 4.

Successful six-membered ring formations using nucleophiles attached to sp2-hybridized carbons. Reagents and conditions: a) Amberlyst 15, CH2Cl2, rt; b) BF3•OEt2, CH2Cl2, −78 °C to −50 °C, 73% from 15.
This discovery led to a revised strategy toward 11 (Scheme 5). In this new route, cyclization to form the tricyclic portion of the molecule would precede fragment coupling along the C15–C16 bond. A Lewis acid-initiated cyclization of triepoxide 31 with a tBu ester trapping nucleophile attached would give 29.10a–b The other fragment of the molecule would come from diepoxide 32,15 which could undergo a Payne rearrangement to give 30.16
Scheme 5.

A new retrosynthetic disconnection toward 11.
Triepoxide 31 was synthesized in four steps from commercially available material as outlined in Scheme 6. Starting from (E,E)-farnesol, Sharpless asymmetric epoxidation followed by a Shi epoxidation gave triepoxy alcohol 33.6 Conversion of the alcohol to an iodide and displacement with an enolate derived from tbutyl acetate provided 31. Upon exposure of this substrate to BF3•OEt2, the desired tricyclic lactone 35 was obtained in up to 25% over two steps after TES protection of the resultant secondary alcohol. Analysis of the X–ray crystal structure confirmed structure of tricyclic alcohol 34.
Scheme 6.

Synthesis and cyclization of triepoxide 31. Reagents and conditions: a) L-(+)-DET, Ti(OiPr)4, tBuOOH, 4Å MS, CH2Cl2, −50 °C to −40°C, 95%, 87% ee; b) Shi ketone (36), Oxone, nBu4NHSO4, aq. K2CO3, Na2B4O7 buffer, pH 10.5, (CH3O)2CH2/CH3CN/H2O, 0 °C, 88%; c) I2, PPh3, imidazole, CH2Cl2, 0 °C to rt, 87%; d) tBuOAc, LDA, HMPA, THF, −78 °C, 88%; e) BF3•OEt2, 1,2,3-(MeO)3-C6H3, CH2Cl2, −78 °C; f) TESCl, imid, DMF, 45 °C, 25% from 31.
With lactone 35 in hand, we envisioned forming the C15–C16 bond of ent-1 via a Suzuki–Miyaura fragment coupling. Toward this goal, 35 was elaborated to alkenyl triflate 39 as delineated in Scheme 7: first a DIBAL-H reduction gave a lactol, which underwent TMSCN displacement to give nitrile 37.17 This was then followed by a MeMgBr addition to furnish methyl ketone 38. Conversion to alkenyl triflate 39 (used without purification in the coupling reaction) was achieved through reaction with Comins’ reagent in the presence of LHMDS.18
Scheme 7.
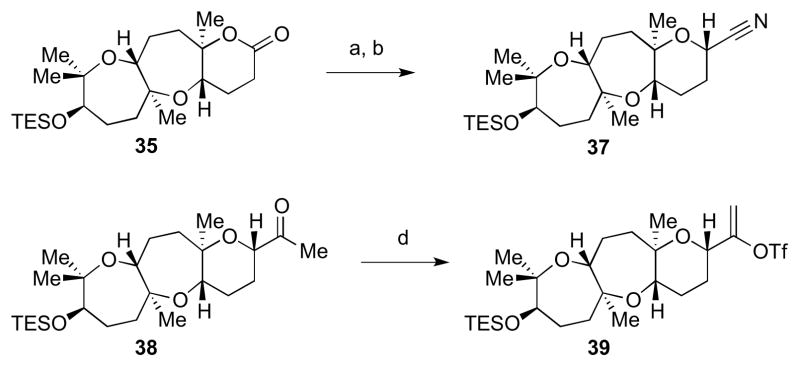
Synthesis of alkenyl triflate 39. Reagents and conditions: a) DIBAL-H, toluene, −78 °C; b) TMSCN, BF3•OEt2, −12 °C to −5 °C, 51% from 35, 78:22 dr; c) Ni(acac)2, MeMgBr, toluene, −15 °C to −8 °C, 50%, 93:7 dr; d) (SO2CF3)2NC5H3NCl, LHMDS, THF, −78 °C to 0 °C.
Access to the other coupling partner started with geraniol, which was exposed to a Sharpless asymmetric epoxidation followed by a Shi epoxidation to give 32 (Scheme 8).15 The diepoxide was then subjected to NaOH in THF/H2O to effect a Payne rearrangement, giving 30. Reaction with an ylide derived from trimethylsulfonium iodide followed by bis-TIPS protection gave 40.19
Scheme 8.
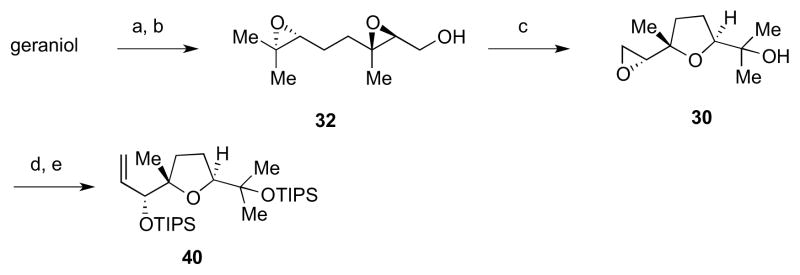
Synthesis of alpha olefin 40. Reagents and conditions: a) L-(+)-DET, Ti(OiPr)4, tBuOOH, 4Å MS, CH2Cl2, −50 °C to −40 °C, 84% ee; b) (i) Shi ketone (36), Oxone, nBu4NHSO4, aq. K2CO3, Na2B4O7 buffer, pH 10.5, (CH3O)2CH2/CH3CN/H2O, 0 °C, 83% (from geraniol); (ii) pNO2BzCl, Et3N, CH2Cl2, rt,; (iii) 1 M NaOH, THF/H2O, rt, 95:5 dr, 95% ee (after recrystallization of the pNO2benzoate derivative); c) NaOH, THF/H2O, rt, 59%; d) (i) (CH3)3SI, nBuLi, THF, −15 °C to rt; (ii) TIPSCl; e) TIPSOTf, Et3N, CH2Cl2, 45 °C, 64% from 30.
The Suzuki–Miyaura fragment coupling commenced with hydroboration of 40 to 41; treatment with aqueous Cs2CO3, Pd(dppf)Cl2, and crude alkenyl triflate 39 at 55 °C provided the desired coupled product (Scheme 9). Deprotection with TBAF gave a mixture of 42 and 43 in approximately 46% combined yield over two steps from 39, providing the full carbon skeleton of ent-1.
Scheme 9.
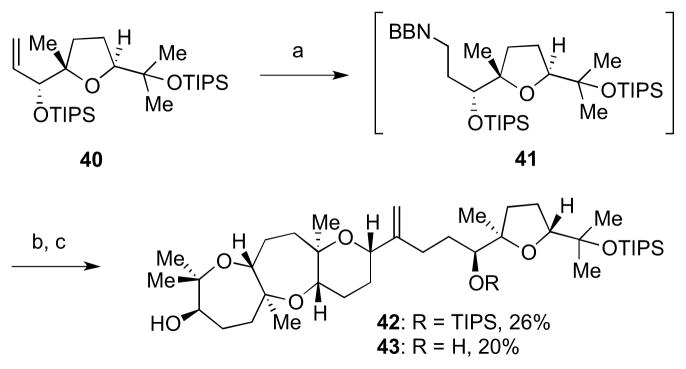
Suzuki cross-coupling to access the carbon skeleton of ent-1. Reagents and conditions: a) 9-BBN dimer, THF, 55 °C; b) 39, PdCl2(dppf), aq. Cs2CO3, THF/DMF/H2O, 55 °C; c) TBAF, THF, rt, 26% 42 and 20% 43 from 39.
Model system 44 was used to investigate the next key transformation, installation of the neopentyl bromide moiety.13 Oxepene 46 was often the main product observed upon conversion of the alcohol to a good leaving group followed by treatment with various bromide sources. Examination of X–ray structural data of 34 (Scheme 6, might be representative of 44) showed a β-hydrogen antiperiplanar to the alcohol, hence allowing facile elimination to occur. Attempts to convert oxepene 46 to the desired bromide 45 under acidic conditions led only to decomposition.
A new strategy was necessary to prepare ent-1 and we turned to the current proposed biosynthesis of 1 for inspiration (Scheme 11).2 Starting from (6S,7S,10R,11R,14R,15R,18S,19S)-squalene tetraepoxide 47, the proposed bioprecursor to 1, an acid-initiated epoxide-opening cascade could give bicyclic intermediate 48. This is followed by a discrete bromoetherification step to complete the fused tricyclic portion of 1 bearing a bromo-oxepane. Support for this mechanism comes from the isolation of predehydrovenustatriol acetate, a metabolite containing the entire carbon skeleton of venustatriol (2), but lacking the bromo-oxane ring.2 Bromoetherification to form a single bromo-oxane or bromo-oxepane ring has been used widely in the syntheses of various bromotriterpenes.20
Scheme 11.

Current proposed biosynthesis of 1.
A biogenesis in which an epoxide-opening cascade is initiated by formation of a bromonium ion has been proposed for thyrsiferol,20b venustatriol,3 and enshuol,5 but yet to be demonstrated chemically. McDonald21 and Holton22 demonstrated that an epoxide-opening event can be initiated by electrophilic activation of an alkene (using a bromenium or phenylselenium ion, respectively) to afford two rings simultaneously. We surmised that a bromonium-initiated cascade involving a multiepoxide chain likely to be operative in the biosynthesis of 1 (Scheme 12) and decided to pursue such a strategy to construct ent-1.
Scheme 12.
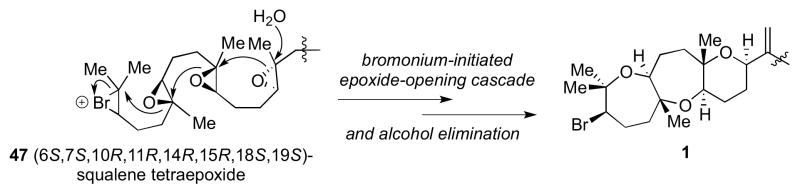
A bromonium-initiated epoxide-opening cascade to access 1.
2.2. Model studies of the bromonium-initiated cyclizations
Initial cyclization studies were conducted using monoepoxide model system 50 with a tBu ester trapping nucleophile (Table 1).13 Using the conditions reported by McDonald et al. (using Br(coll)2ClO4 in CH2Cl2),21 no desired product was obtained (entry 1). However, using the highly polar non-nucleophilic solvent 1,1,1,3,3,3-hexafluoro-iso-propanol (HFIP),22,23 the desired bicyclic lactones 52 and 52′ were obtained in 73–76% combined yield (as a 1:1 mixture of C3 diastereomers, entry 2). This transformation could be conducted using either NBS or Br(coll)2ClO4 as the reagent for presumed bromonium formation.
Table 1.
Bromonium-initiated cyclization studies using monoepoxide model systems.
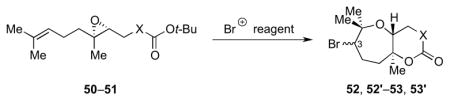
| ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | X | Solvent | Product | Yield (%)a | |
| NBS | Br(coll)2ClO4 | |||||
| 1 | 50 | CH2 | CH2Cl2 | 52, 52′ | na | -- |
| 2 | 50 | CH2 | HFIP | 52, 52′ | 76 | 73 |
| 3 | 51 | O | HFIP | 53, 53′ | 91 | 99 |
Isolated as a 1:1 mixture of diastereomers in all cases.
When the new conditions were applied to monoepoxide substrate 51 bearing a tBu carbonate trapping nucleophile, carbonates 53 and 53′ were obtained in nearly quantitative yields, significantly improved from the previously reported value (entry 3).21
Encouraged by this result, we applied the method to diepoxide substrates containing a tBu ester, carbonate, or alcohol trapping nucleophiles (Table 2, 54–56).7 Generally, yields of desired products did not depend significantly on the reagent used for bromonium formation. A trapping nucleophile attached to an sp2-hybridized carbon usually gave higher yields than that attached to an sp3-hybridized carbon.
Table 2.
Bromonium-initiated cyclization studies using diepoxide model systems.
| Substrate | Product | Yield (%)a | |
|---|---|---|---|
| NBS | Br(coll)2ClO4 | ||
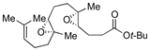
|
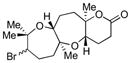
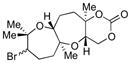
|
73 | 61 |
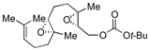
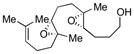
|
|
66 | 65 |
|
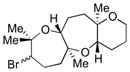
|
58 | 52 |
Isolated as a 1:1 mixture of diastereomers in all cases.
The bromonium-initiated epoxide-opening cascades could also incorporate intermolecular trapping nucleophiles when a substrate lacking an intramolecular nucleophile such as 60 was used (Table 3).13 Potentially, this could enable an approach to the fused tricyclic portion of ent-1 closer to the proposed biogenesis, through incorporation of a water molecule (Scheme 12). As shown in Table 3, an ester (entry 1), primary alcohol (entry 2), or even water (entry 3) afforded bicyclic products 61, 61′–63, 63′ in good yields. If no exogenous nucleophiles were added, HFIP itself was incorporated to form 64, 64′ (entry 4). Use of a hydroxide base gave both alcohols 63, 63′ and hexafluorinated products 64, 64′, perhaps arising from initial deprotonation of the solvent (entry 5).
Table 3.
Incorporation of exogenous trapping nucleophiles.
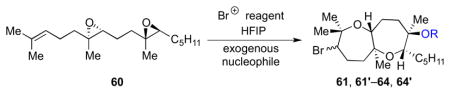
| ||||
|---|---|---|---|---|
| Entry | Nucleophile (equiv) | R | Product | Yield (%)a |
| 1 | Bu4NOAc (1.5) | Ac | 61, 61′ | 74b |
| 2 | EtOH (56) | Et | 62, 62′ | 56c |
| 3 | H2O (125) | H | 63, 63′ | 41b |
| 4 | -- | CH(CF3)2 | 64, 64′ | 33c |
| 5 | CsOH•H2O (1.5) | H | 63, 63′ | 45b,d |
Isolated as a 1:1 mixture of diastereomers in all cases.
NBS used.
Br(coll)2ClO4 used.
19% of 64, 64′ also isolated.
Though a 1:1 mixture of diastereomers was isolated in each of these cyclizations, when one such mixture (57, 57′) was subjected to base, the diastereomer with relative configuration found in ent-1 (57) was more resistant toward E2 elimination (Scheme 13). Unsurprisingly, the diastereomer that underwent facile elimination (57′) was of the same relative configuration as model system 44 (Scheme 10). Observation of the three-dimensional structures of 57 and 57′ explained the differential aptitutes of these intermediates toward E2 elimination.24 As shown in Figure 2, there was a β-hydrogen antiperiplanar to the bromine in 57′, which was not found in 57. Hence, we have shown that elimination of either a bromide (57′) or an alcohol derivative (Scheme 10) could give rise to an oxepene, which is found in the related natural product armatol A (5, Figure 1).
Scheme 13.
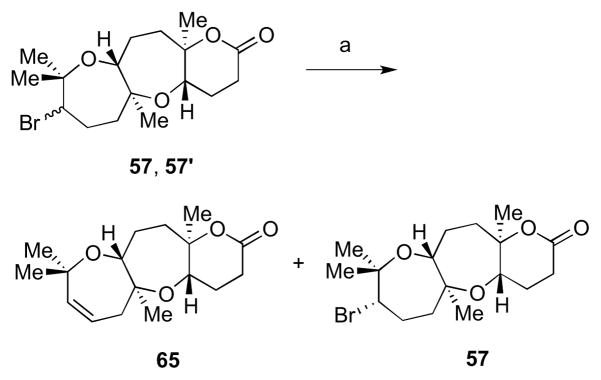
Differential aptitudes of diastereomers 57 and 57′ toward E2 elimination. Reagents and conditions: a) DBU, DMSO, 80 °C.
Scheme 10.
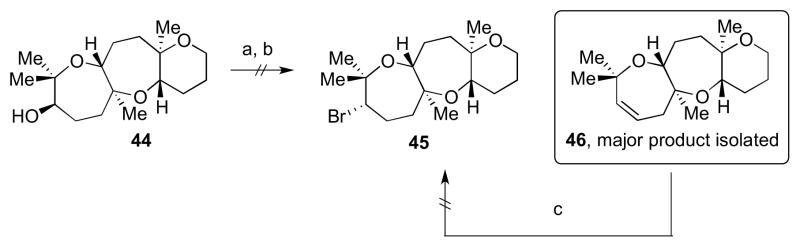
Representative bromine installation studies. Reagents and conditions: a) ClCH2SO2Cl, pyr, DMAP; b) LiBr, DMPU; c) HBr, AcOH or PBr3, SiO2.
Figure 2.
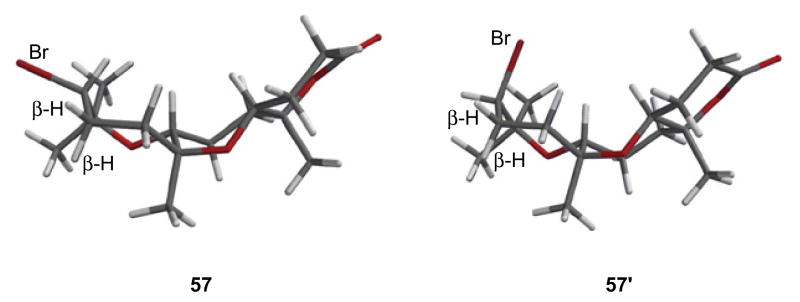
Computed three-dimensional structures of 57 and 57′ (Spartan, HF/6-31G*).
2.3. A new cascade to construct the tricyclic fragment of ent-1
Though 57 could potentially be carried forward to complete the synthesis (Scheme 7), we proposed a new cascade to construct the tricyclic portion of ent-1: utilizing triepoxide 66 to give tetracycle 67 (Scheme 14).7 An advantage to this new strategy would be construction of the C14 stereocenter in the cascade, which was previously unattainable (Scheme 3) and required a number of steps to construct (Scheme 7). A tBu carbonate was chosen as the intramolecular trapping nucleophile in place of a tBu ester previously used in 31, for more straightforward elaboration toward fragment coupling (vide infra).10a–b The fragment coupling partner 30 would be constructed using our previous strategy (Scheme 8).
Scheme 14.

A new cascade for the construction of the tricyclic fragment of ent-1.
Upon exposure of 66 to NBS in HFIP at 0 °C for 15 min, tetracycles 67 and 67′ were obtained in 72% combined yield as a 1:1 mixture of C3 epimers (Scheme 15).7 After chromatographic separation, basic hydrolysis of the cyclic carbonate moiety of 67 was followed by diol cleavage with NaIO4 to give methyl ketone 68. This was converted to alkenyl triflate 69 using Comins’ reagent and LHMDS as before.
Scheme 15.
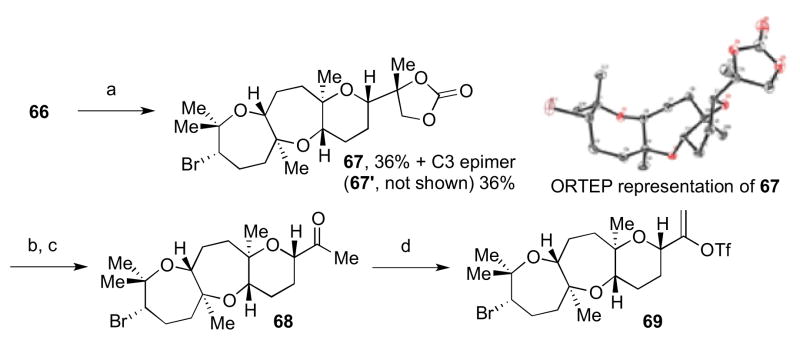
Access to the tricyclic fragment of ent-1. Reagents and conditions: a) NBS, 4Å MS, HFIP, 0 °C, 36% (+C3 epimer, 36%); b) NaOH, MeOH, rt, 83%; c) NaIO4, THF/H2O, rt, 96%; d) (SO2CF3)2NC5H3NCl, LHMDS, THF, −78 °C, quant.
Compared to the previous condition, the Suzuki–Miyaura fragment coupling utilizing 69 was carried out at a lower temperature to avoid complications involving the sp3 C–Br atom (Scheme 16). Smaller silyl groups on alpha olefin 70 facilitated the final deprotection step using TBAF. This concluded the first total synthesis of ent-dioxepandehydrothyrsiferol (ent-1). Discovery of a bromonium-initiated epoxide-opening cascade enabled construction of the unique trans-anti-trans 7,7,6-fused polyether framework containing a bromo-oxepane in a single step.7
Scheme 16.
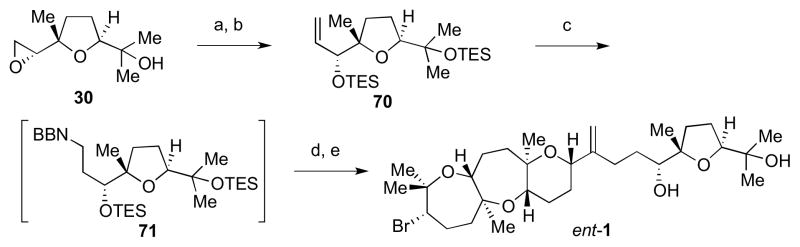
Completion of the synthesis of ent-1. Reagents and conditions: a) (CH3)3SI, nBuLi, THF, −13 °C to 5 °C, 73%; b) TESCl, imid, DMF, rt, 95%; c) 9-BBN dimer, THF, 60 °C; d) 69, PdCl2(dppf), aq. Cs2CO3, THF/DMF/H2O, 40 °C, 78%; e) TBAF, THF, rt, 83%.
2.4. Total synthesis and structural elucidation of armatol A
The armatol family of natural products was isolated in 2000,8 and there have been only two synthetic studies reported toward a single member of this class, armatol F.25 In these communications, the authors reported approaches toward the proposed structure of the natural product.8 However, the absolute and relative configurations of armatols A–F had actually never been determined, and such was our goal through de novo synthesis.
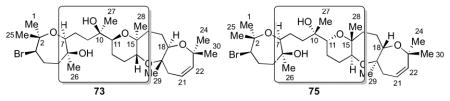
Though the absolute stereochemistry of the lone bromo-oxepane ring has been deduced by degradation studies followed by Mosher analysis,8,26 the absolute stereochemistry of the C10 tetrasubstituted center had yet to be determined. In addition, though the relative stereochemistry of the 6,7,7-fused tricycle was known to be trans-syn-trans (deduced from NOE, NOESY and other NMR techniques),8 the absolute stereochemistry of this fragment relative to the rest of the molecule was unknown. Hence, four possible diastereomers (72–75) were consistent with the published data (Scheme 18).
Scheme 18.

The four possible diastereomers of armatol A.
Another intriguing feature found in armatols A, B, D, and F is the cis relationship between the H and Me groups found on the lone bromo-oxepane moiety (Scheme 18, shown for 72), as opposed to the more common trans relationship.
In order to determine the absolute and relative configuration of armatol A, a convergent approach to access diastereomers 72–75 was devised (Scheme 19, shown for 72). Key features of the route included: 1) installation of the C21–C22 olefin via elimination of a suitably activated alcohol and (2) late stage installation of the C10 stereocenter via methylmetal addition to a ketone such as 76. Lastly, (3) the C9–C10 bond could be formed from alkyne 77 and tricyclic aldehyde 78.
Scheme 19.

Retrosynthetic disconnection of one possible diastereomer of armatol A (72).
Based on our study with model system 51 (Table 1), cis-fused bicyclic carbonate 79 could be derived from nerol derivative 80 via a bromonium-initiated 7-endo-trig cyclization. The fused trans-syn-trans tricycle 78 could be accessed by a Lewis acid-initiated epoxide-opening cascade of triepoxide 14. Since both enantiomers of fragment 14 would be readily accessible, this approach could provide 72–75 in a rapid and convergent manner.
Access to each enantiomer of the key fused tricyclic fragment is shown in Scheme 20. From aldehyde 82,27 vinylmetal addition followed by enzymatic resolution provided tetraenes 83 and 84 with high enantioselectivities.28 Shi asymmetric epoxidation was followed by deacetylation to give triepoxide 14. Exposure to BF3•OEt2 gave the trans-syn-trans tricycle 85 in 18% yield (two steps from 14 after acetylation). Notably, an allylic alcohol trapping nucleophile was tolerated in this case, as opposed to our previous observation in Scheme 3. This is perhaps due to the absence of any unfavorable diaxial interactions in the transition state of this diastereomer. Ozonolysis of alpha olefin 85 gave 78, ready for fragment coupling. Tetraene 84, on the other hand, was exposed to ent-Shi catalyst (ent-36) under a similar epoxidation condition to give ent-14.29 Cyclization as before provided ent-85, which was oxidized to ent-78.
Scheme 20.

Syntheses of fused tricycles 78 and ent-78. Reagents and conditions: a) CH2CHMgBr, THF, −10 °C to rt, 73%; b) Novozyme 435, vinyl acetate, Et2O, 4 °C, 40%, 99% ee (for 83), 33%, 98% ee (for 84); c) Shi ketone (36), Oxone, nBu4NHSO4, aq. K2CO3, Na2B4O7 buffer, pH 10.5, (CH3O)2CH2/CH3CN/H2O, rt, 88%, 3.5:1 dr; d) LiOH, THF/MeOH/H2O, 0 °C, 93%; e) BF3•OEt2, CH2Cl2, −78 °C; f) Ac2O, Et3N, DMAP, CH2Cl2, rt, 18% from 14; g) (i) O3, NaHCO3, CH2Cl2/MeOH, −78 °C; (ii) PPh3, −78 °C to rt, 65%; h) ent-Shi ketone (ent-36), Oxone, nBu4NHSO4, aq. K2CO3, Na2B4O7 buffer, pH 10.5, (CH3O)2CH2/CH3CN/H2O, rt, 61%, 3.5:1 dr; i) BF3•OEt2, CH2Cl2, −78 °C; j) Ac2O, Et3N, DMAP, CH2Cl2, rt, 25% from ent-14; k) (i) O3, NaHCO3, CH2Cl2/MeOH, −78 °C; (ii) PPh3, −78 °C to rt, 74%.
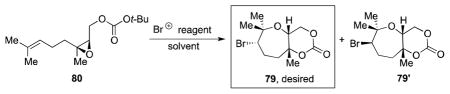
Studies toward formation of the bromo-oxepane fragment of the natural product started with 80 (Table 4).13 As with trans analogue 51, cyclization under literature condition using Br(coll)2ClO4 in CH2Cl2 resulted in a modest combined yield (25%) of diastereomers 79 and 79′ in a 2:3 ratio (Table 4, entry 1).21 Consistent with our earlier findings, employing the polar non-nucleophilic solvent HFIP significantly increased the yield (entry 2). Altering the counterion of the reagent and performing the cyclization in MeNO2 were found to give comparable yields (entries 3 and 4). Bromodiethylsulfonium bromopentachloroantimonate (BDSB), a reagent recently reported by Snyder et al., also provided similar yields for this cyclization (entries 5 and 6).30 The decreased yields, as compared to 51, may result from a non-optimal positioning of the carbonate trapping nucleophile. Hence, the unstable epoxonium ion intermediate would be prone to undergo undesirable side reactions more readily.31
Table 4.
Cyclization studies of nerol-derived carbonate 80.
| |||||
|---|---|---|---|---|---|
| Entry | Bromonium source | Solvent | T (°C) | Yield 79 (%) | Yield 79′ (%) |
| 1 | Br(coll)2ClO4 | CH3CN | −40 | 10 | 15 |
| 2 | Br(coll)2ClO4 | HFIP | 0 | 18 | 32 |
| 3 | Br(coll)2BF4 | HFIP | 0 | 17 | 31 |
| 4 | Br(coll)2BF4 | MeNO2 | 0 | 22 | 36 |
| 5 | BrSEt2SbCl5 | MeNO2 | 0 | 22 | 34 |
| 6 | BrSEt2SbCl5 | HFIP | 0 | 23 | 36 |
With the optimum cyclization conditions in hand, access to the bromo-oxepane fragment of 5 proceeded as shown in Scheme 21. Starting from nerol, Sharpless asymmetric epoxidation installed the epoxide in a modest enantioselectivity. This could be enhanced by enzymatic resolution,32 which was then followed by installation of the tBu carbonate group to give ent-80. The bromonium-initiated cyclization was conducted using Br(coll)2BF4 in MeNO2 to give the desired diastereomer ent-79 in 22% yield. Hydrolysis under basic condition followed by oxidation provided aldehyde 86, which was converted via Seyferth–Gilbert homologation to alkyne 77 after TMS protection.33
Scheme 21.
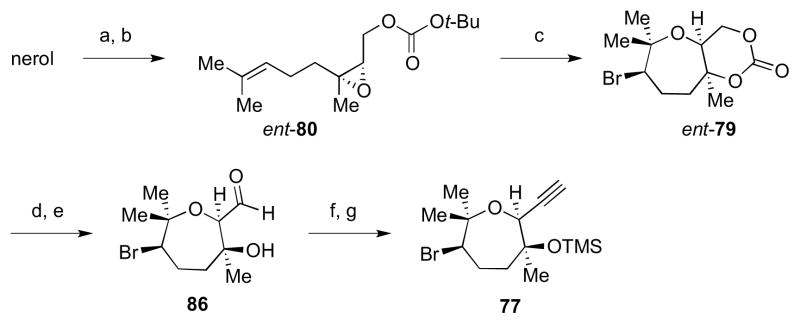
Elaboration of the bromo-oxepane moiety for fragment coupling. Reagents and conditions: a) (i) L-(+)-DET, Ti(OiPr)4, tBuOOH, 4 Å MS, CH2Cl2, −23 °C; (ii) Amano lipase-PS, vinyl acetate, Et2O, 0 °C, 99% ee, 41% from nerol; (iii) K2CO3, MeOH, rt, 99%; b) Boc2O, 1-Meimid, 0 °C to rt, 77%; c) Br(coll)2BF4, 4 Å MS, MeNO2, 0 °C, 22%; d) NaOH, MeOH, rt; e) SO3•pyr, DMSO, Et3N, CH2Cl2, 0 °C to rt; 74% (2 steps from ent-79); f) dimethyl-1-diazo-2-oxopropylphosphate, K2CO3, MeOH, rt; g) TMSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 59% (2 steps from 86).
With an ample supply of both coupling partners in hand, we turned our attention to the backbone of armatol A, as shown in Scheme 22. Alkyne 77 was treated with Cp2Zr(H)Cl, followed by transmetalation to the organozinc intermediate, and subsequently treated with aldehyde 78 to give allylic alcohol 87.34 Hydrogenation in EtOH gave the saturated alkane with concomitant TMS ether cleavage. Finally, oxidation to ketone 76 was accomplished using TPAP and NMO. An analogous sequence of steps carried out with 77 and ent-78 led to ketone 89. Starting with ketones 76 and 89, all four possible diastereomers of armatol A (72–75) could be accessed.
Scheme 22.

Fragment coupling to access the backbone of armatol A. Reagents and conditions: a) (i) [Cp2Zr(H)Cl], CH2Cl2, rt; (ii) Me2Zn, CH2Cl2, −78 °C; (iii) 78, CH2Cl2, −78 °C to rt, 64%; b) H2 (1 atm), Pd/C, EtOH, rt; c) TPAP, NMO, 4ÅMS, CH2Cl2, 0 °C to rt, 53% (from 87); d) (i) [Cp2Zr(H)Cl], CH2Cl2, rt; (ii) Me2Zn, CH2Cl2, −78 °C; (iii) ent-78, CH2Cl2, −78 °C to rt; e) H2 (1 atm), Pd/C, EtOH, rt, 52% (from ent-78); f) TPAP, NMO, 4Å MS, CH2Cl2, 0 °C to rt, 90%.
Treatment of 76 with methyllithium at −78 °C in THF led to a 1:1 ratio of C10 epimers (90 and 91, Scheme 23). The relative configuration of each product was assigned after reaction of 76 with methylmagnesium bromide in THF, which gave the (R)-diastereomer 90 selectively after acetate deprotection. This is due to addition of MeMgBr to the less hindered face of the chelate formed between the ketone and the oxygen of the THP ring.35
Scheme 23.

Access to two diastereomeric carbon skeletons of armatol A. Reagents and conditions: a) MeLi, THF, −78 °C, 84%, 1:1 dr; b) MeMgBr, THF, −78 °C to 0 °C; c) K2CO3, MeOH, rt, 67%.
Similarly, reaction of 89 with MeLi yielded 92 and 93, whilst reaction with MeMgBr gave 92 only after deacetylation (Scheme 24). Gratifyingly, no Br or acetate elimination was observed under these conditions.
Scheme 24.

Access to the remaining diastereomeric carbon skeletons of armatol A. Reagents and conditions: a) MeLi, THF, −78 °C, 75%, 1:1 dr; b) MeMgBr, THF, −78 °C, 64%; c) K2CO3, MeOH, rt.
At this point, the 13C NMR shifts between the four diastereomers 90–93 and natural armatol A were compared (Table 5).8 It was observed that the three carbons directly attached to the C10 stereocenter (C9, C11, and C27) in 90 and 92 were much closer to the assigned carbon shifts of the natural product (within 0.4 ppm). Conversely, the 13C NMR shifts of 91 and 93 differ by as much as 2.3 ppm from the reported data, allowing us to rule out 91 and 93 as potential structures leading to armatol A.
Table 5.
Comparison of 13C NMR shifts of 90–93 against natural armatol A.8
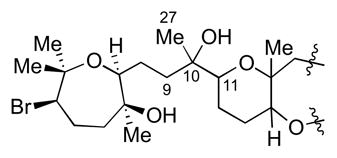
| |||||
|---|---|---|---|---|---|
| Position | δ 90a | δ 91a | δ 92a | δ 93a | δ natural 5a,8 |
| 9 | 34.4 | 36.8b | 34.7 | 36.8b | 34.5 |
| 10 | 73.3 | 73.4 | 73.3 | 73.4 | 73.4 |
| 11 | 76.3 | 74.4b | 76.3 | 75.4b | 76.1 |
| 27 | 24.0 | 23.0b | 24.3 | 21.7b | 23.9 |
13C NMR data taken in C6D6, referenced to 128.0 ppm.
|δ|>0.4 ppm relative to natural 5.
The final challenge involved elimination of the neopentyl alcohol moiety to install the oxepene, which was studied using model system 94. Standard elimination conditions (Martin’s sulfurane, Grieco’s protocol, and Burgess’s reagent) all failed to provide the desired alkene.36 Conversion to the activated chloromesylate 95 followed by heating with base led to ring contraction products 97 and 98 (Scheme 25). These outcomes were unexpected, given our previous experience with model system 44 (Scheme 10). Products 97 and 98 could arise from an intermediate such as 96, which resulted from anchimeric assistance of one of the oxepane’s oxygen in displacing the leaving group. The desired oxepene 99 was not observed.
Scheme 25.

Oxepene installation studies using model system 94. Reagents and conditions: a) DBU, THF, reflux.
An alternative strategy was needed for oxepene installation. Using model system 100, oxidation of either diastereomer of the secondary alcohol with Dess-Martin periodinane gave the desired ketone in 64% yield (Scheme 26). This could be converted to hydrazone 101 using H2NNHTs in MeOH in 43% yield.
Scheme 26.
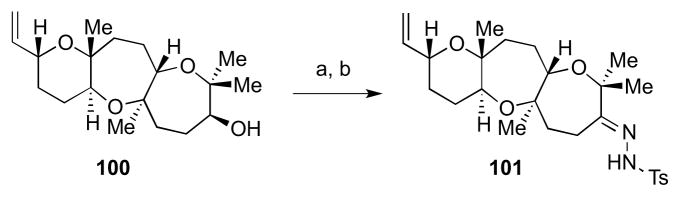
Elaboration to hydrazone 101. Reagents and conditions: a) Dess-Martin periodinane, NaHCO3, CH2Cl2, 0 °C to rt, 64%; b) H2NNHTs, MeOH, 40 °C, 43%.
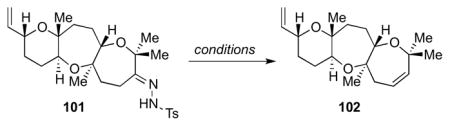
After hydrazone formation, 101 was subjected to an array of conditions as shown in Table 6.37 Only sodium hydride in toluene or THF led to the desired olefin 102 in up to 16% yield.38 With a method to form the oxepene in hand, we were poised to complete the synthesis and assign the absolute configuration of armatol A.
Table 6.
Model studies for oxepene installation from hydrazone 101.
| ||||
|---|---|---|---|---|
| Entry | Base | T (°C) | Solvent | Result |
| 1 | MeLi | −78 °C to rt | THF | NR |
| 2 | nBuLi | −78 °C to rt | THF | Decomp. |
| 3 | LHMDS | −78 °C to rt | THF | NR |
| 4 | NaH | reflux | THF | 12% yield |
| 5 | NaH | 90 °C | PhMe | 16% yield |
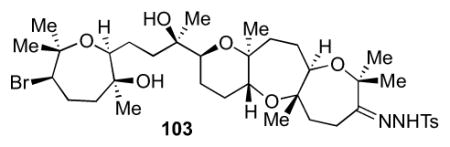
The oxidation-elimination strategy was applied to late-stage intermediate 90. Ley oxidation followed by condensation with TsNHNH2 led to hydrazone 103 in a good yield (Scheme 27). Treatment with sodium hydride in THF at 80–100 °C led to the desired alkene 73 in 6% yield. A similar sequence carried out with 92 gave alkene 75 (Scheme 28).
Scheme 27.
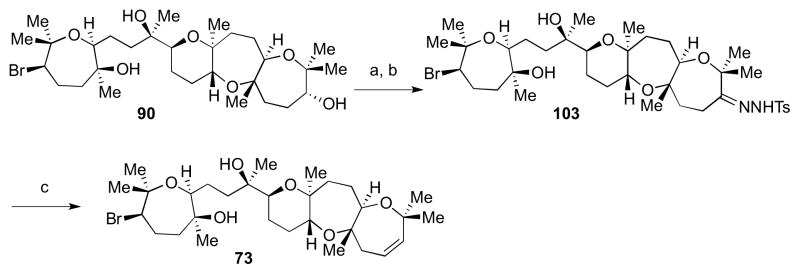
Access to a possible diastereomer of 5. Reagents and conditions: a) TPAP, NMO, 4Å MS, CH2Cl2, 0 °C to rt, 82%; b) H2NNHTs, MeOH, 45 °C, 63%; c) NaH, THF, 90 °C to 110 °C, 6%.
Scheme 28.
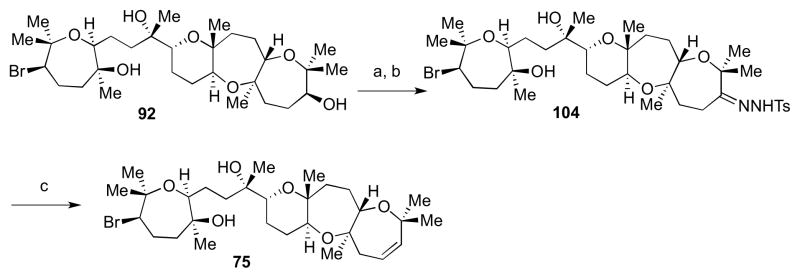
Access to alkene 75. Reagents and conditions: a) TPAP, NMO, 4Å MS, CH2Cl2, 0 °C to rt, 97%; b) H2NNHTs, MeOH, 45 °C, 63%; c) NaH, THF, 100 °C, 6%.
Table 7 summarizes selected 13C NMR data for 73 and 75 compared to natural armatol A.8 All signals in 73 are within 0.1 ppm of the reported data, with the exception of C11 (in C6D6), which differs by 0.2 ppm. Hydrogen bonding of the tertiary alcohol with the adjacent THP ring might account for this difference. Conversely, 13C NMR data for 75 in C6D6 differ by at least 0.2 ppm, specifically in the C7 to C13 portion of the molecule. Even more significantly, the optical rotation of 73 in CHCl3 is +33.9, while that of 75 is −17.9. The reported optical rotation of armatol A in CHCl3 is +43.4. Taken together, these data confirm the correct structure of armatol A (5) as 73.
Table 7.
Tabulation of 13C NMR data of diastereomers 73 and 75 as compared to natural armatol A.8
| |||||||
|---|---|---|---|---|---|---|---|
| Position | δ 73a | δ 75a | δ natural 5a,8 | Position | δ 73b | δ 75b | δ natural 5b,8 |
| 7 | 76.4 | 76.0c | 76.4 | 7 | 76.6 | 76.1c | 76.6 |
| 8 | 23.4 | 23.7c | 23.4 | 8 | 23.9 | 24.0c | 23.7 |
| 9 | 33.4 | 33.4 | 33.4 | 9 | 34.1 | 34.3c | 34.1 |
| 10 | 73.1 | 73.0 | 73.1 | 10 | 73.1 | 72.8 | 73.0 |
| 11 | 74.9 | 75.0 | 74.9 | 11 | 75.5 | 75.4c | 75.7 |
| 12 | 27.0 | 27.1 | 26.9 | 12 | 25.6 | 25.8 | 25.6 |
| 13 | 27.2 | 27.2 | 27.3 | 13 | 27.5 | 27.6c | 27.3 |
| 14 | 73.5 | 73.5 | 73.5 | 14 | 74.2 | 74.2 | 74.1 |
| 15 | 77.2 | 77.2 | 77.2 | 15 | 77.4 | 77.4 | 77.4 |
| 26 | 26.0 | 25.5c | 25.9 | 26 | 25.3 | 25.2 | 25.2 |
| 27 | 23.4 | 23.8c | 23.4 | 27 | 23.5 | 24.1c | 23.5 |
| 28 | 26.0 | 25.0c | 25.9 | 28 | 17.5 | 17.5 | 17.5 |
| 29 | 18.0 | 18.0 | 18.0 | 29 | 18.1 | 18.2 | 18.1 |
| 30 | 29.4 | 29.4 | 29.3 | 30 | 29.5 | 29.5 | 29.5 |
13C NMR data taken in CDCl3, referenced to 77.0 ppm.
13C NMR data taken in C6D6, referenced to 128.0 ppm.
|δ|>0.2 ppm.
Upon completion of this synthesis, comparisons were made for 13C and 1H NMR data of structure 73 against the reported data for armatols B–F (6–10, table 8).8
Table 8.
Select tabulation of NMR data of 73 compared to the reported data for armatols B–F (6–10).8
| Position | δ 73a,b | δ 6b,8 | δ 7b,8 | δ 8b,8 | δ 9b,8 | δ 10b,8 |
|---|---|---|---|---|---|---|
| C9 | 33.4 | 33.5 | 33.4 | 34.0c | 33.7 | 33.1 |
| C10 | 73.1 | 73.2 | 73.4 | 73.7c | 73.7c | 72.9 |
| C11 | 74.9 | 75.4c | 75.5c | 75.7c | 75.3 | 76.6c |
| C27 | 23.4 | 23.5 | 23.5 | 25.5c | 23.5 | 23.4 |
| H27 | 1.09 | 1.09 | 1.09 | 1.09 | 1.10 | 1.10 |
Synthetic data for armatol A.
13C NMR data taken in CDCl3, referenced to 77.0 ppm. 1H NMR data taken in CDCl3, referenced to 7.26 ppm.
|δ|>0.4 ppm relative to 73.
The data is consistent with the hypothesis that the natural products share identical quaternary stereochemistry at C10. We believe this also applies to armatol D, despite the marked difference in the 13C NMR shift at C27 of this natural product as compared to 73. We have shown the absolute stereochemistry of armatol A (73) through de novo synthesis and have support that our proposed structure might apply for the remaining members of the armatol family to give structures 105–109 (Scheme 29).
Scheme 29.

Proposed structures of armatols A–F.
2.5. Proposed biogenesis for the armatol family of natural products
Our proposed biogenesis for the armatol natural products (73 and 105–108) is shown in Scheme 30. In line with our previous proposal for dioxepandehydrothyrsiferol (1), a bromonium-initiated epoxide-opening cascade of a polyepoxide precursor such as 110 could give tricycles 112 and 112′, common intermediates to the natural products. Bromonium formation at C22–C23 alkene would not have to be facially-selective, as both C22 diastereomers are found in nature. A stereoselective bromonium formation at the C2–C3 alkene could initiate a cyclization event with the remaining epoxide. Opening of this epoxonium at C6 by a water molecule with inversion would give armatol C (106) and E (108) while opening with retention would give armatol B (105) and D (107). Armatol A (73) could then arise from elimination of an HBr equivalent from 105 or 107. A different polyepoxide precursor might give rise to 109, given the unique cis ring junction between two fused oxepanes found in this natural product.
Scheme 30.

Proposed biogenesis for 73 and 105–108.
3. Conclusion
In summary, we have completed the first total syntheses of ent-dioxepandehydrothyrsiferol (ent-1) and armatol A (73). Through our studies of ent-1, we have discovered a biomimetic bromonium-initiated epoxide-opening cascade that proceeded optimally in a polar non-nucleophilic solvent. This methodology was used to construct the fused trans-anti-trans tricyclic portion of ent-1, bearing a bromo-oxepane in one step from a polyepoxide precursor, previously unattainable utilizing the corresponding Lewis acid-initiated epoxide-opening cascade. The bromo-oxepane moiety in 73 was also constructed using this method from a nerol derivative, representing one of the first examples of such a transformation utilizing a cis epoxide.
The convergent route developed toward armatol A allowed rapid access to all four possible diastereomers of this natural product (72–75). A Lewis acid-initiated epoxide-opening cascade constructed both enantiomers of the fused trans-syn-trans tricycle. After fragment coupling and installation of the C10 quaternary center, a Bamford–Stevens reaction constructed the oxepene (as part of the tricycle) and confirmed the relative and absolute configuration of armatol A (73). It is also possible that the structure we propose applies to the other members of the armatol family. Finally, through our work with ent-1 and the discovery of the bromonium-initiated epoxide-opening cascade, we have proposed a unified biogenesis for this natural product as well as members of the armatol family.
4. Experimental
4.1 General
Experimental procedures and characterization data for selected compounds are included in this text. Additional experimental procedures, characterization data, and spectra for all new compounds can be found in the Supporting Information submitted along with this manuscript and the Supporting Information of a previous communication.7
Unless otherwise noted, all reactions were performed under an oxygen-free atmosphere of argon with rigid exclusion of moisture from reagents and glassware. Teflon stir bars were oven or flame-dried prior to use. Except where noted, all solvents and triethylamine used in the reactions were purified via an SG Water USA solvent column system. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) (≥ 99%) was purchased from Aldrich Chemical Company and was used without further purification. Nitromethane was dried at 90 °C overnight with CaH2 before being distilled into and stored in an airtight Schlenk tube. 4Å MS were activated by flame drying under high vacuum three times (with cooling in between) immediately before use. HN(i-Pr)2, hexamethylphosphoramide (HMPA), and 1,3-dimethyltetrahydropyrimin-2(1H)-one (DMPU) were distilled from CaH2 before use. BF3•OEt2 and Ti(Oi-Pr)4 were distilled from CaH2 before use. EtOH was distilled from 4Å MS before use. Solvents for the Suzuki cross-coupling reaction were rigorously degassed through freeze-pump-thaw cycles within a week before use and kept in an airtight Schlenck tube. Cs2CO3 used in the Suzuki cross-coupling reaction was pumped on under high vacuum overnight, kept and used inside a glovebox. 9-Borabicyclo[3.3.1]nonane (9-BBN) dimer was pumped on under high vacuum overnight, kept and used inside a glovebox. CuI and the CH2Cl2 adduct of PdCl2(dppf) was purchased from Strem chemical company and kept and used inside a glovebox. Bis(cyclooctadienyl)nickel(0) (Ni(cod)2) and tricyclopentylphosphine (Cyp3P) were purchased from Strem Chemicals, Inc., stored under nitrogen atmosphere and used without further purification. Tetrabutylammonium acetate was pumped under vacuum overnight, kept and used inside a glovebox. Trimethylsulfonium iodide was azeotropically dried from toluene three times before use. SO3•pyr and (CH3)3N•HCl were pumped on under high vacuum overnight before use. NaIO4 adsorbed on SiO2 was made as follows: 2.57 g of NaIO4 was dissolved in 5 mL of H2O at 70–80 °C. It was then poured into 10.0 g of silica. The resulting mixture was stirred well for 30 min until homogenous.39 Shi ketone 36 was prepared from D-fructose according to the procedure of Vidal-Ferran and coworkers and was used without recrystallization.40 t-BuOOH was purchased from Fluka as a ~5.5 M solution in decane stored over activated 4Å MS. MsCl was distilled from calcium hydride before use. [(Ph3P)CuH]6 was purchased from Fluka (brick red powder), pumped on under high vacuum overnight, kept and used inside a glovebox. N-Bromosuccinimide (NBS) was recrystallized from H2O before use and kept at 0 °C in the absence of light.
Br(coll)2ClO4 was prepared according to the previously reported procedure,41 and kept at 0 °C in the absence of light.
Analytical thin layer chromatography was performed using EM Science silica gel 60 F254 plates. The developed chromatogram was analyzed by UV lamp (254 nm) and Ceric Ammonium Molybdate (CAM) or ethanolic phosphomolybdic acid (PMA) solution. Liquid chromatography was performed using flash chromatography of the indicated solvent system on Silicycle Silica Gel (230–400 mesh). Alternatively, flash chromatography was also performed on the Biotage Isolera™ automated purification unit with SNAP columns.™ Analytical HPLC was performed on the column phase indicated on a Hewlett-Packard 1100 Series HPLC. 1H and 13C NMR spectra were recorded on a Varian Inova-500 MHz, Bruker AVANCE-400 MHz, or Bruker AVANCE-600 MHz spectrometer in CDCl3 or C6D6. Chemical shifts in 1H NMR spectra are reported in part per million (ppm) on the δ scale from an internal standard of residual CHCl3 in CDCl3 (7.27 ppm) or residual C6HD5 in C6D6 (7.16 ppm). Data are reported as follows: chemical shift, multiplicity (app = apparent, br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant in hertz (Hz), and integration. Chemical shifts of 13C NMR spectra are reported in ppm from the central peak of CDCl3 (77.23 ppm) or residual C6D6 (128.39 ppm) on the δ scale.19F NMR spectra were recorded on a Varian Inova-300 MHz or Bruker AVANCE-400 MHz spectrometer in C6D6 using either CF3CH2OH (at −77.8 ppm) or C6F6 (at −164.9 ppm) as a reference. Infrared (IR) spectra were recorded on a Perkin-Elmer 2000 FT-IR. High-resolution mass spectra (HRMS) were obtained on a Bruker Daltonics APEXIV 4.7 Tesla Fourier Transform Ion Cyclotron Resonanace Mass Spectrometer by Li Li of the Massachusetts Institute of Technology Department of Chemistry Instrumentation Facility. Optical rotations were measured on a Jasco Model 1010 polarimeter at 589 nm.
4.1 Preparation of lactones S5 and 28
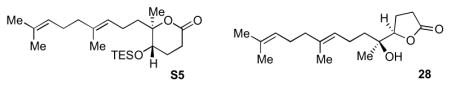
Aldehyde 1513 (600 mg, 2.27 mmol, 100 mol %) was dissolved in 2-methyl-2-butene (60 mL, ~25 mL/mmol of aldehyde) at room temperature in a 250 mL Erlenmeyer flask. tert-Butyl alcohol (60 mL) and water (30 mL) were added. NaH2PO4•H2O (1.252 g, 9.076 mmol, 400 mol%) was added and the mixture was stirred vigorously at rt for 5 min until the mixture became homogeneous. Sodium chlorite (1.23 g, 13.6 mmol, 600 mol %) was added in one portion. The mixture was stirred vigorously at rt for 1 h. The mixture was poured into 5% aqueous NaH2PO4 solution (200 mL) and extracted twice with diethylether (100 mL then 150 mL). The organic fraction was dried with MgSO4. NMR of the crude reaction mixture showed the desired acid 26. The crude mixture was used directly in the next step.
Crude acid 26 (~2.27 mmol, 100 mol %, used directly without purification) was dissolved in dichloromethane (45 mL). The solution was cooled to −78 °C. BF3•OEt2 was diluted in dichloromethane (5 mL) and added to the reaction mixture over 2 min. Temperature of the cold bath was maintained under −70 °C for 8 h. Acetone was added to the cold bath to warm the bath to −50°C over 5 min. The reaction was quenched with saturated NaHCO3 (20 mL). The flask was removed from the cold bath and warmed to room temperature. The mixture was extracted with dichloromethane. The organic layer was dried with MgSO4. 1H NMR of the crude mixture indicated δ-lactone 27:γ-lactone 28 ratio of 95:5. Column chromatography isolated γ-lactone 28. 1H NMR (400 MHz, C6D6): δ 5.23 (t, J = 6.7 Hz, 1H), 5.17 (t, J = 7.0 Hz, 1H), 3.68 (t, J = 7.4 Hz, 1H), 2.20-1.70 (m, 8H), 1.68 (s, 3H), 1.58 (s, 3H), 1.57 (s, 3H), 1.45-1.10 (m, 4H), 1.08 (s, 3H); 13C NMR (100 MHz, C6D6): δ 176.8, 135.8, 131.7, 125.1, 125.0, 85.5, 72.8, 40.5, 37.8, 29.1, 27.5, 26.2, 23.5, 22.5, 22.1, 18.1, 16.4.
δ-Lactone 27 and imidazole (232 mg, 3.40 mmol, 150 mol %) were dissolved in N,N-dimethylformamide (12 mL). Under argon, chlorotriethylsilane (0.460 mL, 2.72 mmol, 120 mol %) was added in one portion at room temperature. The solution was heated at 50 °C for 4h. The reaction was removed from the oil bath and cooled to rt. The reaction was quenched with water (5 mL, exothermic). The mixture was diluted with diethylether (100 mL) and washed twice with saturated NH4Cl. The organic fraction was dried with MgSO4. Column chromatography isolated 658 mg of silylated δ-lactone S5 (73% yield over three steps from aldehyde 15). [α]20D = +18.0 (c = 2.8, CH2Cl2); IR (NaCl, thin film): 2957, 2914, 2877, 1738, 1457, 1378, 1239, 1098, 1006, 745 cm−1; 1H NMR (400 MHz, C6D6): δ 5.23 (t, J = 6.8 Hz, 1H), 5.20 (t, J = 7.1 Hz, 1H), 3.51 (dd, J = 4.6, 7.2 Hz, 1H), 2.42 (dt, J = 7.3, 18.0 Hz, 1H), 2.30-2.00 (m, 7H), 1.68 (s, 3H), 1.60 (s, 3H), 1.57 (s, 3H), 1.60-1.40 (m, 4H), 1.21 (s, 3H), 0.89 (t, J = 8.0 Hz, 9H), 0.44 (q, J = 7.8 Hz, 6H); 13C NMR (100 MHz, C6D6): δ 168.8, 136.1, 131.6, 125.2, 124.5, 85.2, 69.6, 40.52, 40.49, 27.5, 27.1, 26.2, 25.7, 22.6, 21.3, 18.1, 16.4, 7.4, 5.6; HR-MS (ESI) m/z calcd for C23H42O3Si [M+Na]+: 417.2795, found 417.2802.
4.2 Lewis acid-mediated cyclization to prepare 34
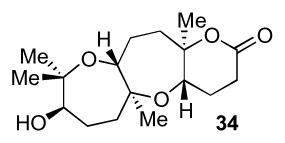
Ester 3113 (5.15 g, 14.0 mmol, 100 mol %) and 1,2,3-trimethoxybenzene (4.70 mg, 27.0 mmol, 200 mol %) were dissolved in CH2Cl2 (280 mL). The mixture was cooled under argon to −78 °C. BF3•OEt2 (1.77 mL, 14.0 mmol, 100 mol %) was added. The mixture was stirred at −78 °C for 1 h and quenched with saturated NaHCO3 (50 mL) at −78 °C. The cold bath was removed and the mixture was warmed to rt. Layers were separated and the aqueous layer was extracted two times with CH2Cl2 (2 × 100 mL). The extract was dried with MgSO4. Column chromatography isolated cyclization product 34 in a concentrated CH2Cl2 solution (~ 3 to 5 mL) and carried on directly to the next step. [α]20D = −3.1 (c = 1.3, CH2Cl2); IR (NaCl, thin film): 3470, 2976, 2941, 1722, 1381, 1273, 1206, 1083 cm−1; 1H NMR (400 MHz, C6D6): δ 4.14 (d, J = 8.9 Hz, 1H), 3.64 (dd, J = 5.1, 11.7 Hz, 1H), 3.40 (t, J = 5.1 Hz, 1H), 2.27 (dt, J = 3.5, 12.8 Hz, 1H), 2.16-2.00 (m, 2H), 1.90-1.45 (m, 10H), 1.26 (s, 3H), 1.20 (s, 3H), 1.13 (s, 3H), 0.97 (s, 3H); 13C NMR (100 MHz, C6D6): δ 169.3, 84.8, 80.5, 78.4, 76.7, 76.5, 68.7, 42.1, 31.9, 29.2, 29.1, 28.8, 26.1, 25.4, 22.9, 20.9, 20.7; HR-MS (ESI) m/z calcd for C17H28O5 [M+Na]+: 335.1829, found 335.1833.
4.3 Suzuki–Miyaura fragment coupling to prepare 42 and 43
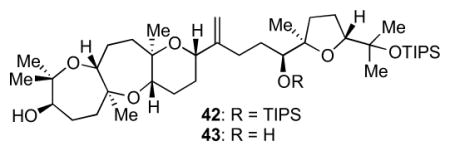
9-BBN dimer (24.4 mg, 0.100 mmol, 110 mol %) was placed in a Schlenk tube. Alkene 4013 (51.3 mg, 0.100 mmol, 110 mol%) in THF (1 mL) was added under argon at rt. More THF (0.2 mL) was used for rinsing. The Schlenk tube was closed and the mixture was heated at 55 °C for 20 h. After the mixture was cooled to rt, cesium carbonate solution (0.20 mL, 0.20 mmol, 220 mol%, 1M in H2O, saturated with nitrogen) was added under argon. Bubbling occurred immediately. The mixture was stirred at rt for 15 min. Crude alkenyl triflate 3913 (~0.0884 mmol, 100 mol %) in THF (1 mL) was added. Pd(dppf)Cl2 (8 mg, 0.01 mmol, 10 mol%) in DMF (1 mL) was added. The Schlenk tube was closed and the mixture was heated at 55 °C for 18 h. The reaction was cooled to room temperature. The crude was diluted with Et2O, washed with 0.5M HCl and brine, and dried with MgSO4. Column chromatography isolated cross coupling products. The mixture of products was dissolved in THF (5 mL) and TBAF (80 μL, 0.08 mmol, 1 M THF) was added at rt. The reaction was stirred 1.25 h. The mixture was diluted with Et2O and washed with H2O. Column chromatography isolated TES deprotected cross coupling product 42 (19 mg, 26%) and also cross coupling product that has both TES and 2° TIPS group deprotected (43) (12 mg, 20%). Data for 42: IR (NaCl, thin film): 3451, 2943, 2866, 1463, 1380, 1082, 883 cm−1; 1H NMR (600 MHz, C6D6): δ 4.96 (s, 1H), 4.93 (s, 1H), 4.24 (m, 2H), 3.93 (t, J = 5.2 Hz, 1H), 3.89 (dd, J = 6.4, 9.0 Hz, 1H), 3.81 (dd, J = 4.3, 11.5 Hz, 1H), 3.20 (d, J = 6.3 Hz, 1H), 2.54 (m, 2H), 2.34 (t, J = 12.8 Hz, 1H), 2.24 (q, J = 10.4 Hz, 1H), 2.15-1.50 (m, 16H), 1.45 (s, 3H), 1.37 (s, 3H), 1.35 (s, 3H), 1.30 (s, 3H), 1.27 (s, 3H), 1.21 (m, 21H), 1.16 (m, 21H), 1.09 (s, 3H), 0.93 (s, 3H); 13C NMR (100 MHz, C6D6): δ 152.9, 109.2, 87.8, 87.1, 80.1, 78.9, 78.7, 78.0, 77.2, 76.8, 74.9, 71.7, 70.8, 42.4, 36.0, 34.3, 32.4, 30.6, 29.9, 29.3, 29.0, 27.6, 27.5, 26.2, 25.42, 25.35, 23.1, 22.6, 21.1, 20.1, 19.11, 19.05, 14.12, 14.08; HR-MS (ESI) m/z calcd for C48H92O7Si2 [M+Na]+: 859.6274, found 859.6277.
4.4 Model studies of the bromonium-initiated cyclization using monoepoxides 50 and 51
4.4.1 Representative procedure for the bromonium-initiated cyclization in HFIP
4Å MS was activated as described in the General Experimental Methods. The substrate was added in HFIP. Either NBS or Br(coll)2ClO4 was added in one portion under Ar at the specified temperature. The reaction mixture was stirred in the absence of light. It was then filtered through celite, eluting with Et2O. After concentrating the filtrate, the residue was repartitioned between Et2O and H2O. The aqueous layer was extracted with Et2O (2X) and the combined organic layers were dried over MgSO4, filtered, and concentrated. Column chromatography (25% EtOAc in hexanes) isolated the products together as a 1:1 mixture of C3 epimers as colorless oils. In few cases, second column chromatography (50% to 60% Et2O in hexanes) was performed to separate the two diastereomers for characterization purposes. Relative configurations of the diastereomers were determined by nOe studies.
4.4.2 Preparation of 52 and 52′ using NBS as the bromenium source
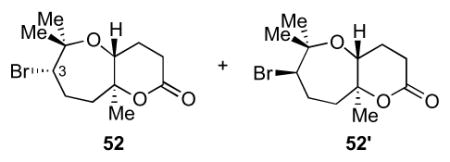
The general procedure was followed with ester 5013 (25.4 mg, 0.0946 mmol, 100 mol %), 4Å MS (234 mg, unactivated mass), HFIP (1.8 mL), and NBS (50.5 mg, 0.284 mmol, 300 mol %). The reaction was performed at 0 °C for 1 h. Lactones 52, 52′ were isolated together as a 1:1 mixture of C3 epimers (20.9 mg, 0.0718 mmol, 76%) as a colorless oil, which was further separated using column chromatography with a Et2O/hexanes solvent system.
4.4.3 Preparation of 52 and 52′ using Br(coll)2ClO4 as the bromenium source
The general procedure was followed with ester 5013 (50.0 mg, 0.187 mmol, 100 mol %), 4Å MS, HFIP (3.7 mL), and Br(coll)2ClO4 (236 mg, 0.560 mmol, 300 mol %). The reaction was performed at rt for 15 min. Lactones 52, 52′ were isolated together as a 1:1 mixture of C3 epimers (39.6 mg, 0.136 mmol, 73%) as a colorless oil. Data for 52: [α]22D = −7.6 (c = 0.085, CHCl3); IR (thin film, NaCl): 2977, 2936, 1773, 1734, 1700, 1457, 1384, 1263, 1208, 1134, 1077 cm−1; 1H NMR (600 MHz, CDCl3): δ 4.09-4.07 (m, 1H), 3.66 (dd, J = 11.8, 5.2 Hz, 1H), 2.71 (ddd, J = 18.7, 7.5, 1.8 Hz, 1H), 2.59 (dd, J = 11.4, 8.2 Hz, 1H), 2.19-2.16 (m, 2H), 2.03-1.99 (m, 1H), 1.96-1.92 (m, 1H), 1.85-1.80 (m, 1H), 1.79-1.75 (m, 1H), 1.40 (s, 3H), 1.39 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 170.1, 84.4, 79.1, 69.5, 59.3, 43.8, 31.4, 29.3, 26.3, 25.2, 24.9, 20.9; HR-MS (ESI) m/z calcd for C12H19BrO3 [M+H]+: 291.0590, found 291.0593. Data for 52′: [α]22D = + 1.7 (c = 0.23, CHCl3); IR (thin film, NaCl): 2979, 2937, 1773, 1734, 1701, 1457, 1269, 1242, 1193, 1150, 1100, 1084 cm−1; 1H NMR (600 MHz, CDCl3): δ 4.43 (dd, J = 6.2, 2.6 Hz, 1H), 4.11 (dd, J = 11.9, 5.3 Hz, 1H), 2.71 (ddd, J = 18.8, 7.7, 2.1 Hz, 1H), 2.63 (dd, J = 11.0, 8.3 Hz, 1H), 2.43 (ddd, J = 14.5, 9.6, 4.9 Hz, 1H), 2.10-2.07 (m, 2H), 1.91-1.85 (m, 2H), 1.83-1.81 (m, 1H), 1.46 (s, 3H), 1.41 (s, 3H), 1.33 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 170.7, 84.9, 78.3, 69.5, 64.7, 38.9, 30.7, 29.3, 28.7, 27.5, 24.8, 21.8; HR-MS (ESI) m/z calcd for C12H19BrO3 [M+H]+: 291.0590, found 291.0597.
4.4.4 Preparation of 53 and 53′ using NBS as the bromenium source
The general procedure was followed with carbonate 51 (35.1 mg, 0.130 mmol, 100 mol %), 4Å MS (241 mg, unactivated mass), HFIP (1.9 mL), and NBS (69.2 mg, 0.390 mmol, 300 mol %). The reaction was performed at 0 °C for 1 h. Lactones 53, 53′ were isolated together as a 1:1 mixture of C3 epimers (34.5 mg, 0.118 mmol, 91%) as a colorless oil.42
4.4.5 Preparation of 53 and 53′ using Br(coll)2ClO4 as the bromenium source
The general procedure was followed with carbonate 51 (25.5 mg, 0.0944 mmol, 100 mol %), 4Å MS, HFIP (1.5 mL), and Br(coll)2ClO4 (117.0 mg, 0.277 mmol, 300 mol %). The reaction was performed from 0 °C to rt for 3 h. Lactones 53, 53′ were isolated together as a 1:1 mixture of C3 epimers (27.3 mg, 0.0931 mmol, 99%) as a colorless oil.42
4.5 Model studies of the bromonium-initiated cyclization using diepoxide 60
4.5.1 Representative procedure for the bromonium-initiated cyclization in HFIP
To diepoxide 6013 was added the specified amount of trapping nucleophile in HFIP. Either NBS or Br(coll)2ClO4 was added in one portion under Ar at the specified temperature. The reaction mixture was stirred in the absence of light. It was then quenched by the addition of satd. aq. Na2S2O3 and filtered through celite, eluting with Et2O. After concentrating the filtrate, the residue was repartitioned between Et2O and a 1:1 mixture of satd. aq. Na2S2O3/NaCl. The aqueous layer was extracted with Et2O (2x). The combined organic layers were dried over MgSO4, filtered, and concentrated. Column chromatography isolated the products.
4.5.2 Preparation of 61 and 61′
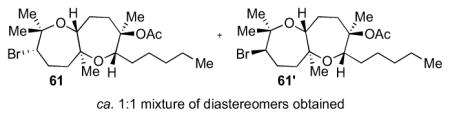
The general procedure was followed with 6013 (50.0 mg, 0.170 mmol, 100 mol %), 4Å MS (250 mg, activated mass), n-Bu4NOCOMe (76.9 mg, 0.301 mmol, 175 mol %), HFIP (2.5 mL), and NBS (60.5 mg, 0.340 mmol, 200 mol %). The reaction was performed at 0 °C for 15 min. Column chromatography (10 to 20% Et2O in hexanes) isolated the following as colorless oils: dioxepane 61 (21.8 mg), 61′ (23.9 mg) and a mixture of the two (9.00 mg) for a total of 0.126 mmol (74% yield) of products. Relative configurations of the diastereomers were determined by nOe studies. Data for 61: [α]22D = −25.3 (c = 0.91, CHCl3); IR (thin film, NaCl): 2930, 2858, 1734, 1700, 1653, 1559, 1457, 1379 cm−1; 1H NMR (500 MHz, C6D6): δ 4.063 (dd, J = 10.0, 1.5 Hz, 1H), 3.76 (dd, J = 10.5, 1.7 Hz, 1H), 3.28 (d, J = 10.5 Hz, 1H), 2.15 (td, J = 13.5, 3.1 Hz, 1H), 2.06 (ddd, J = 13.0, 5.5, 2.6 Hz, 1H), 2.02-1.89 (m, 2H), 1.87-1.79 (m, 1H), 1.69 (s, 3H), 1.67-1.49 (m, 6H), 1.43 (s, 3H), 1.41-1.27 (m, 8H), 1.16 (s, 6H), 0.95 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, C6D6): δ 169.9, 86.6, 78.8, 78.0, 76.6, 73.9, 60.3, 41.0, 37.6, 33.0, 32.3, 30.8, 29.1, 27.2, 26.2, 24.9, 23.5, 22.4, 21.4, 19.5, 14.7; HR-MS (ESI) m/z calcd for C21H37BrO4 [M+Na]+: 455.1767, found 455.1752. Data for 61′: [α]22D = −24.5 (c = 0.89, CHCl3); IR (thin film, NaCl): 2932, 2860, 1734, 1718, 1653, 1559, 1457, 1377 cm−1; 1H NMR (500 MHz, C6D6): δ 4.39 (dd, J = 10.0, 1.5 Hz, 1H), 4.29 (dd, J = 10.5, 1.1 Hz, 1H), 4.04 (dd, J = 6, 2.2 Hz, 1H), 2.62 (m, 1H), 2.31-2.23 (m, 1H), 1.94-1.86 (m, 1H), 1.78-1.72 (m, 2H), 1.70-1.64 (m, 6H), 1.54-1.46 (m, 6H), 1.40-1.32 (m, 7H), 1.28 (s, 3H), 1.18-1.12 (m, 1H), 0.94 (t, J = 7.0 Hz, 3H), 0.90 (s, 3H); 13C NMR (100 MHz, C6D6): δ 169.7, 86.4, 79.3, 76.7, 76.3, 74.3, 66.6, 38.0, 36.0, 32.8, 30.9, 29.9, 29.3, 28.7, 28.5, 27.1, 23.5, 22.4, 21.8, 19.4, 14.8; HR-MS (ESI) m/z calcd for C21H37BrO4 [M+Na]+: 455.1767, found 455.1748.
4.5.3 Preparation of 62 and 62′
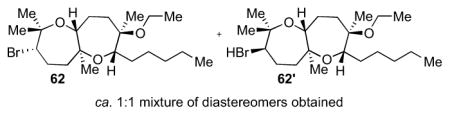
The general procedure was followed with 6013 (25.2 mg, 0.0856 mmol, 100 mol %), EtOH (0.28 mL), HFIP (1.4 mL), and Br(coll)2ClO4 (72.2 mg, 0.171 mmol, 200 mol %). The reaction was performed at 0 °C for 17 h. Column chromatography (5% to 10% Et2O in hexanes) isolated bicycle 62 (10.0 mg) and 62′ (10.0 mg) for a total of 0.0479 mmol (56% yield) of products. Relative configurations of the diastereomers were determined by nOe studies. Data for 62: [α]22D = −29.8 (c = 0.17, CHCl3); IR (thin film, NaCl): 2929, 2856, 1457, 1377, 1139, 1116, 1105, 1064 cm−1; 1H NMR (600 MHz, CDCl3): δ 4.13-4.11 (m, 1H), 3.48-3.44 (m, 2H), 3.42-3.36 (m, 2H), 2.18-2.16 (m, 2H), 1.93 (ddd, J = 13.2, 6.0, 2.4 Hz, 1H), 1.86-1.79 (m, 1H), 1.78-1.72 (m, 2H), 1.63-1.59 (m, 8H), 1.57 (dd, J = 12.6, 2.4 Hz, 1H), 1.54-1.49 (m, 1H), 1.47-1.43 (m, 1H), 1.37 (s, 6H), 1.35-1.30 (m, 2H), 1.16 (s, 3H), 1.14 (s, 3H), 0.92 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 79.1, 78.6, 78.3, 77.0, 76.6, 60.5, 57.1, 41.1, 39.3, 32.9, 32.4, 30.7, 29.0, 27.5, 26.2, 25.3, 23.4, 21.5, 17.8, 16.9, 14.8; HR-MS (ESI) m/z calcd for C21H39BrO3 [M+H]+: 419.2155, found 419.2155. Data for 62′: [α]22D = −21.7 (c = 0.15, CHCl3); IR (thin film, NaCl): 2929, 2858, 1457, 1378, 1117, 1073, 1063 cm−1; 1H NMR (600 MHz, CDCl3): δ 4.56 (d, J = 6.0 Hz, 1H), 4.05 (d, J = 10.2 Hz, 1H), 3.56 (d, J = 10.2 Hz, 1H), 3.48 (pentet, J = 6.6 Hz, 1H), 3.39 (pentet, J = 7.2 Hz, 1H), 2.42 (td, J = 12.6, 3.6 Hz, 1H), 2.07-2.00 (m, 2H), 1.86 (ddd, J = 12.6, 5.4, 1.8 Hz, 1H), 1.82-1.74 (m, 2H), 1.66 (dd, J = 13.2, 2.4 Hz, 1H), 1.56-1.53 (m, 2H), 1.45 (s, 3H), 1.39-1.33 (m, 5H), 1.29 (s, 3H), 1.28-1.26 (m, 5H), 1.20 (s, 3H), 1.15 (s, 3H), 0.92 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ79.2, 79.1, 77.1, 76.7, 76.2, 67.0, 57.0, 39.0, 35.8, 32.9, 30.6, 30.5, 29.2, 29.0, 28.8, 27.2, 23.5, 21.8, 18.2, 17.0, 14.9; HR-MS (ESI) m/z calcd for C21H39BrO3 [M+H]+: 419.2155, found 419.2165.
4.5.4 Preparation of 63 and 63′
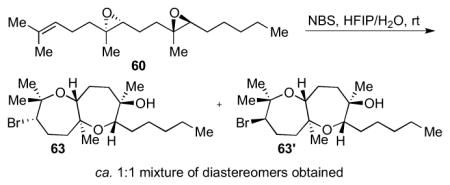
The general procedure was followed with 6013 (36.8 mg, 0.125 mmol, 100 mol %), H2O (0.28 mL), HFIP (0.42 mL), and NBS (44.5 mg, 0.250 mmol, 200 mol %). The reaction was performed at rt for 22 h. Column chromatography (20% to 30% Et2O in hexanes) isolated bicycle 63 (8.80 mg), 63′ (8.60 mg), and a mixture of the two (2.80 mg) for a total of 0.0513 mmol (41% yield) of products. Relative configurations of the diastereomers were determined after deacetylation of bicycles 61 and 61′ (DIBAL-H, CH2Cl2, −30 °C). Data for 63: [α]22D = −31.0 (c = 0.29, CHCl3); IR (thin film, NaCl): 3420, 2927, 2856, 1653, 1559, 1457, 1437, 1378 cm−1; 1H NMR (500 MHz, C6D6): δ 3.80 (dd, J = 10.5, 2.0 Hz, 1H), 3.03 (dd, J = 10.0, 1.8 Hz, 1H), 3.02 (d, J = 10.0 Hz, 1H), 2.03-1.91 (m, 2H), 1.84-1.74 (m, 2H), 1.68-1.59 (m, 1H), 1.55 (ddd, J = 12.5, 4.9, 1.9 Hz, 1H), 1.45-1.28 (m, 12H), 1.27-1.20 (m, 1H), 1.18 (s, 3H), 1.16 (s, 3H), 1.07 (s, 3H), 0.97 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, C6D6): δ 78.5, 77.9, 77.0, 76.9, 74.4, 60.4, 44.3, 41.0, 33.1, 32.3, 30.5, 29.3, 27.5, 26.1, 25.0, 23.5, 21.9, 21.5, 14.8; HR-MS (ESI) m/z calcd for C19H35BrO3 [M+Na]+: 413.1662, found 413.1657. Data for 63′: [α]22D = −33.4 (c = 0.42, CHCl3); IR (thin film, NaCl): 3395, 2930, 2858, 1457, 1653, 1559, 1540, 1507, 1378 cm−1; 1H NMR (500 MHz, C6D6); δ 4.13 (d, J = 10.5 Hz, 1H), 4.06 (dd, J = 6.0, 2.1 Hz, 1H), 3.51 (dd, J = 10.0, 1.8 Hz, 1H), 2.59-2.53 (m, 1H), 1.92-1.82 (m, 2H), 1.80-1.24 (m, 1H), 1.71-1.61 (m, 3H), 1.54-1.33 (m, 12H), 1.28 (s, 3H), 1.15 (s, 3H), 0.95 (t, J = 7.0 Hz, 3H), 0.90 (s, 3H); 13C NMR (100 MHz, C6D6): δ 79.0, 77.4, 76.9, 76.6, 74.5, 67.2, 44.8, 36.1, 32.9, 30.6, 29.9, 29.7, 28.9, 28.6, 27.4, 23.5, 21.9, 21.7, 14.8; HR-MS (ESI) m/z calcd for C19H35BrO3 [M+Na]+: 413.1662, found 413.1653.
4.5.5 Preparation of 64 and 64′
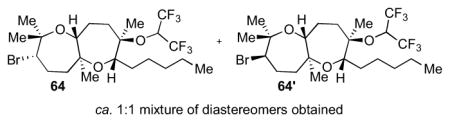
The general procedure was followed with 6013 (20.8 mg, 0.0706 mmol, 100 mol %), HFIP (1.1 mL), and Br(coll)2ClO4 (59.5 mg, 0.141 mmol, 200 mol %). The reaction was performed at 0 °C for 20 h. Column chromatography (packed with 2% Et3N in hexanes, flushed with hexanes, then 2% to 10% Et2O in hexanes) isolated bicycle 64 (6.00 mg) and 64′ (6.50 mg) for a total of 0.0233 mmol (33% yield) of products. Relative configurations of the diastereomers were determined by nOe studies. Data for 64: [α]22D = −23.1 (c = 0.15, CHCl3); IR (thin film, NaCl): 2956, 2927, 2857, 1653, 1559, 1457, 1383, 1355,1284, 1227, 1196 cm−1; 1H NMR (500 MHz, C6D6): δ 3.91 (septet, J = 6.0 Hz, 1H), 3.63 (dd, J = 10.0, 3.3 Hz, 1H), 3.22 (dd, J = 10.0, 2.0 Hz, 1H), 2.92 (d, J = 10.5 Hz, 1H), 1.98-1.88 (m, 3H), 1.60-1.52 (m, 2H), 1.41-1.33 (m, 7H), 1.32-1.25 (m, 7H), 1.11 (s, 3H), 1.10 (s, 3H), 0.94 (t, J = 7.0 Hz, 3H), 0.92 (s, 3H); 13C NMR (125 MHz, C6D6): δ 85.8, 78.8, 78.1, 76.6, 75.9, 70.2 (t, J = 31.3 Hz), 59.9, 40.6, 39.7, 32.9, 32.1, 30.4, 28.7, 27.2, 26.0, 25.0, 23.5, 21.3, 16.1, 14.7; 19F NMR (300 MHz, C6D6): δ −70.9 (q, J = 6.9 Hz), −70.8 (q, J = 6.6 Hz, 6F). (Referenced with C6F6 at −164.9 ppm). Data for 64′: [α]22D = −13.7 (c = 0.25, CHCl3); IR (thin film, NaCl): 2956, 2930, 2859, 1653, 1559, 1465, 1457, 1437, 1385, 1356, 1284 cm−1; 1H NMR (500 MHz, C6D6): δ 4.14 (dd, J = 10.5, 1.1 Hz, 1H), 3.97 (septet, J = 6.0 Hz, 1H), 3.97 (d, J = 6.0 Hz, 1H), 3.73 (dd, J = 10.0, 1.8 Hz, 1H), 2.49 (ddd, J = 14.0, 11.0, 4.3 Hz, 1H), 2.05-2.00 (m, 1H), 1.73-1.58 (m, 5H), 1.48-1.44 (m, 2H), 1.42-1.28 (m, 12H), 1.24 (s, 3H), 1.02 (s, 3H), 0.90 (t, J = 7.0 Hz, 3H), 0.89 (s, 3H); 13C NMR (125 MHz, C6D6): δ 85.7, 79.3, 76.8, 76.4, 76.3, 70.3 (t, J = 32.5 Hz), 66.2, 40.0, 35.8, 32.7, 30.6, 29.8, 29.0, 28.8, 28.4, 27.1, 23.5, 21.8, 16.6, 14.7; 19F NMR (300 MHz, C6D6): δ −71.0 (q, J = 6.6 Hz), −70.9 (q, J = 6.6 Hz, 6F). (Referenced with C6F6 at −164.9 ppm).
4.5.6 Preparation of 63, 63′–64, 64′ using reaction of 60 with CsOH•H2O
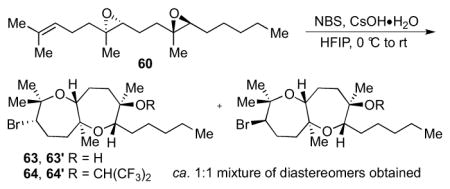
The general procedure was followed with 6013 (26.8 mg, 0.0910 mmol, 100 mol %), CsOH•H2O (24.5 mg, 0.146 mmol, 150 mol %), HFIP (1.4 mL), and NBS (34.7 mg, 0.195 mmol, 200 mol %). The reaction was performed at 0 °C while warming to rt over 15 h. Column chromatography (5 to 50% Et2O in hexanes) isolated 63, 63′ (16.0 mg total, 0.0409 mmol, 45%) and 64, 64′ (9.20 mg, 0.0170 mmol, 19%) as colorless oils.
4.6 Preparation of tricycle 85
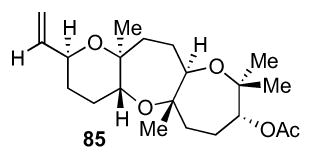
To a flame-dried 100 ml round-bottom flask equipped with a stir bar was added triepoxide 1413 (1.56 g, 4.81 mmol) in 100 mL of dichloromethane and cooled to −78 °C. BF3•OEt2 (210 μL, 1.70 mmol) was added dropwise and the reaction was stirred at the same temperature for 15 min. Saturated NH4Cl was added to quench the reaction and it was warmed to room temperature. The reaction was poured into a seperatory funnel and extracted with CH2Cl2 (3x) from brine. The organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude reaction mixture was dissolved in 50 mL of dichloromethane in a 100 mL flame-dried round-bottom flask equipped with a stir bar. Acetic anhydride (950 μL, 10.0 mmol), triethylamine (2.80 mL, 20.1 mmol), and DMAP (59.8 mg, 0.490 mmol) were all added and reaction was stirred at room temperature overnight. Saturated NH4Cl was added and the reaction was transferred to a separatory funnel and extracted with CH2Cl2 (3x). The organics were dried over Na2SO4, filtered, and concentrated in vacuo. Column chromatography provided colorless oil 85 (325 mg, 0.886 mmol, 18.4 % yield). [α]24D = +22.9 (c = 0.06, CHCl3); IR (NaCl, thin film): 2936, 2361, 2339, 1734, 1653, 1540, 1457, 1241, 1078 cm−1; 1H NMR (500 MHz, CDCl3): δ 5.84-5.77 (m, 1H), 5.21 (dt, J = 17.3, 1.4 Hz, 1H), 5.09 (ddd, J = 10.5, 1.6, 1.1 Hz, 1H), 4.92 (d, J = 6.7 Hz, 1H), 4.01-3.97 (m, 1H), 3.62 (dd, J = 11.1, 5.0 Hz, 1H), 3.46 (dd, J = 11.5, 2.5 Hz, 1H), 2.12 (s, 3H), 2.10-2.04 (m, 2H), 2.00-1.81 (m, 3H), 1.81-1.61 (m, 5H), 1.54-1.43 (m, 2H), 1.30 (s, 3H), 1.26 (s, 3H), 1.16 (s, 3H), 1.14 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 208.5, 139.6, 115.3, 78.8, 78.6, 78.4, 77.4, 77.1, 71.1, 70.2, 40.8, 36.8, 32.1, 29.2, 28.9, 27.8, 23.2, 21.8, 21.3, 20.1, 16.4; HR-MS (ESI) m/z calcd for C21H34O5 [M+Na]+: 389.2298, found 389.2290.
4.7 Representative procedure for the bromonium-initiated cyclization of 80 in HFIP using Br(coll)2BF4 to prepare 79 and 79′
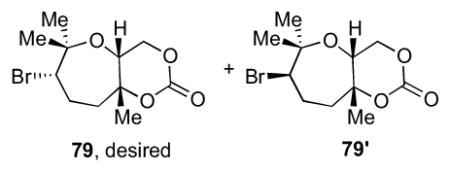
To a 500 mL round-bottom flask equipped with a stir bar was added 4Å molecular sieves (27 g) followed by carbonate 8013 (3.87 g, 14.3 mmol) and 278 mL of HFIP. The reaction was cooled to 0 °C and Br(coll)2BF4 (17.6 g, 43.0 mmol) was added in one portion and the reaction was stirred for 30 min. The reaction was filtered through Celite and then added to 150 mL of brine and 150 mL of saturated Na2S2O3. The aqueous layer was extracted with CH2Cl2 (3x) and then the organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude solid was purified by column chromatography (20 to 50 to 100% EtOAc in hexanes) to furnish 79 (1.31 g, 4.48 mmol, 31% yield) and 79′ (693 mg, 2.36 mmol, 17% yield) as white solids. Data for 79: [α]24D = −23.0 (c = 0.1, CHCl3); IR (NaCl, thin film): 2937, 1732, 1159, 1540, 1457, 1298, 1208, 1120, 1086, 1040 cm−1; 1H NMR (600 MHz, CDCl3): δ 4.52 (dd, J = 11.5, 2.9 Hz, 1H), 4.22 (dd, J = 11.4, 1.8 Hz, 1H), 3.88 (dd, J = 10.6, 1.9 Hz, 1H), 3.77 (dd, J = 2.7, 1.9 Hz, 1H), 2.50 (dtd, J = 14.5, 10.6, 3.6 Hz, 1H), 2.20 (ddd, J = 15.0, 6.9, 3.3 Hz, 1H), 2.03-1.99 (m, 1H), 1.72-1.67 (m, 1H), 1.43 (s, 3H), 1.42 (s, 3H), 1.39 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 149.2, 83.7, 79.2, 71.2, 66.8, 57.1, 39.9, 30.3, 25.7, 25.5, 24.4; HR-MS (ESI) m/z calcd for C11H17O4Br [M+Na]+: 315.0202, found 315.0214. Data for 79′: [α]24D = −9.7 (c = 1.2, CHCl3); IR (NaCl, thin film): 2981, 1750, 1457, 1388, 1292, 1247, 1218, 1122, 1091, 1042 cm−1; 1H NMR (600 MHz, CDCl3): 4.46 (dd, J = 11.6, 2.8 Hz, 1H), 4.18 (dd, J = 11.6, 2.2 Hz, 1H), 4.13 (dd, J = 10.3, 2.5 Hz, 1H), 3.94 (t, J = 2.5 Hz, 1H), 2.35 (ddt, J = 14.9, 10.2, 2.2 Hz, 1H), 2.14 (ddd, J = 14.8, 10.1, 1.5 Hz, 1H), 2.03-1.99 (m, 1H), 1.86-1.80 (m, 1H), 1.46 (s, 3H), 1.43 (s, 3H), 1.41 (s, 3H); 13C NMR (125 MHz, CDCl3): 149.4, 86.6, 79.5, 70.4, 67.8, 61.9, 38.4, 30.4, 29.2, 25.8, 22.4; HR-MS (ESI) m/z calcd for C11H17O4Br [M+H]+: 293.0389, found 293.0374.
4.8 Fragment coupling to access 87
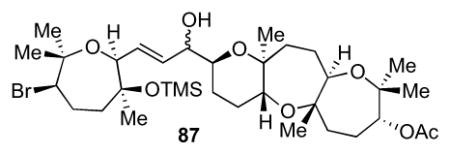
To a 25 mL round-bottom flask equipped with a stir bar was added [Cp2Zr(H)Cl] (256 mg, 0.990 mmol) and alkyne 7713 (321 mg, 96.0 mmol) and placed under nitrogen in the dark. 9.6 mL of CH2Cl2 was added and stirred for 30 min at room temperature. The flask was then cooled to −78 °C and Me2Zn (2.0 M in toluene, 505 μL, 1.01 mmol) was added dropwise. After stirring for 15 min, a solution of aldehyde 7813 (306 mg, 0.830 mmol) in 8 mL of CH2Cl2 was added and the reaction was allowed to slowly warm to room temperature overnight. A saturated solution of NH4Cl was added and a white solid formed. The aqueous layer was extracted with CH2Cl2 (3x) and then the organic layers were dried over Na2SO4, filtered, and concentrated in vacuo to provide a colorless oil. The crude was purified by column chromatography (5 to 50% EtOAc in hexanes) to provide allylic alcohol 87 (373 mg, 0.530 mmol, 64% yield) as a colorless oil. [α]24D = +28.2 (c = 0.4, CHCl3); IR (NaCl, thin film): 3583, 2939, 1739, 1442, 1379, 1248, 1067, 840, 753 cm−1; 1H NMR (600 MHz, CDCl3): δ 5.85 (dd, J = 15.6, 7.5 Hz, 1H), 5.50 (dd, J = 15.6, 6.7 Hz, 1H), 4.91 (d, J = 6.8 Hz, 1H), 3.96 (d, J = 10.4 Hz, 1H), 3.86-3.84 (m, 1H), 3.63 (t, J = 6.8 Hz, 1H), 3.58 (dd, J = 9.7, 6.1 Hz, 1H), 3.45 (dd, J = 11.3, 2.0 Hz, 1H), 3.34 (ddd, J = 11.8, 7.2, 1.9 Hz, 1H), 2.69 (d, J = 2.4 Hz, 1H), 2.54 (td, J = 12.9, 10.1 Hz, 1H), 2.12 (s, 3H), 2.08-2.01 (m, 2H), 1.94-1.86 (m, 4H), 1.79 (dd, J = 14.4, 13.6 Hz, 2H), 1.70 (t, J = 14.0 Hz, 1H), 1.62 (dt, J = 6.8, 3.1 Hz, 2H), 1.52-1.44 (m, 4H), 1.41 (s, 3H), 1.33 (s, 3H), 1.25 (s, 3H), 1.24 (s, 3H), 1.16 (s, 3H), 1.15 (s, 3H), 1.06 (s, 3H), 0.11 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 170.3, 131.6, 131.0, 78.8, 78.6, 78.3, 78.3, 77.7, 77.6, 77.4, 75.9, 75.5, 73.2, 70.3, 60.8, 44.2, 40.6, 36.8, 31.0, 29.1, 28.9, 28.1, 27.8, 27.4, 26.2, 24.4, 23.2, 21.8, 21.3, 20.1, 16.4, 2.8.
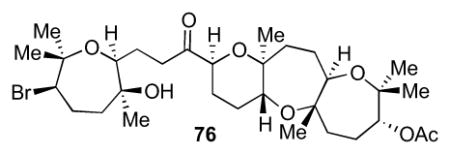
4.9 Preparation of 76
To a 25 mL round-bottom flask equipped with a stir bar was added allylic alcohol 87 (373 mg, 0.530 mmol) and 5.2 mL of EtOH. Palladium on carbon (218 mg, 10% by weight) was added and placed under vacuum. Hydrogen gas was added and the reaction was stirred at room temperature for 90 min. The solution was filtered through Celite (eluted with EtOAc) to remove the palladium. The crude reaction mixture was purified by the Biotage purification system to provide the intermediate alcohol (223 mg, 0.352 mmol, 66% yield) as a colorless oil.
To a 10 mL round-bottom flask equipped with a stir bar were added 4Å molecular sieves (194 mg) followed by alcohol from the previous step (223 mg, 0.352 mmol) in 3.5 mL of CH2Cl2. N-Methylmorpholine-N-oxide (131 mg, 1.12 mmol) was added and the reaction was cooled to 0 °C. Tetrapropylammonium perruthenate (29.0 mg, 82.5 mmol) was added and the reaction was allowed to warm to room temperature over 2 h. The crude reaction was filtered through Celite using a CH2Cl2 and EtOAc wash and concentrated in vacuo. The reaction was purified by the Biotage purification system to provide 76 (180 mg, 0.285 mmol, 81% yield) as a colorless oil. [α]24D = −4.8 (c = 0.24, CHCl3); IR (NaCl, thin film): 3584, 2976, 2938, 1736, 1445, 1379, 1243, 1138, 109, 1069, 1029 cm−1; 1H NMR (600 MHz, CDCl3): δ 4.92 (d, J = 6.6 Hz, 1H), 3.89 (td, J = 7.5, 4.0 Hz, 2H), 3.60 (dd, J = 13.8, 7.0 Hz, 1H), 3.47 (dd, J = 11.4, 2.3 Hz, 1H), 3.41 (dd, J = 10.3, 2.8 Hz, 1H), 2.96 (s, 1H), 2.69-2.67 (m, 2H), 2.33 (qd, J = 13.0, 1.6 Hz, 1H), 2.12 (s, 3H), 2.04-1.60 (m, 13H), 1.53-1.46 (m, 4H), 1.41 (s, 3H), 1.31 (s, 3H), 1.26 (s, 3H), 1.25 (s, 3H), 1.17 (s, 3H), 1.15 (s, 3H), 1.14 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 212.1, 170.2, 78.9, 78.6, 78.2, 78.0, 77.8, 77.4, 75.8, 74.6, 72.3, 69.8, 68.1, 59.1, 44.5, 43.0, 40.5, 36.7, 34.1, 30.5, 30.4, 28.9, 25.8, 25.7, 25.6, 25.0, 24.5, 23.2, 21.8, 21.3, 19.6, 16.4.
4.10 Preparation of 90 and 91 by the addition of MeLi to 76
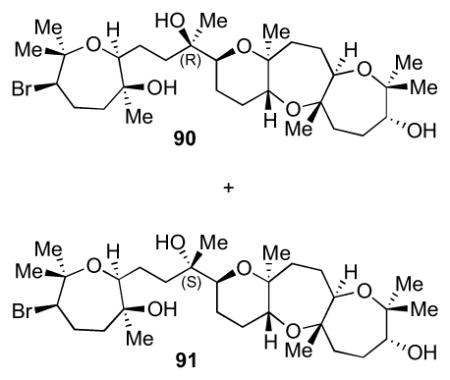
To a 20 mL vial with stir bar was added ketone 76 (180 mg, 0.285 mmol) and 3.5 mL of THF and the reaction was cooled to −78 °C. MeLi (950 μL, 1.60 M in Et2O, 1.52 mmol) was added dropwise and the reaction was stirred for 1 h. The reaction solidified and eventually stirring ceased. Saturated NH4Cl was added, and the reaction was allowed to warm to rt. The aqueous layer was extracted with Et2O (3x), dried over Na2SO4, filtered, and the solvent was removed in vacuo. The crude was redissolved in 10 mL of THF and cooled to −78 °C. MeLi (1.0 mL, 1.6 M in Et2O, 1.6 mmol) was added dropwise and the reaction was stirred at this temperature for 1 h. The reaction stayed in solution and went to completion by TLC. Saturated NH4Cl was added and allowed to warm to rt. The aqueous layer was extracted with Et2O (3x) and then the organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by careful column chromatography (0.1% to 2% MeOH in Et2O) on the Biotage purification system to provide clean alcohol 91 (29.7 mg, 49.3 mmol, 17% yield) and the remaining material as a mixture of alcohol 91 and alcohol 90 (117 mg, 0.192 mmol, 67% yield). Data for 90: [α]24D = +40.9 (c = 0.2, CHCl3). For reference, ent-90: [α]24D = −41.7 (c = 0.1, CHCl3); IR (NaCl, thin film): 3583, 3452, 2932, 1659, 1641, 1462, 1451, 1378, 1137, 1071, 891 cm−1; 1H NMR (500 MHz, C6D6): δ 3.81 (dd, J = 11.4, 2.4 Hz, 1H), 3.65 (dd, J = 10.6, 5.4 Hz, 1H), 3.51 (d, J = 11.1 Hz, 1H), 3.23 (td, J = 8.5, 3.5 Hz, 2H), 2.92 (s, 1H), 2.78 (dd, J = 9.9, 3.0 Hz, 1H), 2.24 (s, J = 5.2 Hz, 1H), 2.24-2.14 (m, 3H), 1.95-1.43 (m, 17H), 1.34 (s, 3H), 1.33 (s, 3H), 1.25 (s, 6H), 1.14 (s, 6H), 1.01 (s, 3H), 0.98 (s, 3H); 13C NMR (125 MHz, C6D6): δ 79.6, 78.2, 78.1, 78.0, 77.9, 76.9, 76.7, 76.3, 73.4, 72.4, 71.2, 59.7, 44.7, 41.1, 36.8, 34.4, 31.0, 30.0, 29.4, 28.3, 26.3, 26.1, 26.0, 25.7, 25.6, 24.1, 24.0, 22.1, 20.2, 17.1; HR-MS (ESI) m/z calcd for C30H53BrO7 [M+H]+: 605.3047, found 605.3059. Data for 91: 1H NMR (400 MHz, C6D6): δ 3.80 (dd, J = 11.4, 2.3 Hz, 1H), 3.62 (dd, J = 10.2, 6.0 Hz, 1H), 3.50 (d, J = 11.0 Hz, 1H), 3.29-3.19 (m, 2H), 2.90 (bs, 1H), 2.78 (dd, J = 9.3, 3.2 Hz, 1H), 2.30 (bs, 1H), 2.27-2.12 (m, 3H), 1.92-1.75 (m, 4H), 1.72-1.53 (m, 8H), 1.53-1.40 (m, 5H), 1.33 (s, 6H), 1.25 (s, 3H), 1.24 (s, 3H), 1.13 (s, 3H), 1.10 (s, 3H), 1.02 (s, 3H), 0.97 (s, 3H); 13C NMR (100 MHz, C6D6): δ 79.6, 78.1, 78.1, 77.9, 77.9, 76.7, 76.6, 74.4, 73.4, 72.4, 71.0, 59.8, 44.7, 41.1, 36.9, 36.8, 31.0, 30.1, 29.4, 28.3, 26.3, 26.1, 25.9, 25.6, 25.6, 24.7, 23.0, 22.0, 20.3, 17.1; HR-MS (ESI) m/z calcd for C30H53BrO7 [M+H]+: 605.3047, found 605.3051.
4.11 Preparation of 90 by the addition of MeMgBr to 76 and deacetylation
To a 5 mL round-bottom flask equipped with a stir bar was added ketone 76 (65.2 mg, 103 μmol) and 1.6 mL of THF. The reaction was cooled to −78 °C and MeMgBr (200 μL, 3.0 M in Et2O, 600 μmol) was added and the reaction was allowed to warm to 0 °C over 1 h. The reaction was quenched with sat. NH4Cl and the aqueous layer was extracted with Et2O (3x). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude oil was purified by column chromatography (20 to 50% EtOAc in hexanes) to provide the acetate intermediate (19.6 mg, 30.3 μmol, 29% yield) and alcohol 90 (9.60 mg, 15.9 μmol, 15% yield) along with recovered ketone 76 (35.0 mg, 55.4 μmol, 54% yield). The recovered starting material was redissolved in 3.2 mL of THF and MeMgBr (200 μL, 3.0 M in Et2O, 600 μmol) was added at −78 °C and allowed to stir for 1 h. The reaction was quenched with saturated NH4Cl and extracted with Et2O (3x). The organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue, along with the acetate intermediate (19.6 mg, 30.3 μmol) was dissolved in 3.4 mL of MeOH and K2CO3 (18.3 mg, 132 μmol) was added and stirred at room temperature for 4 h. The reaction was then quenched with saturated NH4Cl and extracted with Et2O (5x). The organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude was purified by column chromatography (50% EtOAc in hexanes) to provide alcohol 90 (34.6 mg, 57.1 umol, 67% yield) as a colorless oil.
4.12 Preparation of 103
To a 5 mL round-bottom flask equipped with a stir bar was added 15 mg of 4Å MS and heated under vacuum for 5 min. The flask was cooled to 0 °C and alcohol 90 (18.0 mg, 0.0298 mmol) was added in 2 mL of CH2Cl2. NMO (15.0 mg, 0.128 mmol) was added and then TPAP (~2 mg, 0.006 mmol) was added and the reaction was allowed to slowly warm to room temperature for 2 h. The crude material was purified by the Biotage purification system to furnish the intermediate ketone (14.7 mg, 0.0244 mmol, 82% yield).
To a 5 mL flask equipped with a stir bar was added ketone from the previous step (25.8 mg, 42.7 μmol) and tosyl hydrazone (19.3 mg, 0.104 mmol) in 300 μL of MeOH. The reaction was heated to 45 °C with a reflux condenser for 18 h. The reaction was cooled to room temperature and concentrated in vacuo and loaded directly onto a silica gel column. The crude was purified by column chromatography (50 to 100% EtOAc in hexanes) to furnish hydrazone 103 (20.7 mg, 26.8 μmol, 63% yield) as a white solid. [α]24D = −15.4 (c = 1.0, CHCl3); IR (NaCl, thin film): 3535, 3220, 2977, 2937, 1644, 1598, 1463, 1381, 1342, 1262, 1169, 1088, 1031, 814, 756 cm−1; 1H NMR (600 MHz, C6D6): δ 8.02 (d, J = 8.2 Hz, 2H), 6.79 (d, J = 8.2 Hz, 2H), 3.54 (d, J = 11.1 Hz, 2H), 3.50 (t, J = 8.0 Hz, 2H), 3.28 (dd, J = 11.7, 2.2 Hz, 1H), 2.96 (s, 1H), 2.75 (dd, J = 9.4, 2.9 Hz, 1H), 2.54 (dd, J = 11.3, 2.1 Hz, 1H), 2.24-2.17 (m, 1H), 2.13-2.05 (m, 2H), 2.02-1.97 (m, 1H), 1.92 (s, 3H), 1.91-1.80 (m, 6H), 1.65-1.57 (m, 4H), 1.53-1.48 (m, 3H), 1.48-1.38 (m, 4H), 1.35 (s, 3H), 1.29 (s, 3H), 1.25 (s, 3H), 1.17 (s, 3H), 1.12 (s, 3H), 1.05 (s, 3H), 1.01 (s, 3H), 0.99 (s, 3H); 13C NMR (150 MHz, C6D6): δ 164.1, 143.9, 136.8, 129.9, 128.8, 80.8, 80.2, 78.1, 78.0, 77.4, 76.3, 76.20, 73.3, 72.4, 71.1, 59.7, 44.7, 41.2, 40.8, 37.1, 34.7, 31.0, 29.5, 27.4, 26.1, 26.0, 25.56, 25.5, 24.4, 24.3, 23.3, 21.7, 20.0, 16.2; HR-MS (ESI) m/z calcd for C37H59BrSN2O8 [M+H]+: 771.3248, found 771.3245.
4.13 Preparation of 73
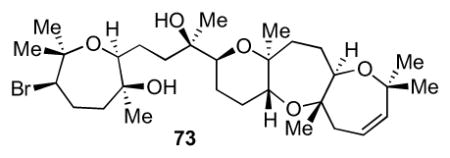
To a 500 μL sealed tube equipped with a stir bar was added sodium hydride (6.50 mg, 0.285 mmol, 95% in mineral oil) in the glove box and then transferred to the hood under nitrogen. The reaction was purged while a solution of hydrazone 103 (16.6 mg, 21.5 μmol) in 500 μL of THF was added by syringe. The reaction was sealed and heated to 90 °C for 100 min then to 110 °C for 70 min. The reaction turned brown and was then cooled to room temperature. H2O was added to quench remaining sodium hydride, followed by addition of 1 mL of saturated NH4Cl. The aqueous layer was extracted with Et2O (5x) and then dried over Na2SO4, filtered, and concentrated in vacuo. Careful column chromatography (20 to 50% HPLC-grade EtOAc in hexanes) provided a small amount of analytically clean 73 (0.8 mg, 1.3 μmol, 6% yield) as colorless oil. [α]24D = +33.9 (c = 0.04, CHCl3); IR (NaCl, thin film): 3432, 2922, 2851, 1727, 1463, 1378, 1261, 1088 cm−1; 1H NMR (600 MHz, C6D6): δ 5.46-5.42 (m, 4H), 5.21 (d, J = 11.7 Hz, 1H), 3.85 (dd, J = 11.4, 4.6 Hz, 1H), 3.53 (d, J = 11.0 Hz, 1H), 3.48 (dd, J = 11.5, 4.5 Hz, 1H), 3.26 (dd, J = 7.7, 4.3 Hz, 1H), 3.22 (dd, J = 10.9, 3.2 Hz, 1H), 2.89 (d, J = 0.5 Hz, 1H), 2.81 (dt, J = 6.4, 3.4 Hz, 1H), 2.58 (ddd, J = 15.4, 5.7, 1.7 Hz, 1H), 2.25-2.14 (m, 2H), 2.14-2.10 (m, 1H), 2.03 (t, J = 7.5 Hz, 1H), 2.01-1.95 (m, 1H), 1.84-1.67 (m, 5H), 1.65-1.46 (m, 6H), 1.34 (s, 3H), 1.29 (s, 3H), 1.25 (s, 6H), 1.22 (s, 3H), 1.15 (s, 3H), 1.10 (s, 3H), 1.02 (s, 3H), 0.96-0.90 (m, 1H); 13C NMR (125 MHz, C6D6): δ 137.0, 122.8, 80.7, 78.3, 77.9, 77.8, 77.0, 76.9, 75.8, 74.5, 73.4, 72.4, 59.7, 44.7, 42.6, 39.1, 34.4, 31.0, 29.9, 28.1, 27.9, 26.5, 26.1, 26.0, 25.6, 25.4, 24.1, 23.9, 18.5, 17.9; HR-MS (ESI) m/z calcd for C30H51O6Br [M+H]+: 587.2942, found 587.2930.
Supplementary Material
Scheme 17.

The armatols family of natural products (proposed strucutres).
Acknowledgments
This work was supported by the NIGMS (GM-72566). We thank Dr. Jeffrey H. Simpson for helpful discussions regarding NMR experiments, Li Li for mass spectrometry data, and Dr. Peter Müller for the crystal structures of 34 and 67. J.T. would like to thank Novartis and Amgen for graduate fellowships.
Footnotes
Experimental procedures and characterization data for compounds 14–15, ent-14, 17–18, 20, 22, 26, 31, 33, 35, 37–40, 44, 46, 50, 60, 75, 78, ent-79, 80, ent-80, 83–84, 89, 92–93, and 101–102 are provided. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers 931730 (tricycle 34) and 755014 (tetracycle 67). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or deposit@ccdc.cam.ac.Uk). Experimental procedures and characterization data for compounds 30, 32, 54–56, 57–59, 66–67, 67′, 68–70, and ent-1 can be found in the Supporting Information of a previous communication.7
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Fernandez JJ, Souto ML. Nat Prod Rep. 2000;17:235. doi: 10.1039/a909496b. [DOI] [PubMed] [Google Scholar]
- 2.Manriquez CP, Souto ML, Gavin JA, Norte M, Fernandez JJ. Tetrahedron. 2001;57:3117. [Google Scholar]
- 3.Sakemi S, Higa T. Tetrahedron Lett. 1986;27:4287. [Google Scholar]
- 4.Blunt JW, Hartshorn MP, McLennan TJ, Munro MHG, Robinson WT, Yorke SC. Tetrahedron Lett. 1978:69. [Google Scholar]
- 5.Matsuo Y, Suzuki M, Masuda M. Chem Lett. 1995;24:1043. [Google Scholar]
- 6.We selected ent-1 as the target of our synthetic study to use the more readily available enantiomer of fructose-derived ketone 36 to establish the configuration of the isolated epoxides (in intermediate 33, Scheme 6): Tu Y, Wang ZX, Shi Y. J Am Chem Soc. 1996;118:9806.Wang ZX, Tu Y, Frohn M, Zhang JR, Shi Y. J Am Chem Soc. 1997;119:11224.Shi Y. Acc Chem Res. 2004;37:488. doi: 10.1021/ar030063x.
- 7.Tanuwidjaja J, Ng SS, Jamison TF. J Am Chem Soc. 2009;131:12084. doi: 10.1021/ja9052366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciavatta ML, Wahidulla S, D’Souza L, Scognamiglio G, Cimino G. Tetrahedron Lett. 2000;57:617. [Google Scholar]
- 9.For our group’s previous reports on epoxide-opening cascades for potential applications in natural products syntheses: Morten CJ, Byers JA, Van Dyke AR, Vilotijevic I, Jamison TF. Chem Soc Rev. 2009;38:3175. doi: 10.1039/b816697h. and references therein.
- 10.For previous use of Me directing groups in epoxide-opening events, see: Valentine JC, McDonald FE. Synlett. 2006:1816.McDonald FE, Tong R, Valentine JC, Bravo F. Pure Appl Chem. 2007;79:281. and references therein.Kumar VS, Aubele DL, Floreancig PE. Org Lett. 2002;4:2489. doi: 10.1021/ol0261074.Kumar VS, Wan S, Aubele DL, Floreancig PE. Tetrahedron: Asym. 2005;16:3570.Wan S, Gunaydin H, Houk KN, Floreancig PE. J Am Chem Soc. 2007;129:7915. doi: 10.1021/ja0709674.
- 11.For precedence of bromine installation from hindered alcohols with similar substitution pattern, see: Boger DL, Mathvink RJ. J Org Chem. 1990;55:5442.Su WS, Fang JM, Cheng YS. Tetrahedron Lett. 1995;36:5367.Kim D, Kim IH. Tetrahedron Lett. 1997;38:415.Ranu BC, Jana R. Eur J Org Chem. 2005;4:755. doi: 10.1021/jo051373r.
- 12.a) Ng S-S, Jamison TF. Tetrahedron. 2005;61:11405. [Google Scholar]; b) Ng S-S, Jamison TF. J Am Chem Soc. 2005;127:7320. doi: 10.1021/ja0521831. [DOI] [PubMed] [Google Scholar]; c) Ng S-S, Jamison TF. Tetrahedron. 2006;62:11350. [Google Scholar]
- 13.Please refer to the Supporting Information for syntheses of these substrates.
- 14.González IC, Forsyth CJ. Tetrahedron Lett. 2000;41:3805. [Google Scholar]
- 15.McDonald FE, Bravo F, Wang X, Wei X, Toganoh M, Rodriguez JR, Do B, Neiwert WA, Hardcastle KI. J Org Chem. 2002;67:2515. doi: 10.1021/jo0110092. [DOI] [PubMed] [Google Scholar]
- 16.a) Payne GB. J Org Chem. 1962;27:3819. [Google Scholar]; b) Hanson RM. Org React. 2002;60:2. [Google Scholar]
- 17.a) Booth H, Dixon JM, Khedhair KA. Tetrahedron. 1992;48:6161. [Google Scholar]; b) Shenoy SR, Smith DM, Woerpel KA. J Am Chem Soc. 2006;128:8672. doi: 10.1021/ja061110u. [DOI] [PubMed] [Google Scholar]
- 18.Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299. [Google Scholar]
- 19.Alcaraz L, Harnett JJ, Mioskowski C, Martel JP, Le Gall T, Shin DS, Falck JR. Tetrahedron Lett. 1994;35:5449. [Google Scholar]
- 20.a) Corey EJ, Ha DC. Tetrahedron Lett. 1988;29:3171. doi: 10.1016/0040-4039(88)85122-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hashimoto M, Kan T, Nozaki K, Yanagiya M, Shirahama H, Matsumoto T. J Org Chem. 1990;55:5088. [Google Scholar]; c) González IC, Forsyth CJ. J Am Chem Soc. 2000;122:9099. [Google Scholar]; d) Morimoto Y, Nishikawa Y, Takaishi M. J Am Chem Soc. 2005;127:5806. doi: 10.1021/ja050123p. [DOI] [PubMed] [Google Scholar]; e) Morimoto Y, Yata H, Nishikawa Y. Angew Chem Int Ed. 2007;46:6481. doi: 10.1002/anie.200701737. [DOI] [PubMed] [Google Scholar]
- 21.Bravo F, McDonald FE, Neiwert WA, Hardcastle KI. Org Lett. 2004;6:4487. doi: 10.1021/ol048212e. [DOI] [PubMed] [Google Scholar]
- 22.Zakarian A, Batch A, Holton RA. J Am Chem Soc. 2003;125:7822. doi: 10.1021/ja029225v. [DOI] [PubMed] [Google Scholar]
- 23.Schadt FL, Schleyer PVR, Bentley TW. Tetrahedron Lett. 1974:2335.HFIP has the following Hansen Solubility Parameters (MPa0.5): δd = 14.17, δp = 2.41, δh = 13.89: Pospiech D, Gottwald A, Jehnichen D, Friedel P, John A, Harnisch C, Voigt D, Khimich G, Bilibin AY. Colloid Polym Sci. 2002;280:1027.
- 24.Spartan. Wavefunction, Inc; Irvine, CA: 2010. Image generated using Hartree-Fock 6-31G* basis set. [Google Scholar]
- 25.a) Fujiwara K, Hirose Y, Sato D, Kawai H, Suzuki T. Tetrahedron Lett. 2010;51:4263. [Google Scholar]; b) Fujiwara K, Tanaka K, Katagiri Y, Kawai H, Suzuki T. Tetrahedron Lett. 2010;51:4543. [Google Scholar]
- 26.Mosher ester analysis was conducted on the resultant secondary alcohol formed after treatment of the bromo-oxepane moiety with zinc and ammonium chloride: Suenaga K, Shibata T, Takada N, Kigoshi H, Yamada K. J Nat Prod. 1998;61:515.
- 27.Coates RM, Ley DA, Cavender PL. J Org Chem. 1978;43:4915. [Google Scholar]
- 28.Quirin C, Kazmaier U. Eur J Org Chem. 2009;3:371. [Google Scholar]
- 29.A procedure to ent-36 was first reported in reference 6(b). This procedure was not amenable to large-scale synthesis; Yutaka Ikeuchi provided a convenient new route that will be disclosed in due course.
- 30.a) Snyder SA, Treitler DS. Angew Chem Int Ed. 2009;48:7899. doi: 10.1002/anie.200903834. [DOI] [PubMed] [Google Scholar]; b) Snyder SA, Treitler DS, Brucks AP. J Am Chem Soc. 2010;132:14303. doi: 10.1021/ja106813s. [DOI] [PubMed] [Google Scholar]
- 31.Upon completion of this work, bromonium-initiated cyclization of substrates containing an epoxide with cis configuration was reported: Bonney KJ, Braddock DC. J Org Chem. 2012;77:9574. doi: 10.1021/jo301580c.
- 32.Tashiro T, Mori K. Tetrahedron: Asymmetry. 2005;16:1801. [Google Scholar]
- 33.a) Seyferth D, Marmor RS, Hilbert P. J Org Chem. 1971;36:1379. [Google Scholar]; b) Gilbert JC, Weerasooriya U. J Org Chem. 1979;44:4997. [Google Scholar]; c) Roth GJ, Liepold B, Müller SG, Bestmann HJ. Synthesis. 2004:59. [Google Scholar]
- 34.a) Hart DW, Schwartz J. J Am Chem Soc. 1974;96:8115. [Google Scholar]; b) Schwartz J, Labinger JA. Angew Chem Int Ed Engl. 1976;15:333. [Google Scholar]; c) Wipf P, Jahn H. Tetrahedron. 1996;52:12853. [Google Scholar]
- 35.For selected precedence of this observation: Cram DJ, Abd Elhafez FA. J Am Chem Soc. 1952;74:5828.Nakata T, Kishi Y. Tetrahedron Lett. 1978;19:2745.Still WC, McDonald JH., III Tetrahedron Lett. 1980;21:1031.Corey EJ, Ha DC. Tetrahedron Lett. 1988;29:3171. doi: 10.1016/0040-4039(88)85122-0.Little RD, Nishiguchi GA. In: In Studies in Natural Products Chemistry. Attaur-Rahman, editor. Vol. 35. Elsevier; Amsterdam: 2008. pp. 3–56.
- 36.a) Burgess EM, Penton HR, Jr, Taylor EA. J Am Chem Soc. 1970;92:5224. [Google Scholar]; b) Arhart RJ, Martin JC. J Am Chem Soc. 1972;94:5003. [Google Scholar]; c) Martin JC, Franz JA, Arhart RJ. J Am Chem Soc. 1974;96:4604. [Google Scholar]; d) Grieco PA, Gilman S, Nishizawa M. J Org Chem. 1976;41:1485. [Google Scholar]
- 37.a) Bamford WR, Stevens TS. J Chem Soc. 1952:4735. [Google Scholar]; b) Shapiro RH, Heath MJ. J Am Chem Soc. 1967;89:5734. [Google Scholar]; c) Adlington RM, Barrett AGM. Acc Chem Res. 1983;16:55. [Google Scholar]
- 38.a) Piers E, Abeysekera BF, Herbert DJ, Suckling ID. Can J Chem. 1985;63:3418. [Google Scholar]; b) Sasaki M, Murae T, Takahashi T. J Org Chem. 1990;55:528. [Google Scholar]; c) Nan F, Liu L, Xiong Z, Chen Y, Li T, Li Y. Collect Czech Chem Commun. 1997;62:1089. [Google Scholar]; d) Maimone TJ, Shi J, Ashida S, Baran PS. J Am Chem Soc. 2009;131:17066. doi: 10.1021/ja908194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong YL, Shing TKM. J Org Chem. 1997;62:2622. doi: 10.1021/jo9621581. [DOI] [PubMed] [Google Scholar]
- 40.Nieto N, Molas P, Benet-Buchholz J, Vidal-Ferran A. J Org Chem. 2005;70:10143. doi: 10.1021/jo051682h. [DOI] [PubMed] [Google Scholar]
- 41.a) Neverov AA, Brown RF. J Org Chem. 1998;63:5977. doi: 10.1021/jo980627o. [DOI] [PubMed] [Google Scholar]; b) Neverov AA, Feng HX, Hamilton K, Brown RS. J Org Chem. 2003;68:3802. doi: 10.1021/jo020750m. [DOI] [PubMed] [Google Scholar]
- 42.For syntheses and characterization of carbonates 51, 53, and 53′, please see: Bravo F, McDonald FE, Neiwert WA, Hardcastle KI. Org Lett. 2004;6:4487. doi: 10.1021/ol048212e.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.