

Abstract
Having a theoretical understanding of the orientation of immunoglobulin on an immobilized solid surface is important in biomedical pathogen-detecting systems and cellular analysis. Despite the stable adsorption of immunoglobulin on a polystyrene (PS) surface that has been applied in many kinds of immunoassays, there are many uncertainties in antibody-based clinical and biological experimental methods. To understand the binding mechanism and physicochemical interactions between immunoglobulin and the PS surface at the atomic level, we investigated the binding behavior and interactions of the monoclonal immunoglobulin G (IgG) on the PS surface using the computational method. In our docking simulation with the different arrangement of translational and rotational orientation of IgG onto the PS surface, three typical orientation patterns of the immunoglobulin G on the PS surface were found. We precisely analyzed these orientation patterns and clarified how the immunoglobulin G interacts with the PS surface at atomic scale in the beginning of the adsorption process. Major driving forces for the adsorption of IgG onto the PS surface come from serine (Ser), aspartic acid (Asp), and glutamic acid (Glu) residues.
Keywords: bionano interface, immunoassay, polystyrene, IgG, physical adsorption, simulation
Video abstract
Introduction
The molecular-level specific recognition of biomolecules plays a fundamental role in the biological system. Over the last few decades, various assays and biosensors have been successfully developed, based on the multiple noncovalent specific bonds between biomolecules, eg, electrostatic, electrodynamic, hydrogen, and hydrophobic interactions.1 The immunoassay is an antibody-based detecting technique for a specific antigen;2 it exploits highly sensitive and specific binding interactions between an antigen and an antibody.3,4 Depending on the assay format, immunoassays can be used qualitatively and quantitatively, and the application of immunoassays has been extended to various research fields, such as environmental monitoring, medical diagnostics, proteomics, pharmaceutical drug screening research, and basic cellular analytical research.5,6
Enzyme-linked immunosorbent assays (ELISAs) are the most fundamental and basic immunoassay for clinical diagnostic and biological research fields, due to their high sensitivity and versatility.6–8 In conventional ELISA, the antibody or antigen is usually immobilized on a polystyrene (PS) substrate by physical adsorption. The adsorption of proteins on PS surfaces has been studied extensively. In particular, the adsorption of immunoglobulin G (IgG) molecules onto PS substrates is of considerable interest in medical and biological fields, as the IgG system is widely used for micro- and nanoscale detection of an antigen-antibody reaction. Svensson et al investigated the adsorption of IgG onto a PS surface using ellipsometry and obtained the thickness of a layer of the antibody.9 They also proposed some orientation patterns of the IgG and discussed the efficiency of hydrophobic interactions between the antibody and the surface. The interaction of adsorption of IgG onto the PS surface was revealed that the major adsorption force comes from the hydrophobic interaction between the protein and the polymer surface.10 In the present study, the physicochemical interactions between immunoglobulin and the PS surface have been investigated at an atomic level by using the human IgG molecule as an example, and the binding mechanism and the orientation patterns of the antibody on the PS surface are discussed in detail.
PS has been widely used in industrial and medical fields because of its low cost, durability, hydrophobicity, nontoxicity, and optical transparency. IgG is a predominant immunoglobulin in the serum with molecular weight of about 150 kilodaltons.11 It can bind many kinds of antigens, such as virus and bacteria, by the antigen-binding sites of IgG. Moreover, IgG has another binding site at the Fc (fragment crystallizable) region of IgG, and it can bind to the cell surface through Fcγ receptors (FcγRs). FcγR is a membrane glycoprotein, and it can make a complex of IgG–FcγR. However, the exact information of the binding site of IgG on the PS surface is still a controversial one.12–14 The binding ability of IgG has been considered applicable to various fields of research and industry. Experimentally, the dynamic analysis of IgG molecules on PS-coated quartz crystal microbalances15 has been studied for the quantitative analysis of antibody immobilization and for immunological activity of proteins.16–18 However, the interaction of IgG with the PS surface has not been fully investigated yet in the atomic level. In this work, we performed the molecular mechanics calculation to investigate the orientation and mechanism of the binding interaction of IgG onto the PS surface. In our docking analysis, we obtained a plausible conformation of the orientation of IgG on the PS surface with strong interaction and discussed the major interactions in these orientations in detail.
Materials and methods
Modeling of IgG
The most abundant isotope of antibodies is human IgG. Among these families, immunoglobulin G1 (IgG1) is the most typical type, and it was used in our simulation. The structure of IgG1 is shown in Figure 1. It consists of two heavy (H) and two light (L) chains and is divided into three main regions. Those are one Fc and two Fab (fragment antigen-binding) domains, as shown in Figure 1A. Each Fab–domain is further categorized into variable (V) and constant (C) parts. Each H-chain is composed of one variable (HV) domain and three constant (HC1, HC2, HC3) domains. Each L-chain links to the H-chain by one interdisulfide bond. Fc–domain is a stem of the IgG, and it links to the next H-chain with three interdisulfide bonds of the hinge. As a total, the H and L chains consist of four and two intradisulfide bonds, respectively. IgG1’s structure was determined by X-ray crystallography,19 and the coordinate of the crystallographic structure is available in the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB entry 1IGY). In this work, the amino acid residues were renumbered sequentially from 1 to 434 for each H chain and from 1 to 213 for each L chain from the original numbering of 1IGY, respectively, for clarity. In the simulation, IgG1 was constructed with protonated form at N-terminus and unprotonated form at C-terminus in all the chains, as it exists in solution with neutral pH. All histidine residues were modeled in the unionized state by being singly protonated at the N-position.
Figure 1.

Schematic representation (A) and minimized conformation (B) of the structure of IgG1.
Notes: Two heavy (H) and two light (L) chains are shown by mauve, orange and green, blue, respectively. The H-chains are composed of one variable domain (HV) and three constant domains (HC1, HC2, HC3); L-chains are composed of one variable domain (LV) and one constant domain (LC), respectively. Minimized conformation of IgG1 is shown with face (top) and side (bottom) views in (B).
Abbreviations: H, heavy chain; L, light chain; C, constant domain; V, variable; Fab, fragment antigen-binding; Fc, fragment crystallizable; S, sulfur atom.
The crystallographic structure of 1IGY was first minimized to relieve any undesirable strains existing in the original PDB data by using the Chemistry at HARvard Molecular Mechanics34 (CHARMM)20 software with its allatom force field.21 The energy minimization was performed using conjugate gradient22 (CONJ) technique in implicit water environment with dielectric constant 80.23 The energy minimized conformation of IgG1 is shown in Figure 1B. The root mean square deviation for the backbone of minimized conformation of IgG1 (Figure 1B) was calculated to be 3.1 Å, which was close to the crystal structure of IgG1. This indicates that the crystal structure of IgG1 keeps well after the energy minimization in implicit water environment.
Modeling of PS surface
To make a PS amorphous surface, ethylbenzene molecules were used as a model to represent the styrene monomer, according to the previous report.24 A model of a PS box was constructed by randomly filling the ethylbenzene molecules in a cubic box that measured 25 nm × 25 nm × 25 nm. That is, we filled the molecules in the box with arbitrary orientations with the density of 1.05 grams per mL, which corresponds to the experimentally observed value of the bulk density of PS.25 Then, molecular dynamics (MD) simulation was performed on this PS box with periodic boundary condition for 0.2 ns to equilibrate the surface. The details of the MD simulation are described in the next paragraph. To reduce the computational expense in the next docking simulation, the sheet with 2.0 nm thickness was cut out from the cubic box, and the surface was used as a model of the PS surface in this work. The experimental atomic force microscopy images of the PS surface show that it has a roughness with around 2 μm periodical odd-shaped morphology.18 According to this, it can be considered that our narrow simulation surface with the area of 25 nm × 25 nm could be reasonably assumed as a flat surface.
MD simulation was performed using CHARMM3420 software with its all-atom force field.21 A three-dimensional periodic boundary condition was assigned to the box. The MD simulation was carried out with isobaric-isothermal (NPT) ensemble with 1 femtosecond time step. The details of the calculation procedure were the same as our previous simulations;26,27 ie, nonbonded interactions were calculated using a group-based cutoff with a switching function20 and were updated every five time steps. The switching function was turned on at 1.2 nm and turned off at 1.35 nm. All the bonds containing hydrogen were constrained using the SHAKE BONH algorithm.28 Electrostatic interaction was calculated using the Ewald summation method.29 The dielectric constant was set at 1.0. The temperature was set at 25°C, which was controlled by a Nosé-Hoover thermostat.30,31 All data visualization was done using Visual Molecular Dynamics 1.8.7 software (University of Illinois and the Beckman Institute, Champaign, IL, USA).32
Simulation procedure
Figure 2 shows the initial conformation of the orientation of IgG1 on the PS surface to investigate the interaction of IgG1 with the PS surface. In this conformation, the first and second principal axes of inertia (X and Y direction in the Figure 2) of IgG1 were set parallel to the PS surface, and the third principal axis (Z direction) of IgG1. The distance between the center of mass of IgG1 and the PS surface is arbitrarily set at 4.5 nm as an initial value along the Z direction where the IgG molecule is slightly overlapped with the PS surface. The docking simulation of the system was divided into two steps. First, from the above initial point, the various different arrangements with relative orientations of the IgG1 against the PS surface were generated, and the interaction energy was evaluated at each orientation with their fixed conformations using CHARMM3420 software with its all-atom force field.21 Those different orientations of IgG1 on the PS surface were constructed by rotating IgG1 around the Y and Z axes at an interval of 10 degrees in the range as shown in Table 1. At each rotation, IgG1 was translated along the X–Y area on the PS surface and the Z axes from the PS surface within the range, as also shown in Table 1. After the calculation of the interaction energies for all these conformations, the conformations of the IgG1 on the PS surface, which had strong interactions between them, were selected. The docking simulations were performed with the dielectric constant of 1.0, at which it was considered that no bulk water existed between the IgG1 and the PS surface interacting area.
Figure 2.
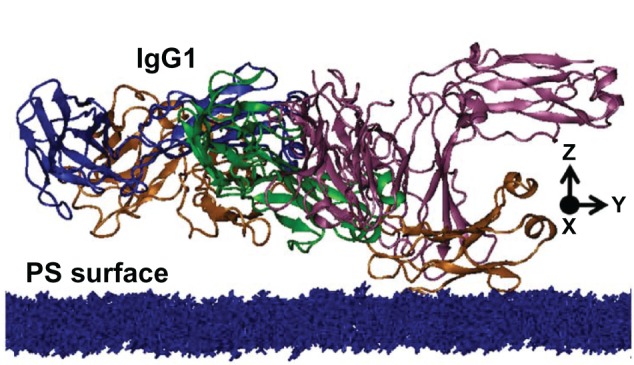
Initial structure of IgG1, on the PS surface in the docking simulation.
Note: The PS surface is shown with blue representation.
Abbreviations: IgG1, immunoglobulin G1; PS, polystyrene.
Table 1.
Range of the rotation angles and translation distances of IgG1 on the PS surface used for the docking simulation are listed
| Axis | Rotation (degree)
|
Translation (nm)
|
||
|---|---|---|---|---|
| Interval | Range | Interval | Range | |
| X | – | – | 0.05 | 0~0.5 |
| Y | 10 | −90 to 80 | 0.05 | 0~0.5 |
| Z | 10 | 0~350 | 0.05 | 0~5.0 |
Abbreviations: IgG1, immunoglobulin G1; PS, polystyrene.
While the conformations obtained in the above docking simulation were calculated using the single-point energy calculation with a fixed structure of the PS surface and the IgG1 molecule, they were, as a second step, further energy minimized without any constraint on the molecular systems to avoid the undesired effect of the initial distribution of the monomer in the PS surface model.
Results and discussion
Orientation of IgG1 onto PS surface
Docking simulation between IgG1 and the PS surface was performed with various relative orientations, according to the procedure discussed in the previous section. The results are sorted with the values of the interaction energies between the IgG1 and the PS surface; three groups of the orientation patterns with high interaction energies were found. From each group, the conformation with the highest interaction energy was selected. As these three conformations were obtained from the single point energy calculation in the docking simulation, they were further energy minimized without any constraint on the molecules. The conformations of these three orientations after the minimization procedure are shown in Figure 3. To clarify how IgG1 interacts with the PS surface, a schematic representation of the orientation behaviors are shown in the insets. The total interaction energies of these orientations in Figure 3A–C were −81.2, −57.3, and −56.8 kilocalorie per mole (kcal/mol), respectively. Svensson et al proposed some possible interaction patterns of the IgG1 on the PS surface with their schematic illustrations.9 The three conformations obtained in this work look similar to the typical orientation behaviors of their end-on, flat-on, and side-on orientations.9 However, the details of the orientation patterns and the interaction sites are somewhat different from their illustrations. We discussed the interaction behaviors between IgG1 and the PS surface for these conformation patterns in detail.
Figure 3.

Snapshots of three different relative orientations of IgG1 on the PS surface obtained from the docking simulation.
Notes: The orientations of Fab-Fab-on (A), Fc-on (B), and Fab-Fc-on (C) are shown in the top row. Schematic representations are shown in the insets of the top row. The interacting regions between IgG1 and the PS surface are indicated in the figures from (a) to (e). The interacting amino acid residues are shown by vdW representations with yellow color. Interacting chains in each of the three orientations were extracted and shown in the bottom row, where the interacting amino residues with the PS surface are again represented by vdW spheres with yellow color. Equivalent amino residues in the other pair chains of IgG1 are shown in the same figure by vdW spheres with green and blue colors.
Abbreviations: IgG1, immunoglobulin G1; PS, polystyrene; H, heavy chain; L, light chain; C, constant domain; V, variable; vdW, van der Waals interaction; ter, terminal.
The orientation of IgG1 in Figure 3A indicated that IgG1 interacts with the PS surface with two Fab domains of IgG1 molecule. We denoted this orientation as Fab–Fab–on, and their two interaction sites are shown in Figure 3A as (a) and (b). In these sites, the interacting amino acid residues are shown as van der Waals (vdW) radius representation with with yellow color. The interaction energies were separately evaluated in both (a) and b-region, and their values were determined to be −16.8 and −72.3 kcal/mol, respectively. In this orientation, it was found that the LV part of the L chain in the Fab domain was involved in both interactions. As the region of Lv is known as the antigen-binding site in the antigen–antibody reaction, this indicates that the same part of the antigen-binding region of Fab works as an interaction site of IgG1 on the PS surface. Although the interaction energy of the a-region (Figure 3A) is smaller than that of the b-region in this calculation, countertype-binding surface in the a-region, and green spheres interact with the PS surface in the b-region (Figure 3A).
In the case of Figure 3B and C, it can be seen that the H chains of IgG1 interact with the PS surface. The orientation in Figure 3B indicates that the HC2 domain in the Fc part of IgG1 interacts with the PS surface. Therefore, we denote this orientation as Fc–on in the following section. This interaction site is shown as (c) in Figure 3B. Kato et al indicated from their nuclear magnetic resonance experiments that the Fc region binds to FcγR at the negatively charged area of the HC2 domain.14 This indicates that the same HC2 domain works as the binding site in both PS and FcγR cases. Vice versa, a countertype binding orientation should also exist where the green spheres in the other HC2 domain in Fc part interacts with PS surface (Figure 3B, bottom).
On the other hand, in the case of Figure 3C, IgG1 binds to the PS surface through two sites in the H chains. One site locates at the HC3 domain in the Fc region of one H chain, and the other locates at the HV domain of the other H chain. These two interaction sites are shown as (d) and (e) in Figure 3. We denote this orientation as Fab–Fc–on in the following section. The analysis of these two interaction sites indicated that larger numbers of amino residues in the HV domain were involved in the interaction with the surface compared to the HC3 domain.
To investigate the interaction between IgG1 and the PS surface in more detail, we extracted the amino acid residues, which were strongly interacting with the PS surface in these three orientations. The residue numbers and their interaction energy of the strongly interacting amino acid residues, which interact with the PS surface with the energy more than −2.0 kcal/mol, were selected and shown in Tables 2–4. Among the amino residues listed in these tables, the strongest attracting interaction can be found between the aspartic acid (Asp) residues and the PS surface in all cases. Serine (Ser) residue works as a strongly interacting residue in Fab–Fab–on and Fab–Fc–on orientations (Tables 2 and 4), although its contribution is weak in Fc–on (Table 3). Instead, Glutamic acid (Glu) works as a strongly interacting residue in the Fc–on orientation. Nevertheless, most types of the interacting amino acid residues listed in Tables 2–4 are similar in all these three cases. They are Ser, threonine (Thr), and asparagine (Asn) in the uncharged hydrophilic amino residues, and Asp, Glu, and arginine (Arg) in the charged amino acid residues, respectively. That is, the similar residues are involved in the interaction with the PS surface in these three cases and only the extent of the strength would change, depending on the difference of the arrangement in these three orientations.
Table 2.
Amino acid residues and their interaction energies of IgG1 with the PS surface for Fab-Fab-on
| Residues | a-region
|
Residues | b-region
|
||||
|---|---|---|---|---|---|---|---|
| vdW | Elec | Total | vdW | Elec | Total | ||
| Uncharged hydrophilic amino acids | |||||||
| Ser51 | −5.0 | −3.3 | −8.3 | ||||
| Ser64 | −3.3 | −0.3 | −3.6 | ||||
| Ser66 | −1.4 | −0.6 | −2.0 | ||||
| Ser75 | −5.5 | −3.2 | −8.7 | ||||
| Ser76 | −2.5 | −0.6 | −3.1 | ||||
| Thr62 | −4.5 | 0.7 | −3.8 | ||||
| Charged amino acids | |||||||
| Arg17 | −10.2 | 0.4 | −9.8 | ||||
| Arg53 | −4.0 | −5.4 | −9.4 | ||||
| Asp69 | −0.2 | −7.1 | −7.3 | Asp59 | −4.8 | −5.9 | −10.7 |
| Asp69 | −0.2 | −2.8 | −3.0 | ||||
| Glu26 | −2.8 | −2.4 | −5.2 | Glu16 | −1.2 | −2.5 | −3.7 |
| Total | −3.0 | −9.5 | −12.5 | Total | −42.6 | −23.5 | −66.1 |
Note: The energies are shown in kcal/mol.
Abbreviations: PS, polystyrene; Ser, serine; Thr, threonine; Arg, arginine; Asp, aspartic acid; Glu, glutamic acid; IgG1, immunoglobulin G1; vdW, van der Waals interaction; elec, electrostatic; Fab, fragment antigen-binding; kcal/mol, kilocalorie per mole.
Table 4.
Amino acid residues and their interaction energies of IgG1 with PS surface for Fab–Fc–on
| Residues | d-interacting
|
Residues | e-interacting
|
||||
|---|---|---|---|---|---|---|---|
| vdW | Elec | Total | vdW | Elec | Total | ||
| Uncharged hydrophilic amino acids | |||||||
| Ser53 | −3.4 | −0.6 | −4.0 | ||||
| Ser74 | −6.8 | −2.0 | −8.8 | ||||
| Thr27 | −3.8 | 1.3 | −2.5 | ||||
| Thr29 | −5.4 | −2.5 | −7.9 | ||||
| Thr30 | −4.1 | −0.2 | −4.3 | ||||
| Asn76 | −2.0 | 0.0 | −2.0 | ||||
| Charged amino acids | |||||||
| Glu409 | −0.2 | −1.8 | −2.0 | Arg73 | −11.4 | 1.2 | −10.2 |
| Asp72 | −1.9 | −8.1 | −10.0 | ||||
| Total | −0.2 | −1.8 | −2.0 | Total | −38.8 | −10.9 | −49.7 |
Note: The energies were shown in unit of kcal/mol.
Abbreviations: IgG1, immunoglobulin G1; PS, polystyrene; vdW, van der Waals interaction; elec, electrostatic; Ser, serine; Thr, threonine; Asn, asparagine; Glu, glutamic acid; Arg, arginine; Asp, aspartic acid.
Table 3.
Amino acid residues and their interaction energies of IgG1 with the PS surface for Fc–on
| Residues | c-region
|
||
|---|---|---|---|
| vdW | Elec | Total | |
| Uncharged hydrophilic amino acids | |||
| Asn272 | −6.7 | 0.7 | −6.0 |
| Asn306 | −0.7 | −1.8 | −2.5 |
| Charged amino acids | |||
| Asp271 | −4.0 | −12.7 | −16.7 |
| Asp303 | −0.1 | −6.3 | −6.4 |
| Glu309 | −4.6 | −12.1 | −16.7 |
| Glu324 | −1.9 | −7.7 | −9.6 |
| Glu274 | −0.3 | −2.8 | −3.1 |
| Total | −18.3 | −42.7 | −61.0 |
Note: The energies were shown in kcal/mol.
Abbreviations: IgG1, immunoglobulin G1; PS, polystyrene; Fc, fragment crystallizable; vdW, van der Waals interaction; elec, electrostatic; Asn, asparagine; Asp, aspartic acid; Glu, glutamic acid; kcal/mol, kilocalorie per mole.
The interaction energies were divided into two terms of van der Waals and electrostatic terms and were shown in Tables 2–4. The way of dividing interaction force into these force terms may somewhat depend on the force fields used in the molecular mechanics calculation. Although the interaction force should, in principle, be described by the electronic theory of quantum mechanics, the force fields of molecular mechanics were determined to maximally reproduce the interaction force using these two terms of van der Waals and electrostatic terms. From this point of view, the division and the meaning of these two terms is a little artificial. However, the qualitative interpretation of the nature of the origin of the interaction, which has van der Waals nature and/or electrostatic nature, can be possible and useful, to know the types of the interaction mechanisms. It can be seen from Tables 2–4 that the major driving force for adsorption of IgG1 onto the PS surface comes from the van der Waals interaction terms. These results are reasonable because the total charge of the PS molecule is electronically neutral. However, the amino acid residues of Asp and Glu with a negatively charged side chain interact with the PS surface by electrostatic natures. Positively charged amino acid residue of Arg has a strong interaction with the PS surface by van der Waals force. However, it also shows a strong electrostatic interaction. It may depend on the distance between Arg and the PS surface. The individual interaction behaviors of these amino residues with the PS surface are discussed in the next paragraphs in detail.
Interactions in Fab–Fab–on orientation
Amino acid residues in Fab–Fab–on orientation, which strongly interact with the PS surface, are shown in Table 2. It can be seen that there are five Ser residues in the B site. To investigate the interaction between the Ser residues and the PS surface, the orientation behavior of them is shown in Figure 4. Among them, Ser75 showed the strongest interaction, and its orientation behavior is shown in Figure 4D. In this orientation, the aliphatic CH group of the PS molecule interacts with the oxygen atoms of the carbonyl group in the peptide main chain and the hydroxyl group of Ser75 simultaneously with the total interaction energy of −8.7 kcal/mol (Figure 4D).
Figure 4.
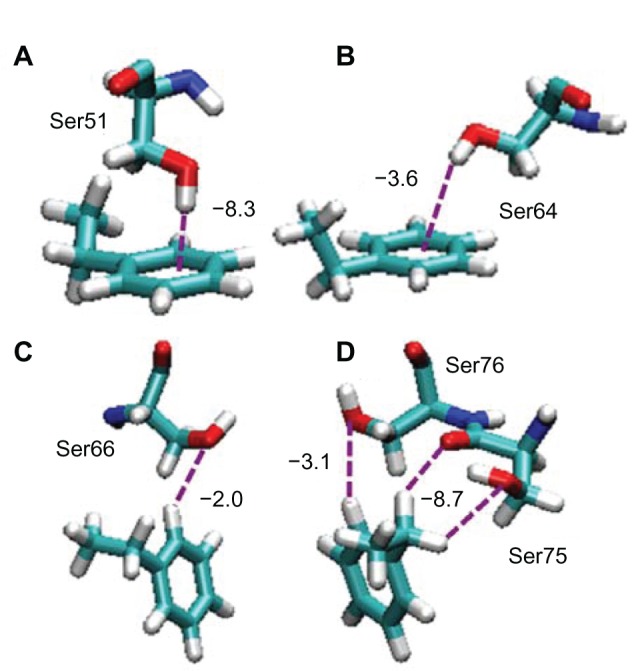
Snapshots of the interactions between Ser residues and the PS surface included in the Fab–Fab–on orientation.
Notes: Only the interacting pairs of the amino residue and the ethylbenzene molecule are shown for simplicity. Ser51, Ser64, Ser66, Ser75, and Ser76 in b-region are shown in (A–D), and (D), respectively. Oxygen, nitrogen, carbon, and hydrogen atoms are shown by red, blue, green, and gray colors, respectively.
Abbreviations: Ser, serine; PS, polystyrene; Fab, fragment antigen-binding.
These interactions are categorized into CH/O interactions, the nature of which is precisely discussed by Takahashi et al.33 Secondary strong interaction was Ser51 (Figure 4A). The hydroxyl hydrogen of the Ser51 orients to the benzene ring perpendicularly with interaction energy of −8.3 kcal/mol (Figure 4A). This interaction is categorized into OH/π interaction. Ser64 also takes a similar orientation with the Ser51, but its interaction energy is smaller than Ser51 because the OH/π orientation is a little bent from the preferable perpendicular orientation (Figure 4B). The nature of the OH/π interaction was investigated by Suzuki et al using ab initio calculation with a benzene and water molecule system, and they showed that the hydrogen atom of water points toward the π-cloud of benzene.34,35 The above mentioned interactions have been parameterized in the CHARMM force field.36,37 The parameter sets of the CHARMM force field were confirmed to reproduce the binding mode with excellent agreement to the ab initio calculation. In Figure 4C and D, the hydroxyl oxygens of the Ser66 and Ser76 interact with the proton at the edge of the benzene ring. These interactions are again another type of CH/O interaction. Although each of the interactions did not provide a dominant energy with the PS surface interaction, these multiple interactions of Ser residues could work as about one-third of the total interaction energy of the IgG molecule onto the PS surface, as shown in Table 2.
Arg17 and Arg53 show larger interaction energy than those of Ser (Table 2). These interaction behaviors are shown in Figure 5. It can be seen that the positively charged guanidine group closely contacts with the benzene ring of the PS. This orientation indicates the existence of cation-π interaction between them. This interaction has been investigated theoretically38 and experimentally,39 using the ammonium ion and benzene derivative system. Interactions of Arg with the aromatic ring in proteins were also reported when some of the strong cation-π interactions were involved in perpendicular orientation in the protein.40 The cation-π interaction has been parameterized in the CHARMM force field.41 The parameter sets of CHARMM force field were confirmed with the quantum chemistry approaches.
Figure 5.
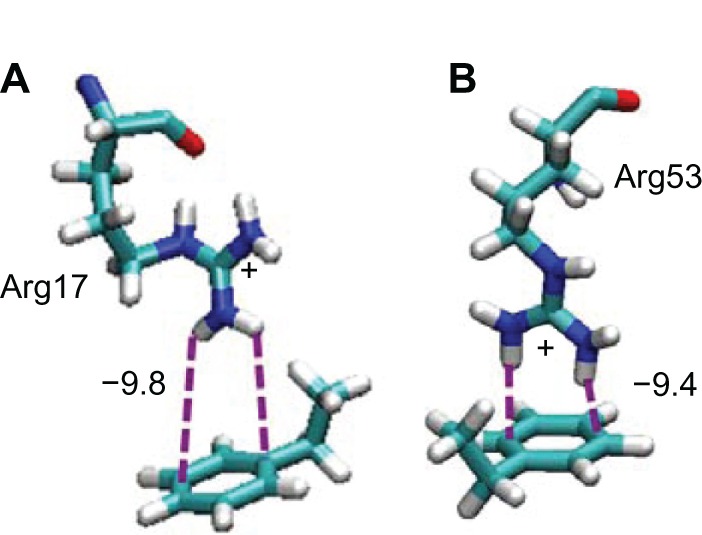
Snapshots of the interactions between Arg residues and the PS surface included in the Fab–Fab–on orientation.
Notes: Arg17 and Arg53 in b-region are shown in (A) and (B), respectively.
Abbreviations: Arg, arginine; PS, polystyrene; Fab, fragment antigen-binding.
Another strong interaction found in the Fab–Fab–on conformation is the Asp interaction with the PS surface molecule, listed in Table 2. They are Asp69 in a-region and Asp59, Asp69 in b-region, which are shown in Figure 6A–C, respectively. These are the interactions between the carboxylate anion and the benzene ring, which are also frequently found in biological systems. Jackson et al showed the interaction between the anions and the electropositive ring edge proton of an aromatic group by the theoretical calculations.42 This is known as the anion–quadrupole interaction. In our calculation, it can be seen that the hydrogens at the edge of aromatic groups orient toward the carboxylate oxygens to show the interaction between them. Amino acid residue of Glu includes carboxylate anions as well as Asp. Although the anion–quadrupole interaction for Glu residues was not found in the Glu26 and Glu16 orientation in Fab–Fab–on conformation, another possible interaction of CH/π and CH/N can be seen in Figure 6D and E.
Figure 6.
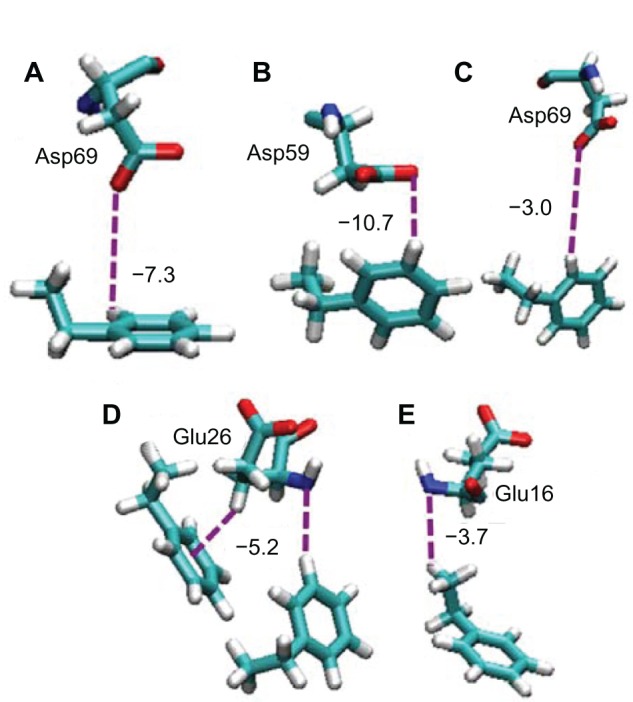
Snapshots of the interactions among Asp and Glu residues with the PS surface included in the Fab–Fab–on orientation.
Notes: Asp69 and Glu26 in a-region are shown in (A) and (D), respectively. Asp59, Asp69, and Glu16 in b-region are shown in (B), (C), and (E), respectively.
Abbreviations: Asp, aspartic acid; Glu, glutamic acid; PS, polystyrene; Fab, fragment antigen-binding.
Interactions in Fc–on and Fab–Fc–on orientation
In Fc–on orientation shown in Figure 3B, amino acid residues in HC2 domain in the Fc part of IgG1 interact with the PS surface. It was reported previously that the Fc region binds to receptors on the cell FcγR at a negatively charged area of the HC2 domain.13 In our obtained Fc–on orientation, the Asp and Glu residues in HC2 domain were found to interact strongly with a surface of PS (Table 3). The orientation behavior of these residues is shown in Figure 7A–E. The negatively charged carboxylate oxygens of residues of Glu309 and Glu324 are considered to interact with the hydrogen at the edge of the aromatic groups of PS molecules by the anionquadrupole interaction. The negatively charged residues of Asp271 and Asp303, which are shown in Table 3 but not shown in Figure 7, are also considered to have a similar interaction of anion–quadrupole interaction as was already discussed in Figure 6. On the other hand, Glu274 showed the possible orientation to have a CH/N interaction with the PS surface. The side chains of Asn272 and Asn306 are found to interact with the benzene group of the PS surface by NH/π and CH/O type interactions, respectively. There are some reports on the NH/π interaction that work between amide NH and the π electron system of the benzene group.35,43–45 All these multiple interactions, not only the anion–quadrupole interaction but also the weak NH/π and CH/O interactions, would cooperatively work as a force to bind the IgG molecule with Fc–on orientation onto the PS surface.
Figure 7.
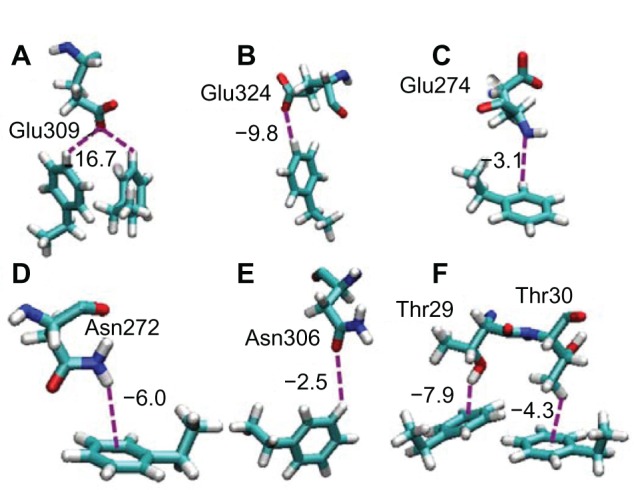
Snapshots of the interactions among Glu, Asn, and Thr residues with the PS surface included in the Fc–on and Fab–Fc–on orientations.
Notes: Glu309, Glu324, Glu274, Asn272, and Asn306 in c-region are shown in (A) to (E), respectively. Thr29 and Thr30 in E region are shown in (F).
Abbreviations: Glu, glutamic acid; Asn, asparagine; Thr, threonine; Fab, fragment antigen-binding; Fc, fragment crystallizable.
The Fab –Fc–on orientation is shown in Figure 3C and the typical interacting residues in the binding sites of (d) and (e) were investigated, and the results are shown in Table 4. The residues of Ser, Asn, Arg, and Asp, which were found in the previous Fab–Fab–on and Fc–on orientations, are also found in the interaction in this case. The remaining amino residue with large interaction energy in Table 3 is Thr. The interacting behaviors of the residues of Thr29 and Thr30 with benzene ring are shown in Figure 7F. The carboxyl group of Thr29 residue orients toward the benzene ring with OH/π interaction. On the other hand, methyl proton of Thr30 interacts with the benzene ring, which is known as CH/π interaction.
To summarize, it can be said again that the binding of the IgG1 molecule onto the PS surface can be considered as a cooperative work of weak interactions, such as anion–quadrupole, NH/π, CH/O interactions, and so on. The adsorption of the antibody onto the surface has been studied, depending on the surface properties of wettability,46 pH,47 physical and chemical treatments,48 and others. In this study, we investigated the orientation and mechanism of the binding of IgG1 onto an untreated PS surface in the neutral environment. The main orientation of IgG1 onto the PS surface was found to be Fab–Fab–on type. The major interaction comes from Ser residues through van der Waals interaction, which is the same as the results of IgG and Fab fragment adsorptions at hydrophobic surfaces, mainly in an end-on orientation.49 However, it is known that the amino sequences in the Fab region will be variable. Therefore, strictly speaking, our result is valid for the specific IgG1, which we used as a model. Although our result implies that a similar interaction would be expected when the interacting residues, such as Arg, Ser, and Arg, are included in the Fab region, we need to investigate further the interactions using other IgGs to generalize the result.
From the experimental point of view, it is important to know the influence of the experimental conditions, such as the temperature, pH, and so on. However, our docking simulation cannot evaluate the effect of the temperature. This is because the docking simulation is based on the lowest energy minimum conformation, which means that the obtained structure corresponds to the one at 0°K. Therefore, in future work, we will plan to perform the MD simulation where we can observe the absorption behavior of the IgG with temperature influence. Although we have not discussed the effect of the pH of the experimental condition in this work, a decrement of the adsorption at a low pH environment would be expected, because the interaction energies of acidic amino acid residues would reduce, as Asp and Glu would be protonated and have neutral states.
Conclusion
The interaction between IgG1 and the PS surface was investigated using the computational method. Docking simulation found three conformations with different orientations which have strong interaction between IgG1 and the PS surface. They are named as Fab–Fab–on, Fc–on, and Fab–Fc–on orientations in this work, according to the interacting sites of IgG1. The main driving force for the adsorption of IgG1 onto the PS surface comes from Ser residues by the OH/π interaction, which originally comes from the mixture of the van der Waals and electrostatic interaction natures. The amino acid residues of Asp and Glu with negatively charged side chains were found to interact with the PS surface by anion–quadrupole interactions. Although the anion–quadrupole interaction showed relatively large value compared to the CH/O, CH/N, CH/π, and NH/π interactions in these three orientations, all these multiple interactions would work cooperatively as a force to bind the IgG1 molecule onto the PS surface.
Acknowledgments
This research was supported by The Asian Research Center in Mongolia and the Korean Foundation for Advanced Studies within the framework of the Project #1 (2013–2014). NJ is grateful to the School of Chemistry and Chemical Engineering, National University of Mongolia, and the Graduate School of Engineering, Yokohama National University, for the help of our collaboration under the research agreement between them. The authors thank the Research Center for Computational Science, Okazaki, Japan, for the use of a computer to perform part of the calculation.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Turner AP. Biosensors – sense and sensitivity. Science. 2000;290(5495):1315–1317. doi: 10.1126/science.290.5495.1315. [DOI] [PubMed] [Google Scholar]
- 2.Sano T, Smith CL, Cantor CR. Immuno-PCR: very sensitive antigen detection by means of specific antibody-DNA conjugates. Science. 1992;258(5079):120–122. doi: 10.1126/science.1439758. [DOI] [PubMed] [Google Scholar]
- 3.Koivunen ME, Krogsrud RL. Principles of immunochemical techniques used in clinical laboratories. Lab Medicine. 2006;37(8):490–497. [Google Scholar]
- 4.Saleh OA, Sohn LL. Direct detection of antibody–antigen binding using an on-chip artificial pore. Proc Natl Acad Sci U S A. 2003;100(3):820–824. doi: 10.1073/pnas.0337563100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng AH, Uddayasankar U, Wheeler AR. Immunoassays in microfluidic systems. Anal Bioanal Chem. 2010;397(3):991–1007. doi: 10.1007/s00216-010-3678-8. [DOI] [PubMed] [Google Scholar]
- 6.Engvall E, Perlmann P. Enzyme-linked immunosorbent assay (ELISA) Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8(9):871–874. doi: 10.1016/0019-2791(71)90454-x. [DOI] [PubMed] [Google Scholar]
- 7.Hage DS. Immunoassays. Anal Chem. 1993;65(12):420R–424R. [Google Scholar]
- 8.Porstmann T, Kiessig ST. Enzyme immunoassay techniques. An overview. J Immunol Methods. 1992;150(1–2):5–21. doi: 10.1016/0022-1759(92)90061-w. [DOI] [PubMed] [Google Scholar]
- 9.Svensson O, Arnebrant T. Antibody–antigen interaction on polystyrene: an in situ ellipsometric study. J Colloid Interface Sci. 2012;368(1):533–539. doi: 10.1016/j.jcis.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 10.Galisteo-González F, Martín-Rodríguez A, Hidalgo-Alvarez R. Adsorption of monoclonal IgG on polystyrene microspheres. Colloid Polym Sci. 1994;272(3):352–358. [Google Scholar]
- 11.Junqueira LCU, Carneiro J. Basic Histology. 10th ed. New York: McGraw-Hill Medical Pub; 2003. [Google Scholar]
- 12.Sondermann P, Huber R, Jacob U. Crystal structure of the soluble form of the human fcgamma-receptor IIb: a new member of the immunoglobulin superfamily at 1.7 Å resolution. EMBO J. 1999;18(5):1095–1103. doi: 10.1093/emboj/18.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maxwell KF, Powell MS, Hulett MD, et al. Crystal structure of the human leukocyte Fc receptor, Fc gammaRIIa. Nat Struct Biol. 1999;6(5):437–442. doi: 10.1038/8241. [DOI] [PubMed] [Google Scholar]
- 14.Kato K, Sautès-Fridman C, Yamada W, et al. Structural basis of the interaction between IgG and Fcgamma receptors. J Mol Biol. 2000;295(2):213–224. doi: 10.1006/jmbi.1999.3351. [DOI] [PubMed] [Google Scholar]
- 15.Okahata Y, Kawase M, Niikura K, Ohtake T, Furusawa H, Ebara Y. Kinetic measurement of DNA hybridization on an oligonucleotide-immobilized 27-MHz quartz crystal microbalance. Anal Chem. 1998;70(7):1288–1296. doi: 10.1021/ac970584w. [DOI] [PubMed] [Google Scholar]
- 16.Muramatsu H, Dicks JM, Tamiya E, Karube I. Piezoelectric crystal biosensor modified with protein A for determination of immunoglobulins. Anal Chem. 1987;59(23):2760–2763. doi: 10.1021/ac00150a007. [DOI] [PubMed] [Google Scholar]
- 17.Caruso F, Rodda E, Furlong DN. Orientational aspects of antibody immobilization and immunological activity on quartz crystal microbalance electrodes. J Colloid Interface Sci. 1996;178(1):104–115. [Google Scholar]
- 18.Atashbar MZ, Bejcek B, Vijh A, Singamaneni S. QCM biosensor with ultra thin polymer film. Sens Actuators B Chem. 2005;107(2):945–951. [Google Scholar]
- 19.Harris LJ, Skaletsky E, McPherson A. Crystallographic structure of an intact IgG1 monoclonal antibody. J Mol Biol. 1998;275(5):861–872. doi: 10.1006/jmbi.1997.1508. [DOI] [PubMed] [Google Scholar]
- 20.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculatons. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 21.Mackerell AD, Jr, Bashford D, Bellot M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 22.Fletcher R, Reeves CM. Function minimization by conjugate gradients. The Computer Journal. 1964;7(2):149–154. [Google Scholar]
- 23.Spassov VZ, Ya n L, Szalma S. Introducing an implicit membrane in generalized born/solvent accessibility continuum solvent models. J Phys Chem B. 2002;106(34):8726–8738. [Google Scholar]
- 24.Boughton AP, Andricioaei I, Chen Z. Surface orientation of magainin 2: molecular dynamics simulation and sum frequency generation vibrational spectroscopic studies. Langmuir. 2010;26(20):16031–16036. doi: 10.1021/la1024394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang CSC, Wilson PT, Richter LJ. Structure of polystyrene at the interface with various liquids. Macromolecules. 2004;37(20):7742–7746. [Google Scholar]
- 26.Javkhlantugs N, Naito A, Ueda K. Molecular dynamics simulation of Bombolitin II in the dipalmitoylphosphatidylcholine membrane bilayer. Biophys J. 2011;101(5):1212–1220. doi: 10.1016/j.bpj.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayakawa D, Ueda K, Yamane C, Miyamoto H, Horii F. Molecular dynamics simulation of the dissolution process of a cellulose triacetate-II nano-sized crystal in DMSO. Carbohydr Res. 2011;346(18):2940–2947. doi: 10.1016/j.carres.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 28.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):3, 27–341. [Google Scholar]
- 29.Ewald PD. Die berechnung optischer und elektrostatischer gitterpotentiale. [The calculation of optical and electrostatic lattice potentials] Ann Phys. 1921;369(3):253–287. German. [Google Scholar]
- 30.Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81(1):511–520. [Google Scholar]
- 31.Hoover WG. Canonical dynamics: Equilibrium phase-space distribution. Phys Rev A. 1985;31(3):1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 32.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi O, Kohno Y, Nishio M. Relevance of weak hydrogen bonds in the conformation of organic compounds and bioconjugates: evidence from recent experimental data and high-level ab initio MO calculations. Chem Rev. 2010;110(10):6049–6076. doi: 10.1021/cr100072x. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki S, Green PG, Bumgarner RE, Dasgupta S, Goddard WA, 3rd, Blake GA. Benzene forms hydrogen bonds with water. Science. 1992;257(5072):942–945. doi: 10.1126/science.257.5072.942. [DOI] [PubMed] [Google Scholar]
- 35.Tsuzuki S. Interactions with aromatic rings. Intermolecular Forces and Clusters I Structure and Bonding. 2005;115:149–193. [Google Scholar]
- 36.Jiang L, Lai L. CH…O hydrogen bonds at protein–protein interfaces. J Biol Chem. 2002;277(40):37732–37740. doi: 10.1074/jbc.M204514200. [DOI] [PubMed] [Google Scholar]
- 37.Macias AT, Mackerell AD., Jr CH/pi interactions involving aromatic amino acids: refinement of the CHARMM tryptophan force field. J Comput Chem. 2005;26(14):1452–1463. doi: 10.1002/jcc.20281. [DOI] [PubMed] [Google Scholar]
- 38.Basch H, Stevens WJ. Hydrogen bonding between aromatics and cationic amino groups. J Mol Struct: Theochem. 1995;338(1–3):303–315. [Google Scholar]
- 39.Meot-Ner M, Deakyne CA. Unconventional ionic hydrogen bonds. 2. NH+.cntdot..cntdot..cntdot..pi.. Complexes of onium ions with olefins and benzene derivatives. J Am Chem Soc. 1985;107(2):474–479. [Google Scholar]
- 40.Gallivan JP, Dougherty DA. Cation-pi interactions in structure biology. Proc Natl Acad Sci U S A. 1999;96(17):9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilis S, Biot C, Buisine E, Dehouck Y, Rooman M. Development of novel statistical potentials describing cation-pi interactions in proteins and comparison with semiempirical and quantum chemistry approaches. J Chem Inf Model. 2006;46(2):884–893. doi: 10.1021/ci050395b. [DOI] [PubMed] [Google Scholar]
- 42.Jackson MR, Beahm R, Duvvuru S, et al. A preference for edgewise interactions between aromatic rings and carboxylate anions: the biological relevance of anion–quadrupole interactions. J Phys Chem B. 2007;111(28):8242–8249. doi: 10.1021/jp0661995. [DOI] [PubMed] [Google Scholar]
- 43.Burley SK, Petsko GA. Amino-aromatic interactions in proteins. FEBS Lett. 1986;203(2):139–143. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- 44.Imai YN, Inoue Y, Nakanishi I, Kitaura K. Amide-pi interactions between foramide and benzene. J Comput Chem. 2009;30(14):2267–2276. doi: 10.1002/jcc.21212. [DOI] [PubMed] [Google Scholar]
- 45.Ottiger P, Pfaffen C, Leist R, Leutwyler S, Bachorz RA, Klopper W. Strong N–H…pi hydrogen bonding in amide-benzene interactions. J Phys Chem B. 2009;113(9):2937–2943. doi: 10.1021/jp8110474. [DOI] [PubMed] [Google Scholar]
- 46.Bouafsoun A, Ponsonnet L, Kerkeni A, Jaffrézic N, Othmane A. Comparative wettability study of polystyrene functionalized with different proteins. Materials Science and Engineering: C. 2007;27(4):709–715. [Google Scholar]
- 47.Song D, Forciniti D. Effects of cosolvents and pH on protein adsorption on polystyrene latex: A dynamic light scattering study. J Colloid Interface Sci. 2000;221(1):25–37. doi: 10.1006/jcis.1999.6560. [DOI] [PubMed] [Google Scholar]
- 48.Qian W, Yao D, Yu F, et al. Immobilization of antibodies on ultraflat polystyrene surfaces. Clin Chem. 2000;46(9):1456–1463. [PubMed] [Google Scholar]
- 49.Buijs J, Lichtenbelt JWT, Norde W, Lyklema J. Adsorption of monoclonal IgGs and their F(ab′)2 fragments onto polymeric surfaces. Colloids Surf B Biointerfaces. 1995;5(1–2):11–23. [Google Scholar]