

Summary
The β-defensins are a class of small, cationic proteins first recognized as antimicrobial components of the innate and adaptive immune system. More recently, one of the major β-defensins produced in skin, β-defensin 3, has been discovered to function as a melanocortin receptor ligand in vivo and in vitro, but its biophysical and pharmacological basis of action has been enigmatic. Here we report functional and biochemical studies focused on human β-defensin 3 (HBD3) and melanocortin receptors 1 and 4. Genetic and pharmacologic studies indicate that HBD3 acts as a neutral melanocortin receptor antagonist, capable of blocking the action of either stimulatory agonists, such as α-melanocyte stimulating hormone, or inhibitory inverse agonists such as Agouti signaling protein (Asip) and Agouti-related protein (Agrp). A comprehensive structure-function analysis demonstrates that two patches of positively charged residues, located on opposite poles of HBD3 and spatially organized by the compact β-defensin fold, are primarily responsible for high affinity binding to melanocortin receptors. These findings identify a distinct mode of melanocortin receptor-ligand interactions based primarily on electrostatic complementarity, with implications for designing ligands that target melanocortin and potentially other seven transmembrane receptors.
Introduction
β-defensins are a rapidly evolving family of small secreted proteins thought to mediate a response to diverse and changing environmental stress (Pazgier et al., 2006). Named by analogy to the α-defensins, which are key antimicrobial components of neutrophil granules, mammalian β-defensins are expressed primarily in epithelial tissues, often inducible upon exposure to proinflammatory agents (Lehrer, 2004). Initially recognized for their potential as “endogenous antibiotics,” some β-defensins can act as ligands for G protein-coupled receptors, including those of the chemokine and melanocortin systems (Candille et al., 2007; Rohrl et al., 2010; Yang et al., 2000a; Yang et al., 1999).
We became interested in β-defensins as seven transmembrane receptor ligands based on the observation that a common Mendelian trait in domestic dogs, dominant inheritance of a black coat color, was caused by mutation of the β-defensin gene, canine β-defensin 103 (CBD103)(Candille et al., 2007). In laboratory mice and many other mammals, dominant inheritance of a black coat color is caused by mutations that constitutively activate the melanocortin 1 receptor (Mc1r) (Barsh et al., 2000). Melanocortin receptors are named for their ability to stimulate cAMP production in response to small peptide agonists such as α-melanocyte stimulating hormone (α-MSH). In vivo, however, the Mc1r and Mc4r exhibit high levels of basal activity in the absence of agonists, and are often regulated by production of the endogenous inverse agonists Agouti signaling protein (ASIP) and Agouti-related protein (AGRP), respectively, which elicit biological effects through their ability to depress cAMP levels by inhibiting melanocortin receptor activity (Ollmann and Barsh, 1999).
Our earlier studies suggested that CBD103 acted as a novel ligand for the MC1R, because CBD103 and its human ortholog, human β-defensin 3 (HBD3), bound specifically to the MC1R expressed in cultured cells, and because transgenic mice that overexpressed CBD103 exhibited dominant inheritance of a black coat color. Although overexpression of CBD103 mimics the coat color effects of α-MSH in vivo, CBD103 did not stimulate cAMP production in vitro, which suggested that CBD103 blocked the ability of ASIP to bind to the MC1R, thereby maintaining high basal levels of MC1R signaling. In these initial studies, HBD3 and CBD103 also bound to the MC4R, a brain melanocortin receptor that controls feeding and body weight, which suggested an additional molecular route for these and possibly other β-defensins to modulate a variety of physiologic processes.
The relationship between CBD103 and pigmentation in dogs and in transgenic mice has led to interest in the possibility that HBD3 might affect MC1R signaling in humans; however, recent studies have yielded somewhat conflicting results. In cultured human melanocytes, Swope et al. (Swope et al., 2012) reported that HBD3 had no effect on cAMP levels, but blocked the ability of α-MSH to stimulate cAMP accumulation, leading these investigators to describe HBD3 as an MC1R antagonist. By contrast, Beaumont et al. (Beaumont et al., 2012) observed that HBD3 promoted cAMP accumulation and MAP kinase activation in HEK cells transfected with the MC1R, and described HBD3 as a partial MC1R agonist.
To better understand the molecular basis for β-defensin action, we have now carried out a series of additional genetic and pharmacologic studies to probe the actions of HBD3 in vivo, and to characterize the structural determinants of HBD3 that are required for melanocortin receptor binding. Despite little sequence homology among members of the β-defensin family, those that have been structurally characterized all share a remarkably similar secondary and tertiary architecture (Bauer et al., 2001; Hoover et al., 2001; Hoover et al., 2000; Sawai et al., 2001; Schibli et al., 2002). Common features include three anti-parallel beta-strands stabilized into a compact fold by three conserved disulfide bridges in the topology CysI-CysV, CysII-CysIV, and CysIII-CysVI that distinguish them from other classes of defensins (Pazgier et al., 2006) (Figure 1A). Generally they possess a high net positive charge and are amphiphilic, features that contribute to their ability to disrupt microbial membrane structure (Kluver et al., 2005). However, β-defensins share little sequence or structural similarities with all previously known melanocortin ligands.
Figure 1.
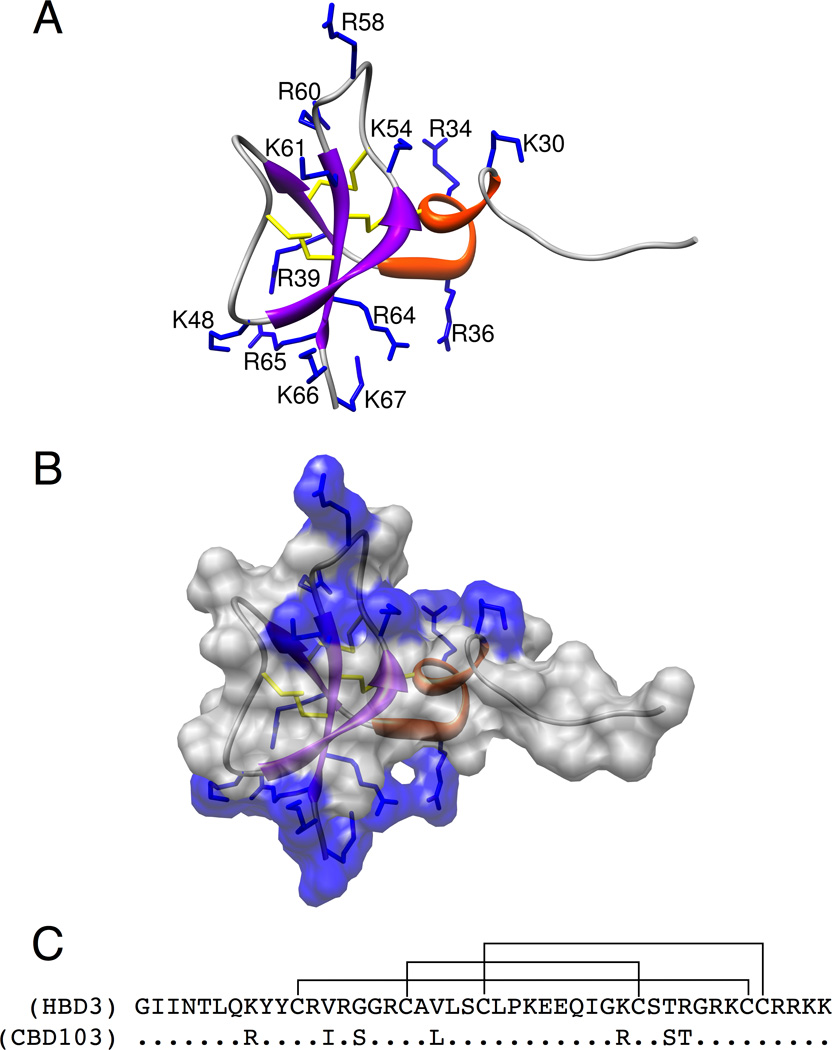
Structure and sequence of HBD3. (A) HBD3 structure is represented by ribbon diagram with helices in red and β-strands in purple. Positively charged residue sidechains are numbered and displayed in blue. The three disulfide bridges are displayed in yellow. (B) HBD3 structure is represented by ribbon diagram and displays the solvent exposed surface area, colored by sidechain charge. Patches of positively charged residues are found on opposite sides of the peptide structure. These were deemed patch 1, the grouping on the top of the structure as it is oriented here, and patch 2 on the bottom of the structure grouped near the C-terminus. (C) The primary sequence of mature HBD3 and CBD103 (residues 23–67) with the native disulfide connectivity shown. See also Figure S3 and Table S1.
As described below, we find that HBD3 behaves as a neutral antagonist at both the MC1R and the MC4R, blocking the effects of the inverse agonists ASIP and AGRP, respectively. Our structural studies indicate that receptor binding is mediated by two solvent exposed patches of positively charged residues located on opposite poles of HBD3. This binding interaction appears to be distinct from other known melanocortin ligand-receptor interactions, and allows HBD3 to achieve high affinity binding through its diffuse charged surface and electrostatic characteristics. Together, these findings define a unique mode of seven transmembrane receptor binding and signaling.
Results
β-defensin relationships and nomenclature
Our original studies on β-defensins were based on dog coat color genetics and used both dog and human peptides. For the genetic work described below, we made use of mice that carry a CBD103 transgene (a dog cDNA) controlled by a strong, widely expressed promoter (Candille et al., 2007). However, for the structure-function work that follows, we have focused exclusively on the human peptide. There is an orthologous relationship (suggesting conservation of function) between the CBD103 (dog) and DEFB103 (human) genes, whose protein products are referred to as CBD103 and HBD3, respectively (Patil et al., 2005). The mature peptides (after signal sequence cleavage) are both 45 residues in length, and there are 7 conservative substitutions between the dog and human peptides (Figure 1C); as shown below, HBD3 has nearly identical properties to CBD103 (Candille et al., 2007) in terms of binding to the MC1R and MC4R.
β-defensin pharmacology and genetic interaction with melanocortin systems
Even though CBD103 mimics the effect of MC1R agonists in vivo, neither CBD103 (Candille et al., 2007) nor HBD3 (Figure S1A) stimulate or reduce cAMP levels in Melan-a cells, which represent a physiologically relevant and sensitive system for measuring MC1R activation. We also examined β-arrestin recruitment by the MC1R, which provides a sensitive test for receptor activation via an alternative effector (Ferguson et al., 1996; Lohse et al., 1990). Melanotan-II, a synthetic melanocortin agonist peptide, promoted robust MC1R/ β-arrestin coupling (EC50 = 5.4×10−10 M), but there was no response to CBD103 or to ASIP in this assay (Figure S1B).
To investigate the interaction between CBD103 and Mc1r signaling in vivo, we extended earlier work in which Tg.CBD103 was shown to cause a black coat color in mice whose Asip genotype is A/a (Candille et al., 2007). The A allele is characterized by transient expression of Asip during the hair growth cycle, which, via transient inhibition of Mc1r signaling, gives rise to hairs that contain a band of yellow pigment on an otherwise black background. In mice carrying the Ay allele, Asip is expressed throughout the entire hair growth cycle, giving rise to animals that are entirely yellow; in this background (Ay/a), we observed that Tg.CBD103 also caused a black coat (Figure 2A, Table 1). By contrast, in mice carrying an Mc1r loss-of-function mutation that, like Ay, gives rise to mice that are entirely yellow, Tg.CBD103 has no effect on coat color (Figure 2B, Table 1). Thus, CBD103 is genetically downstream of Asip but upstream of the Mc1r.
Figure 2.
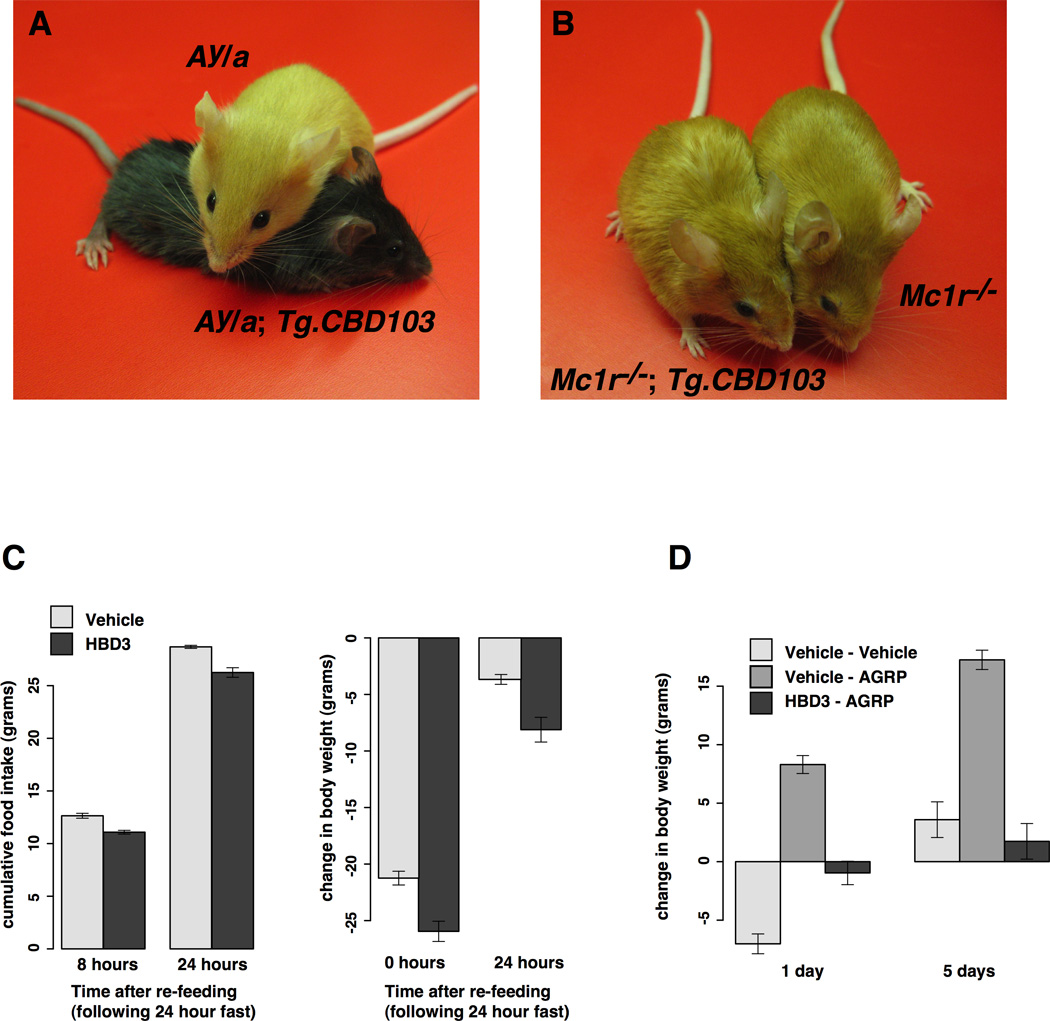
Transgenic mouse models and ICV experiments. (A) Ay/a background mice with and without CBD103 transgene expression. (B) Mc1r−/− background mice with and without CBD103 transgene expression. (C) Effects of β-defensin ICV injection on food intake and body weight change over time during re-feeding experiments. (D) Effects of simultaneous ICV injection of AgRP and HBD3 on change in body weight over time during free-feeding. Each data point is represented as the mean of 6–7 animals +/−SEM. Note: all mice shown are less than eight weeks of age.
Table 1.
Epistasis of CBD103 transgene and pigment-type switching genes
| Genotype | ||
|---|---|---|
| Melanocortin system | CBD103 transgene | Coat color |
| a/a; Mc1r−/−; Pomc+/+ | +/+ | Yellow |
| a/a; Mc1r−/−; Pomc+/+ | Tg/+ | Yellow |
| Ay/a; Mc1r+/+; Pomc+/+ | +/+ | Yellow |
| Ay/a; Mc1r+/+; Pomc+/+ | Tg/+ | Black |
| A/A; Mc1r+/+; Pomc−/− | +/+ | Agouti |
| A/A; Mc1r+/+; Pomc−/− | Tg/+ | Black |
The results described above could also be explained if Tg.CBD103 caused increased expression of Proopiomelanocortin (Pomc), the precursor to α-MSH. Therefore, we asked whether the coat color effects of Tg.CBD103 were dependent on Pomc. In our genetic background (a mixture of three inbred strains, 129, C57BL/6J, and FVB/N), A/A mice homozygous for a Pomc loss-of-function allele have banded hairs, and the addition of Tg.CBD103 converts the coat color to black (Table 1). Thus, the effects of CBD103 on coat color depend on the Mc1r but not on endogenous melanocortins, which provides strong genetic support for the conclusion that the pigmentary effects of CBD103 result from a direct interaction with the Mc1r, despite the inability of CBD 103 to modulate receptor coupling.
Effect of HBD3 on CNS melanocortin signaling
A shift in the balance between black and yellow pigment synthesis provides a very sensitive readout for melanocortin activity. However, a disadvantage of the pigmentary system is that hair follicle melanocytes—the Mc1r-expressing cells engaged by Asip and by CBD103—do not maintain a pigment type-switching response in culture, and the tissue compartment in which those cells reside is relatively inaccessible, which makes pharmacologic studies difficult.
As an alternative approach to study the pharmacology of β-defensin action in vivo, we studied the feeding behavior of rats in response to intracerebroventricular (ICV) injection of melanocortin receptor ligands. Previous studies have established that α-MSH and AgRP are potent central nervous system (CNS) modulators of energy balance when injected ICV, causing inhibition and stimulation of feeding, respectively, mediated largely through the Mc4r. In pilot studies of free-feeding rats implanted with ICV cannulas, we observed that injection of 0.01 – 2 nmol of HBD3 had no effect on behavior or food intake; we chose a dose of 7 ug (~1.2 nmol) for subsequent studies because AgRP injected ICV stimulates feeding at doses of 0.01 – 0.2 nmol, and the affinity of HBD3 for Mc4r is 50 – 100 fold less than AgRP (see below).
We first tested the effects of HBD3 on “re-feeding” after 24 hours of food deprivation, which provides a sensitive assay for alterations in endogenous Mc4r signaling. Animals (n = 6–7/group) were injected either with water or with HBD3 at the onset of fasting, and then allowed free access to food during subsequent monitoring. Under these conditions, when food is restored, rats exhibit a compensatory hyperphagia and recover most of their pre-fasting body weight by 24 hours; HBD3 treatment had a small but significant inhibitory effect on both the food intake and body weight response (Figure 2C, p=0.0065 for food intake p=0.023 for body weight).
We then tested whether HBD3 would affect the response to a submaximal dose (0.01 nmol) of exogenous AgRP in free-feeding animals. In this paradigm, ICV injection of AgRP has no detectable effect on food intake in the first 24 hours (data not shown) but nevertheless elicits a positive and persistent energy balance response with increased weight gain (Figure 2D). ICV injection of HBD3 one hour after AgRP completely blocked this response (p=0.0009) such that animals injected with Vehicle+Vehicle exhibited a body weight response that was not significantly different (p=0.56) from animals injected with AgRP+HBD3 (Figure 2D). In summary, HBD3 has a small but significant effect on the physiologic response to endogenous AgRP (Figure 2C), and a dramatic effect on the pharmacologic response to administered AgRP (Figure 2D).
Structure-function analysis of HBD3 binding to melanocortin receptors
Taken together, the results described above suggest that HBD3 (or CBD103) can act as a neutral antagonist at both the Mc1r and Mc4r, blocking the ability of either the agonist, α-MSH, or the endogenous inverse agonists, Asip and AgRP, to signal through their cognate receptors. Because of the distinct ligand properties of HBD3, we sought to further characterize the molecular determinants responsible for its interaction with melanocortin receptors.
Effects of mutations that impair HBD 3 charged and aromatic residues
Previously characterized melanocortin ligands share a similar binding motif comprised of cationic and aromatic residues, including the RFF sequence in inverse agonists and the HFRW sequence in agonists (Bolin et al., 1999; Hruby et al., 1987; Kiefer et al., 1998; Tota et al., 1999). Substitution of any of these residues individually or in groups greatly reduces melanocortin receptor affinity and function; for example, the RFF→AFF mutation in Asip reduces MC1R affinity by over 760 fold (Ki=2.6nM → Ki>1900nM) (Kiefer et al., 1998). The N-terminal helix of HBD3 has a single linear sequence of positively charged and aromatic residues −K30Y31Y32- similar to RFF motif in the inverse agonists. However, we found that substituting either K30 or Y32 with alanine residues had little overall effect on binding, reducing MC1R/ MC4R affinity ~1.5 fold (Table 2). We also systematically replaced all of the lysines and arginines present in the HBD3 sequence with alanine or glutamine in a series of single, double, and multiple point mutations. Alanine was primarily used for single and double residue substitutions while glutamine was utilized for multiple substitutions to better reflect side chain structure and bulk, while maintaining peptide solubility. Mutating any single or group of two or three charged residues had little effect on receptor binding; these peptide variants bound to MC1R and MC4R with affinities comparable to HBD3, exhibiting remarkable resilience to modification (Figure 3 and Table 2).
Table 2.
Sequence, charge, and binding constants (Ki) for synthetic Human β-defensin peptides
| Peptide Mutant | Sequence | Charge | hMc1ra Ki (nM) |
hMc4ra Ki(nM) |
|---|---|---|---|---|
| Native HBD3 | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK | +11 | 42 | 110 |
| Cys->Serb | GIINTLQKYYSRVRGGRSAVLSSLPKEEQIGKSSTRGRKSSRRKK | +11 | 297 | 436 |
| CysT-V | GIINTLQKYYCRVRGGRSAVLSSLPKEEQIGKSSTRGRKCSRRKK | +11 | 243 | 206 |
| CysTT-TV | GIINTLQKYYSRVRGGRCAVLSSLPKEEQIGKCSTRGRKSSRRKK | +11 | 373 | 567 |
| CysTTT-VT | GIINTLQKYYSRVRGGRSAVLSCLPKEEQIGKSSTRGRKSCRRKK | +11 | 198 | 292 |
| N-term Truncate | GIINTLQKYYSRVRGGRSAV | +4 | NBc | 744 |
| C-term Truncate | LSSLPKEEQIGKSSTRGRKSSRRKK | +7 | NB | NB |
| Middle Truncate | RGGRSAVLSSLPKEEQIG | +1 | NB | NB |
| K30A | GIINTLQAYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK | +10 | 75 | 79 |
| Y32A | GIINTLQKYACRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK | +11 | 55 | 135 |
| R58A | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTAGRKCCRRKK | +10 | 81 | 117 |
| R60A | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGAKCCRRKK | +10 | 38 | 106 |
| E49A | GIINTLQKYYCRVRGGRCAVLSCLPKAEQIGKCSTRGRKCCRRKK | +12 | 146 | 222 |
| R58A, R60A | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTAGAKCCRRKK | +9 | 48 | 131 |
| R64A, K66A | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCARAK | +9 | 99 | 178 |
| R65A, K67A | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRAKA | +9 | 45 | 83 |
| Patch 1 Mutants | ||||
| Neut 1d | GIINTLQQYYCQVRGGRCAVLSCLPKEEQIGQCSTQGQQCCRRKK | +5 | >5000 | 923 |
| Neut 1 Cys->Ser | GIINTLQQYYSQVRGGRSAVLSSLPKEEQIGQSSTQGQQSSRRKK | +5 | NB | NB |
| Neut 1a | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGQCSTQGQQCCRRKK | +7 | 189 | 105 |
| Neut 1b | GIINTLQQYYCQVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK | +9 | 216 | 109 |
| Patch 2 Mutants | ||||
| Neut 2d | GIINTLQKYYCRVQGGQCAVLSCLPQEEQIGKCSTRGRKCCQQQQ | +4 | 422 | 1568 |
| Neut 2 Cys->Ser | GIINTLQKYYSRVQGGQSAVLSSLPQEEQIGKSSTRGRKSSQQQQ | +4 | NB | NB |
| Neut 2a | GIINTLQKYYCRVQGGQCAVLSCLPQEEQIGKCSTRGRKCCRRKK | +8 | 137 | 147 |
| Neut 2b | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCQQQQ | +7 | 193 | 254 |
| Neut 1+2 | GIINTLQQYYCQVQGGQCAVLSCLPQEEQIGQCSTQGQQCCQQQQ | −2 | NB | NB |
Figure 3.
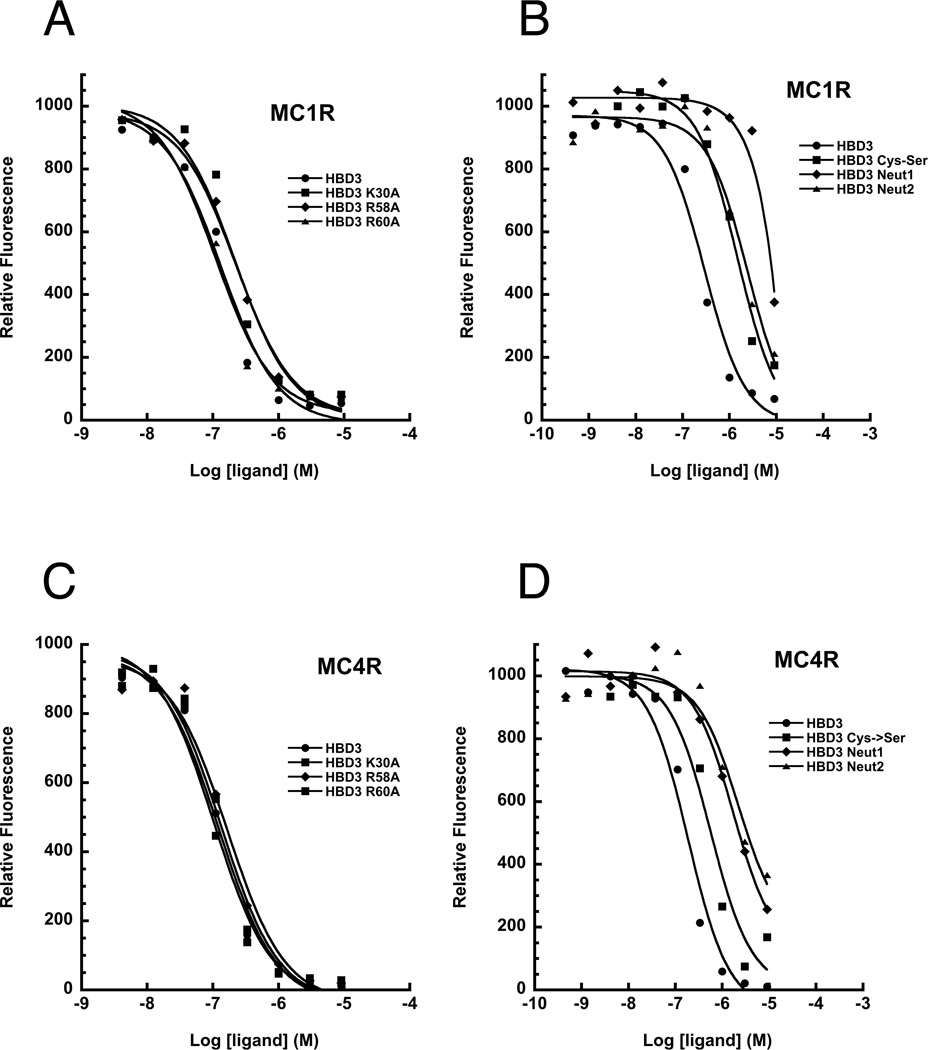
Competitive binding assays with MC1R and MC4R. Subset of competition binding assay curves for synthetic HBD3 variants at either MC1R (A & B) or MC4R (C & D). The logarithm of competing ligand concentration is plotted on the abscissa while the amount of Eu-NDP-MSH bound, measured as relative fluorescence, is plotted on the ordinate. See also Figure S1.
The effects of mutations that impair HBD3 tertiary structure
A hallmark of the β -defensins is their conserved and compact secondary and tertiary structure (Figure 1), stabilized by disulfide bonding. Reduction of the disulfides can significantly compromise β-defensin action, for example in the case where β-defensin 3 suppresses proinflammatory pathways (Semple et al., 2011). We synthesized a series of peptides in which disulfide bonding is disrupted by replacing some or all of the six cysteine residues with serine residues (Table 2). Surprisingly, elimination of all disulfide bonds from HBD3 (HBD3-Cys→Ser) reduced but did not abolish HBD3 binding to MC1R or MC4R, with a 7-fold reduction at MC1R (Ki=42nM to Ki=297nM) and a 4-fold reduction at MC4R (Ki=110nM to Ki=436nM) relative to HBD3 (Figure 3 and Table 2). Peptides in which two of the three disulfide bonds were eliminated behaved similarly to the complete HBD3-Cys→Ser peptide (Table 2). We considered whether or not the HBD3-Cys→Ser mutant might still fold normally, however natural abundance 15N HSQC on HBD3-Cys→Ser exhibited very little amide proton dispersion compared to the fully folded native HBD3 spectra (Figure S2). Thus, like other β-defensins for which disulfide bonding is critical for stabilizing the defensin fold, HBD3-Cys→Ser has little or no regular structure (Bauer et al, 2001).
In addition to disulfide bonding, several other residues that are well conserved throughout the β-defensin family may also play a role in stabilizing the compact fold of β-defensins. Glutamate 49 for example is thought to form an intramolecular salt bridge with R39 that both stabilizes the fold and assists dimer formation (Boniotto et al., 2003; Schibli et al., 2002). We synthesized an HBD3 E49A variant, and observed a 2–3 fold reduction in binding affinity at MC1R and MC4R (Ki=146nM and 222nM, respectively) (Table 2). This reduction in affinity is the largest observed for any of the single point mutants, and is consistent with the conclusion that the compact fold of the protein facilitates but is not absolutely required for receptor binding. As controls, we designed three additional HBD3 peptides in which large regions were eliminated, and in which all cysteine residues in these truncated variants were substituted with serines. None of these peptides (N-term Truncate, C-term Truncate, Middle Truncate, Table 2) exhibited significant receptor binding.
Mutations of positively charged patches disrupts receptor binding
Given that none of the individual aromatic, charged, or conserved HBD3 residues is essential for MCR binding, we investigated a potential requirement for additional binding determinants based on regional physiochemical properties. Inspection of the NMR structure of HBD3 shows an asymmetric distribution of positively charged residues grouped into two patches (Figure 1B). One patch is primarily located on the central loop of the folded peptide (patch 1, represented by 6 residues) while the other larger patch is found on the opposite side, localized on the C-terminal tail (patch 2, represented by 7 residues). We confirmed this arrangement of positive charges and disulfide bond connectivity by solving the structure of an “in-house” HBD3 preparation. The 20 best structures from our CYANA calculation ranked according to target function, which were all below 10 Å2, possessed a backbone R.M.S.D of 0.38 Å between residues 33 and 66 and were consistent with the known native fold of HBD3 (Figure S3 and Table S1).
The presence and arrangement of the positively charged patches on the surface of HBD3 suggests the possibility that HBD3-melanocortin receptor binding is mediated by electrostatic interaction domains. To test this hypothesis, we measured melanocortin receptor affinity for three charge neutral HBD3 variants, for which all charged residues from patch 1 (HBD3-Neut1), patch 2 (HBD3-Neut2), or both patch 1 and 2 (HBD3-Neut1+2) were substituted with glutamine residues (Table 2, Figure S4). To ensure that these more extensively modified HBD3 variants were folded into the native defensin conformation, we synthesized them using a full orthogonal cysteine protection scheme which allowed sequential formation of individual disulfide bonds. After the multi-step folding process was complete, we confirmed by analytical HPLC and mass spectrometry that we obtained one fully folded conformer for each variant. Additionally, we synthesized native HBD3 using the same orthogonal folding scheme, producing one final conformer, which had identical MCR affinity constants and analytical HPLC retention time as HBD3 folded by conventional oxidation.
Substituting both charged patches simultaneously (HBD3-Neut1+2) completely eliminated binding at both MC1R and MC4R. When we independently substituted either patch 1 or patch 2, however, we observed receptor specific differences in binding affinity.
At MC1R, elimination of patch 1 was sufficient to severely impair binding (HBD3-Neut1 Ki > 5uM) whereas elimination of patch 2 reduced binding but did not eliminate it completely (HBD3-Neut2 Ki = 422nM). However, affinity for MC1R was completely lost by the HBD3-Neut2 peptide when we disrupted its structure through elimination of disulfide bonding (HBD3-Neut2-Cys-Ser, Table 2). In contrast, elimination of patch 1 had a significant effect on MC4R binding but did not entirely eliminate it (HBD3 Ki = 110nM; HBD3-Neut1 Ki = 923nM). Eliminating patch 2 reduced affinity for MC4R by nearly 15-fold but did not abolish it (HBD3 Ki = 110nM; HBD3-Neut2 Ki = 1568nM). However, affinity for MC4R was completely lost for either patch variant by disruption of disulfide bonding (HBD3-Neut1-CysSer and HBD3-Neut2-CysSer, Table 2). Thus, the positively charged surface patches of HBD3 contribute both to receptor binding, and to binding specificity.
To interrogate the functional importance of smaller charge clusters within each positively charged patch, we synthesized four additional peptides: HBD3-Neut1a and HBD3-Neut1b eliminated four and two charged residues, respectively, within patch 1; and HB3-Neut2a and HBD3-Neut2b eliminated three and four charged residues, respectively, within patch 2. None of these alterations completely disrupted MC1R or MC4R binding, and instead yielded a range of affinities that does not point to a critical role for any single charge cluster (Table 2). However, plotting receptor affinity as a function of charge for all 14 variants with intact disulfide bonds reveals a strong trend (Figure 4), which highlights the importance of charge as a binding determinant and suggests that regional charge patches rather than specific side chains mediate receptor binding.
Figure 4.

Correlation between overall charge of HBD3 variants and MCR binding affinity. Peptide charge (X-axis) is plotted against the log of the measured binding affinity (Ki) at MC1R (black diamonds) and MC4R (open black squares). Peptides lacking some or all disulfide connections were omitted, and low affinity HBD3 variants (with Ki values outside the detectable range of our assay) were set to Ki=5µM.
Regional electrostatic interactions: Informatic analysis
As described further below, we propose that the manner in which β-defensins bind melanocortin receptors is fundamentally distinct from that of “classical” melanocortin ligands (such as α-MSH and ASIP or AgRP), and, instead, is based on regional electrostatic interactions between positively charged patches on the surface of the ligand with negatively charged regions on the surface of the receptor. Previous structure-function studies of MC1R and MC4R identified several critical acidic residues found grouped together that are essential for NDP-MSH and ASIP binding (Haskell-Luevano et al., 2001; Yang et al., 1997). However, MC1R and MC4R structural models also reveal several negatively charged residues on the extracellular surface of each receptor besides those comprising the ligand binding pocket.
We used the Adaptive Poisson-Boltzmann Solver (Baker et al., 2001) to model potential electrostatic interactions between HBD3 or ASIP and melanocortin receptors, which revealed a striking charge complementarity between the peptide ligands and each receptor (Figure 5). HBD3 exhibits a positive potential distributed almost entirely over its solvent exposed surface, whereas ASIP shows positive potential specifically on the loop portion known to interact with the MCR binding pocket (Figure 5A and B). Both receptors exhibit a large negative surface potential owing to the preponderance of acidic residues (Figure 5C and D), which overlaps with but is not restricted to the critical residues of the melanocortin binding pocket. We also compared the charged distribution of melanocortin receptor segments (the predicted transmembrane helices, extracellular, and intracellular regions) with those of all known GPCRs in the human genome. Remarkably, the melanocortin receptors possessed among the greatest numbers of acidic residues in their exo-segments: MC4R with a −8 net charge (five Asp, three Glu) was among the top 3% of most negatively charged GPCR exo-segments, and MC1R with a −6 net charge (four Asp, four Glu, one Lys, one Arg) was among the top 10% (Figure S5). Together with the experimental results, observations from receptor-ligand modeling and comparative GPCR analysis support a model whereby HBD3 can competitively prevent binding of other melanocortin ligands via diffuse electrostatic interactions with receptor exo-segments, without affecting the melanocortin binding pocket or receptor conformation.
Figure 5.
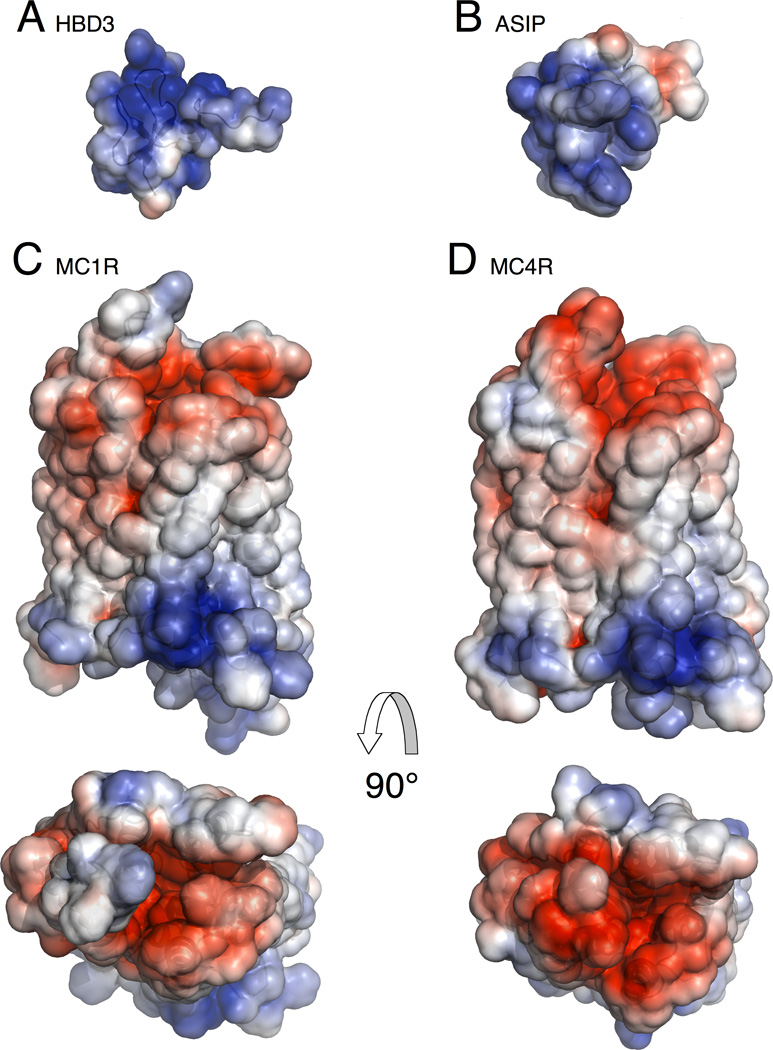
Electrostatic potential of melanocortin system ligands and receptors. Positive potential is displayed in blue, negative potential is displayed in red, and neutral potential displayed in white. (A) HBD3 (PDB ID: 1KJ6) (B) ASIP (PDB ID: 1Y7K) (C) Side and top (extracellular) view of MC1R structural model highlighting the purported ligand binding pocket (D) Side and top (extracellular) view of MC4R structural model (PDB ID: 2IQV). Receptor models were obtained from http://mosberglab.phar.umich.edu/resources/ (Chai et al., 2005; Pogozheva et al., 2005). PDB files were prepared for electrostatic calculations using the PDB2PQR webserver with an AMBER force field (Dolinsky et al., 2007). Electrostatic calculations were performed with the Adaptive-Poisson Boltzmann Solver and visualized using PyMol (Baker et al., 2001). Electrostatic potentials were displayed on the solvent accessible surface using a −3 to +3 range (units of kbT/ec). No ligand binding orientation is implied by the display of HBD3 or ASIP relative to the melanocortin receptors. See also Figure S5.
Discussion
Melanocortin receptor agonists, such as α-MSH, are evolutionarily unrelated to inverse agonists, such as ASIP and AGRP, but both have a common structural feature: a short linear motif (HFRW in α-MSH and RFF in ASIP and AGRP) comprised of cationic and aromatic sidechains that serves as a point of receptor interaction. Both motifs are found on exposed loops extending from the ligand core that interact with corresponding acidic and aromatic residues found on the extracellular receptor-binding surface (Chen et al., 2007; Hruby et al., 1987; Kiefer et al., 1998; Yang et al., 2000b). Mutation of these linear motifs (for instance, RFF ->AFF in ASIP or AGRP) severely reduces receptor affinity and ligand efficacy thereby defining a precise and specific interaction with the receptor-binding pocket (Bolin et al., 1999; Kiefer et al., 1998).
HBD3 (and other β -defensins) often exhibit function away from the cell surface, share no sequence similarity with these previously characterized melanocortin receptor ligands, and our structure-function analysis indicates that the mechanism of HBD3 binding is also different. In contrast to ASIP and AgRP, HBD3 binding depends primarily on two positively charged patches located distally from each other on the protein scaffold. While HBD3 charge alone confers MCR binding affinity, the degree of binding is clearly enhanced by the compact HBD3 fold, stabilized by its unique network of disulfide bonds.
Additional biophysical and pharmacologic studies will be necessary to rigorously test our proposed model of HBD3-MCR binding, but we note that physiologically significant protein-protein binding mediated largely by regional electrostatic interactions is not without precedent (Sinha and Smith-Gill, 2002). Complementary patches of charged residues facilitate sub-picomolar binding (Kd~10−14M) between a ribonuclease and an inhibitor protein of the Barnase-Barstar complex (Buckle et al., 1994). Multiple studies on this pair, including mutational analysis and electrostatic modeling of interaction surfaces, indicate the importance of large groupings of positive residues on Barnase and corresponding acidic as well as non-polar residues on its inhibitor, Barstar (Hartley, 1993; Schreiber and Fersht, 1993). In addition, the metal-ion Zn2+ is also capable of acting as a direct agonist for the MC1R and MC4R (Holst et al., 2002). This interaction is most likely dependent on acidic residues in and around the receptor-binding pocket, which may also be the key residues involved in the electrostatically driven HBD3-MCR interaction.
Biophysical differences in the way that MCR interacts with HBD3 compared to Asip, Agrp, or α-MSH may help to explain how HBD3 can block either a stimulatory (α -MSH) or an inhibitory (Asip, Agrp) ligand. Inverse agonists are thought to act by stabilizing the inactive conformation of seven transembrane domain receptors, i.e., one that has reduced affinity for the Got subunit; our results suggest that HBD3 has no effect on the relative balance between active and inactive conformations, but causes the melanocortin binding pocket to be less accessible to either stimulatory or inhibitory ligands. This perspective can also explain the apparent paradox between our initial description of β-defensin 3 as a peptide whose effects mimicked that of a melanocortin agonist (Candille et al., 2007), and that of Swope et al. (Swope et al., 2012), who refer to HBD3 as an α-MSH antagonist. In vivo (in dogs or in transgenic mice), melanocortin peptides derived from POMC have no detectable effect on pigmentation (Slominski et al., 2005), and the effects of β-defensin 3 are brought about by inhibition of Asip. But HBD3 can also block the effect of exogenous α-MSH added to cultured melanocytes (Swope et al, 2012). Thus, the functional outcome of an HBD3-MCR interaction with regard whether signaling is promoted or inhibited will depend on the specific physiologic context and the relative balance between agonist and inverse agonist (Figure 6).
Figure 6.

Modulation of melanocortin receptor signaling by HBD3. Because MC1R and MC4R have both stimulatory and inhibitory ligands, the effects of a neutral antagonist are context-dependent. Schemes on the left show the response of the indicated ligand; those on the right show how the response is altered by HBD3, which restores basal cAMP levels. (A) In mammalian skin, the primary ligand for MC1R is Agouti protein (there is very little α-MSH); consequently, β-defensin 3 promotes MC1R signaling by inhibiting inverse agonism. (B) In cultured melanocytes stimulated by exogenous α-MSH, HBD3 inhibits MC1R signaling. (C) In the brain, HBD3 can either promote or inhibit MC4R signaling depending on the balance between inverse agonist (Agrp) and agonist (α-MSH, not shown).
The perspective of HBD3 as a new type of melanocortin receptor ligand—one that interacts with the receptor in a way that is fundamentally different from that of α-MSH, Asip, or Agrp—can also help to account for the observations of Beaumont et al. (Beaumont et al., 2012), since the behavior of a ligand as partial agonist in heterologous cells could be influenced by the cell type and assay conditions, including receptor number, local accessibility of different G proteins, and presence of allosteric modulators (Low, 2011). In our work, neither CBD103 nor HBD3 affected cAMP levels in cultured mouse melanocytes; similarly, Swope et al. (Swope et al., 2012) observed no effect of HBD3 on cAMP levels in cultured human melanocytes. Notably, the preparation and source of HBD3 for all of these studies is the same. It is also important to note that our studies of HBD3 and the MC4R are motivated by the opportunity to examine what can happen in an in vivo pharmacologic paradigm, not what normally happens during physiologic regulation of MC4R signaling. Some β-defensins have been reported to be expressed in the rat brain (Froy et al., 2005), but additional studies will be necessary to explore the extent to which β-defensins might normally modulate CNS physiology. By contrast, a physiologic (and/or pathophysiologic) HBD3-MC1R interaction is more well-grounded, since HBD3 is expressed at very high levels by keratinocytes from psoriatic skin (Harder et al., 2001).
Finally, our model for HBD3-MCR binding may also help to explain the promiscuity of β-defensin action at other GPCRs. In particular, the C-C chemokine receptor-2 (CCR2), which also interacts with HBD3, has an electrostatic distribution similar to MCRs and binds to its native ligand, monocyte chemotactic protein-1 (MCP1), primarily through the interaction of negatively charged residues on CCR2 and positively charged clusters on MCP1 (Hemmerich et al., 1999; Rohrl et al., 2010). Similarly, the interaction between HBD1 and C-C chemokine receptor-6 is mediated by a spatially diffuse set of residues (more than half of which are positively charged) on the HBD1 surface. (Pazgier et al., 2007). From a general perspective, acquisition of charge clusters by soluble peptides could drive ligand promiscuity, while acquisition of charge clusters within GPCR exoloops could drive receptor promiscuity. The HBD3-MCR interaction was originally recognized via a phenotype-driven approach, but the model described here suggests a route for identifying analogous interactions based on a sequencedriven approach.
Significance
An unusual feature of melanocortin receptors is their physiologic modulation in opposite directions by endogenous agonists (i.e. α-MSH) and inverse agonists (i.e. ASIP and AgRP). Identification of HBD3 as a third type of melanocortin receptor ligand adds an additional layer of complexity, and the ability to dampen receptor signaling regardless of its direction. Our biochemical studies uncover a fundamentally new mode of MCR binding and regulation that may help in drug design and in the identification of additional GPCR ligands.
Experimental Procedures
Cyclic-AMP and Beta-arrestin Studies
Melan-A cells (Bennett et al., 1987) were grown in RPMI supplemented with 200nM TPA at 37°C and 10% CO2. For cyclic-AMP (cAMP ) assays, the cells were plated onto 96-well half-area plates (Corning #3885) at a density of 20,000 cells/well and grown for 24 hours. After a single wash with phosphate buffered salin, pH7.4 (PBS), peptides (diluted in PBS, pH7.4, containing 0.2% BSA and 0.5 mM IBMX) were added and the plate was incubated at 37°C and 10% CO2 for 40 minutes. cAMP levels were measured using the Hithunter cAMP XS kit (DiscoveRx) with a Centro LB 960 plate reader (Berthold Technologies). The assay was performed in triplicate and analyzed using Graphpad (Prism).
β-arrestin recruitment was measured using a commercial assay system (PathHunter, Discoverx), based on an enzyme complementation strategy that measures MC1R specific β-arrestin recruitment in an engineered Chinese Hamster Ovary cell line. Cells were seeded at a density of 10,000 cells/well and grown for 48 hours in Optimized Cell Culture media (DiscoveRx). 10µl of each ligand, diluted in media, was added to the cells at a final concentrations ranging from 1 .0µM – 5.7pM (MT-II and CBD103) or 0.1 µM – 0.57pM (ASIP-YY). The plate was incubated for 90 minutes at 37°C. 54µl of PathFinder detection reagent was added to each well, and the plate was incubated for 90 minutes at room temperature before reading with a Centro LB 960 plate reader (Berthold Technologies). The assay was performed in triplicate and repeated twice for CBD103.
Experimental Animals
All study protocols were approved by the Animal Care and Use Committee at Stanford University (transgenic mouse studies) or the University of Washington (energy balance studies) and conducted in accordance with NIH guidelines for the care and use of animals.
The transgenic mouse line, Tg.CBD103GΔ23, in which the Dominant black allele of CBD103 is expressed under regulation of a cytomegalovirus/beta-actin hybrid (CAGGS) promoter, has been described previously (Candille et al., 2007; Okabe et al., 1997). Two of the mouse strains, C57BL/6J-Mc1re/J and C57BL/6J-Ay/J, were originally obtained from Jackson Laboratories (Bar Harbor) and maintained in house on C57BL/6J background. Pomc− mice, obtained from Yaswen et. al., were on a mixed genetic background (Yaswen et al., 1999).
Tg.CBD103GΔ23/−;Ay/− mice were recovered from F1 crosses. Tg.CBD103GΔ23/−;Mc1re/e and Tg.CBD103GΔ23/−;Pomc−/− mice were recovered from F2 crosses in which either Tg.CBD103GΔ23/−;Mc1re/+ or Tg.CBD103GΔ23/−;Pomc−/+ individuals were backcrossed to Mc1re/e or Pomc−/+mice, respectively. Coat color was recorded at weaning and genotyping performed with the appropriate primers (Genotyping primers available upon request).
Energy balance studies
Adult male Wistar rats were housed individually in a specific pathogen-free environment, maintained in a temperature-controlled room with a 12-12h light:dark cycle and provided with ad libitum access to water and standard laboratory chow (PMI Nutrition International Inc., MO), unless otherwise stated. For the ICV injection studies, rats were first implanted with an indwelling stainless steel cannula to the third cerebral ventricle (3V) under isoflurane anesthesia as previously described (Schwartz et al, 1992). Buprenorphine hydrochloride (0.3 mg/kg; Rickett Colman Pharmaceuticals, VA) was administered at the completion of the surgery. Rats recovered for at least seven days after surgery while daily food intake and body weight were recorded. Cannula placement was verified before the start of the experiment through the measurement of a sympathetically mediated increase in plasma glucose 60-min following 3rd-icv injection of 210µg of 5-thio-D-glucose.
Animals were habituated to regular handling and received a mock injection prior to the commencement of experiments. To determine whether central administration of HBD3 attenuates the re-feeding response following a fast, animals received an ICV injection of either vehicle (water) or HBD3 at a dose of 7 ug (n=6–7/group). Injections were administered using an injector (33-gauge) needle that extended 1mm beyond the tip of the cannula over a period of 60s in a final volume of 2ul. Food was immediately withdrawn from the animals and replaced again 24 hours later. Food intake and body weight were measured prior to the fast, at the completion of the fast and at 24 hours following re-feeding. To determine whether HBD3 attenuates AgRP-induced weight gain, animals received an ICV pre-treatment injection of either vehicle or HBD3, 1 hour prior to ICV administration of AgRP (0.01nmol) or its vehicle to create three groups: 1) Veh-Veh; 2) Veh-AgRP and 3) HBD3-AgRP (n=8–10/group). Body weight gain was measured over a 5-day period following injections.
Peptide Synthesis
Peptides were produced on either an Applied Biosystems 433A peptide synthesizer or a CEM Liberty1 microwave peptide synthesizer using standard Fmoc chemistry. Amino acids were purchased from NovaBiochem and were assembled on Rink-amide-MBHA resin. For peptides produced on the Applied Biosystems 433A synthesizer, pre-activated Fmoc-Cys(Trt)-OPfp was used to avoid enantiomerization. HBTU was purchased from Advanced Chemtech, all other reagents were purchased from Sigma-Aldrich. Fmoc protecting groups were removed using a 1% DBU/HMI mixture in DMF while 4 equivalents of an amino acid/HBTU/DIEA mixture was used for coupling (except pre-activated Fmoc-Cys(Trt)-Opfp, which was only coupled in DMF solution). For peptides produced on the CEM Liberty1 synthesizer, Fmoc protecting groups were removed using 20% piperidine with 0.1M HOBt in DMF. Amino acids were coupled using 5 molar equivalents of Diisopropylcarbodiimide (DIC) and 10 molar equivalents of hydroxybenzotriazole (HOBt) in DMF. Cleavage of all peptides was performed in a TFA/TIS/EDT/Phenol (90:4:4:2) solution for 90min. Standard oxidative folding was achieved by dissolving peptides to a concentration of 0.1mg/mL in folding buffer (0.5–1.0M GuHCl, 0.1M Tris, 1mM GSH, 0.1mM GSSG, pH 8.5) and stirring for 48 hours. Folding was monitored by HPLC, which, in each case, revealed one major species that was used in subsequent experiments. For peptides produced with full-orthogonal cysteine protection, disulfides were formed according to previously published protocols (Schulz et al., 2005). Briefly, peptides were synthesized as outlined above except for the cysteine precursor where we utilized three different kinds of side-chain protecting groups. Cysteines 1 and 5 were protected with the standard Trityl protecting group, which is removed during the TFA cleavage step. Cysteines 2 and 4 were protected using the acetamidomethyl (Acm) protecting group, and cysteines 3 and 6 were protected with the tert-butyl protecting group. The first disulfide (CysI-CysV) was formed using our standard oxidative folding buffer. The second disulfide (CysII-CysIV) was formed using a solution of molecular iodine in glacial acetic acid which simultaneously removes the Acm protecting group and oxidizes the disulfide. The third and final disulfide (CysIII-CysVI) was formed using a DMSO/Anisole mixture in neat TFA, which simultaneously removes the tert-butyl protecting group and oxidizes the disulfide. The folded products were purified by C18 column HPLC and identified as fully oxidized peptides by mass spectrometry. Quantitative concentrations were determined with amino acid analysis at the molecular structure facility at UC Davis.
NMR Spectroscopy and Structure Calculations
NMR samples were prepared in pH 5.0 buffer consisting of 10% D20, 50mM CD3COOD, 0.1% w/v NaAzide, 200uM TMSP, and NaOD for pH adjustment. Susceptibility-matched NMR tubes (Shigemi Inc.) were used to accommodate the small sample volumes (~350uls), which resulted in working peptide concentrations between 1–2mM. The NOESY, TOCSY, and DQF-COSY spectra were acquired on a 900-MHz Bruker Avance II NMR spectrometer equipped with a TCI cryoprobe (HCN) housed in the UC Berkeley NMR facility. The HSQC experiments were performed on a 600-MHz Varian Unity+ spectrometer equipped with a cryoprobe at UCSC. Water suppression for NOESY, TOCSY, and DQF-COSY spectra was achieved using the WATERGATE sequence while HSQC water suppression utilized standard TN sequences. The NOESY and TOCSY spectra were obtained at 25°C with 2048 complex points in the t2 dimension, 400 t1 increments, and a spectral width of 10,901Hz. The DQF-COSY spectra were obtained with 8192 complex points in the t2 dimension and 256 t1 increments. The HSQC spectra were acquired with 1024 complex points in the t2 dimension, 128 t1 increments, and a spectral width of 8,426 Hz. All data was processed using the NMRPipe software package (Delaglio et al., 1995).
Peak assignment and integration was performed manually using SPARKY after which peak and chemical shift lists were converted to XEASY format for use in CYANA structure calculations (Goddard and Kneller, 2008). Automated NOE peak assignment and structure calculations were carried out using CYANA 2.1 on a Macintosh G4 (OS version 10.5) and an Intel Core2 Quad running Linux openSUSE 11.1 (Guntert, 2004). Data inputs included a list of chemical shifts, an integrated NOE peak list, dihedral angle constraint files, and disulfide bond constraints in the form of pseudo-NOE upper-distance limits between cysteine pairs. Two dihedral angle constraint files were generated separately prior to use in the structure calculations. We used 30 3JHα-HN coupling constants obtained from the DQF-COSY spectra to generate one dihedral angle restraint file using the macro GRIDSEARCH in CYANA. A second dihedral angle constraint file was generated using the output from the online program PREDITOR which uses backbone alpha and amide chemical shift values and the primary sequence as input data to predict φ, ψ, χ1, and ω torsion angles (Berjanskii et al., 2006). Calculations utilized 1000 seed structures with 10,000 simulated annealing steps using torsion angle dynamics and the resulting best 20 structures sorted by target function were employed in our structure-function analysis. We performed energy minimization with different disulfide linkages and found that the consensus disulfide pattern gave the lowest target function value. Molecular graphics images were produced using either the UCSF Chimera package or the Pymol Molecular Graphics System, Version 1.3, Schrödinger, LLC (Pettersen et al, 2004).
Competitive Binding Assays
All receptor-ligand binding assays were performed using the DELFIA lanthanide-based detection system on intact Human Embryonic Kidney (HEK) 293T cells transiently transfected with MCR expression constructs. The human MC1R and MC4R constructs were generously provided by Dr. Ying- Kui Yang, Department of Pediatric Surgery, University of Alabama, Birmingham. For binding assays, a 10 cm dish of 293T cells was transfected with 10µg of a melanocortin receptor expression construct using calcium phosphate, with transfection efficiency monitored by parallel transfection with a GFP-tagged MC4R construct. After 6 – 12 hours, the media was replaced. After 20 hours, the cells were dissociated from the plate with an enzyme-free cell dissociation buffer (Life Technologies), washed once with PBS, resuspended in L*R binding buffer (PerkinElmer), and then plated on a 96-well Acrowell filtration plate (Pall). The Europium-labeled NDP-MSH was obtained two different ways; a custom order from Perkin-Elmer and an in-house preparation. For the in-house preparation, NDP-MSH was synthesized with the addition of a C-terminal cysteine, which was subsequently labeled with the Perkin-Elmer DELFIA Eu-N1-iodoacetamide chelate labeling reagent using their recommended protocol (Catalog number AD0002). Dissociation constants (Kd) for both Eu-NDP-MSH preparations at MC1R and MC4R were measured in saturation binding assays, with non-specific Eu-NDP-MSH binding determined by blocking with 1.25µM of unlabeled NDP-MSH.
For displacement binding experiments, Eu-NDP-MSH (ranging from 0.5µM to 2.5µM across experiments) was added together with a 3-fold serial dilution of HBD3 peptides (from 9.0×10−6M to 4.6×10−10M). After a two hour incubation at 37°C, plates were washed three times with ice-cold DELFIA wash buffer (PerkinElmer), and the europium chelate was then dissociated by adding150µl of DELFIA enhancement solution (PerkinElmer) to each well. Plates were then incubated at room temperature for at least 45 minutes and time-resolved fluorescence measured using a FluoStar Optima plate reader (BMG Labtech). The conditions for displacement binding with regard to cell density (10,000 – 70,000 cells/well) and Eu-NDP-MSH concentration (ranging from 0.5µM to 2.5µM across experiments) were based on prior studies (Candille et al, 2007), with adjustments made for variation among different preparations of Eu-NDP-MSH. The time and temperature used for the binding assay were based on prior experiments and recommendations from PerkinElmer. Displacement binding data were analyzed using either Graphpad (Prism) or Kaleidograph (Synergy Software, Version 3.6.4). All affinity measurements in Table 2 are representative of three independent experiments performed in duplicate.
Supplementary Material
Highlights.
HBD3 acts as a neutral antagonist for MC1R and MC4R; modulating the effects of both stimulatory and inhibitory ligands
HBD3 binds MC1R and MC4R primarily through spatially distinct clusters of positively charged residues
A unique ligand-receptor binding interaction facilitates context-dependent seven transmembrane-domain receptor signaling
Acknowledgements
This work was supported by NIH grant DK064265 (GM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsh G, Gunn T, He L, Schlossman S, Duke-Cohan J. Biochemical and genetic studies of pigment-type switching. Pigment Cell Res. 2000;8(13 Suppl):48–53. doi: 10.1034/j.1600-0749.13.s8.10.x. [DOI] [PubMed] [Google Scholar]
- Bauer F, Schweimer K, Kluver E, Conejo-Garcia JR, Forssmann WG, Rosch P, Adermann K, Sticht H. Structure determination of human and murine beta-defensins reveals structural conservation in the absence of significant sequence similarity. Protein science : a publication of the Protein Society. 2001;10:2470–2479. doi: 10.1110/ps.24401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont KA, Smit DJ, Liu YY, Chai E, Patel MP, Millhauser GL, Smith JJ, Alewood PF, Sturm RA. Melanocortin-1 receptor-mediated signalling pathways activated by NDP-MSH and HBD3 ligands. Pigment Cell Melanoma Res. 2012;25:370–374. doi: 10.1111/j.1755-148X.2012.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DC, Cooper PJ, Hart IR. A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int J Cancer. 1987;39:414–418. doi: 10.1002/ijc.2910390324. [DOI] [PubMed] [Google Scholar]
- Berjanskii MV, Neal S, Wishart DS. PREDITOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 2006;34:W63–W69. doi: 10.1093/nar/gkl341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin KA, Anderson DJ, Trulson JA, Thompson DA, Wilken J, Kent SB, Gantz I, Millhauser GL. NMR structure of a minimized human agouti related protein prepared by total chemical synthesis. FEBS Lett. 1999;451:125–131. doi: 10.1016/s0014-5793(99)00553-0. [DOI] [PubMed] [Google Scholar]
- Boniotto M, Antcheva N, Zelezetsky I, Tossi A, Palumbo V, Verga Falzacappa MV, Sgubin S, Braida L, Amoroso A, Crovella S. A study of host defence peptide beta-defensin 3 in primates. Biochem J. 2003;374:707–714. doi: 10.1042/BJ20030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckle AM, Schreiber G, Fersht AR. Protein-protein recognition: crystal structural analysis of a barnase-barstar complex at 2.0-A resolution. Biochemistry. 1994;33:8878–8889. doi: 10.1021/bi00196a004. [DOI] [PubMed] [Google Scholar]
- Candille SI, Kaelin CB, Cattanach BM, Yu B, Thompson DA, Nix MA, Kerns JA, Schmutz SM, Millhauser GL, Barsh GS. A Beta-defensin mutation causes black coat color in domestic dogs. Science. 2007;318:1418–1423. doi: 10.1126/science.1147880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai BX, Pogozheva ID, Lai YM, Li JY, Neubig RR, Mosberg HI, Gantz I. Receptor-antagonist interactions in the complexes of agouti and agouti-related protein with human melanocortin 1 and 4 receptors. Biochemistry. 2005;44:3418–3431. doi: 10.1021/bi0478704. [DOI] [PubMed] [Google Scholar]
- Chen M, Cai M, Aprahamian CJ, Georgeson KE, Hruby V, Harmon CM, Yang Y. Contribution of the conserved amino acids of the melanocortin-4 receptor in [corrected] [Nle4,D-Phe7]-alpha-melanocyte-stimulating [corrected] hormone binding and signaling. J Biol Chem. 2007;282:21712–21719. doi: 10.1074/jbc.M702285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SS, Downey WE, 3rd, Colapietro AM, Barak LS, Menard L, Caron MG. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- Froy O, Hananel A, Chapnik N, Madar Z. Differential expression of rat beta-defensins. IUBMB Life. 2005;57:41–43. doi: 10.1080/15216540500088912. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY 3. San Francisco, CA: University of California, San Francisco; 2008. [Google Scholar]
- Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- Harder J, Bartels J, Christophers E, Schroder JM. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J Biol Chem. 2001;276:5707–5713. doi: 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- Hartley RW. Directed mutagenesis and barnase-barstar recognition. Biochemistry. 1993;32:5978–5984. doi: 10.1021/bi00074a008. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C, Cone RD, Monck EK, Wan YP. Structure activity studies of the melanocortin-4 receptor by in vitro mutagenesis: identification of agouti-related protein (AGRP), melanocortin agonist and synthetic peptide antagonist interaction determinants. Biochemistry. 2001;40:6164–6179. doi: 10.1021/bi010025q. [DOI] [PubMed] [Google Scholar]
- Hemmerich S, Paavola C, Bloom A, Bhakta S, Freedman R, Grunberger D, Krstenansky J, Lee S, McCarley D, Mulkins M, et al. Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor, CCR2. Biochemistry. 1999;38:13013–13025. doi: 10.1021/bi991029m. [DOI] [PubMed] [Google Scholar]
- Holst B, Elling CE, Schwartz TW. Metal ion-mediated agonism and agonist enhancement in melanocortin MC1 and MC4 receptors. J Biol Chem. 2002;277:47662–47670. doi: 10.1074/jbc.M202103200. [DOI] [PubMed] [Google Scholar]
- Hoover DM, Chertov O, Lubkowski J. The structure of human beta-defensin-1: new insights into structural properties of beta-defensins. J Biol Chem. 2001;276:39021–39026. doi: 10.1074/jbc.M103830200. [DOI] [PubMed] [Google Scholar]
- Hoover DM, Rajashankar KR, Blumenthal R, Puri A, Oppenheim JJ, Chertov O, Lubkowski J. The structure of human beta-defensin-2 shows evidence of higher order oligomerization. J Biol Chem. 2000;275:32911–32918. doi: 10.1074/jbc.M006098200. [DOI] [PubMed] [Google Scholar]
- Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, Castrucci AM, Hintz MF, et al. alpha-Melanotropin: the minimal active sequence in the frog skin bioassay. J Med Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- Kiefer LL, Veal JM, Mountjoy KG, Wilkison WO. Melanocortin receptor binding determinants in the agouti protein. Biochemistry. 1998;37:991–997. doi: 10.1021/bi971913h. [DOI] [PubMed] [Google Scholar]
- Kluver E, Schulz-Maronde S, Scheid S, Meyer B, Forssmann WG, Adermann K. Structure-activity relation of human beta-defensin 3: influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry. 2005;44:9804–9816. doi: 10.1021/bi050272k. [DOI] [PubMed] [Google Scholar]
- Lehrer RI. Primate defensins. Nat Rev Microbiol. 2004;2:727–738. doi: 10.1038/nrmicro976. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Low MJ. Agnostic about in vivo inverse agonism of agouti-related peptide. Endocrinology. 2011;152:1731–1733. doi: 10.1210/en.2011-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. 'Green mice' as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Ollmann MM, Barsh GS. Down-regulation of melanocortin receptor signaling mediated by the amino terminus of Agouti protein in Xenopus melanophores. J Biol Chem. 1999;274:15837–15846. doi: 10.1074/jbc.274.22.15837. [DOI] [PubMed] [Google Scholar]
- Patil AA, Cai Y, Sang Y, Blecha F, Zhang G. Cross-species analysis of the mammalian beta-defensin gene family: presence of syntenic gene clusters and preferential expression in the male reproductive tract. Physiol Genomics. 2005;23:5–17. doi: 10.1152/physiolgenomics.00104.2005. [DOI] [PubMed] [Google Scholar]
- Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J. Human beta-defensins. Cell Mol Life Sci. 2006;63:1294–1313. doi: 10.1007/s00018-005-5540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazgier M, Prahl A, Hoover DM, Lubkowski J. Studies of the biological properties of human beta-defensin 1. J Biol Chem. 2007;282:1819–1829. doi: 10.1074/jbc.M607210200. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pogozheva ID, Chai BX, Lomize AL, Fong TM, Weinberg DH, Nargund RP, Mulholland MW, Gantz I, Mosberg HI. Interactions of human melanocortin 4 receptor with nonpeptide and peptide agonists. Biochemistry. 2005;44:11329–11341. doi: 10.1021/bi0501840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J Immunol. 2010;184:6688–6694. doi: 10.4049/jimmunol.0903984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai MV, Jia HP, Liu L, Aseyev V, Wiencek JM, McCray PB, Jr, Ganz T, Kearney WR, Tack BF. The NMR structure of human beta-defensin-2 reveals a novel alpha-helical segment. Biochemistry. 2001;40:3810–3816. doi: 10.1021/bi002519d. [DOI] [PubMed] [Google Scholar]
- Schibli DJ, Hunter HN, Aseyev V, Starner TD, Wiencek JM, McCray PB, Jr, Tack BF, Vogel HJ. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J Biol Chem. 2002;277:8279–8289. doi: 10.1074/jbc.M108830200. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Fersht AR. Interaction of barnase with its polypeptide inhibitor barstar studied by protein engineering. Biochemistry. 1993;32:5145–5150. doi: 10.1021/bi00070a025. [DOI] [PubMed] [Google Scholar]
- Schulz A, Kluver E, Schulz-Maronde S, Adermann K. Engineering disulfide bonds of the novel human beta-defensins hBD-27 and hBD-28: differences in disulfide formation and biological activity among human beta-defensins. Biopolymers. 2005;80:34–49. doi: 10.1002/bip.20193. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Sipols AJ, Marks JL, Sanacora G, White JD, Scheurink A, Kahn SE, Baskin DG, Woods SC, Figlewicz DP, et al. Inhibition of hypothalamic neuropeptide Y gene expression by insulin. Endocrinology. 1992;130:3608–3616. doi: 10.1210/endo.130.6.1597158. [DOI] [PubMed] [Google Scholar]
- Semple F, MacPherson H, Webb S, Cox SL, Mallin LJ, Tyrrell C, Grimes GR, Semple CA, Nix MA, Millhauser GL, et al. Human beta-defensin 3 affects the activity of pro-inflammatory pathways associated with MyD88 and TRIF. Eur J Immunol. 2011;41:3291–3300. doi: 10.1002/eji.201141648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha N, Smith-Gill SJ. Electrostatics in protein binding and function. Curr Protein Pept Sci. 2002;3:601–614. doi: 10.2174/1389203023380431. [DOI] [PubMed] [Google Scholar]
- Slominski A, Plonka PM, Pisarchik A, Smart JL, Tolle V, Wortsman J, Low MJ. Preservation of eumelanin hair pigmentation in proopiomelanocortin-deficient mice on a nonagouti (a/a) genetic background. Endocrinology. 2005;146:1245–1253. doi: 10.1210/en.2004-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope VB, Jameson JA, McFarland KL, Supp DM, Miller WE, McGraw DW, Patel MA, Nix MA, Millhauser GL, Babcock GF, et al. Defining MC1R regulation in human melanocytes by its agonist alpha-melanocortin and antagonists agouti signaling protein and beta-defensin 3. J Invest Dermatol. 2012;132:2255–2262. doi: 10.1038/jid.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tota MR, Smith TS, Mao C, MacNeil T, Mosley RT, Van der Ploeg LH, Fong TM. Molecular interaction of Agouti protein and Agouti-related protein with human melanocortin receptors. Biochemistry. 1999;38:897–904. doi: 10.1021/bi9815602. [DOI] [PubMed] [Google Scholar]
- Yang D, Chen Q, Chertov O, Oppenheim JJ. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J Leukoc Biol. 2000a;68:9–14. [PubMed] [Google Scholar]
- Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dickinson C, Haskell-Luevano C, Gantz I. Molecular basis for the interaction of [Nle4,D-Phe7]melanocyte stimulating hormone with the human melanocortin-1 receptor. J Biol Chem. 1997;272:23000–23010. doi: 10.1074/jbc.272.37.23000. [DOI] [PubMed] [Google Scholar]
- Yang YK, Fong TM, Dickinson CJ, Mao C, Li JY, Tota MR, Mosley R, Van Der Ploeg LH, Gantz I. Molecular determinants of ligand binding to the human melanocortin-4 receptor. Biochemistry. 2000b;39:14900–14911. doi: 10.1021/bi001684q. [DOI] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.