

Abstract
The formation of adducts by the reaction of chemicals with DNA is a critical step for the initiation of carcinogenesis. The structural analysis of various DNA adducts reveals that conformational and chemical rearrangements and interconversions are a common theme. Conformational changes are modulated both by the nature of adduct and the base sequences neighboring the lesion sites. Equilibria between conformational states may modulate both DNA repair and error-prone replication past these adducts. Likewise, chemical rearrangements of initially formed DNA adducts are also modulated both by the nature of adducts and the base sequences neighboring the lesion sites. In this review, we focus on DNA damage caused by a number of environmental and endogenous agents, and biological consequences.
Introduction
DNA Damage, arising from both endogenous and exogenous chemical insults, is believed to represent an initiating step in chemical carcinogenesis. Considerable data exist with regard how specific DNA adducts perturb its structure, and how these structural perturbations interfere with the biological processing of DNA [1]. A major goal is to understand the basis whereby chemical modification of DNA triggers specific mutations. The field has advanced rapidly over the past several decades. One reason has been the development of automated DNA synthesis methodolgies [2], and progress in regioselectively and stereospecificaly incorporating specific types of damage into duplex DNA [1]. Collectively, these approaches enable the construction of site-specifically damaged DNA templates for structural and biological studies. Additionally, advances in applications of NMR spectroscopy [3] and X-ray crystallography [4] to the study of DNA enable high-resolution structures of damaged DNA to be obtained. In this review, we will focus on the conformational and configurational rearrangements of DNA adducts, derived from a number of environmental and endogenous DNA damaging agents, and in the context of biological consequences.
Conformational Interconversions of DNA Adducts
Conformational intercon-versions involve rotation about one or more chemical bonds, but do not require bond breakage. The conformational interconversions of interest here have sufficiently large activation barriers that, under physiologically relevant conditions, interconversion occurs slowly at ms or slower time scales, and it is anticipated that different conformers may elicit differential biological responses. In DNA, a major site of conformational interconverson for deoxynucleosides involves the N-glycosyl bond connecting the deoxyribose sugar to the heteroaromatic nucleobase. In B-form DNA, the N-glycosyl bond is maintained in the anti-conformation, thus orienting the sterically bulky nucleobase away from the deoxyribose sugar. In duplex DNA, this orients complementary nucleobases to form the canonical Watson–Crick base pairing interactions. Often, however, the introduction of sterically bulky substituents to DNA disrupts this conformational equilibrium, and shifts the N-glycosyl bond into the syn-conformation, in which the nucleobase is oriented toward the deoxyribose. In duplex DNA, this shifts the Watson–Crick H-bonding edge of the base toward the major groove, and hinders canonical Watson–Crick H-bonding. Characteristic examples of bulky adducts that undergo such conformational interconversions include those arising from C8-dG alkylation by aromatic amines, C8-dG oxidative-damage products 8-oxo-dG and 8-oxo-dA, 1,N2-etheno and -propano annelation products of dA and dG, and 4-hydroxy-equilenin.
Aromatic Amines
Early evidence that conformational heterogeneity modulated the structures of DNA adducts came from studies of arylamine adducts. The subject has been reviewed by Cho [5] and Patel et al. [6]. Arylamines are environmental carcinogens; human exposures are associated with the etiology of bladder cancer [7]. The most studied example is N-acetyl-2-aminofluorene. 2-Aminofluorene (AF) is acetylated in vivo; the acetylated species then reacts with DNA [8–10] to give 2-amino-N-(deoxyguanosin-8-yl)fluorene (C8-dG AF) (Fig. 1). This has been detected in mammalian cells [11]. When the C8-dG AF adduct is placed opposite dC in the 5′-AXG-3′ sequence, it exists in two conformations, referred to as the external-AF and inserted-AF conformations [12]. The external-AF conformation features the anti-conformation, while the inserted-AF conformation features the syn conformation about the glycosyl bond (Fig. 2). In the external-AF conformation, the AF moiety is in the major groove. The X(anti)·C(anti) base pair exhibits Watson–Crick H-bonding [12] (Fig. 2). In the inserted-AF conformation, the syn glycosyl bond places the AF moiety into the DNA helix and displaces the damaged guanine base and the complementary cytosine [12], resulting in the disruption of Watson–Crick H-bonding at the X(syn)·C(anti) base pair (Fig. 2). A similar equilibrium is observed between external- and inserted-AF in the 5′-CXC-3′ sequence [13]. When the C8-dG AF adduct is placed opposite dA in the 5′-CXC-3′ sequence, modeling the intermediate associated with G→ T mutations observed for AF [14], the damaged nucleotide also adopts the syn conformation about the glycosyl bond, but the AF moiety orients in the minor groove [15]. The AF-dG adduct has also been examined in sequences derived from c-Ha-ras-protooncogenes with modification at codon 61, a site of G→T mutations, by Cho et al. [16], and Eckel and Krugh [12][17]. The amount of the major conformation of the AF-dG adduct is ca. 60% [16]; the aminofluorene moiety rotates toward the major groove. The major conformer adopted by the corresponding 4-aminobiphenyl-modified base [18] is similar.
Fig. 1.

DNA Adducts derived from aromatic amines and heterocyclic amines
Fig. 2. Stereoview of duplexes containing a C8-dG AF adduct.
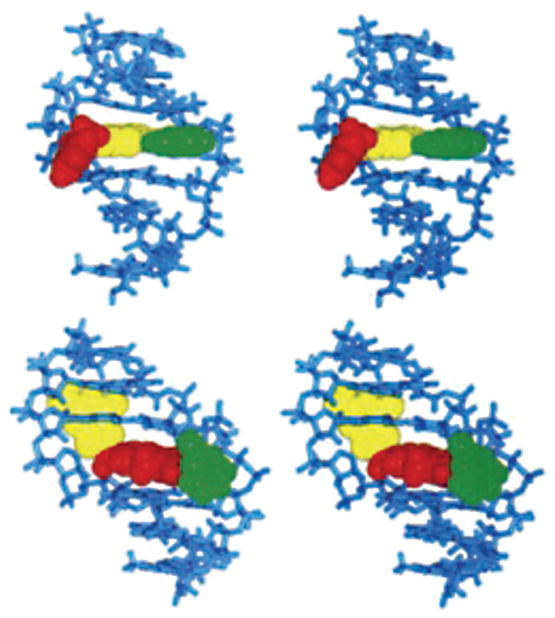
The top panel shows the external-AF conformation, and the bottom panel shows the inserted-AF conformation as viewed from the major groove. The AFadduct is in red, the adducted guanine is in yellow, and the complementary dC is in green.
Reproduced with permission from [12] (© 1994 American Chemical Society).
Base substitutions are associated with these adducts, especially G→T mutations [14], although the correct incorporation of dCTP predominates during translesion bypass [19]. Frameshift mutations induced by AF adducts are associated with the E. coli NarI hot spot sequence (C-G1-G2-C-G3-C-C), in which −2 base deletions occur at G3 [20–25]. The NarI frameshift pathway is SOS-dependent but umuDC-independent, and DNA Pol II is responsible for the frameshifts [25]. When the AF-dG adduct is placed into the E. coli NarI sequence (C-G1-G2-C-G3-C-C), associated with −2 base deletions at G3 [20–25], its conformation is sequence-dependent [26]. The adduct favors the external-AF conformation when placed at G1 and G2, respectively, while an equal mixture of both conformers exists when AF is placed at G3. The conformational equilibrium is also affected by the next nearest neighbors [6]. Structures of the C8-dG adduct placed opposite to 3′-terminal primer cytosine have been obtained in a ternary complex with the Dpo4 polymerase [27]. The C8-dG AF adduct remained in the anti-conformation about the glycosyl bond with the AF moiety positioned in the major groove [27]. Overall, the conformational equilibrium of the C8-dG AF adduct, and the concomitant distortion of DNA structure, may contribute to both base substitution and frameshift mutations during DNA replication, in a sequence-dependent manner [12].
The heterocyclic amines 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) [28–31] and 2-amino-1-methyl-6-phenyllimidazo[4,5-b]pyridine (PhIP) are produced, when protein-rich foods are cooked. IQ is activated to an N-hydroxy oxidation product [32–35] and reacts with DNA to produce C8-dG adducts that are observed in rodents and primates, as measured by 32P post-labeling [36]. A minor adduct forms at N2-dG [37]. In nucleosides, the C8-dG IQ adduct (Fig. 1) exists in the syn-conformation about the glycosyl bond [38], while the N2-dG IQ adduct adopts the anti conformation. When placed opposite cytosine in a duplex containing the E. coli NarI sequence, the C8-dG IQ adduct also adopts the syn-conformation (Fig. 3) [40], as does the C8-dG PhIP adduct (Fig. 4) [41]. This places the Watson–Crick edge of the modified dG into the major groove. The IQ moiety intercalates between the flanking C·G base pairs. These studies corroborated molecular-mechanics analysis of the C8-dG IQ-modified duplex [42]. As for the C8-dG AF adduct, the syn conformation of the C8-dG IQ adduct may contribute to error-prone replication, in a sequence-dependent manner. The structure of the N2-dG IQ adduct has not yet been examined in an oligodeoxynucleotide duplex. It has been proposed that differences in the accumulation and rates of removal of C8-dG IQ vs. N2-dG IQ adducts in rodents and non-human primates may be attributable to differences in conformation about the glycosyl bond in these two adducts. Adducts in the syn-conformation may be more easily recognized and excised as they induce greater distortion in the DNA [43]. Turesky et al. [43] showed that C8-dG IQ adduct was removed, whereas the N2-dG IQ adduct was persistent. In bacteria, mutations occur primarily at G·C base pairs [44][45], and IQ gives frameshift mutations in (CG)n repeats. Similar levels of mutations are observed in mammalian hprt [46] and ef-2 [47] gene assays.
Fig. 3. Structure of the C8-dG IQ (green)-modified three-base-pair duplex, looking into the major groove and normal to the helix axis.
The IQ ring is in red and is inserted between flanking base pairs. The dotted line indicates a H-bond formed between the amino group of modified dG and the O-atom of the 5′ phosphodiester linkage. This illustration was prepared from PDB entry 2 HKC using PyMol [39] software.
Fig. 4. Structure of a C8-dG PhIP (green)-modified three-base-pair duplex, looking into the major groove and normal to the helix axis.
This illustration was prepared from PDB entry 1 HZO using PyMol [39] software.
8-Oxo-dG and 8-Oxo-dA
Deoxyadenosine and deoxyguanosine are hydroxylated at C(8) to form 7,8-dihydro-8-oxoadenine (8-oxo-dA) and 7,8-dihydro-8-oxoguanine (8-oxo-dG) (Fig. 5) [48][49]. These lesions tautomerize [50][51]; the 6-amino-8-oxo and 6,8-dioxo [50–52] species predominate for 8-oxo-dA and 8-oxo-dG, respectively, in oligodeoxynucleotides [53][54]. DNA Synthesis proceeds past both lesions. High-fidelity polymerases insert dCTP or dATP opposite 8-oxo-dG in varying proportions dependent upon the polymerase, with extension preferentially occurring from 8-oxo-dG·A base pairs [55–58]. Consistent with this observation, 8-oxo-dG induces primarily G→T transversions in human cells [59]. The Y-family polymerases Dpo4 [60–62] and Pol η [63][64] preferentially insert dCTP over dATP opposite 8-oxo-dG, but favor extension from the 8-oxo-dG·C pair, thus allowing error-free bypass.
Fig. 5.
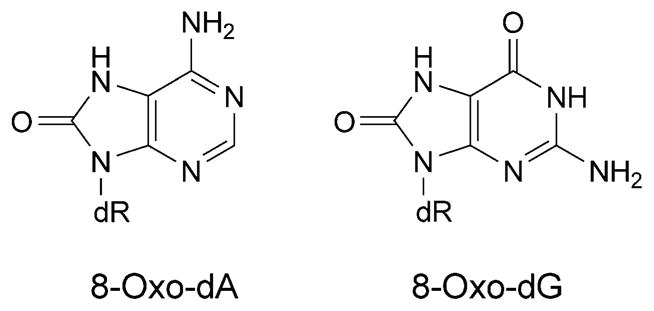
8-Oxo-dG and 8-oxo-dA adducts
The 8-oxo-dG lesion perturbs base interactions and backbone conformations in single-stranded DNA [65]. In vitro, 8-oxo-dG pairs with all four dNTPs [66], but preferentially with A and C [58]. Studies of 8-oxo-dG and different complementary bases indicate that one base generally adopts the syn-conformation in the purine· purine pairs [67–69]. Thus, 8-oxo-dG mismatched with A adopts the syn-conformation, and the complementary A adopts the anti-conformation about the glycosyl bond [70][71], similar to G·A mispairs [69][72] (Fig. 6). This could explain the mis-insertion of dATP opposite 8-oxo-dG, yielding G→T transversions. In contrast, when 8-oxo-dG pairs with C, both 8-oxo-dG and C adopt the anti-conformation about the glycosyl bond, and the 8-oxo-dG·C pair forms Watson–Crick H-bonds [53][73]. Template distortion associated with 8-oxo-dG (anti) complementary to primer terminus dC has been observed for T7 [74] and for the Bacillus Pol I fragment BF [75]. In contrast, neither the template nor the polymerases were affected by 8-oxo-dG (syn) opposite primer terminus dA, possibly enabling the 8-oxo-dG·A base pair to evade proofreading by T7 [74] or Bacillus Pol I fragment BF [75]. This may also provide an explanation for extension by these polymerases from the 8-oxo-dG·A base pair [55–58]. Structures of 8-oxo-dG opposite A, C, T, or G, and the next nascent base pair in ternary complexes with the Dpo4 polymerase show that neither the template backbone nor the structure of the active site are perturbed by the 8-oxo-dG·C or 8-oxo-dG·A pairs [76]. However, the 8-oxo-dG·A pair adopts both the 8-oxo-dG (syn)·A (anti) and 8-oxo-dG (anti)·A (syn) alignments. This may explain the poor primer extension from the 3′-terminal primer base A by the Dpo4 polymerase. In the case of the 8-oxo-dG·C pair, the unperturbed Dpo4-active site explains the efficient primer extension. The OGG-2 glycosylase repairs 8-oxo-dG paired with G or A [77]. The E. coli glycoslyase MutY repairs 8-oxo-dG·G mispairs [78]. A duplex containing the 8-oxo-dG·G mispair has been examined. The 8-oxo-dG lesion adopts the syn-conformation about the glycosyl bond. The damaged base is inserted into the helix (Fig. 7) [79]. With 8-oxo-dA, dTTP is predominantly incorporated by Y-family polymerases, yielding error-free bypass [80]. In COS-7 cells, the mutagenicity of 8-oxo-dA is four times lower than that of 8-oxo-dG [81]; it induces mainly A→C transversions. The mutation frequency and spectrum associated with 8-oxo-dA depends on the sequence context of the lesion. In the 5′-TXG-3′ sequence, the mutation frequency was 1.2%. In the 8-oxo-dA·G mispair 8-oxo-dA adopts the syn-conformation, while the complementary G adopts the anti-conformation [82] (Fig. 8).
Fig. 6.
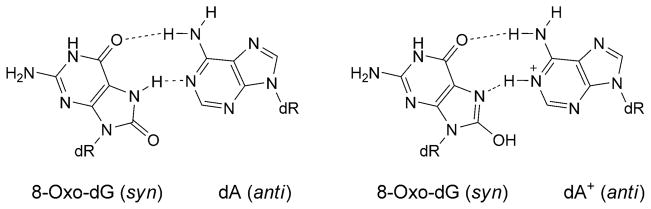
H-Bonding patterns involving the 8-oxo-dG (syn)·A (anti) and 8-oxo-dG (syn)·A+ (anti) base pairs
Fig. 7.

Base pair showing the three-centered H-bonding between C(8)=O of 8-oxo-dG and the imino, and one of the amino H-atoms of the complementary dG
Fig. 8.
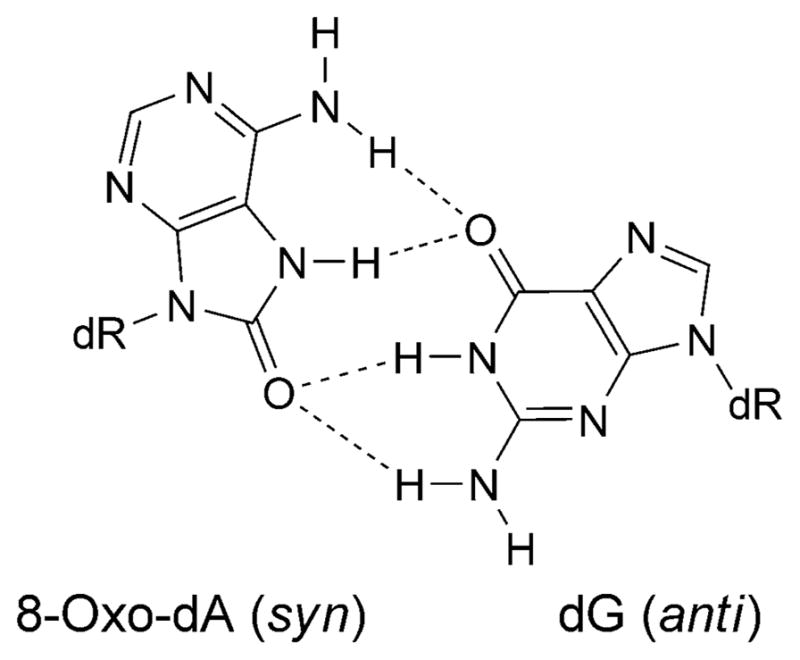
Base pair showing the proposed reverse three-center H-bonding for the 8-oxo-dA (syn)·G (anti) base pair
Etheno Adducts
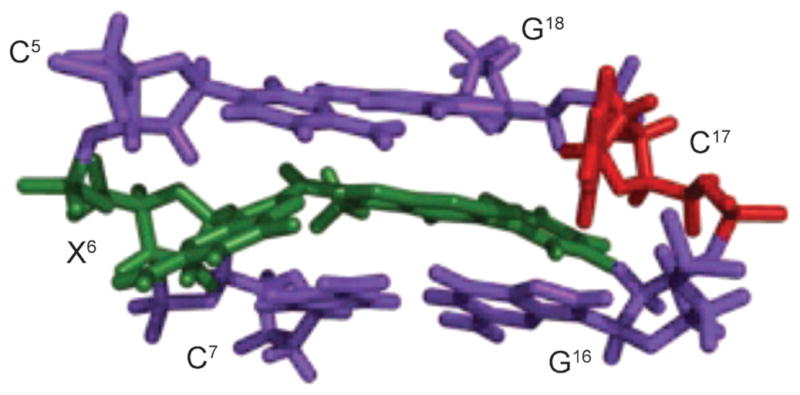
These arise from the reaction of vinyl chloride with DNA. The 3,N4-εdC adduct (Fig. 9) is mutagenic. In vitro studies showed that the mammalian polymerases α and δ predominantly incorporate dATP or dTTP opposite 3,N4-εdC, while polymerase β incorporates primarily dCTP [83]. In vitro replication studies with a DNA template containing 3,N4-εdC using the Klenow exo− fragment of DNA polymerase I showed primarily dATP and dTTP incorporation opposite the lesion [84–86]. The structures of 3,N4-εdC opposite dA, dT, dG, or dC have been determined [87–90]. When placed opposite dT in duplex DNA, 3,N4-εdC adopts the syn-conformation about the glycosyl bond, while the complementary dT adopts the anti-conformation [88] (Fig. 10, a). The 3,N4-εdC (syn)·T (anti) alignment is stabilized by a H-bond between T N(3)H and 3,N4-εdC C(4)–N. This base pair stacks with the flanking base pairs. The conformational change of 3,N4-εdC from the anti- to syn-orientation and maintaining coplanar alignment may explain the facilitation of misincorporation of dTTP opposite 3,N4-εdC by DNA polymerases.
Fig. 9.
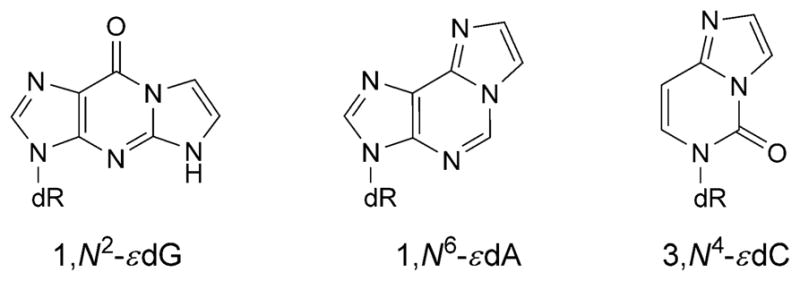
Structures of etheno adducts
Fig. 10. Structures of the 3,N4-εdC adduct (X6) opposite dT, dA, and dG in the 5′-C5X6C7-3′ sequence.

a) The 3,N4-εdC (syn)·T (anti) base pair. The etheno moiety is in the major groove. b) The 3,N4-εdC (anti)· A (anti) base pair, showing partial intercalation within the 3,N4-εdC (anti)·A (anti) alignment. c) The 3,N4-εdC (anti)·G (anti) base pair. The 3,N4-εdC and complementary base are shown in green and red, respectively. These illustrations were prepared from PDB entries 1B5K and 1B6Y using PyMol [39] software.
When placed opposite dA, 3,N4-εdC and the complementary dA remain in the anti-conformation about the glycosyl bond [87]. The 3,N4-εdC (anti)·dA (anti) pair adopts a staggered conformation in which each nucleotide displaces 5′-side and intercalates between the bases on the complementary strand (Fig. 10, b). The partial intercalation of 3,N4-εdC (anti) and dA (anti) bases produces stacking between the aromatic rings of 3,N4-εdC and dA, and with flanking base pairs. Steric factors preclude a coplanar alignment between 3,N4-εdC and dA. Nevertheless, dATP is preferentially incorporated opposite 3,N4-εdC in vitro [86], and both dATP and dGTP are incorporated opposite this lesion in cells [91]. The structural data suggest that the partially intercalated structure may promote translesion synthesis past this lesion.
When placed opposite dG, both 3,N4-εdC and dG adopt the anti conformations [89]. The 3,N4-εdC is displaced and shifts towards major groove, while the complementary dG remains stacked (Fig. 10, c). The 3,N4-εdC (anti)·G (anti) alignment is stabilized by H-bonds involving dG. The structural studies corroborate the low mutagenicity of 3,N4-εdC in E. coli, suggesting that 3,N4-εdC (anti)·G (anti) pairing occurs during replication.
The 1,N6-εdA adduct (Fig. 9) induces A→C transversion. When placed opposite dG, 1,N6-εdA adopts the syn-conformation about the glycosyl bond, positioning the exocyclic ring toward the major groove, while dG adopts the anti-conformation [92]. The 1,N6-εdA (syn)·G (anti) alignment is stabilized by two H-bonds from the N(1)H imino H-atom and one H-atom from NH2 of dG to N(9) and N(1) of 1,N6-εdA, respectively (Fig. 11). The syn conformation of 1,N6-εdA is accommodated without disruption of flanking base pairs and may account for the incorporation of dGTP opposite 1,N6-εdA during replication. In mammalian cells, the alkylpurine-DNA-N-glycosylase recognize and repair 1,N6-εdA by a similar mechanism as proposed for 1,N2-εdG adduct (for a review, see [93]).
Fig. 11. Structure of the 1,N6-εdA adduct (X14) opposite dG, in the 5′-C13X14C15-3′ sequence.

The top panel shows the three base pairs with the 1,N6-εdA adduct (syn conformation) located in the major groove. The bottom panel shows the 1,N6-εdA (syn)·G (anti) base pair alignment viewed down the axis and the two H-bonds that stabilize this base pair. Reproduced with permission from [92] (© 2002 American Chemical Society).
The 1,N2-εdG lesion (Fig. 9) is released from DNA by both the E. Coli mismatch-specific uracil DNA glycosylase and the human alkylpurine-DNA-N-glycosylase [94]. The flipping of damaged nucleotides out of the helix and into the glycosylase-active site provides a mechanism by which the DNA glycosylases interact with damaged DNA [93]. At neutral pH, 1,N2-εdG equilibrates between the syn- and anti-conformations about the glycosyl bond. At acidic pH, it adopts the syn-conformation, and the complementary dC adopts the anti conformation (Fig. 12, a) [95]. This 1,N2-εdG (syn)· dC (anti) pair is stabilized by Hoogsteen H-bonds. 1,N2-εdG introduces a localized perturbation involving the modified base pair and its 3′- and 5′-neighbor base pairs. The 3′-neighbor dG·dC base pair also equilibrates between Watson–Crick and Hoogsteen pairs (Fig. 12, b). At basic pH, both 1,N2-εdG and the complementary dC adopt the anti-conformation about the glycosyl bond (Fig. 12, c) [96]. The etheno moiety is inserted into the duplex, and dC is displaced. No H-bonding is observed between the base pairs. A similar conformational transition is observed for 1,N2-εdG opposite dC in the 5′-CXC-3′ sequence [97]. The decreased melting temperature of the DNA containing the 1,N2-εdG adduct [95][96] and its conformational exchange in duplex DNA, at neutral pH, may facilitate damage recognition [94].
Fig. 12. Structures of the 5′-T5X6G7-3′·5′-C18C19A20-3′ duplex containing the 1,N2-εdG adduct (X6), opposite dC, as a function of pH.

a) At pH 5.2, the major conformation features the 1,N2-εdG (syn)·C (anti) pair in the Hoogsteen conformation. b) At pH 5.2, a second conformation exists in which both the 1,N2-εdG (syn)·C (anti) base pair and its 3′-neighbor dG (syn)·C (anti) base pair are in Hoogsteen conformation. c) At pH 8.6, the 1,N2-εdG lesion (green) adopts the anti-conformation. The X6, G7, and C19 nucleotides are in green, orange, and red, respectively. These illustrations were prepared from PDB files used in [95][96], using PyMol [39] software.
Product analysis from the primer extension studies suggests that the Dpo4 polymerase uses several mechanisms, including dATP incorporation and also a variation of dNTP-stabilized misalignment to bypass 1,N2-εdG lesions [98]. Insertion of the correct nucleotide might be facilitated by the syn-conformation of 1,N2-εdG, which would allow Hoogsteen pairing with incoming dCTP. However, in structures of 1,N2-εdG inserted into a template containing the 5′-TXG-3′ sequence, and the formation of either binary or ternary complexes with the S. solfataricus DNA polymerase Dpo4, 1,N2-εdG adopted the anti-conformation about the glycosyl bond [98], as does the structurally similar 1,N2-propanodeoxyguanosine (PdG) adduct [99].
1,N2-Propanodeoxyguanosine
When incorporated into the 5′-(CpG)4-3′ frameshift hot-spot of the hisD3052 gene carried on an M13 vector, PdG (Fig. 13) induces frameshift mutations [100]. In both E. coli and simian kidney COS-7 cells, G→T transversions are observed; in SOS-induced E. coli, G→A transitions are also observed. The mutation frequency for single-stranded DNA that contains PdG is 100% in non-SOS-induced E. coli, 68% in SOS-induced cells, but only 8% in COS-7 cells [91]. The structure of PdG adduct has been studied in a variety of sequence contexts [101–110]. Its characterization paired to dC has been performed in two sequences, within a d(CG)3-iterated repeat and in the 5′-TXT-3′ sequence. In both, it adopts the syn conformation about the glycosyl bond, forming a PdG(syn)·C+(anti) Hoogsteen-like pair (Fig. 14). In the 5′-(CG)3-3′ iterated repeat, the 3′-neighbor dG also interconverts between syn- and anti-conformations, generating multiple structures. A correlation between the formation of tandem Hoogsteen base pairs and the two-base deletion mutations observed in the d(CG)3 context has been proposed. PdG also displays conformational exchange about the glycosyl bond when placed opposite dA. The syn-conformer forms two H-bonds with protonated dA; the anti-conformer partially intercalates between its partner dA and the 5′-neighbor nucleotide (Fig. 15). The PdG· G pair is similar to the PdG·A pair in that PdG adopts the syn-conformation about the glcyosyl bond and forms two H-bonds with the complementary dG.
Fig. 13.

Structures of M1dG, PdG, γ-OH-PdG, and α-OH-PdG
Fig. 14. Structure of the PdG adduct (green) in the 5′-T5X6T7-3′ sequence.

The PdG adduct is in the syn-conformation, and the propano moiety faces into the major groove. The dotted lines represent the NOEs observed between PdG and flanking bases T5 and T7. Reproduced with permission from [102] (© 2002 American Chemical Society).
Fig. 15. Structures of the PdG adduct (X5) opposite dA, in the 5′-G4X5G6-3′ sequence.

a) At basic pH, the PdG (anti)·A (anti) base pair alignment as viewed from major groove (top) and down the axis (bottom). b) At acidic pH, the PdG (syn)·A (anti) base pair alignment as viewed from major groove (top) and down the axis (bottom), showing the two H-bonds that stabilize this base pair. Reproduced with permission from [105] (© 2002 American Chemical Society).
4-Hydroxyequilenin
A metabolic product of equilin and equilenin, formulated in the hormone replacement therapy drug Premarin [111][112], 4-hydroxyequilenin (4-OHEN), auto-oxidizes to form cytotoxic quinoids [111]. These alkylate DNA [113–118] to form cyclic dC, dA, and dG adducts [112][115][119][120]; structures of four stereoisomeric adducts have been identified [119][120] for each modified base [121]. The 4-OHEN-dC (Fig. 16) adduct predominates. Two stereoisomeric dA and three dG 4-OHEN adducts have been found in the mammary fat pads of rats upon 4-OHEN injection [115], and dA, dG, and dC adducts have been detected in breast tissues of patients [122]. The Watson–Crick edge of 4-OHEN-derived adducts is obstructed by the formation of the cyclic ring (Fig. 16).
Fig. 16.

Chemical structures and configurational characteristics of the 4-OHEN-dA and dC adducts
The conformations of the four 4-OHEN-C stereoisomers have been examined in the 5′-GXT-3′ sequence by computational approaches [123][124]. The calculations suggest that the lesions are located in the major or minor groove with the modified cytosines adopting the syn- or anti-conformations, respectively. The configuration of the 4-OHEN-dC adducts orients the equilenin rings with respect to the 5′→3′ direction of the modified strand, and positions the equilenin CH3 and OH groups. Calculations have also been performed on stereosiomeric 4-OHEN-dA adducts [124][125]. The 4-OHEN-dC adducts differ structurally as compared to 4-OHEN-dA adducts in terms of H-bonding, stacking, bending, groove dimensions, solvent exposure, and hydrophobic interactions.
The 4-OHEN-dC adduct induces C→G and C→A transversions when a supF shuttle vector plasmid system is propagated in human cells [126]. Polymerases bypass the 4-OHEN-dC and dA lesions with efficiencies depending upon configuration and the identity of the damaged base [127–129]. Primer extension conducted with the polymerases Dpo4, Pol η, and Pol κ indicate that 4-OHEN-dC is bypassed with insertion of an incorrect dNTP or by strand slippage [128][130]. With Pol κ, both dCTP and dATP are inserted opposite stereoisomeric 4-OHEN-dC adducts, and primer extension with complementary dC is greater than that with complementary dA [128]. The insertion of dGTP is inefficient. With Pol η, the bypass frequencies of 4-OHEN-dC stereoisomers [128] differ by two orders of magnitude. With Pol η, both insertion of dATP and extension are greater than those for dGTP, the correct nucleotide. For 4-OHEN-dA, the bypass frequency also depends upon configuration [129]. However, both pols η and κ insert the correct nucleotide dTTP opposite 4-OHEN-dA [129]. Mismatched dATP and dCTP products are also observed for Pol κ and Pol η, respectively.
Polycyclic Aromatic Hydrocarbons
DNA Adducts arising from polycyclic aromatic hydrocarbons (PAHs) undergo more extensive conformational rearrangements, allowing the large planar aromatic moiety to intercalate into the DNA helix, or orient in the minor or major groove of the DNA. The complex subject of PAH structure–activity relationships has been reviewed by Geacintov et al. [131][132], and Broyde et al. [133]. Exposures to PAH are associated with cancer etiology [134–137]. Benzo[a]pyrene (B[a]P) is one of the most common PAHs [138]. It is metabolized to 9,10-epoxy-7,8-diol stereoisomers, principally the (+)-(7R,8S,9S,10R)-enantiomer (B[a]PED), although minor amounts of the (−)-(7S,8R,9R,10S)-enantiomer also form [139]. These diols alkylate the N2-dG position by trans-addition to C(10) of B[a]PED, but minor amounts of cis-addition are observed [140][141]. The lesions form efficiently in the presence of m5dC in 5′-CpG-3′ sequences that are recognized by DNA methyltransferases [142–144]. With respect to structure–activity relationships, the most studied adducts are (+)-trans-B[a]P-N2-dG, (+)-cis-B[a]P-N2-dG, (−)-trans-B[a]P-N2-dG, and (−)-cis-B[a]P-N2-dG [131]. For the (+)-trans- and (−)-trans-B[a]P-N2-dG adducts, the pyrenyl moieties orient in the minor groove, pointing either into the 5′- or the 3′-directions of the modified strand, respectively, relative to the modified guanine [145][146]. The (+)-cis-B[a]P-N2-dG adduct differs, having a base-displaced intercalative conformation with the modified guanine and the complementary cytosine displaced into the minor and major grooves, respectively [147]. Sequence-dependent and stereospecific conformational differences play an important role in the structural biology of PAH adducts [148].
If not repaired, the B[a]P-N2-dG adducts (Fig. 17) induce mutations [149–152]. In cell-free extracts, removal of the (+)-trans-B[a]P-N2-dG lesion by the NER apparatus depends on the base sequence in which it is embedded. The rate of incision of the lesion by the prokaryotic UvrABC system is two-fold greater in the 5′-TXT-3′ than in the 5′-CXC-3′ sequence. The former sequence exhibits a lower thermal stability [153]. In the 5′-CXC-3′ sequence, the pyrenyl moiety of (+)-trans-B[a]P-N2-dG resides in the minor groove in the 5′-direction along the modified strand [145]. In the 5′-TXT-3′ sequence, a similar motif is accompanied by increased conformational heterogeneity [154]. Furthermore, the 5′-CXC-3′ sequence is characterized by a rigid bend, whereas the 5′-TXT-3′ sequence is characterized by a more flexible bend [155]. The two sequences have also been investigated using molecular dynamics (MD) calculations [156][157]. The MD results highlight the importance of local dynamics in the vicinity of the lesion and show that the 5′-TXT-3′ sequence is more flexible, and exhibits weaker Watson–Crick H-bonding adjacent to the lesion, poorer stacking interactions, local roll/bending dynamics, and minor groove flexibility. In the 5′-TXC-3′ sequence, a similar minor groove conformation is observed, but a minor conformation involving insertion of the BP moiety into the duplex with disruption of Watson–Crick H-bonding at the lesion site is proposed [158]. Sequence effects on the minor groove conformations of this adduct in the 5′-CXG-3′ and 5′-GXC-3′ sequences have also been reported [159] in the 5′-GG-3′ mutation hotspot context. Cai et al. [157] have proposed that the amino groups in tandem 5′-GG-3′ sequences modulate the efficiency of NER.
Fig. 17.

Structures of dG and dA adducts derived from B[a]P
Structures of DNA template·primers containing the (+)-trans-B[a]P N2-dG adduct, complexed with the S. sulfataricus Dpo4 polymerase, have been examined [160]. In one, the adducted base mispairs with adenine at the template·primer junction, and an incoming dATP is opposite template dT 5′ to the lesion. This corresponds to efficient incorporation of dATP; moreover, the X·A mispair is efficiently extended by the polymerase. Significantly, the B[a]P intercalates, occupying space corresponding to one base pair between the last two bases at the primer strand terminus [160]. The damaged base is in the syn-conformation about the glycosyl bond and shifts into the minor groove. Base pairing is disrupted both at the X·A mispair and the T·dATP insertion complex 5′ to the lesion. The 3′ orientation of B[a]P relative to the modified base allows B[a]P to stack with the neighboring bases with its long axis at an angle of 40° with respect to the DNA.
The B[a]P epoxy diols also react with DNA to form N6-dA adducts (Fig. 17) [161][162]. Repair studies of N6-dA adducts formed by fjord-region B[a]P epoxy diols vs. bay region epoxy-diol metabolites of benzo[c]phenanthrene (B[c]P) show that the bay region adducts are removed, while fjord region adducts are refractory to repair [163]. If not repaired, these N6-dA adducts are mutagenic, correlating with reports on B[a]P-induced mutagenesis at adenines [164–166]. The mutagenic spectra of N6-dA B[a]P adducts depend upon configuration at C(10) position, configurations of the OH groups at C(7), C(8), and C(9), and depend upon DNA sequence [167–169]. For N6-dA B[a]P adducts, C(10) (C(10) is the site of DNA alkylation) (R)-stereoisomers intercalate in the 5′-direction [170–173], whereas C(10) (S)-stereoisomers intercalate in the 3′ direction [174]. The structure of the (+)-cis-B[a]P-N6-dA adduct in a primer extension complex with the Dpo4 polymerase has been obtained, in which the adduct is paired with primer terminus dT, and the incoming dATP pairs with the next undamaged templating base, dT. Two conformations of the (+)-cis-B[a]P-N6-dA adduct are observed. In the first, the B[a]P is intercalated. In the other, it orients in the major groove perpendicular to the DNA base pairs. The (+)-trans-B[a]P-N6-dA adduct, mismatched opposite dG, adopts the syn conformation about the glycosyl bond [175] (Fig. 18). This could explain A→C transversions induced by the (+)-trans-B[a]P-N6-dA adduct.
Fig. 18.

Structure of the N6-dA (RSRS)-B[a]P adduct (green) opposite dG (red) in a three-base-pair segment of duplex DNA.
This illustration was prepared from PDB entry 1DXA using PyMol [39] software.
Other N6-dA PAH adducts follow a similar pattern with respect to the configuration at the adducted C-atoms. For benz[a]anthracene, this is the C(1)-atom. The (1R)-adduct intercalates to the 5′-side of the damaged base [176], while the (1S)-adduct intercalates to the 3′-side [177]. Likewise, for trans-B[c]P, configuration at C(1) is crucial [178][179]. The (1S)-adduct permits the B[c]P ring to intercalate 3′ to lesion site without disrupting Watson–Crick H-bonding. The (1R)-adduct intercalates to the 5′-side of the modified base pair [178][179]. The greater thermal stabilities of duplexes containing fjord region N6-dA lesions correlate with lower susceptibilities of excision by NER [163]. Computational studies of a template·primer containing the (−)-(1S)-trans-B[c]P-N6-dA adduct in a complex with the Dpo4 polymerase suggest that B[c]P intercalation likely impedes replication [180]. The (+)-trans-benzo[g]chrysene-dA adduct also follows the pattern whereby (R)-configuration at C(14) correlates with 5′-intercalation [181].
Configurational Rearrangements of DNA Adducts
Increasingly, the importance of interconversions of DNA adducts involving bond breakage and rearrangement has become recognized. Such rearrangements can be either chemically reversible, i.e., existing at equilibrium, or irreversible. Examples of specific adducts include abasic sites, the epimerization of thymine glycol lesions, deamination and the Dimroth rearrangement of N1-dA lesions, the rearrangement of 1,N2-dG lesions arising from malondialdehyde and α,β-unsaturated enals, and α/β anomerization of formamidopyr-imidine (FAPY) lesions.
Deamination of Butadiene-Derived N1-dA Adducts
Buta-1,3-diene (BD) is extensively used in the polymer industry, e.g., in the manufacture of styrene–butadiene rubber [182][183]. The metabolism of BD is depicted in Scheme 1. Reaction of butadiene epoxides with dA results in the formation of N1-dA adducts. These may undergo hydrolysis of the 6-NH2 group to give the analogous inosine derivatives [184] (Fig. 19). Deamination of dA represents a promutagenic event, because, during DNA replication, the resulting dI nucleotide is recognized as dG and preferentially pairs with incoming dCTP. When ligated into the single-stranded vector M13mp7L2 and transfected into repair-deficient E. coli, the 2′-deoxy-N1-[(2S)-1-hydroxybut-3-en-2-yl]inosine adduct codes for incorporation of dCTP [185]. Similar results are observed with COS-7 cells [186]. Structural analyses reveal that, following deamination of the N1-dA lesion, the glycosyl bond of the (S)-N1-BDO-dI adduct rotates into the syn-conformation, placing the BD moiety into the major groove. The complementary dT (anti) remains intrahelical at the adduct site. The results suggest that the tendency of the (S)-N1-BDO-dI adduct to code for incorporation of dCTP may be attributed to the propensity of this adduct to form a protonated Hoogsteen-pairing interaction with dCTP during replication [186].
Scheme 1.
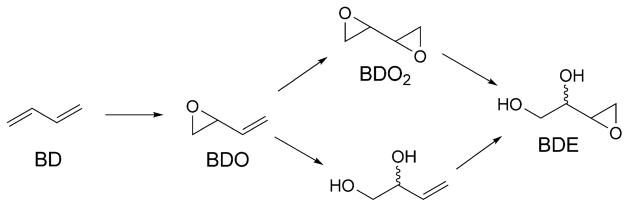
Reactive Metabolites of BD
Fig. 19.
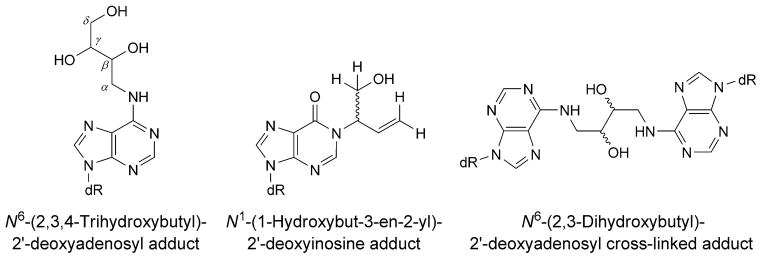
Chemical structures of BD-derived adenine adducts
Dimroth Rearrangement of N1-dA Adducts. Alternatively, N1-dA adducts of BD undergo chemical rearrangement to isomeric N6-dA adducts via a Dimroth rearrangement [187], in which ring cleavage between the N(1)- and C(2)-atoms of dA is followed by 180° internal rotation [188]. The consequence is that the initially formed N1-dA lesion, which is anticipated to be highly mutagenic due to the fact that it precludes Watson–Crick bonding during DNA replication, is converted to an N6-dA lesion (Fig. 19), which is projected to orient into the major groove of DNA, resulting in minimal structural perturbation and be less mutagenic. Structural studies of a series of N6-dA adducts of styrene oxide and butadiene epoxides suggests that, indeed, they induce minimal structural perturbation of DNA and are contained within the major groove [189–194]. Likewise, site-specific mutagenesis conducted in E. coli revealed that these adducts are only weakly mutagenic [195][196].
Malondialdehyde
Malondialdehyde (MDA), produced by lipid peroxidation and prostaglandin biosynthesis [197][198], is mutagenic in bacterial and mammalian cells [199–201] and is carcinogenic in rats [202]. In Salmonella typhimurium, MDA induces insertions and deletions as well as base substitutions [200][203][204]. Replication of MDA-modified single-stranded M13 genomes in E. coli causes G→T, A→G, and C→ T mutations [199]. Adduction of MDA to deoxyguanosine produces 3-(2′-deoxy-β-δ-erythro-pentofuranosyl)pyrimido[1,2-a]purin-10(3H)-one (M1dG; Fig. 13) [205][206]. Like PdG, discussed above, M1dG is a 1,N2-dG annelation product, but chemically and biologically, it behaves much differently in DNA. Reddy and Marnett [207] incorporated M1dG into an oligodeoxynucleotide. The spectrum of mutations induced by M1dG using M13 vectors replicated in E. coli showed M1dG →A and M1dG →T mutations, and low levels of M1dG →C mutations; however, the mutation frequency was ca. 1%, when cytosine was placed opposite the lesion [208]. M1dG also induced −1 and −2 base deletions when positioned in an iterated 5′-(CpG)4-3′ sequence, but not when positioned in a non-iterated sequence in both E. coli and in COS-7 cells [209].
An explanation as to why M1dG is weakly mutagenic was elucidated when it was discovered that when M1dG is placed opposite dC it rearranges to N2-(3-oxoprop-1- enyl)-dG (OPdG; Scheme 2) [210]. Riggins et al. [211][212] concluded that ring opening of M1dG as a nucleoside or in oligodeoxynucleotides occurs via a reversible second-order reaction with hydroxide, catalyzed by the complementary dC. The closure of the resulting N2-(3-oxo-1-propenyl)-dG anion is pH-dependent and under neutral and acidic conditions ring-closure is biphasic, leading to the rapid formation of intermediates that slowly convert to M1dG in a general-acid-catalyzed reaction, in the presence of dC in the complementary strand. Structural studies in the 5′-(CpG)3-3′ sequence show that the OPdG propenyl chain is located in the minor groove, facilitating Watson–Crick H-bonding with dC [213]. Structural studies with OPdG adduct located in the 5′-TXT-3′ sequence lead to a similar conclusion [214].
Scheme 2.

Reorganization of M1dG and γ-HO-PdG to the Ring-Opened Forms in DNA
Structural studies have also been conducted when M1dG and OPdG adducts are placed opposite a 2-base deletion (2BD) complementary strand in the 5′-(CpG)3 sequence [215–217]. M1dG is stable in the 2BD duplex and remains ring-closed when it is the 5′-nucleotide of a two-nucleotide bulge [215]. Both bulged nucleotides are in the anti conformation about the glycosyl bond and appear inside the helix, but lack H-bonding interactions [216]. On the other hand, the OPdG-2BD duplex undergoes bulge migration from the 3′-neighbor base pairs to the adduct region [217]. The bulge migration transiently positions OPdG opposite dC in the complementary strand and, consequently, hinders the conversion of OPdG to M1dG. Thus, in contrast to the rapid OPdG→M1dG conversion during denaturation [210], the ring-closure of OPdG in this 2-base deletion 5′-(GpC)3 sequence requires 140 days at room temperature. When the M1dG-containing primer-template was crystallized with the polymerase Dpo4 and dNTP, M1dG maintained the ring-closed form. It intercalated into the duplex and displaced the complementary dC to the minor groove [218].
Enal-Derived Adducts
Acrolein is mutagenic in bacterial and mammalian cells [204][219–221], and is carcinogenic in rats [222]. The binding pattern of acrolein-DNA adducts is similar to the p53 mutational pattern in human lung cancer, implicating acrolein as a major cigarette-related lung cancer-inducing agent [223]. Acrolein causes tandem G→T transversions in the supF gene on the shuttle vector plasmid pMY189. Crotonaldehyde is genotoxic and mutagenic in human lymphoblasts [224] and induces liver tumors in rodents [225]. 4-Hydroxynonenal (HNE) is produced from the metabolism of membrane lipids [226] and is a major in vivo peroxidation product of ω–6 polyunsaturated fatty acids [197][227]. HNE induces a DNA damage response in Salmonella typhimurium [228][229]. It also causes mutations in V79 CHO cells, and DNAs from liver specimens from individuals suffering from Wilson’s disease and hemochromatosis contain mutations attributed to HNE-dG adducts [230]. Acrolein and related α,β-unsaturated aldehydes undergo Michael addition to dG, also yielding 1,N2-dG ring-annelation products. Like the M1dG lesion, these also undergo further downstream chemistry in DNA, in a sequence-dependent manner. Enal-derived adducts have been detected in human and rodent DNA [231–235]. Harris, Rizzo, and co-workers site-specifically incorporated acrolein, crotonaldehyde, and HNE derived γ-OH-PdG adducts into oligodeoxynucleotides [236–238].
i) Acrolein
Acrolein reacts with dG to produce γ-OH-PdG and α-OH-PdG (Fig. 13). Correct replication across γ-OH-PdG is efficient in both mammalian and bacterial cells [239–241]. In DNA, γ-OH-PdG undergoes ring opening when placed opposite dC, forming N2-(3-oxopropyl)-dG (Scheme 2) [242]. This facilitates Watson–Crick H-bonding with the complementary dC and could explain the weak mutagenicity of γ-OH-PdG. Nair et al. [243] showed that yeast Rev1 DNA polymerase incorporates the correct nucleotide dC opposite PdG, a model for γ-OH-PdG, with nearly the same efficiency as opposite an undamaged dG. But it cannot extend the primer. However, Pol ζ can carry out the subsequent extension reaction. The crystal structure, in complex with Rev1, showed that PdG rotated to the syn conformation about the glycosyl bond, and the incoming dCTP did not pair with PdG, but instead paired with Arg324 from yeast Rev1 polymerase.
Unlike γ-OH-PdG, α-OH-PdG (Fig. 13) blocks DNA replication in human cells, and it codes for dCTP incorporation, with minor G→A and G→T base substitution mutations when bypassed by Y-family polymerases [241]. The α-OH-PdG lesion is stable in DNA when placed opposite dC [244]. It adopts the syn-conformation around the glycosyl bond, forming a Hoogsteen-like pair to its complementary cytosine (Fig. 20). In vitro replication using Y-family DNA polymerases showed that Pol η and Pol κ catalyze mutagenic replication across α-OH-PdG, while Rev1 and Pol τ mediate accurate replication, with the later incorporating dATP and dTTP at low frequencies [245]. As for γ-OH-PdG, the ternary complex containing Rev1, PdG-modified DNA, and dCTP showed that PdG rotates to the syn conformation about the glycosyl bond, which positions PdG into a small hydrophobic cavity, while the incoming dCTP interacts with an Arg residue by forming two H-bonds [243]. Thus, the structure of α-OH-PdG reported by Zaliznyak et al. [244] identified the bonds that would keep the α-OH-PdG in the syn-conformation at the replication fork of Pol τ. This supports the formation of a non-mutagenic α-OH-PdG (syn)·C (anti) replication intermediate.
Fig. 20. Structures of diastereoisomeric α-OH-PdG adducts (X6; green) in the 5′-C5X6C7-3′ sequence.
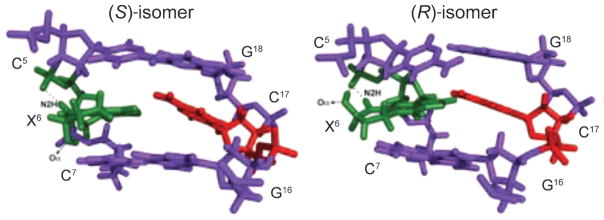
The α-OH-PdG adducts are in the syn conformation with the propano ring facing the major groove. The dotted lines represent H-bonds that are proposed to stabilize the syn conformations of the (R)- and (S)-diastereoisomers of the α-OH-PdG adduct. These illustrations were prepared from PDB entries 2KD9 and 2KDA using PyMol [39] software.
ii) Crotonaldehyde
Similar methodology has been used to study the crotonaldehyde-derived γ-OH-PdG adduct, in which the α-CH3 group creates a new stereogenic center at C(6) [246]. Four stereoisomers are possible for the crotonaldehyde-derived γ-OH-PdG adducts; those with trans-configuration of γ-OH and α-CH3 predominate. In single-strand DNA, the major and minor epimers at C(8) interconvert. The ring-opened intermediate is undetectable in single-strand DNA. The diastereoisomeric adducts have been placed opposite dC and dT in DNA. The crotonaldehyde-derived γ-OH-PdG adducts exhibit higher stability than does the acrolein-derived adduct. When placed opposite dT, the ring-opened species is undetectable. On the other hand, both (R)- and (S)-α-CH3-γ-OH-PdG adducts undergo ring-opening to the N2-dG aldehydes and corresponding N2-dG aldehydrols when placed opposite dC (Scheme 2). However, the ring-opening is incomplete. Higher pH and temperatures favor the N2-dG aldehyde adducts. The structure of the (S)-α-CH3-γ-OH-PdG adduct shows the ring-opened (S)-α-CH3-N2-dG aldehyde adduct forms Watson–Crick pairing with the complementary dC, leaving the aldehyde moiety within the minor groove. The aldehyde of the N2-dG aldehyde adduct orients in the 3′-direction, while the (S)-α-CH3 group orients in the 5′-direction [247].
iii) 4-Hydroxynonenal
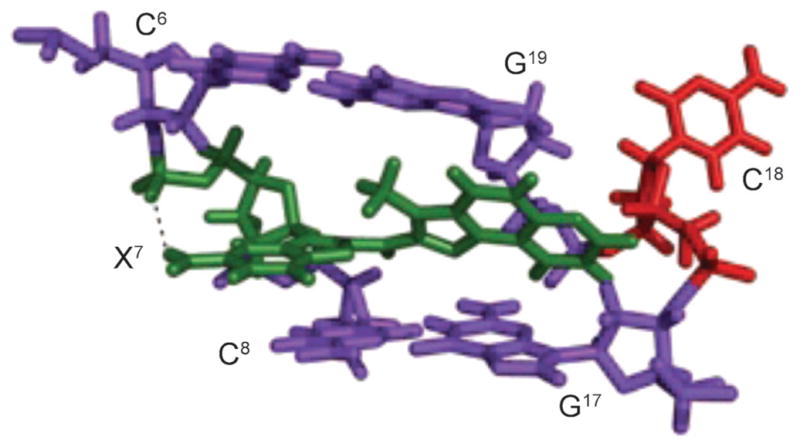
HNE reacts with the N2-amino group of dG to give four diasteromeric 1,N2-dG adducts (Fig. 21) [248–250], and all of them are detected in cellular DNA [231][251–256]. HNE is mutagenic [227] and carcinogenic in rodent cells [257]. HNE induces primarily G→T transversions, accompanied by lower levels of G→A transitions, in the supF gene of shuttle vector pSP189 replicated in human cells [258]. The site-specific mutagenesis studies showed that the (6S,8R,11S)- and (6R,8S,11R)-1,N2-HNE-dG adducts are mutagenic, inducing low levels of G→T transversions and G→T transitions. The initial studies revealed that, when 1,N2-HNE-dG adducts are placed opposite dC in duplex DNA, the exocyclic ring opens, leaving intact Watson–Crick base pairing for the coding face of the adducted dG (Scheme 2) [259]. This accounts for the low levels of mutations associated with these adducts. When mismatched with dA in DNA, (6S,8R,11S)-1,N2-HNE-dG maintains its exocyclic structure [260]. This duplex mimics the situation following incorrect incorporation of dATP opposite the (6S,8R,11S)-1,N2-HNE-dG adduct (G→T transversion). The adduct undergoes a conformational equilibrium between the syn- and anti-conformations about the glycosyl bond. At basic pH, the equilibrium shifts toward the anti-conformation where 1,N2-HNE-dG intercalates and displaces the complementary dA in the 5′-direction (Fig. 22). The HNE aliphatic chain is oriented toward the minor groove of the DNA. At acidic pH, the equilibrium shifts toward the syn-conformation in which the HNE moiety is located in the major groove (Fig. 22). The complementary adenine is protonated, and the (6S,8R,11S)-1,N2-HNE-dG (syn)·dA+ (anti) base pair is stabilized by Hoogsteen type H-bonding. Thus, at neutral pH, both the syn- and anti-conformations are present. Xing et al. [248] attributed the low levels of G→T transversions to the reorientation of the (6S,8R,11S)-1,N2-HNE-dG adduct into the syn conformation around the glycosyl bond, which might allow misincorporation of dATP opposite the lesion. The results confirm that such reorientation happens, when (6S,8R,11S)-1,N2-HNE-dG is mismatched with dA in DNA.
Fig. 21.

Four stereoisomers of HNE-derived exocyclic 1,N2-dG adducts
Fig. 22. Structures of the 1,N2-dG (6S,8R,11S)-HNE-adduct (X7; green) opposite dA (red), in the 5′-C6X7A8-3′ sequence context.
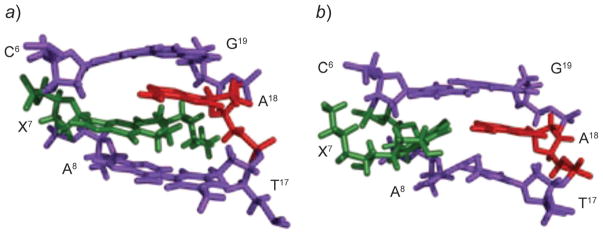
a) At basic pH, X7 adopts the anti-conformation and intercalates. The complementary dA is displaced in the 5′-direction, and the HNE moiety is oriented toward the minor groove. b) At acidic pH, X7 adopts the syn-conformation allowing formation of H-bonds with protonated adenine, and the HNE moiety is placed into the major groove. These illustrations were prepared from PDB entries 2KAS and 2KAR using PyMol [39] software.
Abasic Sites
Hydrolytic cleavage of the nucleobase from the 2′-deoxyribose creates the abasic site, or apurinic and apyrimidinic sites (AP; Scheme 3). There probably is a constant level of ca. 10,000 abasic sites in typical human cells [261]. Most AP sites result from spontaneous depurination [261], but deamination of cytosine to uracil, which is then eliminated by uracil glycosylases, also occurs [262]. The abasic site is also an intermediate in the base excision repair process [263]. AP Sites are mutagenic [264] and cause mis-incorporation in bacterial and mammalian cells [265–271]. DNA Polymerases preferentially incorporate dATP opposite AP sites (sometimes referred to as the 3A-rule3) [272–274]. During translesion DNA synthesis by Y-family DNA polymerases, AP sites also cause frameshifts [275–278].
Scheme 3.
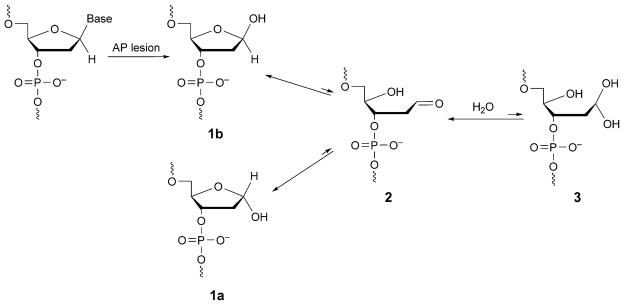
Epimerization of Abasic Sites
The AP site exists in three configurations, the cyclic hemiacetal 1 (a and b), the aldehyde 2, and the hydrated aldehyde 3 (Scheme 3). The equilibria of these species in different sequences, when AP sites are placed opposite all four 2′-deoxynucleotides [279][280], show that cyclic hemiacetal 1 predominates and constitutes over 99% of the population. Less than 1% of aldehyde 2 is also observed, whereas 3 is undetectable.
The equilibrium of AP species leads to the anomeric configuration interconversion of the deoxyribose. Stereoisomers 1a and 1b exist in equilibrium, when the AP site is placed opposite dA, dC, dG, or dT [279]. Beger and Bolton [281] reported that the β-anomer exists predominantly, when the AP site is placed opposite dC in the 5′-AXA-3′ sequence (X =AP site), whereas both α- and β-anomers exist, when AP is placed opposite dA. They proposed that a H2O-bridged H-bond with the complementary dC might contribute to the stereoselectivity. Based on the structure of the human ApeI endonuclease/THF-containing duplex, from which it was concluded that the β-anomer could not be accommodated in the active site of the enzyme, Mol et al. [282] proposed that the endonuclease only incises the α-anomer. However, other binding studies of AP surrogates with this enzyme show no differences for the two anomers and lead to the opposite conclusion that the configuration at C(1′) of deoxyribose is not important for enzymatic recognition [283].
AP Sites decrease the stability of DNA, depending especially on the identities of the flanking base pairs but only mildly on the orphan base [284][285]. The structures of α- and β-anomer of the AP site have been compared. Structural and dynamic studies have been performed, when AP or AP surrogates are placed opposite dA, dC, dG, or dT in different sequences [286]. The conformation around the AP site is more perturbed, when the base opposite to the AP is a pyrimidine than a purine [287][288]. The deoxyribose position depends on the type of orphan base, the configuration at C(1′) of the deoxyribose, and the flanking base pairs. Goljer et al. [289] reported that, when dA is the orphan base, the α-anomer of the natural AP site adopts an extrahelical conformation, whereas the β-anomer adopts an intrahelical conformation. Further studies by the same group, however, showed that both AP anomers stack interhelically in different sequences [281]. When AP is placed opposite dA, a H2O-mediated H-bond has been proposed [281]. This forms between the orphan dA and the hemiacetal OH group of the β-anomer, and maintains the deoxyribose moiety inside the helix. Formation of a H2O bridge is impossible for the α-anomer, which then adopts an extruded conformation [281]. When the AP site was placed opposite dC, the α-anomer was not present. Rather, two conformations of the β-anomer were observed, depending on the H2O-mediated H-bond between the OH group and the orphan cytosine N(3) or C(2)=O. Both H-bonds keep the β-anomer inside the helix [281]. Recent structural and dynamic studies with AP and AP surrogates confirm that both AP anomers are inside the helix [283][287][288]. However, the OH group of both α-AP and β-AP is more likely to form H-bonds directly with opposite dA, dC, and dT [287][288]. The biological effects of the anomers of the AP lesion remain unclear.
Thymine Glycol
The oxidation of thymine and 5-methylcytosine produces 5,6-dihydro-5,6-dihydroxy-2′-thymine, thymine glycol (Tg) [290][291]. Tg has been detected in animal and human urine; human cells probably repair hundreds of these lesions per day [292][293]. Tg blocks DNA replication [294–296] and induces base-substitution mutations [297]. It inhibits DNA synthesis by many prokaryotic and eukaryotic DNA polymerases one nucleotide before and opposite the lesion site. Several DNA polymerases lacking 3′,5′ exonuclease activity can bypass the Tg lesion, albeit slowly [298–300].
Tg exists in DNA as two diastereoisomeric pairs of epimers, the (5R)-cis,trans-pair (5R,6S);(5R,6R) and the (5S)-cis,trans-pair (5S,6R);(5S,6S) (Scheme 4) [301–303]. The (5R) pair is more abundant and more stable [302]. For both the (5R)- and (5S)-pairs, the cis-epimers predominate at the nucleoside level [302]. The biological responses to Tg adducts are modulated by configuration. For example, the Y-family polymerase Pol η bypasses the (5R)-epimers more efficiently [304], whereas Pol κ bypasses the (5S)-epimers more efficiently [305]. The human hNTH1 glycosylase shows a greater preference for excising the (5R)-epimers [306], whereas the hNEIL1 glycosylase shows a greater preference for excising the (5R)-epimers [307][308]. Similar observations have been made for prokaryotic, yeast, and murine glycosylases [309]. The base excision repair of Tg lesion by DNA N-glycosylases/AP lyases is modulated by the cis/trans-epimerization of these two sets of diastereoisomers [310].
Scheme 4.
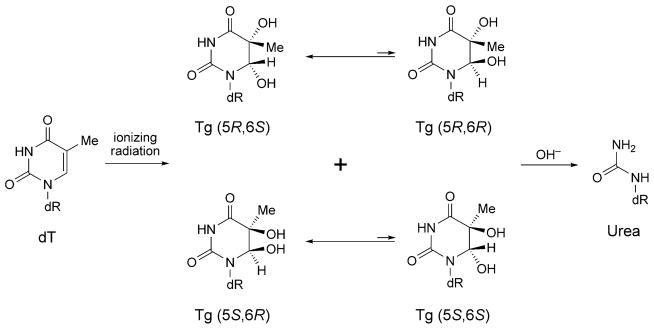
Production of Thymine Glycol (Tg) and Urea Adducts by Exposure of Thymine to Ionizing Radiation
The (5R) epimers have been studied, when Tg is placed opposite dA or dG in the 5′-GXG-3′ (X=Tg) sequence [311]. For the duplex containing the X·A pair, the ratio cis-(5R,6S)/trans-(5R,6R) is 7 :3 at 30°. In contrast, for the duplex containing the X·G pair, the cis-(5R,6S)-epimer predominates; the trans-(5R,6R) epimer is undetectable. The introduction of (5R)-Tg in the 5′-AXA-3′ and 5′-GXC-3′ sequence contexts when paired opposite dA induces localized structural perturbations with the loss of H-bonding at the lesion sites [312][313]. Tg is displaced toward the major groove, increasing its exposure to the solvent. In contrast, when paired opposite dA in the 5′-GXG-3′ sequence, the cis-(5R,6S)-Tg lesion only minimally distorts the helical backbone [314]. Both Tg and the complementary dA insert into the helix and remain in a Watson–Crick alignment (Fig. 23,a and b). However, stacking between Tg and the 3′-neighbor G·C base pair is disrupted. Two conformations are obtained for the cis-(5R,6S)-Tg, depending on the axial or equatorial conformations of the Me group. The NMR-based MD simulations predict that the axial conformation of the cis-(5R,6S)-Tg is favored. An intrastrand H-bond observed between the Tg C(6)–OH and the N(7) position of a 3′-purine may account for the structural differences the cis-(5R,6S)-Tg in the 5′-GXG-3′ and 5′-GXC-3′ sequence contexts. Consistent with the structure in the 5′-GXG-3′ sequence, when (5R)-Tg-containing binary primer-template complex is co-crystallized with the replicative RB69 DNA polymerase, the cis-(5R,6S)-Tg epimer is intrahelical and forms a Watson–Crick base pair with the dA at the primer 3′-terminus. The Tg Me group is in the axial conformation, hindering stacking of the adjacent 5′-template guanine. These results provide a rationale for the observation that extension past the (5R)-Tg lesion by the Klenow fragment of E. coli DNA polymerase I or T4 DNA polymerase is prohibited.
Fig. 23. Structures of Tg (green)-containing duplexes.
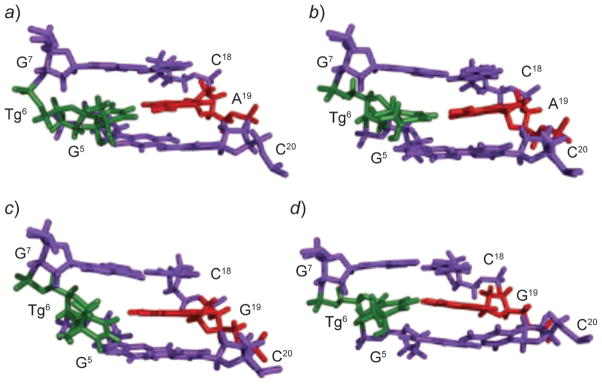
a) The axial conformation of the Tg moiety when placed opposite dA. b) The equatorial conformation of the Tg moiety when placed opposite dA. c) The axial conformation of the Tg moiety when placed opposite dG. d) The equatorial conformation of the Tg moiety when placed opposite dG. These illustrations were prepared from PDB entries 2KH5, 2KH6, 2KH7, and 2KH8 using PyMol [39] software.
The base excision repair of Tg lesions also depends upon the identity of the opposing base. The hNth glycosylase repairs the cis-(5R,6S)-Tg more efficiently when it is placed opposite adenine than when placed opposite guanine [310]. Thus, the solution structure of cis-(5R,6S)-Tg-containing oligodeoxynucleotide has also been refined in the 5′-GXG-3′ sequence when mismatched with dG (Fig. 23,c and d) [315]. Both Tg and the opposite dG remain stacked in the helix. The Tg·G base pair is in the wobble orientation. The Tg shifts toward the major groove and stacks below the 5′-neighbor base, its complement dG also stacks below the 5′-neighbor. Stacking between the Tg and the 3′-neighbor base pair is disrupted. In contrast to when placed opposite dA, the cis-(5R,6S)-Tg does not form strong intrastrand H-bonds with the imidazole N(7)-atom of the 3′-neighbor purine.
Alkaline hydrolysis of Tg produces urea adduct (Scheme 4), which exists as the minor species of thymine oxidation by exposure to the ionizing radiation [290][316]. Urea adducts constitute replicative blocks for DNA polymerases and subsequently inhibit chain elongation [295][317]. It also preferentially promotes misincoporation of dGTP [318]. To explain the biological effects, a structure of the nucleotide containing urea adduct has been studied, when dT or dG is placed opposite [319]. Whereas the conformation is undeterminable due to the broad NMR resonances when placed opposite dG, the urea deoxyribose exists as two conformations when placed opposite dT, depending on the cis/trans-orientation of the urea unit with respect to the deoxyribose. The trans-anti-isomer, which exists as the major species, is expected to be placed intrahelically and form H-bonds with opposite thymine. The cis-isomer is unable to form these H-bonds and exists as the minor species.
The urea adduct has also been placed in a −1 frame shift sequence [320]. Again, at equilibrium, two species are found in slow exchange. The urea adduct is either intra- or extrahelical within the right-handed DNA duplex, determined by the H-bonds formed by the cis/trans-isomers. In the minor intrahelical species, the cis-isomer forms two H-bonds with the bases in the opposite strand, whereas the trans-isomer does not. For the major extrahelical species, the trans-isomer forms two H-bonds, one is intrastrand with the guanine in the modified strand, the other is interstrand with the adenine in the opposite strand, whereas the cis-isomer only forms one H-bond. In the major species, the urea residue lies in the minor groove, and the neighboring bases are stacked over each other in a way similar to a normal B-DNA structure.
Oxidation of 8-Oxo-dG to Guanidinohydantoin and Spiroiminodihyantoin
The fact that initially formed 8-oxo-dG lesions, discussed above, can undergo further oxidation, has become appreciated in recent years. The 8-oxo-dG is more prone to oxidation than is guanine [321], its downstream oxidation products are guanidinohydantoin (Gh), and two stereoisomers of spiroiminodihydantoin ((R)-Sp and (S)-Sp) (Fig. 24) [322–324]. Gh predominates in duplex DNA, while Sp predominates at the nucleoside level [325]. Sp has been detected in DNA from E. coli cells [326]. Gh-Containing DNA is bypassed by E. coli DNA pol I, which inserts dATP or dGTP opposite the lesion [327]. However, Gh and Sp are strong replication blocks to Pol α and human Pol β [327]. Gh is mutagenic with DNA polymerases incorporating purines, which results in G·C→T·A or G·C→C·G transversions [328][329]. A structure of the replicative RB69 DNA polymerase in complex with DNA containing Gh revealed that Gh is extrahelical and rotated toward the major groove [330]. In contrast to 8-oxoG, Gh in this structure was in a high syn-conformation and presented the same H-bond donor and acceptor pattern as thymine, which may explain why polymerases incorporate a purine opposite Gh during error-prone bypass [330].
Fig. 24.

Chemical structures of DNA adducts derived from oxidation of 8-oxo-dG
Aflatoxin B1
Aflatoxin B1 (AFB) is a mycotoxin that contaminates agricultural products [331]. It is a mutagen in bacteria [332–334], a carcinogen in fish [335][336], and a carcinogen in rodents [337][338]. AFB is mutagenic, with G→T transversions being observed in a variety of prokaryotic and eukaryotic systems [334][339][340]. Aflatoxin exposures are implicated in mutations to the p53 tumor suppressor gene [341–344], and the mycotoxin is implicated in the etiology of human liver cancer [345–348].
The reaction of N7-dG at C(8) of the electrophilic AFB-exo-8,9-epoxide yields the N7-dG cationic adduct (Scheme 5) [349–352]. This adduct yields the characteristic G→T mutations but is only moderately mutagenic in E. coli [353]. The N7-dG cationic adduct is chemically unstable [354][355]. At physiological pH, depurination occurs, to release AFB-guanine and yield an apurinic site [355]. Of greater concern is the potential for base-catalyzed cleavage of the imidazole ring to yield the formamidopyr-imidine (FAPY) derivative. Two equilibrating AFB-FAPY species in duplex DNA exhibit different biological consequences in DNA replication: one of the two species is potently mutagenic, yielding G→T mutations, while the other blocks replication [356]. These have been assigned to the α- and β-anomers of the FAPY adduct [357][358]. In duplex DNA, the β-anomer predominates [358]. In single-strand DNA, however, a 2 :1 mixture of α- and β-anomers is observed [356].
Scheme 5.

Formation of Aflatoxin B1 DNA Adducts
Both the initially formed N7-dG cationic adduct and the FAPY rearrangement products have been examined as to structure in DNA. In all instances, the AFB moiety intercalates above the 5′-face of the damaged guanine [358–362]. When dC is placed opposite the N7-dG cationic adduct, Watson–Crick base pairing is maintained for the AFB-N7-dG·dC base pair. With regard to the highly mutagenic β-anomer of the FAPY species, Watson–Crick H-binding is conserved for the FAPY·dC and the flanking base pairs (Fig. 25). In duplex DNA with a 3′-neighboring adenine, both anomers adopt the (E)-conformation of the formyl O-atom, involving a H-bond between the formyl oxygen and the non-Watson–Crick H-bonded N6-amine H-atom of the neighboring adenine [362].
Fig. 25.
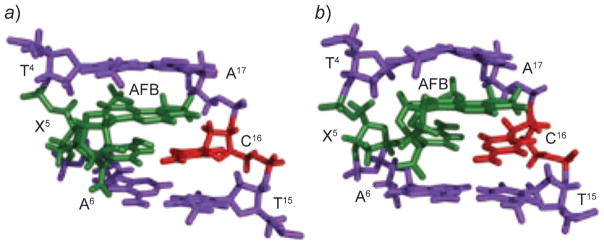
a) Structure of the AFB1-α-FAPY adduct (green) in duplex DNA. b) Structure of the AFB-β-FAPY adduct (green) in duplex DNA. These illustrations were prepared from PDB entries 2KH3 and 1 HM1 using PyMol [39] software.
Significantly, the α-FAPY anomer decreases the stability of the DNA duplex, whereas the β-anomer increases the stability [362]. This suggests that it may be refractory to nucleotide excision repair processes in vivo. Favorable interstrand stacking is considered to be the main factor of the stability of AFB-β-FAPY. In contrast, the α-anomer disrupts the helical structure of DNA [362]. Intercalation of the AFB moiety induces perturbations in the phosphodiester and backbone torsion angles, ε and ζ, respectively, and the deoxyribose shifts to become parallel to the FAPY base and is displaced toward the minor groove. Intrastrand stacking between the AFB moiety and the 5′-neighbor thymine remains, but interstrand stacking is not observed. As a result, compared with the unmodified sequence, the α-FAPY anomer destabilizes the duplex.
Adduct-Induced DNA Cross-Linking
DNA Interstrand cross-links represent one of the most serious types of damage, since replication and transcription require separation of the complementary strands. In eukaryotes, interstrand cross-link repair requires cooperation of multiple proteins that belong to different biological pathways, including NER, homologous recombination, TLS, double-strand break repair, and the Fanconi anemia pathway (reviewed in [363–366]).
The 1,N2-dG enal adducts formed by acrolein, crotonaldehyde, and HNE discussed above are capable of forming interstrand N2-dG:N2-dG cross-links. The formation of the cross-links is sequence dependent. Enals induce interstrand cross-links in the 5′-CpG-3′ sequence, but not in the 5′-GpC-3′ sequence [236–238][367]. The rearrangments of the γ-OH-PdG adducts to the ring-opened N2-dG aldehydes is critical for cross-link formation (Scheme 6). These N2-dG:N2-dG linkages can be chemically reduced [236][237]. This implies that they exist, in part, as imines. When DNA containing the cross-links is enzymatically digested to nucleotides, the cross-links are isolated as pyrimido-purinones [236–238]. However, in duplex DNA, the N2-dG:N2-dG linkages exist predominantly as carbinolamines [246][368][369]. The carbinol-amine linkage maintains Watson–Cricking base pairing [246][369]. The HNE-induced cross-link exhibits extraordinary chemical stability. Whereas less than 50% of acrolein and crotonaldehyde derived γ-HOPdG adducts are converted the cross-links [236][237], the HNE-derived (6S,8R,11S) γ-OH-PdG adduct is fully converted [238].
Scheme 6.

Formation of the N2-dG:N2-dG Interstrand Cross-Link When Enal-Derived γ-HO-PdG Adduct Is Placed Opposite dC in DNA
Formation of N2-dG:N2-dG cross-links is stereoselective. The (R)-carbinolamine linkage constitutes 80–90% of the cross-link induced by the γ-OH-PdG adduct [370]. Its structure has been refined in the 5′-CpG-3′ sequence (Fig. 26) [370]. The carbinolamine linkage is located in the minor groove, maintaining Watson–Crick H-bonding of the cross-linked base pairs. The anti-conformation of the carbinol OH group with respect to Cα of the tether minimized steric interactions, and allowed the formation of a H-bond between the carbinol OH group and the cytosine C(2)=O located in the 5′-neighboring G·C base pair. This H-bond might explain the stability of this cross-link, and the stereochemical preference for the (R)-configuration of the cross-link.
Fig. 26. Structure of the (R)-γ-hydroxytrimethylene-N2-dG:N2-dG interstrand cross-link (green) in a three-base-pair duplex suggests the presence of a H-bond between the OH group and 5′-neighbor C20, which may account for the stability of the interstrand cross-link.

This illustration was prepared from the PDB file used in the article [370] using PyMol software [39].
The preferential formation of the cross-link in the 5′-CpG-3′ sequence as compared to the 5′-GpC-3′ sequence is attributed to the longer distance between the two N2-dG atoms in the 3′-direction than in the 5′-direction, accompanied by greater destabilization of the DNA duplex by the cross-link in the 5′-GpC-3′ sequence [371][372]. Structures of the fully reduced trimethylene cross-links have been refined in both 5′-CpG-3′ and 5′-GpC-3′ sequences [373]. Whereas perturbation caused by the cross-link in the 5′-CpG-3′ sequence is minimal, the perturbation in the 5′-GpC-3′ sequence is significant (Fig. 27). Differential stacking of the base pairs at the cross-linking region explains the difference in stabilities of the trimethylene cross-links in the 5′-CpG-3′ and 5′-GpC-3′ sequence contexts, and, in turn, account for the sequence selectivity of the interstrand cross-link formation induced by the enal-derived 1,N2-dG adducts.
Fig. 27. Differential base stacking at the cross-linking region explains the difference in stabilities of the trimethylene-N2-dG:N2-dG interstrand cross-links in the 5′-CpG-3′ vs. the 5′-GpC-3′ sequences, and, in turn, accounts for the same sequence selectivity of interstrand cross-link formation induced by the enal-derived 1,N2-dG adducts.

a) and b) show base stacking in the 5′-CpG-3′ sequence; c) and d) show base stacking in the 5′-GpC-3′ sequence. This illustration was prepared from PDB entries 2KNK and 2KNL using PyMol software [39] (© 2009 American Chemical Society).
Configuration of the γ-OH-PdG adducts also modulates interstrand cross-link formation. The crotonaldehyde derived (R)-CH3-γ-OH-PdG adduct induces N2-dG:N2-dG interstrand cross-links more efficiently than the (S)-CH3-γ-OH-PdG adduct does [237]. The (S)-CH3-γ-OH-PdG places the aldehyde toward the 3′-neighbor A·T base pair in the 5′-CpG-3′ sequence [247]. Conformational adjustment is required to reorient the aldehyde group to the 5′-neighbor C·G base pair, and consequently results in the slow production of the cross-link. Model studies showed that re-orientation of the aldehyde in the 5′-direction led to the interference of the (S)-CH3 group with the 3′-neighbor A·T base pair and decreased the stability of the DNA duplex. The structures of stereospecific α-CH3-trimethylene cross-links, which are used as surrogates for the crotonaldehyde-derived carbinolamine cross-links, supported this conclusion (Fig. 28) [374]. The (S)-isomer of the α-CH3-trimethylene cross-link exhibits lower thermal stability than the (R)-isomer does. Both isomers of the cross-links are located in the minor groove and retain Watson–Crick H-bonds at the tandem cross-linked C·G base pairs. However, the α-CH3 group of the (R)-isomer is positioned in the center of the minor groove, whereas the α-Me group of the (S)-isomer is positioned in the 3′-direction, showing steric interference with the DNA helix.
Fig. 28.
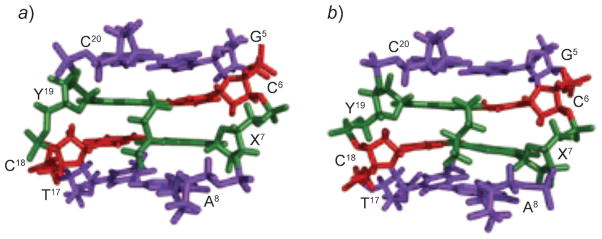
Structures of the (R)- and (S)-γ-CH3-trimethylene-N2-dG:N2-dG interstrand cross-links (green) viewed from the minor groove showing different orientations of the CH3 group. a) (R)-Configuration. b) (S)-Configuration. These illustrations were prepared from PDB entries 2 HMD and 2 HMR using PyMol [39] software.
Of the four stereoisomers of the HNE-derived γ-OH-PdG adducts, only the (6S,8R,11S)-configurated one induces interchain cross-links [238]. All HNE-derived γ-OH-PdG adducts undergo ring-opening to N2-dG aldehydes and derivatives when placed opposite dC [375]. The existence of small amounts of N2-dG aldehyde adducts, which have been detected for the (6S,8R,11S)- and (6R,8S,11R)-stereoisomers [376], accounts for the slow formation of the cross-link. The structures of ring-opened N2-dG cyclic hemiacetals of (6S,8R,11S)- and (6R,8S,11R)-configurations show the HNE moiety is located in the minor groove with the directions of aldehyde group differing for the two stereoisomers (Fig. 29). The (6S,8R,11S)-aldehyde orients to the 5′-neighbor C·G base pair and favors cross-link formation. In contrast, the (6R,8S,11R)-aldehyde orients to the 3′-neighbor A·T base pair. Re-orientation of the aldehyde unit in the 5′-direction to favor the interstrand cross-link formation is disfavored due to the interference of the HNE moiety with the 3′-neighbor [377].
Fig. 29. Structures of the ring-opened 1,N2-dG (6R,8S,11R)- and (6S,8R,11S)-HNE adducts in a three-base-pair duplex.
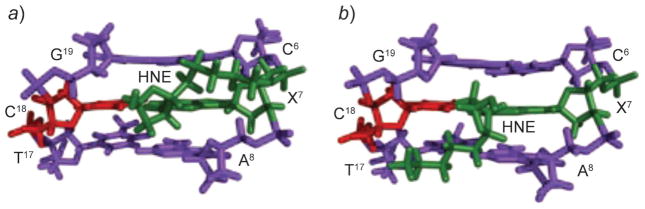
Differential orientations of the HNE moiety in the ring-opened species determine the capabilities to induce N2-dG :N2-dG interstrand cross-links (green) by diastereoisomeric 1,N2-dG HNE adducts. a) (6R,8S,11R)-Configuration. b) (6S,8R,11S)-Configuration. These illustrations were prepared from PDB entries 2K8T and 2K8U using PyMol [39] software.
Butadiene-Mediated Cross-Linking
The bis-electrophile 1,2,3,4-diepoxybutane (DEB) is considered to be the ultimate carcinogenic metabolite of buta-1,3-diene. Initial DNA alkylation by DEB produces 2-hydroxy-3,4-epoxybut-1-yl adducts. These can undergo further reaction to form DNA cross-links. Zhang and Elfarra demonstrated that the reaction of DEB with dG produced nucleoside adducts resulting from alkylation at N(1) and N(7) of dG, 2′-deoxy-1-(2-hydroxyoxiran-2-ylethyl)guanosine and 2′-deoxy-7-(2-hydroxyoxiran-2-ylethyl)guanosine [378]. Incubation of the N1 adducts with dG led to formation of diastereoisomers of 1-[4-(2-amino-1,7-dihydro-6-oxo-6H-purin-7-yl)-2,3-dihydroxybutyl]-2′-deoxyguanosine (N7-dG-N1-dG-BD). Incubation of the N7-dG adducts with dG led to formation of the bis-dG cross-link 7,7′-(2,3-dihydroxybutane-1,4-diyl)bis[2-amino-1,7-dihydro-6H-purin-6-one]. The sequence context of these cross-links is of particular interest, and not well understood. These cross-links predominate within 5′-GNC-3′/3′-CNG-5′ sequences, where N is any nucleotide [379]. The efficiencies of cross-linking are dependent upon configuration, with (S,S)- >(R,R)- >meso-DEB [380]. Other DEB-mediated cross-links have been reported. Tretyakova and co-workers reported regioisomeric dG-dA cross-linking products involving the N7-dG N-atom, and the N1-, N3-, N6-, and N7-dA N-atoms [381]. The 1-(hypoxanthin-1-yl)-4-(guanin-7-yl)butane-2,3-diol (N1 HX-N7-dG-BD) cross-link has also been identified [382]. It was proposed that the latter was formed by the hydrolytic deamination of 1-(adenin-1-yl)-4-(guanin-7-yl)butane-2,3-diol. Alternatively, DEB can form exocyclic lesions by alkylating two sites of the same DNA base. Zhang and Elfarra have identified bis-alkylation products of dG [378][383][384]. Tretyakova and co-workers identified bis-alkylation products of dA [385][386]. They proposed that DEB alkylates the N(1)-position of dA to form N1-(2-hydroxy-3,4-epoxybut-1-yl)-dA adducts, which undergo an SN2-type intramolecular nucleophilic substitution and rearrangement to give 1,N6-[2-hydroxy-3-(hydroxymethyl)propane-1,3-diyl]-2′-dA and 1,N6-[2-hydroxy-1-(hydroxymethyl)propane-1,3-diyl]-2′-dA. Both annelation products were identified in DNA treated with DEB in vitro and in liver DNA of mice exposed to BD by inhalation. Their formation provides a possible mechanism of mutagenesis at A:T base pairs. The initially formed 2-hydroxy-3,4-epoxybut-1-yl adducts may also interact with nucleophilic side chains within DNA-binding proteins to form DNA–protein conjugates, e.g., with DNA repair proteins [387]. Structural studies of these adducts have not been reported but will be of considerable interest. However, the synthetically accessible and stable N6,N6-dA intrastrand cross-links have been used as model systems to probe the structures of DEB-induced cross-links in the major groove of DNA, and their consequent biological processing. The structures reveal that the major conformational difference between the (R,R)- and (S,S)-BD cross-links (Fig. 19) regards the conformation of the C4 butadiene chain (27,28). The (R,R)-BD cross-link exists in the extended chain conformation with minimal perturbation of the DNA (27), while the (S,S)-BD cross-link creates a greater structural perturbation (28). Although both (R,R)- and (S,S)-BD cross-links were mutagenic in both E. coli and COS-7 cells, the (S,S)-BD cross-link exhibited a lower overall mutagenic frequency (20%) than that of the (R,R)-BD cross-link (54%) (29).
Summary
Early on, it was recognized that the alkylation of DNA, particularly when forming sterically bulky lesions, can be accommodated by conformational rearrangements at the damage sites, which do not involve bond breaking. Numerous examples now exist, particularly involving conformational rearrangement about the glycosyl torsion angle. These conformational interconversions have sufficiently large activation barriers that, under physiologically relevant conditions, interconversion occurs slowly at ms or slower time scales, and it is anticipated that different conformers may elicit differential biological responses. It has also been increasingly recognized that DNA damage may result in configurational rearrangements involving bond breakage; such rearrangements may be either reversible or irreversible and may alter the biological response to the damage. Finally, the ability of bis-electrophiles, such as α,β-unsaturated aldehydes or diepoxybutane, to cross-link DNA has been of considerable interest. Such cross-links, if not repaired, are anticipated to be highly genotoxic, interfering both with replication and repair processes.
Acknowledgments
We acknowledge research grant support from the NIH, R01 CA-55678, R01 ES-05509, and P01 ES-05355. The Vanderbilt University Center in Molecular Toxicology is funded by NIH grant P30 ES-00267. The Vanderbilt-Ingram Cancer Center is funded by NIH grant P30 CA-68485. Vanderbilt University and NIH grant RR-05805 provided additional funding for NMR instrumentation.
References
- 1.Delaney JC, Essigmann JM. Chem Res Toxicol. 2008;21:232. doi: 10.1021/tx700292a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caruthers MH, Barone AD, Beaucage SL, Dodds DR, Fisher EF, McBride LJ, Matteucci M, Stabinsky Z, Tang JY. Methods Enzymol. 1987;154:287. doi: 10.1016/0076-6879(87)54081-2. [DOI] [PubMed] [Google Scholar]
- 3.Feigon J, Sklenar V, Wang E, Gilbert DE, Macaya RF, Schultze P. Methods Enzymol. 1992;211:235. doi: 10.1016/0076-6879(92)11015-b. [DOI] [PubMed] [Google Scholar]
- 4.Egli M. Curr Opin Chem Biol. 2004;8:580. doi: 10.1016/j.cbpa.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Cho BP. J Environ Sci Health, Part C – Environ Carcinog Ecotoxicol Rev. 2004;22:57. doi: 10.1081/LESC-200038217. [DOI] [PubMed] [Google Scholar]
- 6.Patel DJ, Mao B, Gu Z, Hingerty BE, Gorin A, Basu AK, Broyde S. Chem Res Toxicol. 1998;11:391. doi: 10.1021/tx9702143. [DOI] [PubMed] [Google Scholar]
- 7.Talaska G. J Environ Sci Health, Part C – Environ Carcinog Ecotoxicol Rev. 2003;21:29. doi: 10.1081/GNC-120021372. [DOI] [PubMed] [Google Scholar]
- 8.Beland FA, Kadlubar FF. Metabolic Activation and DNA Adducts of Aromatic Amines and Nitroaromatic Hydrocarbons. Springer-Verlag; Heidelberg: 1990. [Google Scholar]
- 9.Miller JA, Miller EC. Prog Exp Tumor Res. 1969;11:273. doi: 10.1159/000391399. [DOI] [PubMed] [Google Scholar]
- 10.Singer B, Grunberger D. Molecular Biology of Mutagens and Carcinogens. Plenum; New York: 1983. [Google Scholar]
- 11.Gupta RC, Dighe NR. Carcinogenesis. 1984;5:343. doi: 10.1093/carcin/5.3.343. [DOI] [PubMed] [Google Scholar]
- 12.Eckel LM, Krugh TR. Biochemistry. 1994;33:13611. doi: 10.1021/bi00250a012. [DOI] [PubMed] [Google Scholar]
- 13.Mao B, Hingerty BE, Broyde S, Patel DJ. Biochemistry. 1998;37:81. doi: 10.1021/bi972257o. [DOI] [PubMed] [Google Scholar]
- 14.Strauss BS. BioEssays. 1991;13:79. doi: 10.1002/bies.950130206. [DOI] [PubMed] [Google Scholar]
- 15.Norman D, Abuaf P, Hingerty BE, Live D, Grunberger D, Broyde S, Patel DJ. Biochemistry. 1989;28:7462. doi: 10.1021/bi00444a046. [DOI] [PubMed] [Google Scholar]
- 16.Cho BP, Beland FA, Marques MM. Biochemistry. 1994;33:1373. doi: 10.1021/bi00172a013. [DOI] [PubMed] [Google Scholar]
- 17.Eckel LM, Krugh TR. Nat Struct Biol. 1994;1:89. doi: 10.1038/nsb0294-89. [DOI] [PubMed] [Google Scholar]
- 18.Cho BP, Beland FA, Marques MM. Biochemistry. 1992;31:9587. doi: 10.1021/bi00155a011. [DOI] [PubMed] [Google Scholar]
- 19.Lutgerink JT, Retèl J, Westra JG, Welling MC, Loman H, Kriek E. Carcinogenesis. 1985;6:1501. doi: 10.1093/carcin/6.10.1501. [DOI] [PubMed] [Google Scholar]
- 20.Bichara M, Fuchs RPP. J Mol Biol. 1985;183:341. doi: 10.1016/0022-2836(85)90005-1. [DOI] [PubMed] [Google Scholar]
- 21.Mah MC, Maher VM, Thomas H, Reid TM, King CM, McCormick JJ. Carcinogenesis. 1989;10:2321. doi: 10.1093/carcin/10.12.2321. [DOI] [PubMed] [Google Scholar]
- 22.Carothers AM, Urlaub G, Mucha J, Yuan W, Chasin LA, Grunberger D. Carcinogenesis. 1993;14:2181. doi: 10.1093/carcin/14.10.2181. [DOI] [PubMed] [Google Scholar]
- 23.Reid TM, Lee MS, King CM. Biochemistry. 1990;29:6153. doi: 10.1021/bi00478a007. [DOI] [PubMed] [Google Scholar]
- 24.Broschard TH, Koffel-Schwartz N, Fuchs RP. J Mol Biol. 1999;288:191. doi: 10.1006/jmbi.1999.2667. [DOI] [PubMed] [Google Scholar]
- 25.Fuchs RP, Fujii S. DNA Repair (Amst) 2007;6:1032. doi: 10.1016/j.dnarep.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 26.Mao B, Hingerty BE, Broyde S, Patel DJ. Biochemistry. 1998;37:95. doi: 10.1021/bi972258g. [DOI] [PubMed] [Google Scholar]
- 27.Rechkoblit O, Kolbanovskiy A, Malinina L, Geacintov NE, Broyde S, Patel DJ. Nat Struct Mol Biol. 2010;17:379. doi: 10.1038/nsmb.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Layton DW, Bogen KT, Knize MG, Hatch FT, Johnson VM, Felton JS. Carcinogenesis. 1995;16:39. doi: 10.1093/carcin/16.1.39. [DOI] [PubMed] [Google Scholar]
- 29.Wakabayashi K, Nagao M, Esumi H, Sugimura T. Cancer Res. 1992;52:2092s. [PubMed] [Google Scholar]
- 30.Sugimura T. Mutat Res. 1997;376:211. doi: 10.1016/s0027-5107(97)00045-6. [DOI] [PubMed] [Google Scholar]
- 31.Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Cancer Sci. 2004;95:290. doi: 10.1111/j.1349-7006.2004.tb03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammons GJ, Milton D, Stepps K, Guengerich FP, Tukey RH, Kadlubar FF. Carcinogenesis. 1997;18:851. doi: 10.1093/carcin/18.4.851. [DOI] [PubMed] [Google Scholar]
- 33.Yamazoe Y, Shimada M, Kamataki T, Kato R. Cancer Res. 1983;43:5768. [PubMed] [Google Scholar]
- 34.Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, Guengerich FP, Sutter TR. Cancer Res. 1996;56:2979. [PubMed] [Google Scholar]
- 35.Boobis AR, Lynch AM, Murray S, de la Torre R, Solans A, Farre M, Segura J, Gooderham NJ, Davies DS. Cancer Res. 1994;54:89. [PubMed] [Google Scholar]
- 36.Schut HA, Snyderwine EG. Carcinogenesis. 1999;20:353. doi: 10.1093/carcin/20.3.353. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Y, Chladek S, Romano LJ. J Org Chem. 1994;59:556. [Google Scholar]
- 38.Turesky RJ, Rossi SC, Welti DH, Lay JO, Jr, Kadlubar FF. Chem Res Toxicol. 1992;5:479. doi: 10.1021/tx00028a005. [DOI] [PubMed] [Google Scholar]
- 39.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; Palo Alto, CA: 2008. [Google Scholar]
- 40.Wang F, DeMuro NE, Elmquist CE, Stover JS, Rizzo CJ, Stone MP. J Am Chem Soc. 2006;128:10085. doi: 10.1021/ja062004v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown K, Hingerty BE, Guenther EA, Krishnan VV, Broyde S, Turteltaub KW, Cosman M. Proc Natl Acad Sci USA. 2001;98:8507. doi: 10.1073/pnas.151251898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu X, Shapiro R, Broyde S. Chem Res Toxicol. 1999;12:895. doi: 10.1021/tx990108w. [DOI] [PubMed] [Google Scholar]
- 43.Turesky RJ, Box RM, Markovic J, Gremaud E, Snyderwine EG. Mutat Res. 1997;376:235. doi: 10.1016/s0027-5107(97)00048-1. [DOI] [PubMed] [Google Scholar]
- 44.Kosakarn P, Halliday JA, Glickman BW, Josephy PD. Carcinogenesis. 1993;14:511. doi: 10.1093/carcin/14.3.511. [DOI] [PubMed] [Google Scholar]
- 45.Watanabe M, Ohta T. Carcinogenesis. 1993;14:1149. doi: 10.1093/carcin/14.6.1149. [DOI] [PubMed] [Google Scholar]
- 46.Terada M, Nagao M, Nakayasu M, Sakamoto H, Nakasato F, Sugimura T. Environ Health Perspect. 1986;67:117. doi: 10.1289/ehp.8667117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson LH, Tucker JD, Stewart SA, Christensen ML, Salazar EP, Carrano AV, Felton JS. Mutagenesis. 1987;2:483. doi: 10.1093/mutage/2.6.483. [DOI] [PubMed] [Google Scholar]
- 48.Conlay JJ. Nature. 1963;197:555. [Google Scholar]
- 49.Van Hemmen JJ, Bleichrodt JF. Radiat Res. 1971;46:444. [PubMed] [Google Scholar]
- 50.Cho BP, Evans FE. Nucleic Acids Res. 1991;19:1041. doi: 10.1093/nar/19.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Culp SJ, Cho BP, Kadlubar FF, Evans FE. Chem Res Toxicol. 1989;2:416. doi: 10.1021/tx00012a010. [DOI] [PubMed] [Google Scholar]
- 52.Aida M, Nishimura S. Mutat Res. 1987;192:83. doi: 10.1016/0165-7992(87)90101-1. [DOI] [PubMed] [Google Scholar]
- 53.Oda Y, Uesugi S, Ikehara M, Nishimura S, Kawase Y, Ishikawa H, Inoue H, Ohtsuka E. Nucleic Acids Res. 1991;19:1407. doi: 10.1093/nar/19.7.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guy A, Duplaa AM, Harel P, Teoule R. Helv Chim Acta. 1988;71:1566. [Google Scholar]
- 55.Einolf HJ, Guengerich FP. J Biol Chem. 2001;276:3764. doi: 10.1074/jbc.M006696200. [DOI] [PubMed] [Google Scholar]
- 56.Furge LL, Guengerich FP. Biochemistry. 1997;36:6475. doi: 10.1021/bi9627267. [DOI] [PubMed] [Google Scholar]
- 57.Lowe LG, Guengerich FP. Biochemistry. 1996;35:9840. doi: 10.1021/bi960485x. [DOI] [PubMed] [Google Scholar]
- 58.Shibutani S, Takeshita M, Grollman AP. Nature. 1991;349:431. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 59.Tolentino JH, Burke TJ, Mukhopadhyay S, McGregor WG, Basu AK. Nucleic Acids Res. 2008;36:1300. doi: 10.1093/nar/gkm1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eoff RL, Irimia A, Angel KC, Egli M, Guengerich FP. J Biol Chem. 2007;282:19831. doi: 10.1074/jbc.M702290200. [DOI] [PubMed] [Google Scholar]
- 61.Rechkoblit O, Malinina L, Cheng Y, Kuryavyi V, Broyde S, Geacintov NE, Patel DJ. PLoS Biol. 2006;4:e11. doi: 10.1371/journal.pbio.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zang H, Irimia A, Choi JY, Angel KC, Loukachevitch LV, Egli M, Guengerich FP. J Biol Chem. 2006;281:2358. doi: 10.1074/jbc.M510889200. [DOI] [PubMed] [Google Scholar]
- 63.Carlson KD, Washington MT. Mol Cell Biol. 2005;25:2169. doi: 10.1128/MCB.25.6.2169-2176.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haracska L, Yu SL, Johnson RE, Prakash L, Prakash S. Nat Genet. 2000;25:458. doi: 10.1038/78169. [DOI] [PubMed] [Google Scholar]
- 65.Malins DC, Polissar NL, Ostrander GK, Vinson MA. Proc Natl Acad Sci USA. 2000;97:12442. doi: 10.1073/pnas.230438797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuchino Y, Mori F, Kasai H, Inoue H, Iwai S, Miura K, Ohtsuka E, Nishimura S. Nature. 1987;327:77. doi: 10.1038/327077a0. [DOI] [PubMed] [Google Scholar]
- 67.Hunter WN, Brown T, Kennard O. J Biomol Struct Dyn. 1986;4:173. doi: 10.1080/07391102.1986.10506338. [DOI] [PubMed] [Google Scholar]
- 68.Brown T, Hunter WN, Kneale G, Kennard O. Proc Natl Acad Sci USA. 1986;83:2402. doi: 10.1073/pnas.83.8.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leonard GA, Booth ED, Brown T. Nucleic Acids Res. 1990;18:5617. doi: 10.1093/nar/18.19.5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kouchakdjian M, Bodepudi V, Shibutani S, Eisenberg M, Johnson F, Grollman AP, Patel DJ. Biochemistry. 1991;30:1403. doi: 10.1021/bi00219a034. [DOI] [PubMed] [Google Scholar]
- 71.McAuley-Hecht KE, Leonard GA, Gibson NJ, Thomson JB, Watson WP, Hunter WN, Brown T. Biochemistry. 1994;33:10266. doi: 10.1021/bi00200a006. [DOI] [PubMed] [Google Scholar]
- 72.Brown T, Leonard GA, Booth ED, Kneale G. J Mol Biol. 1990;212:437. doi: 10.1016/0022-2836(90)90320-L. [DOI] [PubMed] [Google Scholar]
- 73.Lipscomb LA, Peek ME, Morningstar ML, Verghis SM, Miller EM, Rich A, Essigmann JM, Williams LD. Proc Natl Acad Sci USA. 1995;92:719. doi: 10.1073/pnas.92.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brieba LG, Eichman BF, Kokoska RJ, Doublie S, Kunkel TA, Ellenberger T. EMBO J. 2004;23:3452. doi: 10.1038/sj.emboj.7600354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hsu GW, Ober M, Carell T, Beese LS. Nature. 2004;431:217. doi: 10.1038/nature02908. [DOI] [PubMed] [Google Scholar]
- 76.Rechkoblit O, Malinina L, Cheng Y, Geacintov NE, Broyde S, Patel DJ. Structure. 2009;17:725. doi: 10.1016/j.str.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hazra TK, Izumi T, Maidt L, Floyd RA, Mitra S. Nucleic Acids Res. 1998;26:5116. doi: 10.1093/nar/26.22.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang QM, Ishikawa N, Nakahara T, Yonei S. Nucleic Acids Res. 1998;26:4669. doi: 10.1093/nar/26.20.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thiviyanathan V, Somasunderam A, Hazra TK, Mitra S, Gorenstein DG. J Mol Biol. 2003;325:433. doi: 10.1016/s0022-2836(02)01272-x. [DOI] [PubMed] [Google Scholar]
- 80.Guschlbauer W, Duplaa AM, Guy A, Teoule R, Fazakerley GV. Nucleic Acids Res. 1991;19:1753. doi: 10.1093/nar/19.8.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan X, Grollman AP, Shibutani S. Carcinogenesis. 1999;20:2287. doi: 10.1093/carcin/20.12.2287. [DOI] [PubMed] [Google Scholar]
- 82.Leonard GA, Guy A, Brown T, Téoule R, Hunter WN. Biochemistry. 1992;31:8415. doi: 10.1021/bi00151a004. [DOI] [PubMed] [Google Scholar]
- 83.Shibutani S, Suzuki N, Matsumoto Y, Grollman AP. Biochemistry. 1996;35:14992. doi: 10.1021/bi961446o. [DOI] [PubMed] [Google Scholar]
- 84.Singer B, Spengler SJ. The Role of Cyclic Nucleic Acid Adducts in Carcinogenesis and Mutagenesis. In: Singer B, Bartsch H, editors. IARC Monographs. Vol. 70. International Agency for Research on Cancer; Lyon: 1986. [Google Scholar]
- 85.Simha D, Palejwala VA, Humayun MZ. Biochemistry. 1991;30:8727. doi: 10.1021/bi00100a003. [DOI] [PubMed] [Google Scholar]
- 86.Zhang W, Johnson F, Grollman AP, Shibutani S. Chem Res Toxicol. 1995;8:157. doi: 10.1021/tx00043a021. [DOI] [PubMed] [Google Scholar]
- 87.Korobka A, Cullinan D, Cosman M, Grollman AP, Patel DJ, Eisenberg M, de los Santos C. Biochemistry. 1996;35:13310. doi: 10.1021/bi9605696. [DOI] [PubMed] [Google Scholar]
- 88.Cullinan D, Korobka A, Grollman AP, Patel DJ, Eisenberg M, de los Santos C. Biochemistry. 1996;35:13319. doi: 10.1021/bi9605705. [DOI] [PubMed] [Google Scholar]
- 89.Cullinan D, Johnson F, Grollman AP, Eisenberg M, de los Santos C. Biochemistry. 1997;36:11933. doi: 10.1021/bi9705725. [DOI] [PubMed] [Google Scholar]
- 90.Cullinan D, Johnson F, de los Santos C. J Mol Biol. 2000;296:851. doi: 10.1006/jmbi.1999.3490. [DOI] [PubMed] [Google Scholar]
- 91.Moriya M, Zhang W, Johnson F, Grollman AP. Proc Natl Acad Sci USA. 1994;91:11899. doi: 10.1073/pnas.91.25.11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de los Santos C, Kouchakdjian M, Yarema K, Basu A, Essigmann J, Patel DJ. Biochemistry. 1991;30:1828. doi: 10.1021/bi00221a015. [DOI] [PubMed] [Google Scholar]
- 93.Huffman JL, Sundheim O, Tainer JA. Mutat Res. 2005;577:55. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 94.Saparbaev M, Langouet S, Privezentzev CV, Guengerich FP, Cai H, Elder RH, Laval J. J Biol Chem. 2002;277:26987. doi: 10.1074/jbc.M111100200. [DOI] [PubMed] [Google Scholar]
- 95.Shanmugam G, Kozekov ID, Guengerich FP, Rizzo CJ, Stone MP. Chem Res Toxicol. 2008;21:1795. doi: 10.1021/tx8001466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shanmugam G, Goodenough AK, Kozekov ID, Guengerich FP, Rizzo CJ, Stone MP. Chem Res Toxicol. 2007;20:1601. doi: 10.1021/tx7001788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zaliznyak T, Lukin M, Johnson F, de los Santos C. Biochemistry. 2008;47:4606. doi: 10.1021/bi7022514. [DOI] [PubMed] [Google Scholar]
- 98.Zang H, Goodenough AK, Choi JY, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, Guengerich FP. J Biol Chem. 2005;280:29750. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
- 99.Wang Y, Saleh S, Marnett LJ, Egli M, Stone MP. Biochemistry. 2008;47:7322. doi: 10.1021/bi800152j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Benamira M, Singh U, Marnett LJ. J Biol Chem. 1992;267:22392. [PubMed] [Google Scholar]
- 101.Singh US, Moe JG, Reddy GR, Weisenseel JP, Marnett LJ, Stone MP. Chem Res Toxicol. 1993;6:825. doi: 10.1021/tx00036a012. [DOI] [PubMed] [Google Scholar]
- 102.Weisenseel JP, Reddy GR, Marnett LJ, Stone MP. Chem Res Toxicol. 2002;15:127. doi: 10.1021/tx0101090. [DOI] [PubMed] [Google Scholar]
- 103.Huang P, Patel DJ, Eisenberg M. Biochemistry. 1993;32:3852. doi: 10.1021/bi00066a004. [DOI] [PubMed] [Google Scholar]
- 104.Huang P, Eisenberg M. Biochemistry. 1992;31:6518. doi: 10.1021/bi00143a023. [DOI] [PubMed] [Google Scholar]
- 105.Kouchakdjian M, Eisenberg M, Live D, Marinelli E, Grollman AP, Patel DJ. Biochemistry. 1990;29:4456. doi: 10.1021/bi00470a028. [DOI] [PubMed] [Google Scholar]
- 106.Kouchakdjian M, Marinelli E, Gao X, Johnson F, Grollman A, Patel D. Biochemistry. 1989;28:5647. doi: 10.1021/bi00439a047. [DOI] [PubMed] [Google Scholar]
- 107.Kouchakdjian M, Eisenberg M, Johnson F, Grollman AP, Patel DJ. Biochemistry. 1991;30:3262. doi: 10.1021/bi00227a014. [DOI] [PubMed] [Google Scholar]
- 108.Weisenseel JP, Moe JG, Reddy GR, Marnett LJ, Stone MP. Biochemistry. 1995;34:50. doi: 10.1021/bi00001a007. [DOI] [PubMed] [Google Scholar]
- 109.Moe JG, Reddy GR, Marnett LJ, Stone MP. Chem Res Toxicol. 1994;7:319. doi: 10.1021/tx00039a008. [DOI] [PubMed] [Google Scholar]
- 110.Weisenseel JP, Reddy GR, Marnett LJ, Stone MP. Chem Res Toxicol. 2002;15:140. doi: 10.1021/tx010107f. [DOI] [PubMed] [Google Scholar]
- 111.Zhang F, Chen Y, Pisha E, Shen L, Xiong Y, van Breemen RB, Bolton JL. Chem Res Toxicol. 1999;12:204. doi: 10.1021/tx980217v. [DOI] [PubMed] [Google Scholar]
- 112.Bolton JL, Pisha E, Zhang F, Qiu S. Chem Res Toxicol. 1998;11:1113. doi: 10.1021/tx9801007. [DOI] [PubMed] [Google Scholar]
- 113.Pisha E, Lui X, Constantinou AI, Bolton JL. Chem Res Toxicol. 2001;14:82. doi: 10.1021/tx000168y. [DOI] [PubMed] [Google Scholar]
- 114.Chen Y, Liu X, Pisha E, Constantinou AI, Hua Y, Shen L, van Breemen RB, Elguindi EC, Blond SY, Zhang F, Bolton JL. Chem Res Toxicol. 2000;13:342. doi: 10.1021/tx990186j. [DOI] [PubMed] [Google Scholar]
- 115.Zhang F, Swanson SM, van Breemen RB, Liu X, Yang Y, Gu C, Bolton JL. Chem Res Toxicol. 2001;14:1654. doi: 10.1021/tx010158c. [DOI] [PubMed] [Google Scholar]
- 116.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Chem Res Toxicol. 2000;13:135. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 117.Liu X, Yao J, Pisha E, Yang Y, Hua Y, van Breemen RB, Bolton JL. Chem Res Toxicol. 2002;15:512. doi: 10.1021/tx0101649. [DOI] [PubMed] [Google Scholar]
- 118.Bolton JL. Toxicology. 2002;177:55. doi: 10.1016/s0300-483x(02)00195-6. [DOI] [PubMed] [Google Scholar]
- 119.Shen L, Pisha E, Huang Z, Pezzuto JM, Krol E, Alam Z, van Breemen RB, Bolton JL. Carcinogenesis. 1997;18:1093. doi: 10.1093/carcin/18.5.1093. [DOI] [PubMed] [Google Scholar]
- 120.Shen L, Qiu S, Chen Y, Zhang F, van Breemen RB, Nikolic D, Bolton JL. Chem Res Toxicol. 1998;11:94. doi: 10.1021/tx970181r. [DOI] [PubMed] [Google Scholar]
- 121.Embrechts J, Lemiere F, Van Dongen W, Esmans EL. J Mass Spectrom. 2001;36:317. doi: 10.1002/jms.136. [DOI] [PubMed] [Google Scholar]
- 122.Embrechts J, Lemiere F, Van Dongen W, Esmans EL, Buytaert P, Van Marck E, Kockx M, Makar A. J Am Soc Mass Spectrom. 2003;14:482. doi: 10.1016/S1044-0305(03)00130-2. [DOI] [PubMed] [Google Scholar]
- 123.Ding S, Shapiro R, Geacintov NE, Broyde S. Biochemistry. 2005;44:14565. doi: 10.1021/bi051090t. [DOI] [PubMed] [Google Scholar]
- 124.Ding S, Sharpiro R, Cai Y, Geacintov NE, Broyde S. Chem Res Toxicol. 2008;21:1064. doi: 10.1021/tx800010u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ding S, Shapiro R, Geacintov NE, Broyde S. Biochemistry. 2007;46:182. doi: 10.1021/bi061652o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yasui M, Matsui S, Laxmi YR, Suzuki N, Kim SY, Shibutani S, Matsuda T. Carcinogenesis. 2003;24:911. doi: 10.1093/carcin/bgg029. [DOI] [PubMed] [Google Scholar]
- 127.Chen DD, Oum L, Kolbanovskiy A, Kuzmin V, Shastry A, Chang MS, Bolton JL, Geacintov N. Chem Res Toxicol. 2004;17:1782. doi: 10.1021/tx050190x. [DOI] [PubMed] [Google Scholar]
- 128.Suzuki N, Yasui M, Santosh Laxmi YR, Ohmori H, Hanaoka F, Shibutani S. Biochemistry. 2004;43:11312. doi: 10.1021/bi049273n. [DOI] [PubMed] [Google Scholar]
- 129.Yasui M, Laxmi YR, Ananthoju SR, Suzuki N, Kim SY, Shibutani S. Biochemistry. 2006;45:6187. doi: 10.1021/bi0525324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Geacintov NE, Kolbanovskiy A, Kuzmin K, Chang MS, Bolton JL. Chem Res Toxicol. 2003;16:1665. doi: 10.1021/tx050190x. [DOI] [PubMed] [Google Scholar]
- 131.Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. Chem Res Toxicol. 1997;10:111. doi: 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- 132.Geacintov NE, Broyde S, Buterin T, Naegeli H, Wu M, Yan S, Patel DJ. Biopolymers. 2002;65:202. doi: 10.1002/bip.10239. [DOI] [PubMed] [Google Scholar]
- 133.Broyde S, Wang L, Zhang L, Rechkoblit O, Geacintov NE, Patel DJ. Chem Res Toxicol. 2008;21:45. doi: 10.1021/tx700193x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Likhachev AI, Savochkina IV. Vop Onkol. 1996;42:23. [PubMed] [Google Scholar]
- 135.Kriek E, Rojas M, Alexandrov K, Bartsch H. Mutat Res. 1998;400:215. doi: 10.1016/s0027-5107(98)00065-7. [DOI] [PubMed] [Google Scholar]
- 136.Godschalk RW, Van Schooten FJ, Bartsch H. J Biochem Mol Biol. 2003;36:1. doi: 10.5483/bmbrep.2003.36.1.001. [DOI] [PubMed] [Google Scholar]
- 137.Airoldi L, Pastorelli R, Magagnotti C, Fanelli R. Adv Exp Med Biol. 1994;472:231. doi: 10.1007/978-1-4757-3230-6_20. [DOI] [PubMed] [Google Scholar]
- 138.Luch A. Nat Rev Cancer. 2005;5:113. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 139.Conney AH. Cancer Res. 1982;42:4875. [PubMed] [Google Scholar]
- 140.Cheng SC, Hilton BD, Roman JM, Dipple A. Chem Res Toxicol. 1989;2:334. doi: 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- 141.Lukin M, de los Santos C. Chem Rev. 2006;106:607. doi: 10.1021/cr0404646. [DOI] [PubMed] [Google Scholar]
- 142.Denissenko MF, Chen JX, Tang MS, Pfeifer GP. Proc Natl Acad Sci USA. 1997;94:3893. doi: 10.1073/pnas.94.8.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen JX, Zheng Y, West M, Tang MS. Cancer Res. 1998;58:2070. [PubMed] [Google Scholar]
- 144.Weisenberger DJ, Romano LJ. J Biol Chem. 1999;274:23948. doi: 10.1074/jbc.274.34.23948. [DOI] [PubMed] [Google Scholar]
- 145.Cosman M, de los Santos C, Fiala R, Hingerty BE, Singh SB, Ibanez V, Margulis LA, Live D, Geacintov NE, Broyde S, Patel DJ. Proc Natl Acad Sci USA. 1992;89:1914. doi: 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.de los Santos C, Cosman M, Hingerty BE, Ibanez V, Margulis LA, Geacintov NE, Broyde S, Patel DJ. Biochemistry. 1992;31:5245. doi: 10.1021/bi00138a002. [DOI] [PubMed] [Google Scholar]
- 147.Cosman M, de los Santos C, Fiala R, Hingerty BE, Ibanez V, Luna E, Harvey R, Geacintov NE, Broyde S, Patel DJ. Biochemistry. 1993;32:4145. doi: 10.1021/bi00067a001. [DOI] [PubMed] [Google Scholar]
- 148.Goodsell DS. Stem Cells. 2004;22:873. doi: 10.1634/stemcells.22-5-873. [DOI] [PubMed] [Google Scholar]
- 149.Kaufmann WK. Cell Cycle. 2007;6:1460. [PubMed] [Google Scholar]
- 150.Fernandes A, Liu T, Amin S, Geacintov NE, Grollman AP, Moriya M. Biochemistry. 1998;37:10164. doi: 10.1021/bi980401f. [DOI] [PubMed] [Google Scholar]
- 151.Avkin S, Goldsmith M, Velasco-Miguel S, Geacintov N, Friedberg EC, Livneh Z. J Biol Chem. 2004;279:53298. doi: 10.1074/jbc.M409155200. [DOI] [PubMed] [Google Scholar]
- 152.Seo KY, Jelinsky SA, Loechler EL. Mutat Res. 2000;463:215. doi: 10.1016/s1383-5742(00)00047-8. [DOI] [PubMed] [Google Scholar]
- 153.Ruan Q, Liu T, Kolbanovskiy A, Liu Y, Ren J, Skorvaga M, Zou Y, Lader J, Malkani B, Amin S, Van Houten B, Geacintov NE. Biochemistry. 2007;46:7006. doi: 10.1021/bi700294k. [DOI] [PubMed] [Google Scholar]
- 154.Xu R, Mao B, Amin S, Geacintov N. Biochemistry. 1998;37:769. doi: 10.1021/bi971785x. [DOI] [PubMed] [Google Scholar]
- 155.Tsao H, Mao B, Zhuang P, Xu R, Amin S, Geacintov NE. Biochemistry. 1998;37:4993. doi: 10.1021/bi980291c. [DOI] [PubMed] [Google Scholar]
- 156.Cai Y, Patel DJ, Geacintov NE, Broyde S. J Mol Biol. 2007;374:292. doi: 10.1016/j.jmb.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cai Y, Patel DJ, Geacintov NE, Broyde S. J Mol Biol. 2009;385:30. doi: 10.1016/j.jmb.2008.09.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fountain MA, Krugh TR. Biochemistry. 1995;34:3152. doi: 10.1021/bi00010a004. [DOI] [PubMed] [Google Scholar]
- 159.Rodr%guez FA, Cai Y, Lin C, Tang Y, Kolbanovskiy A, Amin S, Patel DJ, Broyde S, Geacintov NE. Nucleic Acids Res. 2007;35:1555. doi: 10.1093/nar/gkm022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bauer J, Xing G, Yagi H, Sayer JM, Jerina DM, Ling H. Proc Natl Acad Sci USA. 2007;104:14905. doi: 10.1073/pnas.0700717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jennette KW, Jeffery AM, Blobstein SH, Beland FA, Harvey RG, Weinstein IB. Biochemistry. 1977;16:932. doi: 10.1021/bi00624a019. [DOI] [PubMed] [Google Scholar]
- 162.Osborne MR, Jacobs S, Harvey RG, Brookes P. Carcinogenesis. 1981;2:553. doi: 10.1093/carcin/2.6.553. [DOI] [PubMed] [Google Scholar]
- 163.Buterin T, Hess MT, Luneva N, Geacintov NE, Amin S, Kroth H, Seidel A, Naegeli H. Cancer Res. 2000;60:1849. [PubMed] [Google Scholar]
- 164.Wei SJ, Chang RL, Wong CQ, Bhachech N, Cui XX, Hennig E, Yagi H, Sayer JM, Jerina DM, Preston BD, Conney AH. Proc Natl Acad Sci USA. 1991;88:11227. doi: 10.1073/pnas.88.24.11227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wei SJ, Chang RL, Bhachech N, Cui XX, Merkler KA, Wong CQ, Hennig E, Yagi H, Jerina DM, Conney AH. Cancer Res. 1993;53:3294. [PubMed] [Google Scholar]
- 166.Wei SJ, Chang RL, Hennig E, Cui XX, Merkler KA, Wong CQ, Yagi H, Jerina DM, Conney AH. Carcinogenesis. 1994;15:1729. doi: 10.1093/carcin/15.8.1729. [DOI] [PubMed] [Google Scholar]
- 167.Chary P, Latham GJ, Robberson DL, Kim SJ, Han S, Harris CM, Harris TM, Lloyd RS. J Biol Chem. 1995;270:4990. doi: 10.1074/jbc.270.10.4990. [DOI] [PubMed] [Google Scholar]
- 168.Page JE, Zajc B, Oh-hara T, Lakshman MK, Sayer JM, Jerina DM, Dipple A. Biochemistry. 1998;37:9127. doi: 10.1021/bi980273v. [DOI] [PubMed] [Google Scholar]
- 169.Page JE, Pilcher AS, Yagi H, Sayer JM, Jerina DM, Dipple A. Chem Res Toxicol. 1999;12:258. doi: 10.1021/tx980244l. [DOI] [PubMed] [Google Scholar]
- 170.Schurter EJ, Sayer JM, Oh-hara T, Yeh HJ, Yagi H, Luxon BA, Jerina DM, Gorenstein DG. Biochemistry. 1995;34:9009. doi: 10.1021/bi00028a009. [DOI] [PubMed] [Google Scholar]
- 171.Zegar IS, Chary P, Jabil RJ, Tamura PJ, Johansen TN, Lloyd RS, Harris CM, Harris TM, Stone MP. Biochemistry. 1998;37:16516. doi: 10.1021/bi9817616. [DOI] [PubMed] [Google Scholar]
- 172.Mao B, Gu Z, Gorin A, Chen J, Hingerty BE, Amin S, Broyde S, Geacintov NE, Patel DJ. Biochemistry. 1999;38:10831. doi: 10.1021/bi991212f. [DOI] [PubMed] [Google Scholar]
- 173.Volk DE, Thiviyanathan V, Rice JS, Luxon BA, Shah JH, Yagi H, Sayer JM, Yeh HJ, Jerina DM, Gorenstein DG. Biochemistry. 2003;42:1410. doi: 10.1021/bi026745u. [DOI] [PubMed] [Google Scholar]
- 174.Zegar IS, Kim SJ, Johansen TN, Horton PJ, Harris CM, Harris TM, Stone MP. Biochemistry. 1996;35:6212. doi: 10.1021/bi9524732. [DOI] [PubMed] [Google Scholar]
- 175.Yeh HJC, Sayer JM, Liu X, Altieri AS, Byrd RA, Lakshman MK, Yagi H, Schurter EJ, Gorenstein DG, Jerina DM. Biochemistry. 1995;34:13570. doi: 10.1021/bi00041a037. [DOI] [PubMed] [Google Scholar]
- 176.Li Z, Mao H, Kim HY, Tamura PJ, Harris CM, Harris TM, Stone MP. Biochemistry. 1999;38:2969. doi: 10.1021/bi982072x. [DOI] [PubMed] [Google Scholar]
- 177.Li Z, Kim HY, Tamura PJ, Harris CM, Harris TM, Stone MP. Biochemistry. 1999;38:16045. doi: 10.1021/bi9903650. [DOI] [PubMed] [Google Scholar]
- 178.Cosman M, Fiala R, Hingerty BE, Laryea A, Lee H, Harvey RG, Amin S, Geacintov NE, Broyde S, Patel D. Biochemistry. 1993;32:12488. doi: 10.1021/bi00097a029. [DOI] [PubMed] [Google Scholar]
- 179.Cosman M, Laryea A, Fiala R, Hingerty BE, Amin S, Geacintov NE, Broyde S, Patel DJ. Biochemistry. 1995;34:1295. doi: 10.1021/bi00004a024. [DOI] [PubMed] [Google Scholar]
- 180.Wang L, Wu M, Yan SF, Patel DJ, Geacintov NE, Broyde S. Chem Res Toxicol. 2005;18:441. doi: 10.1021/tx049786v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Suri AK, Mao B, Amin S, Geacintov NE, Patel DJ. J Mol Biol. 1999;292:289. doi: 10.1006/jmbi.1999.2974. [DOI] [PubMed] [Google Scholar]
- 182.Bond JA. CRC Crit Rev Toxicol. 1989;19:227. doi: 10.3109/10408448909037472. [DOI] [PubMed] [Google Scholar]
- 183.Himmelstein MW, Acquavella JF, Recio L, Medinsky MA, Bond JA. Crit Rev Toxicol. 1997;27:1. doi: 10.3109/10408449709037482. [DOI] [PubMed] [Google Scholar]
- 184.Selzer RR, Elfarra AA. Chem Res Toxicol. 1996;9:875. doi: 10.1021/tx960039a. [DOI] [PubMed] [Google Scholar]
- 185.Rodriguez DA, Kowalczyk A, Ward JBJ, Harris CM, Harris TM, Lloyd RS. Environ Mol Mutagen. 2001;38:292. doi: 10.1002/em.10026. [DOI] [PubMed] [Google Scholar]
- 186.Kanuri M, Nechev LV, Tamura PJ, Harris CM, Harris TM, Lloyd RS. Chem Res Toxicol. 2002;15:1572. doi: 10.1021/tx025591g. [DOI] [PubMed] [Google Scholar]
- 187.Tretyakova N, Lin YP, Upton PB, Sangaiah R, Swenberg JA. Toxicology. 1996;113:70. doi: 10.1016/0300-483x(96)03429-4. [DOI] [PubMed] [Google Scholar]
- 188.Engel JD. Biochem Biophys Res Commun. 1975;64:581. doi: 10.1016/0006-291x(75)90361-7. [DOI] [PubMed] [Google Scholar]
- 189.Feng B, Zhou L, Passarelli M, Harris CM, Harris TM, Stone MP. Biochemistry. 1995;34:14021. doi: 10.1021/bi00043a008. [DOI] [PubMed] [Google Scholar]
- 190.Stone MP, Feng B. Magn Reson Chem. 1996;34:S105. [Google Scholar]
- 191.Feng B, Voehler M, Zhou L, Passarelli M, Harris CM, Harris TM, Stone MP. Biochemistry. 1996;35:7316. doi: 10.1021/bi952526f. [DOI] [PubMed] [Google Scholar]
- 192.Hennard C, Finneman J, Harris CM, Harris TM, Stone MP. Biochemistry. 2001;40:9780. doi: 10.1021/bi010564v. [DOI] [PubMed] [Google Scholar]
- 193.Merritt WK, Scholdberg TA, Nechev LV, Harris TM, Harris CM, Lloyd RS, Stone MP. Chem Res Toxicol. 2004;17:1007. doi: 10.1021/tx049908j. [DOI] [PubMed] [Google Scholar]
- 194.Scholdberg TA, Nechev LV, Merritt WK, Harris TM, Harris CM, Lloyd RS, Stone MP. Chem Res Toxicol. 2004;17:717. doi: 10.1021/tx034271+. [DOI] [PubMed] [Google Scholar]
- 195.Latham GJ, Zhou L, Harris CM, Harris TM, Lloyd RS. J Biol Chem. 1993;268:23427. [PubMed] [Google Scholar]
- 196.Carmical JR, Nechev LV, Harris CM, Harris TM, Lloyd RS. Mutagenesis. 2000;35:48. [PubMed] [Google Scholar]
- 197.Esterbauer H, Schaur RJ, Zollner H. Free Radical Biol Med. 1991;11:81. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 198.Esterbauer H. Aldehydic Products of Lipid Peroxidation. In: McBrien DCH, Slater TF, editors. Free Radicals, Lipid Peroxidation and Cancer. Academic Press; London: 1982. p. 101. [Google Scholar]
- 199.Benamira M, Johnson K, Chaudhary A, Bruner K, Tibbetts C, Marnett LJ. Carcinogenesis. 1995;16:93. doi: 10.1093/carcin/16.1.93. [DOI] [PubMed] [Google Scholar]
- 200.Mukai FH, Goldstein BD. Science. 1976;191:868. doi: 10.1126/science.766187. [DOI] [PubMed] [Google Scholar]
- 201.Yau TM. Mech Ageing Dev. 1979;11:137. doi: 10.1016/0047-6374(79)90031-9. [DOI] [PubMed] [Google Scholar]
- 202.Spalding JW. NTP Tech Rep. 1988;331:5. [Google Scholar]
- 203.O’Hara SM, Marnett LJ. Mutat Res. 1991;247:45. doi: 10.1016/0027-5107(91)90032-j. [DOI] [PubMed] [Google Scholar]
- 204.Marnett LJ, Huird HK, Hollstein MC, Levin DE, Esterbauer H, Ames BN. Mutat Res. 1985;148:25. doi: 10.1016/0027-5107(85)90204-0. [DOI] [PubMed] [Google Scholar]
- 205.Basu AK, O’Hara SM, Valladier P, Stone K, Mols O, Marnett LJ. Chem Res Toxicol. 1988;1:53. doi: 10.1021/tx00001a010. [DOI] [PubMed] [Google Scholar]
- 206.Seto H, Okuda T, Takesue T, Ikemura T. Bull Chem Soc Jpn. 1983;56:1799. [Google Scholar]
- 207.Reddy GR, Marnett LJ. J Am Chem Soc. 1995;117:5007. [Google Scholar]
- 208.Fink SP, Reddy GR, Marnett LJ. Proc Natl Acad Sci USA. 1997;94:8652. doi: 10.1073/pnas.94.16.8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.VanderVeen LA, Hashim MF, Shyr Y, Marnett LJ. Proc Natl Acad Sci USA. 2003;100:14247. doi: 10.1073/pnas.2332176100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Mao H, Schnetz-Boutaud NC, Weisenseel JP, Marnett LJ, Stone MP. Proc Natl Acad Sci USA. 1999;96:6615. doi: 10.1073/pnas.96.12.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Riggins JN, Pratt DA, Voehler M, Daniels JS, Marnett LJ. J Am Chem Soc. 2004;126:10571. doi: 10.1021/ja040010q. [DOI] [PubMed] [Google Scholar]
- 212.Riggins JN, Daniels JS, Rouzer CA, Marnett LJ. J Am Chem Soc. 2004;126:8237. doi: 10.1021/ja040009r. [DOI] [PubMed] [Google Scholar]
- 213.Mao H, Reddy GR, Marnett LJ, Stone MP. Biochemistry. 1999;38:13491. doi: 10.1021/bi9910124. [DOI] [PubMed] [Google Scholar]
- 214.Hashim MF, Riggins JN, Schnetz-Boutaud N, Voehler M, Stone MP, Marnett LJ. Biochemistry. 2004;43:11828. doi: 10.1021/bi049360f. [DOI] [PubMed] [Google Scholar]
- 215.Schnetz-Boutaud NC, Saleh S, Marnett LJ, Stone MP. Biochemistry. 2001;40:15638. doi: 10.1021/bi011242u. [DOI] [PubMed] [Google Scholar]
- 216.Schnetz-Boutaud NC, Saleh S, Marnett LJ, Stone MP. Adv Exp Med Biol. 2001;500:513. doi: 10.1007/978-1-4615-0667-6_77. [DOI] [PubMed] [Google Scholar]
- 217.Wang Y, Schnetz-Boutaud NC, Saleh S, Marnett LJ, Stone MP. Chem Res Toxicol. 2007;20:1200. doi: 10.1021/tx700121j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Eoff RL, Stafford JB, Szekely J, Rizzo CJ, Egli M, Guengerich FP, Marnett LJ. Biochemistry. 2009;48:7079. doi: 10.1021/bi9003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kawanishi M, Matsuda T, Nakayama A, Takebe H, Matsui S, Yagi T. Mutat Res. 1998;417:65. doi: 10.1016/s1383-5718(98)00093-x. [DOI] [PubMed] [Google Scholar]
- 220.Curren RD, Yang LL, Conklin PM, Grafstrom RC, Harris CC. Mutat Res. 1988;209:17. doi: 10.1016/0165-7992(88)90104-2. [DOI] [PubMed] [Google Scholar]
- 221.Smith RA, Cohen SM, Lawson TA. Carcinogenesis. 1990;11:497. doi: 10.1093/carcin/11.3.497. [DOI] [PubMed] [Google Scholar]
- 222.Cohen SM, Garland EM, St John M, Okamura T, Smith RA. Cancer Res. 1992;52:3577. [PubMed] [Google Scholar]
- 223.Feng Z, Hu W, Hu Y, Tang MS. Proc Natl Acad Sci USA. 2006;103:15404. doi: 10.1073/pnas.0607031103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Czerny C, Eder E, R4nger TM. Mutat Res. 1998;407:125. doi: 10.1016/s0921-8777(97)00069-4. [DOI] [PubMed] [Google Scholar]
- 225.Chung FL, Tanaka T, Hecht SS. Cancer Res. 1986;46:1285. [PubMed] [Google Scholar]
- 226.Benedetti A, Comporti M, Esterbauer H. Biochim Biophys Acta. 1980;620:281. doi: 10.1016/0005-2760(80)90209-x. [DOI] [PubMed] [Google Scholar]
- 227.Burcham PC. Mutagenesis. 1998;13:287. doi: 10.1093/mutage/13.3.287. [DOI] [PubMed] [Google Scholar]
- 228.Cajelli E, Ferraris A, Brambilla G. Mutat Res. 1987;190:169. doi: 10.1016/0165-7992(87)90050-9. [DOI] [PubMed] [Google Scholar]
- 229.Benamira M, Marnett LJ. Mutat Res. 1992;293:1. doi: 10.1016/0921-8777(92)90002-k. [DOI] [PubMed] [Google Scholar]
- 230.Hussain SP, Raja K, Amstad PA, Sawyer M, Trudel LJ, Wogan GN, Hofseth LJ, Shields PG, Billiar TR, Trautwein C, Hohler T, Galle PR, Phillips DH, Markin R, Marrogi AJ, Harris CC. Proc Natl Acad Sci USA. 2000;97:12770. doi: 10.1073/pnas.220416097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chung FL, Nath RG, Ocando J, Nishikawa A, Zhang L. Cancer Res. 2000;60:1507. [PubMed] [Google Scholar]
- 232.Zhang S, Villalta PW, Wang M, Hecht SS. Chem Res Toxicol. 2007;20:565. doi: 10.1021/tx700023z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Chung FL, Zhang L, Ocando JE, Nath RG. IARC Sci Publ. 1999;150:45. [PubMed] [Google Scholar]
- 234.Chung FL, Nath RG, Nagao M, Nishikawa A, Zhou GD, Randerath K. Mutat Res. 1999;424:71. doi: 10.1016/s0027-5107(99)00009-3. [DOI] [PubMed] [Google Scholar]
- 235.Nair U, Bartsch H, Nair J. Free Radical Biol Med. 2007;43:1109. doi: 10.1016/j.freeradbiomed.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 236.Kozekov ID, Nechev LV, Sanchez A, Harris CM, Lloyd RS, Harris TM. Chem Res Toxicol. 2001;14:1482. doi: 10.1021/tx010127h. [DOI] [PubMed] [Google Scholar]
- 237.Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. J Am Chem Soc. 2003;125:50. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- 238.Wang H, Kozekov ID, Harris TM, Rizzo CJ. J Am Chem Soc. 2003;125:5687. doi: 10.1021/ja0288800. [DOI] [PubMed] [Google Scholar]
- 239.Yang IY, Hossain M, Miller H, Khullar S, Johnson F, Grollman A, Moriya M. J Biol Chem. 2001;276:9071. doi: 10.1074/jbc.M008918200. [DOI] [PubMed] [Google Scholar]
- 240.VanderVeen LA, Hashim MF, Nechev LV, Harris TM, Harris CM, Marnett LJ. J Biol Chem. 2001;276:9066. doi: 10.1074/jbc.M008900200. [DOI] [PubMed] [Google Scholar]
- 241.Yang IY, Johnson R, Grollman AP, Moriya M. Chem Res Toxicol. 2002;15:160. doi: 10.1021/tx010123c. [DOI] [PubMed] [Google Scholar]
- 242.de los Santos C, Zaliznyak T, Johnson F. J Biol Chem. 2001;276:9077. doi: 10.1074/jbc.M009028200. [DOI] [PubMed] [Google Scholar]
- 243.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structure. 2008;16:239. doi: 10.1016/j.str.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 244.Zaliznyak T, Bonala R, Attaluri S, Johnson F, de los Santos C. Nucleic Acids Res. 2009;37:2153. doi: 10.1093/nar/gkp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Sanchez AM, Minko IG, Kurtz AJ, Kanuri M, Moriya M, Lloyd RS. Chem Res Toxicol. 2003;16:1019. doi: 10.1021/tx034066u. [DOI] [PubMed] [Google Scholar]
- 246.Cho YJ, Wang H, Kozekov ID, Kurtz AJ, Jacob J, Voehler M, Smith J, Harris TM, Lloyd RS, Rizzo CJ, Stone MP. Chem Res Toxicol. 2006;19:195. doi: 10.1021/tx050239z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Cho YJ, Wang H, Kozekov ID, Kozekova A, Kurtz AJ, Jacob J, Voehler M, Smith J, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chem Res Toxicol. 2006;19:1019. doi: 10.1021/tx0600604. [DOI] [PubMed] [Google Scholar]
- 248.Xing D, Sun L, Cukier RI, Bu Y. J Phys Chem B. 2007;111:5362. doi: 10.1021/jp0673922. [DOI] [PubMed] [Google Scholar]
- 249.Winter CK, Segall HJ, Haddon WF. Cancer Res. 1986;46:5682. [PubMed] [Google Scholar]
- 250.Kowalczyk P, Cieśla JM, Komisarski M, Kuśmierek JT, Tudek B. Mutat Res. 2004;550:33. doi: 10.1016/j.mrfmmm.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 251.Yi P, Zhan D, Samokyszyn VM, Doerge DR, Fu PP. Chem Res Toxicol. 1997;10:1259. doi: 10.1021/tx970100r. [DOI] [PubMed] [Google Scholar]
- 252.Wacker M, Schuler D, Wanek P, Eder E. Chem Res Toxicol. 2000;13:1165. doi: 10.1021/tx000058r. [DOI] [PubMed] [Google Scholar]
- 253.Wacker M, Wanek P, Eder E. Chem Biol Interact. 2001;137:269. doi: 10.1016/s0009-2797(01)00259-9. [DOI] [PubMed] [Google Scholar]
- 254.Chung FL, Zhang L. Methods Enzymol. 2002;353:523. doi: 10.1016/s0076-6879(02)53074-3. [DOI] [PubMed] [Google Scholar]
- 255.Liu X, Lovell MA, Lynn BC. Chem Res Toxicol. 2006;19:710. doi: 10.1021/tx0502903. [DOI] [PubMed] [Google Scholar]
- 256.Pan J, Davis W, Trushin N, Amin S, Nath RG, Salem NJ, Chung FL. Anal Biochem. 2006;348:15. doi: 10.1016/j.ab.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 257.Chung FL, Chen HJ, Guttenplan JB, Nishikawa A, Hard GC. Carcinogenesis. 1993;14:2073. doi: 10.1093/carcin/14.10.2073. [DOI] [PubMed] [Google Scholar]
- 258.Feng Z, Hu W, Amin S, Tang MS. Biochemistry. 2003;42:7848. doi: 10.1021/bi034431g. [DOI] [PubMed] [Google Scholar]
- 259.Huang H, Wang H, Qi N, Kozekova A, Rizzo CJ, Stone MP. J Am Chem Soc. 2008;130:10898. doi: 10.1021/ja801824b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Huang H, Wang H, Lloyd RS, Rizzo CJ, Stone MP. Chem Res Toxicol. 2009;22:187. doi: 10.1021/tx800320m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lindahl T. Nature. 1993;362:709. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 262.Lindahl T. Methods Enzymol. 1980;65:284. doi: 10.1016/s0076-6879(80)65038-1. [DOI] [PubMed] [Google Scholar]
- 263.Baute J, Depicker A. Crit Rev Biochem Mol Biol. 2008;43:239. doi: 10.1080/10409230802309905. [DOI] [PubMed] [Google Scholar]
- 264.Loeb LA, Preston BD. Annu Rev Genet. 1986;20:201. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 265.Gentil A, Renault G, Madzak C, Margot A, Cabral-Neto JB, Vasseur JJ, Rayner B, Imbach JL, Sarasin A. Biochem Biophys Res Commun. 1990;173:704. doi: 10.1016/s0006-291x(05)80092-0. [DOI] [PubMed] [Google Scholar]
- 266.Gentil A, Cabral-Neto JB, Mariage-Samson R, Margot A, Imbach JL, Rayner B, Sarasin A. J Mol Biol. 1992;227:981. doi: 10.1016/0022-2836(92)90513-j. [DOI] [PubMed] [Google Scholar]
- 267.Lawrence CW, Borden A, Banerjee SK, LeClerc JE. Nucleic Acids Res. 1990;18:2153. doi: 10.1093/nar/18.8.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Schaaper RM, Loeb LA. Proc Natl Acad Sci USA. 1981;78:1773. doi: 10.1073/pnas.78.3.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Schaaper RM, Kunkel TA, Loeb LA. Proc Natl Acad Sci USA. 1983;80:487. doi: 10.1073/pnas.80.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Kunkel TA. Proc Natl Acad Sci USA. 1984;81:1494. [Google Scholar]
- 271.Kamiya H, Suzuki M, Ohtsuka E. FEBS Lett. 1993;328:125. doi: 10.1016/0014-5793(93)80979-5. [DOI] [PubMed] [Google Scholar]
- 272.Randall SK, Eritja R, Kaplan BE, Petruska J, Goodman MF. J Biol Chem. 1987;262:6864. [PubMed] [Google Scholar]
- 273.Sagher D, Strauss B. Biochemistry. 1983;22:4518. doi: 10.1021/bi00288a026. [DOI] [PubMed] [Google Scholar]
- 274.Boiteux S, Laval J. Biochemistry. 1982;21:6746. doi: 10.1021/bi00269a020. [DOI] [PubMed] [Google Scholar]
- 275.Potapova O, Grindley ND, Joyce CM. J Biol Chem. 2002;277:28157. doi: 10.1074/jbc.M202607200. [DOI] [PubMed] [Google Scholar]
- 276.Kokoska RJ, McCulloch SD, Kunkel TA. J Biol Chem. 2003;278:50537. doi: 10.1074/jbc.M308515200. [DOI] [PubMed] [Google Scholar]
- 277.Kokoska RJ, Bebenek K, Boudsocq F, Woodgate R, Kunkel TA. J Biol Chem. 2002;277:19633. doi: 10.1074/jbc.M202021200. [DOI] [PubMed] [Google Scholar]
- 278.Boudsocq F, Iwai S, Hanaoka F, Woodgate R. Nucleic Acids Res. 2001;29:4607. doi: 10.1093/nar/29.22.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Manoharan M, Ransom SC, Mazumder A, Gerlt JA. J Am Chem Soc. 1988;110:1620. [Google Scholar]
- 280.Wilde JA, Bolton PH. J Am Chem Soc. 1989;111:1894. [Google Scholar]
- 281.Beger RD, Bolton PH. J Biol Chem. 1998;273:15565. doi: 10.1074/jbc.273.25.15565. [DOI] [PubMed] [Google Scholar]
- 282.Mol CD, Izumi T, Mitra S, Tainer JA. Nature. 2000;403:451. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 283.de los Santos C, El-Khateeb M, Rege P, Tian K, Johnson F. Biochemistry. 2004;43:15349. doi: 10.1021/bi048400c. [DOI] [PubMed] [Google Scholar]
- 284.Ide H, Shimizu H, Kimura Y, Sakamoto S, Makino K, Glackin M, Wallace SS, Nakamuta H, Sasaki M, Sugimoto N. Biochemistry. 1995;34:6947. doi: 10.1021/bi00021a006. [DOI] [PubMed] [Google Scholar]
- 285.Gelfand CA, Plum GE, Grollman AP, Johnson F, Breslauer KJ. Biochemistry. 1998;37:7321. doi: 10.1021/bi9803372. [DOI] [PubMed] [Google Scholar]
- 286.Singh MP, Hill GC, Péoc’h D, Rayner B, Imbach JL, Lown JW. Biochemistry. 1994;33:10271. doi: 10.1021/bi00200a007. [DOI] [PubMed] [Google Scholar]
- 287.Chen J, Dupradeau FY, Case DA, Turner CJ, Stubbe J. Biochemistry. 2007;46:3096. doi: 10.1021/bi6024269. [DOI] [PubMed] [Google Scholar]
- 288.Chen J, Dupradeau FY, Case DA, Turner CJ, Stubbe J. Nucleic Acids Res. 2008;36:253. doi: 10.1093/nar/gkm622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Goljer I, Kumar S, Bolton PH. J Biol Chem. 1995;270:22980. doi: 10.1074/jbc.270.39.22980. [DOI] [PubMed] [Google Scholar]
- 290.Téoule R. Int J Radiat Biol. 1987;51:573. doi: 10.1080/09553008414552111. [DOI] [PubMed] [Google Scholar]
- 291.Teoule R, Bonicel A, Bert C, Cadet J, Polverelli M. Radiat Res. 1974;57:46. [PubMed] [Google Scholar]
- 292.Adelman R, Saul RL, Ames BN. Proc Natl Acad Sci USA. 1988;85:2706. doi: 10.1073/pnas.85.8.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Cathcart R, Schwiers E, Saul RL, Ames BN. Proc Natl Acad Sci USA. 1984;81:5633. doi: 10.1073/pnas.81.18.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Clark JM, Beardsley GP. Nucleic Acids Res. 1986;14:737. doi: 10.1093/nar/14.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ide H, Kow YW, Wallace SS. Nucleic Acids Res. 1985;13:8035. doi: 10.1093/nar/13.22.8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Rouet P, Essigmann JM. Cancer Res. 1985;45:6113. [PubMed] [Google Scholar]
- 297.Basu AK, Loechler EL, Leadon SA, Essigmann JM. Proc Natl Acad Sci USA. 1989;86:1. doi: 10.1073/pnas.86.20.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.McNulty JM, Jerkovic B, Bolton PH, Basu AK. Chem Res Toxicol. 1998;11:666. doi: 10.1021/tx970225w. [DOI] [PubMed] [Google Scholar]
- 299.Clark JM, Pattabiraman N, Jarvis W, Beardsley GP. Biochemistry. 1987;26:5404. doi: 10.1021/bi00391a028. [DOI] [PubMed] [Google Scholar]
- 300.Hayes RC, LeClerc JE. Nucleic Acids Res. 1986;14:1045. doi: 10.1093/nar/14.2.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Wang Y. Chem Res Toxicol. 2002;15:671. doi: 10.1021/tx0155855. [DOI] [PubMed] [Google Scholar]
- 302.Lustig MJ, Cadet J, Boorstein RJ, Teebor GW. Nucleic Acids Res. 1992;20:4839. doi: 10.1093/nar/20.18.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Vaishnav Y, Holwitt E, Swenberg C, Lee HC, Kan LS. J Biomol Struct Dyn. 1991;8:935. doi: 10.1080/07391102.1991.10507858. [DOI] [PubMed] [Google Scholar]
- 304.Kusumoto R, Masutani C, Iwai S, Hanaoka F. Biochemistry. 2002;41:6090. doi: 10.1021/bi025549k. [DOI] [PubMed] [Google Scholar]
- 305.Fischhaber PL, Gerlach VL, Feaver WJ, Hatahet Z, Wallace SS, Friedberg EC. J Biol Chem. 2002;277:37604. doi: 10.1074/jbc.M206027200. [DOI] [PubMed] [Google Scholar]
- 306.Ikeda S, Biswas T, Roy R, Izumi T, Boldogh I, Kurosky A, Sarker AH, Seki S, Mitra S. J Biol Chem. 1998;273:21585. doi: 10.1074/jbc.273.34.21585. [DOI] [PubMed] [Google Scholar]
- 307.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Proc Natl Acad Sci USA. 2002;99:3523. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Bandaru V, Sunkara S, Wallace SS, Bond JP. DNA Repair (Amst) 2002;1:517. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 309.Miller H, Fernandes AS, Zaika E, McTigue MM, Torres MC, Wente M, Iden CR, Grollman AP. Nucleic Acids Res. 2004;32:338. doi: 10.1093/nar/gkh190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Ocampo-Hafalla MT, Altamirano A, Basu AK, Chan MK, Ocampo JE, Cummings AJ, Boorstein RJ, Cunningham RP, Teebor GW. DNA Repair (Amst) 2006;5:444. doi: 10.1016/j.dnarep.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 311.Brown KL, Adams T, Jasti VP, Basu AK, Stone MP. J Am Chem Soc. 2008;130:11701. doi: 10.1021/ja8016544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Kao JY, Goljer I, Phan TA, Bolton PH. J Biol Chem. 1993;268:17787. [PubMed] [Google Scholar]
- 313.Kung HC, Bolton PH. J Biol Chem. 1997;272:9227. doi: 10.1074/jbc.272.14.9227. [DOI] [PubMed] [Google Scholar]
- 314.Brown KL, Roginskaya M, Zou Y, Altamirano A, Basu AK, Stone MP. Nucleic Acids Res. 2010;38:428. doi: 10.1093/nar/gkp844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Brown KL, Basu AK, Stone MP. Biochemistry. 2009;48:9722. doi: 10.1021/bi900695e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Teoule R, Bert C, Bonicel A. Radiat Res. 1977;72:190. [PubMed] [Google Scholar]
- 317.Evans J, Maccabee M, Hatahet Z, Courcelle J, Bockrath R, Ide H, Wallace SS. Mutat Res. 1993;299:147. doi: 10.1016/0165-1218(93)90092-r. [DOI] [PubMed] [Google Scholar]
- 318.Maccabee M, Evans JS, Glackin MP, Hatahet Z, Wallace SS. J Mol Biol. 1994;236:514. doi: 10.1006/jmbi.1994.1162. [DOI] [PubMed] [Google Scholar]
- 319.Gervais V, Guy A, Téoule R, Fazakerley GV. Nucleic Acids Res. 1992;20:6455. doi: 10.1093/nar/20.24.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Gervais V, Cognet JA, Guy A, Cadet J, Téoule R, Fazakerley GV. Biochemistry. 1998;37:1083. doi: 10.1021/bi971202j. [DOI] [PubMed] [Google Scholar]
- 321.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617. doi: 10.1021/ja003823x. [DOI] [PubMed] [Google Scholar]
- 322.Luo WC, Muller JG, Rachlin EM, Burrows CJ. Org Lett. 2000;2:613. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 323.Luo WC, Muller JG, Rachlin EM, Burrows CJ. Chem Res Toxicol. 2001;14:927. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 324.Luo W, Muller JG, Burrows CJ. Org Lett. 2001;3:2801. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 325.Burrows CJ, Muller JG, Kornyushyna O, Luo W, Duarte V, Leipold MD, David SS. Environ Health Perspect. 2002;110(Suppl 5):713. doi: 10.1289/ehp.02110s5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Hailer MK, Slade PG, Martin BD, Sugden KD. Chem Res Toxicol. 2005;18:1378. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Duarte V, Muller JG, Burrows CJ. Nucleic Acids Res. 1999;27:496. doi: 10.1093/nar/27.2.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. Biochemistry. 2003;42:9257. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 329.Neeley WL, Delaney S, Alekseyev YO, Jarosz DF, Delaney JC, Walker GC, Essigmann JM. J Biol Chem. 2007;282:12741. doi: 10.1074/jbc.M700575200. [DOI] [PubMed] [Google Scholar]
- 330.Aller P, Ye Y, Wallace SS, Burrows CJ, Doublie S. Biochemistry. 2010;49:2502. doi: 10.1021/bi902195p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Busby WF, Jr, Wogan GN. Aflatoxins. In: Searle CE, editor. Chemical Carcinogens. American Chemical Society; Washington, D.C: 1984. pp. 945–1136. [Google Scholar]
- 332.McCann J, Spingarn NE, Koburi J, Ames BN. Proc Natl Acad Sci USA. 1975;72:979. doi: 10.1073/pnas.72.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Smela ME, Currier SS, Bailey EA, Essigmann JM. Carcinogenesis. 2001;22:535. doi: 10.1093/carcin/22.4.535. [DOI] [PubMed] [Google Scholar]
- 334.Foster PL, Eisenstadt E, Miller JH. Proc Natl Acad Sci USA. 1983;80:2695. doi: 10.1073/pnas.80.9.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Bailey GS, Williams DE, Wilcox JS, Loveland PM, Coulombe RA, Hendricks JD. Carcinogenesis. 1988;9:1919. doi: 10.1093/carcin/9.11.1919. [DOI] [PubMed] [Google Scholar]
- 336.Bailey GS, Loveland PM, Pereira C, Pierce D, Hendricks JD, Groopman JD. Mutat Res. 1994;313:25. doi: 10.1016/0165-1161(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 337.Soman NR, Wogan GN. Proc Natl Acad Sci USA. 1993;90:2045. doi: 10.1073/pnas.90.5.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.McMahon G, Davis EF, Huber LJ, Kim Y, Wogan GN. Proc Natl Acad Sci USA. 1990;87:1104. doi: 10.1073/pnas.87.3.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Levy DD, Groopman JD, Lim SE, Seidman MM, Kraemer KH. Cancer Res. 1992;52:5668. [PubMed] [Google Scholar]
- 340.Hollstein M, Sidransky D, Vogelstein B, Harris CC. Science. 1991;253:49. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 341.Mace K, Aguilar F, Wang JS, Vautravers P, Gomez-Lechon M, Gonzalez FJ, Groopman J, Harris CC, Pfeifer AM. Carcinogenesis. 1997;18:1291. doi: 10.1093/carcin/18.7.1291. [DOI] [PubMed] [Google Scholar]
- 342.Shen HM, Ong CN. Mutat Res. 1996;366:23. doi: 10.1016/s0165-1110(96)90005-6. [DOI] [PubMed] [Google Scholar]
- 343.Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Nature. 1991;350:427. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 344.Bressac B, Kew M, Wands J, Őztűrk M. Nature. 1991;350:429. doi: 10.1038/350429a0. [DOI] [PubMed] [Google Scholar]
- 345.Katiyar S, Dash BC, Thakur V, Guptan RC, Sarin SK, Das BC. Cancer. 2000;88:1565. doi: 10.1002/(sici)1097-0142(20000401)88:7<1565::aid-cncr10>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 346.Shimizu Y, Zhu JJ, Han F, Ishikawa T, Oda H. Int J Cancer. 1999;82:187. doi: 10.1002/(sici)1097-0215(19990719)82:2<187::aid-ijc6>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 347.Rashid A, Wang JS, Qian GS, Lu BX, Hamilton SR, Groopman JD. Br J Cancer. 1999;80:59. doi: 10.1038/sj.bjc.6690321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Yang M, Zhou H, Kong RY, Fong WF, Ren LQ, Liao XH, Wang Y, Zhuang W, Yang S. Mutat Res. 1997;381:25. doi: 10.1016/s0027-5107(97)00142-5. [DOI] [PubMed] [Google Scholar]
- 349.Essigmann JM, Croy RG, Nadzan AM, Busby WF, Jr, Reinhold VN, Buchi G, Wogan GN. Proc Natl Acad Sci USA. 1977;74:1870. doi: 10.1073/pnas.74.5.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Martin CN, Garner RC. Nature. 1977;267:863. doi: 10.1038/267863a0. [DOI] [PubMed] [Google Scholar]
- 351.Garner RC, Miller EC, Miller JA, Garner JV, Hanson RS. Biochem Biophys Res Commun. 1971;45:774. doi: 10.1016/0006-291x(71)90484-0. [DOI] [PubMed] [Google Scholar]
- 352.Coles BF, Welch AM, Hertzog PJ, Smith JRL, Garner RC. Carcinogenesis. 1980;1:79. doi: 10.1093/carcin/1.1.79. [DOI] [PubMed] [Google Scholar]
- 353.Bailey EA, Iyer RS, Stone MP, Harris TM, Essigmann JM. Proc Natl Acad Sci USA. 1996;93:1535. doi: 10.1073/pnas.93.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Lin JK, Miller JA, Miller EC. Cancer Res. 1977;37:4430. [PubMed] [Google Scholar]
- 355.Groopman JD, Croy RG, Wogan GN. Proc Natl Acad Sci USA. 1981;78:5445. doi: 10.1073/pnas.78.9.5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, Essigmann JM. Proc Natl Acad Sci USA. 2002;99:6655. doi: 10.1073/pnas.102167699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Brown KL, Deng JZ, Iyer RS, Iyer LG, Voehler MW, Stone MP, Harris CM, Harris TM. J Am Chem Soc. 2006;128:5188. doi: 10.1021/ja063781y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Mao H, Deng Z, Wang F, Harris TM, Stone MP. Biochemistry. 1998;37:4374. doi: 10.1021/bi9718292. [DOI] [PubMed] [Google Scholar]
- 359.Jones WR, Johnston DS, Stone MP. Chem Res Toxicol. 1998;11:873. doi: 10.1021/tx980047m. [DOI] [PubMed] [Google Scholar]
- 360.Giri I, Jenkins MD, Schnetz-Boutaud NC, Stone MP. Chem Res Toxicol. 2002;15:638. doi: 10.1021/tx010187n. [DOI] [PubMed] [Google Scholar]
- 361.Gopalakrishnan S, Harris TM, Stone MP. Biochemistry. 1990;29:10438. doi: 10.1021/bi00498a002. [DOI] [PubMed] [Google Scholar]
- 362.Brown KL, Voehler MW, Magee SM, Harris CM, Harris TM, Stone MP. J Am Chem Soc. 2009;131:16096. doi: 10.1021/ja902052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Cell. 2005;123:1191. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 364.Noll DM, Mason TM, Miller PS. Chem Rev. 2006;106:277. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Mirchandani KD, D’Andrea AD. Exp Cell Res. 2006;312:2647. doi: 10.1016/j.yexcr.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 366.Patel KJ, Joenje H. DNA Repair (Amst) 2007;6:885. doi: 10.1016/j.dnarep.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 367.Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ. J Biol Chem. 2003;278:31426. doi: 10.1074/jbc.M212549200. [DOI] [PubMed] [Google Scholar]
- 368.Kim HY, Voehler M, Harris TM, Stone MP. J Am Chem Soc. 2002;124:9324. doi: 10.1021/ja020333r. [DOI] [PubMed] [Google Scholar]
- 369.Cho YJ, Kim HY, Huang H, Slutsky A, Minko IG, Wang H, Nechev LV, Kozekov ID, Kozekova A, Tamura P, Jacob J, Voehler M, Harris TM, Lloyd RS, Rizzo CJ, Stone MP. J Am Chem Soc. 2005;127:17686. doi: 10.1021/ja053897e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Huang H, Kim HY, Kozekov ID, Cho YJ, Wang H, Kozekova A, Harris TM, Rizzo CJ, Stone MP. J Am Chem Soc. 2009;131:8416. doi: 10.1021/ja809543j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Dooley PA, Zhang M, Korbel GA, Nechev LV, Harris CM, Stone MP, Harris TM. J Am Chem Soc. 2003;125:62. doi: 10.1021/ja0207798. [DOI] [PubMed] [Google Scholar]
- 372.Dooley PA, Tsarouhtsis D, Korbel GA, Nechev LV, Shearer J, Zegar IS, Harris CM, Stone MP, Harris TM. J Am Chem Soc. 2001;123:1730. doi: 10.1021/ja003163w. [DOI] [PubMed] [Google Scholar]
- 373.Huang H, Dooley PA, Harris CM, Harris TM, Stone MP. Chem Res Toxicol. 2009;22:1810. doi: 10.1021/tx900225c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Cho YJ, Kozekov ID, Harris TM, Rizzo CJ, Stone MP. Biochemistry. 2007;46:2608. doi: 10.1021/bi061381h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Kurtz AJ, Lloyd RS. J Biol Chem. 2003;278:5970. doi: 10.1074/jbc.M212012200. [DOI] [PubMed] [Google Scholar]
- 376.Huang H, Wang H, Qi N, Kozekova A, Rizzo CJ, Stone MP. J Am Chem Soc. 2008;130:10898. doi: 10.1021/ja801824b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Huang H, Wang H, Qi N, Lloyd RS, Rizzo CJ, Stone MP. Biochemistry. 2008;47:11457. doi: 10.1021/bi8011143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Zhang XY, Elfarra AA. Chem Res Toxicol. 2005;18:1316. doi: 10.1021/tx0500979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Millard JT, Hanly TC, Murphy K, Tretyakova N. Chem Res Toxicol. 2006;19:16. doi: 10.1021/tx050250z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Park S, Anderson C, Loeber R, Seetharaman M, Jones R, Tretyakova N. J Am Chem Soc. 2005;127:14355. doi: 10.1021/ja051979x. [DOI] [PubMed] [Google Scholar]
- 381.Park S, Hodge J, Anderson C, Tretyakova N. Chem Res Toxicol. 2004;17:1638. doi: 10.1021/tx0498206. [DOI] [PubMed] [Google Scholar]
- 382.Tretyakova N, Livshits A, Park S, Bisht B, Goggin M. Chem Res Toxicol. 2007;20:284. doi: 10.1021/tx060204e. [DOI] [PubMed] [Google Scholar]
- 383.Zhang XY, Elfarra AA. Chem Res Toxicol. 2003;16:1606. doi: 10.1021/tx0341355. [DOI] [PubMed] [Google Scholar]
- 384.Zhang XY, Elfarra AA. Chem Res Toxicol. 2004;17:521. doi: 10.1021/tx034243r. [DOI] [PubMed] [Google Scholar]
- 385.Seneviratne U, Antsypovich S, Dorr DQ, Dissanayake T, Kotapati S, Tretyakova N. Chem Res Toxicol. 2010;23:1556. doi: 10.1021/tx100146v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Seneviratne U, Antsypovich S, Goggin M, Dorr DQ, Guza R, Moser A, Thompson C, York DM, Tretyakova N. Chem Res Toxicol. 2010;23:118. doi: 10.1021/tx900312e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Loeber R, Rajesh M, Fang Q, Pegg AE, Tretyakova N. Chem Res Toxicol. 2006;19:645. doi: 10.1021/tx0600088. [DOI] [PMC free article] [PubMed] [Google Scholar]