

Abstract
The secondary ent-beyeran-16-yl carbocation (7) is a key branch point intermediate in mechanistic schemes to rationalize the cyclic structures of many tetra- and pentacyclic diterpenes including ent-beyerene, ent-kaurene, ent-trachylobane, and ent-atiserene, presumed precursors to > 1,000 known diterpenes. (Scheme 1) To evaluate these mechanistic hypotheses, we synthesized the heterocyclic analogues, 16-aza-ent-beyerane (12) and 16-aza-ent-trachylobane (13), by means of Hg(II)- and Pb(IV)-induced cyclizations onto the Δ12 double bonds of tricyclic intermediates bearing carbamoylmethyl and aminomethyl groups at C-8. The 13,16-seco 16-nor carbamate (20a) was obtained from ent-beyeran-16-one oxime (17) by Beckmann fragmentation, hydrolysis, and Curtius rearrangement. The aza analogues inhibited recombinant ent-kaurene synthase from Arabidopsis thaliana (GST-rAtKS) with inhibition constants (IC = 1 × 10 −7 and 1 × 10−6 50 M) similar in magnitude to the pseudo-binding constant of the bicyclic ent-copalyl diphosphate substrate (Km = 3 × 10−7 M). Large enhancements of binding affinities (IC50 = 4 × 10−9 and 2 × 10−8 M) were observed in the presence of 1 mM pyrophosphate which is consistent with a tightly bound ent-beyeranyl+/pyrophosphate− ion pair intermediate in the cyclization-rearrangement catalyzed by this diterpene synthase. The weak inhibition (IC50 = 1 × 10−5 M) exhibited by ent-beyeran-16 exo-yl diphosphate (11), and its failure to undergo bridge rearrangement to kaurene, appear to rule out the covalent diphosphate as a free intermediate. 16-Aza-ent-beyerane is proposed as an effective mimic for the ent-beyeran-16-yl carbocation with potential applications as an active site probe for the various ent- diterpene cyclases, and as a novel, selective inhibitor of gibberellin biosynthesis in plants.
Introduction
The biogenetically related polycyclic diterpene hydrocarbons, ent-beyerene, ent-kaurene, ent-trachylobane, and ent-atiserene (Fig. 1, 1-4) occur widely in plants, where they are likely intermediates in the biosynthesis of numerous functionalized and degraded natural products.2 For example, ent-kaurene is a well-established intermediate in the multi-step pathway to the gibberellin family of plant growth regulators3 as well as the intensely sweet-tasting triglycoside stevioside (14) derived from 13-hydroxykaurenoic acid.4 The related diterpene alcohols, ent-kauran-16α-ol and ent-atiseran-16α-ol, were recently found to be oviposition stimulants for the banded sunflower moth.5
Figure 1.
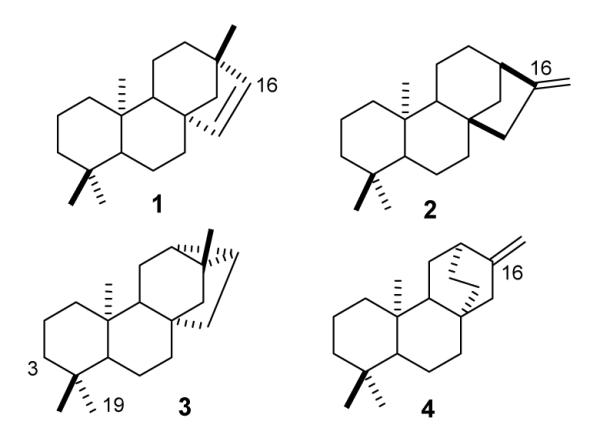
Structures of ent-beyerene, ent-kaurene, ent-trachylobane, and ent-atiserene diterpenes (1-4).
Recent reports document significant bioactivities of oxygenated diterpenes in mammalian cells. ent-Kauren-19-oic acid, an intermediate in gibberellin biosynthesis, exhibits anti-inflammatory activity attributed to inhibition of the nuclear factor κB pathway in macrophages,6a and its 15-oxo derivative blocks mitotic chromosome movement through effects on a kinetochore protein.6b The lethal norkaurane glycoside disulfate, carboxyatractyloside, binds to the mitochondrial ADP/ATP transporter protein.6c The 3β-hydroxy derivative of trachylobane induces apoptosis in human leukemia cells at 50 μM concentration.7
The tetracyclic and pentacyclic structures of these diterpenes are biosynthesized from the bicyclic labdane precursor ent-copalyl diphosphate (5) through a series of cyclizations and rearrangements beginning with an SN’ ring closure that forms the C ring. (Scheme 1)3a,8 Rotation of the C13 vinyl group in the resulting ent-pimaren-8-yl carbocation (6) and a second cyclization generate the ent-beyeran-16-yl ion (7). The four diterpene hydrocarbon end products are then formed from this key branch point intermediate by 1,2- and 1,3-proton eliminations (to 1 and 3), by the Wagner-Meerwein rearrangement 7→ 8 and methyl-methylene elimination (to 2), and by 1,3-hydride shift to the isomeric beyeran-12-yl ion 9, Wagner-Meerwein rearrangement 9→10, and proton elimination (to 4). A covalent beyeranyl diphosphate (PP) 11 is another conceivable intermediate in the multi-step mechanism.
SCHEME 1.

This scheme is supported by the occurrence of similar biomimetic transformations,9 by the results of isotope labeling experiments with crude enzyme extracts containing ent-kaurene synthase (KS) activity,3a,8,10 and by the alteration of product outcomes with mutant KSs.11 An alternative path to the atiserane diterpenes would involve ring opening of a free or enzyme-bound trachylobane intermediate.
Solublization and characterization of KS from the gibberellin-producing fungus G. fujikuroi and various higher plants were reported in pioneering studies by C. A. West and co-workers.8a,12 Separation of distinct ent-beyerene, ent-kaurene, and ent-trachylobane synthases in enzyme extracts from elicited seedlings of Ricinus communis has been recorded.13 KS genes from the fungi G. fujikuroi and Phaeosphaeria Sp L487 and from the higher plants Arabidopsis thaliana, Stevia rebaudiana, Cucuribita maxima (pumpkin), rice (Oryza sativa), and moss have recently been identified and characterized.4c,14
Numerous aza analogues of postulated carbocation intermediates in terpene biogenetic mechanisms have been synthesized and evaluated as inhibitors of specific terpene synthases,15 or for other purposes including sterol biosynthesis inhibitors and anti-fungal agents.16 Although the aza analogue concept has been very useful for inhibitor design, the extent to which tetrahedral ammonium ions with their localized positive charges actually resemble presumably trigonal carbocations with their more dispersed charge is open to question. However, the protonated forms of 2-azabornane and the isomeric 15-azapimarenes are notable as effective mimics for high energy, pyramidalized secondary born-2-yl and pimaren-15-yl carbocation/PP·Mg ion pairs presumably generated at the active sites of bornyl diphosphate and abietadiene synthases.17,18 Azabornane and other aza-terpenes have served as useful active site probes in X-ray crystallography of mono- and sesquiterpene cyclases.19
In this paper we report the synthesis and characterization of 16-aza-ent-beyerane and 16-aza-ent-trachylobane (12 and 13, Fig 2), as well as the potential intermediate ent-beyeran-16-yl PP (11), and a preliminary kinetic evaluation of the compounds as mechanism-based inhibitors of recombinant KS from Arabidopsis thaliana. The nitrogen heterocycle 12 is a novel aza-analogue of the high energy secondary ent-beyeranyl ion 7 in the mechanism in Fig 1, and 16-azatrachylobane (13) resembles a bridged ion (protonated cyclopropane) intermediate or transition state in the Wagner-Meerwein rearrangement step (7 → 8).
Figure 2.
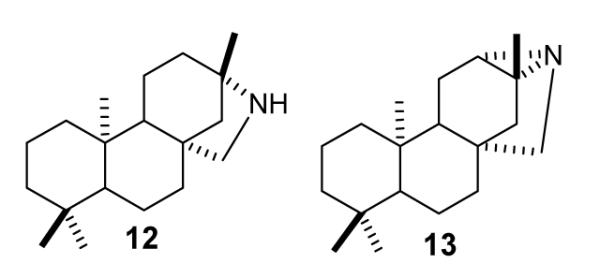
Structures of 16-aza-ent-beyerane (12) and 16-aza-ent-trachylobane (13), proposed aza analogue mimics for the ent-beyeran-16-yl ion 7 (Sch 1) and a protonated ent-trachylobane (3), high energy intermediates in the cyclization of ent-copalyl PP to ent-kaurene.
Results and Discussion
16-Aza-ent-beyerane and 16-Aza-ent-trachylobane
The two heterocyclic analogues (12 and 13) were synthesized from ent-beyeran-16-one (16) by cleavage of ring D, Curtius rearrangement to 13,16-seco nor carbamates 20 and 13,16-seco noramine 21, and recyclizations onto nitrogen as outlined in Schemes 2 - 4. Multi-gram quantities of crude isosteviol (15a) were obtained from stevioside (14) by hydrolysis and pinacol-type rearrangement of the sweet ent-kaurane glycoside20 using either Stevia rebaudiana plant material (2.4%), or better commercial Stevia extract sweetener (24 %). Tetracyclic ketone 16 was prepared from isosteviol methyl ester (15b) through de-oxygenation of the sterically hindered axial carboxyl group at C4 in 6 steps according to previously described procedures: ketalization, LiAlH4 reduction, mesylate formation, thiophenoxide displacement, Li/NH reduction, and hydrolysis (60-62% yield).21
SCHEME 2.

SCHEME 4.
Cleavage of the D ring of ent-beyeranone was accomplished by Beckmann fragmentation of the oxime derivative (17). The oxime was formed as a single isomer in high yield (NH2OH•HCl, pyr) and presumed to have the less sterically crowded E configuration. Fragmentation occurred readily upon reaction of the oxime with tosyl chloride (DMF, 25°C),22 giving rise to a 2.6:1 mixture olefinic nitriles 18a and 18b. Although the double bond isomers had very similar properties and polarities, both were obtained in pure, crystalline form by repeated chromatographies on silica gel. The broadened signal for the vinyl hydrogen (δH 5.33, W1/2 9.5 Hz) in the proton NMR spectrum of the major isomer compared to the much sharper NMR resonance (δH 5.34, narrow q, J = 1 Hz, W1/2 3 Hz) for the vinyl proton of the minor isomer was the basis for preliminary assignment of the double bond positions shown. This assignment was confirmed by X-ray diffraction analysis of a single crystal of the major isomer (See Supporting Information).
Further degradation to the required seco nor-amine carbamates 20 was effected by Curtius rearrangements of the corresponding carboxylic acids 19a and 19b (Scheme 2). The reactions were conducted with the pure Δ12 and Δ13 isomers, and with a 1:4 mixture enriched in the latter. Nitriles 18a and 18b were hydrolyzed to the corresponding 13,16-seco acids (92%) by heating with aq KOH in diethylene glycol in a metal bomb at 195 °C. Curtius degradations of the acids with diphenylphosphoryl azide (Et3N, PhH, reflux, 2h)23,24 gave the isocyanate intermediates that were converted to carbamates 20a and 20b (80 and 82%) by addition of methanol (MeOH, Et3N, reflux, 15h). The isomeric carbamates were readily distinguished by their vinyl hydrogen signals in the respective proton NMR spectra (20a, δH 5.38; 20b, δH 5.08). Hydrolysis (KOH, aq MeOH, reflux, 8h) of the Δ12 seco-nor carbamate (20a) provided the corresponding primary amine 21 in high yield.
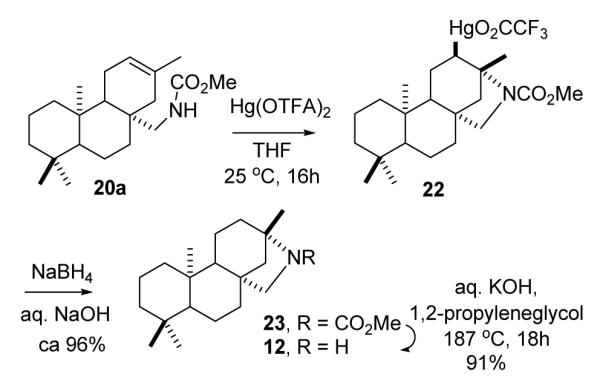
Intramolecular mercury amidation of carbamate 20a with Hg(OAc)2 in THF25a at room temp was quite slow and incomplete after 5 days. However, heterocyclization of 20a (Scheme 3) proceeded smoothly with the more electrophilic trifluoroacetate reagent25b (Hg(O2CCF3)2, THF, 25°C, 16h), and direct reduction of the intermediate mercurial with NaBH4 (aq NaOH) furnished azabeyerane carbamate 23. Cyclization of the Δ13 isomer (20b) with Hg(OTFA)2 in THF proceeded with a similar rate, and NaBH4 reduction afforded 23 with comparable efficiency. The organomercurial intermediate 22 was isolated from a separate run, and the crude, polar solid was characterized by its proton NMR spectrum (See below). Hydrolysis of the cyclized carbamate (KOH, aq 1,2-propylene glycol, reflux 18h) provided azabeyerane (12).
SCHEME 3.
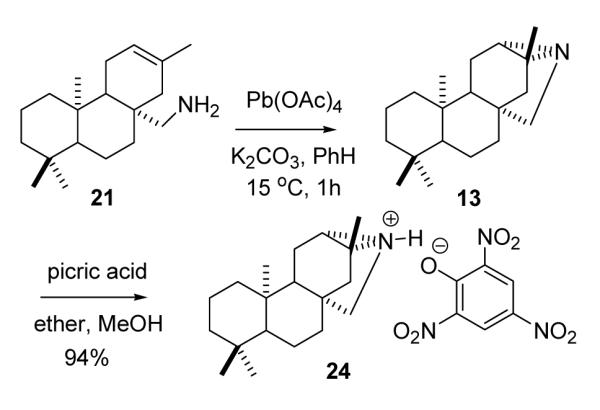
Conversion of seco-nor amine 21 to 16-aza-ent-trachylobane 13 was accomplished by Nagata’s intramolecular aziridination method (Scheme 4)26,27 Oxidation of the primary amine with 1.4 equiv of Pb(OAc)4 in benzene buffered heterogeneously with powdered K2CO3 (15°C, 1h) effected nitrenoid cyclization to the bridged aziridine in quantitative yield. Although azatrachylobane was unstable as a neat liquid at room temp, presumably undergoing polymerization,27 the aza analogue could be stored indefinitely in hexane solution at −20 °C. The picrate derivative (24) was formed readily in ether-methanol at 0 °C in 94% yield. The ORTEP for the X-ray crystal structure of the picrate salt is presented below in Fig 3.
Figure 3.
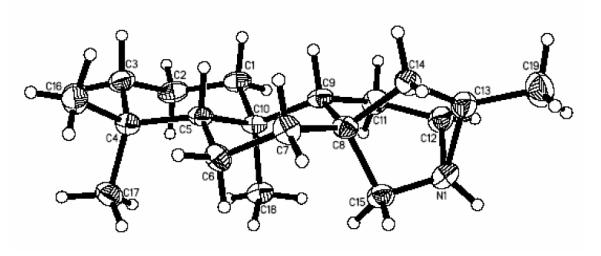
ORTEP diagram from the X-ray crystal structure of 16-aza-ent-trachylobane picrate salt (24). The picrate counterion is omitted.
ent-Beyeran-16 exo-yl Diphosphate
The beyeranyl+ carbocation intermediate (7) shown in Sch 1 is presumably accompanied by the PP·Mg counterion in the active site of the cyclase enzymes. If covalent bond formation occurred as it does in mono and sesquiterpene biosynthetic mechanisms,28-30 the resulting ent-beyeranyl PP (11) would be a new, stable intermediate. Solvolysis of beyeranyl sulfonates in fact leads to bridge rearrangements and formation of kaurane and atiserane derivatives.9a Alternatively, if the beyeranyl+ ion is tightly associated with the PP·Mg anion in the cyclase active site, beyeranyl PP might be an effective inhibitor and active site probe for diterpene synthases. These considerations and the availability of ent-beyeran-16-one (16) were incentives for synthesis of 11 (Scheme 5) and experiments with KS.
SCHEME 5.

Meerwein-Pondorff-Verley reduction of ent-beyeranone in a manner similar to that reported for isosteviol methyl ester (15b)31 afforded an approximately 1:1 mixture of the known endo and exo isomers,32 25a and 25b, which were separated by three successive chromatographies on silica gel in yields of 30 and 36%. The physical and spectral properties agreed well with the literature values. Conversion of the exo isomer to the diphosphate was accomplished by the Cramer-Danilov phosphorylation method (Bu4NH2PO4, CCl3CN, CH3CN, 25° C, 20 min; NH3, MeOH)33,34 and fractionation on a Dowex column with NH4HCO2/MeOH eluent to separate 11 from the monophosphate, and inorganic phosphates. ent-Beyeran-15 exo-yl PP was obtained as the NH4 salt (24%) and characterized by its 1H and 31P NMR spectra.
Proton NMR Spectra
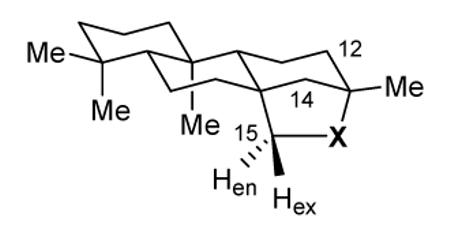
Most of the proton NMR spectra of the tetra- and pentacyclic diterpenes showed distinctive and well-separated signals for H15exo and H15endo with ΔδH ranging from 0.84 to 1.08 ppm. (Table 1) In the cases of exo and endo beyeranols (25a and 25b), the smaller chemical shift of the signals for the protons at C15 resulted in disappearance of H15endo into the broad envelope for the ring protons and only H15exo was readily observable. The consistently higher field positions of H15endo are attributable to the cumulative shielding influence of the C-C bonds in the B and C rings of the cyclic structures. In most cases, the resonance for H15exo displayed additional long range coupling (4JHH = 1.6 - 3.6 Hz), presumeably arising from the W relationship to the axial proton at C14.35 It is interesting that the altered bond angles resulting from aziridine ring formation in azatrachylobane (13) re-orient H15endo into position for W-coupling to H14axial, and consequently the additional coupling appears in the lower field resonance. The W relationship between H15endo and H14axial is apparent in the ORTEP in Fig. 3.
Table 1.
1H NMR chemical shifts (δH), chemical shift differences (ΔδH, exo - endo), multiplicities (m), and coupling constants (J) for the endo and exo protons at C15 of various ent-beyerane derivatives
| Compound No. |
X | H15 δH (ppm) |
ΔδH (ppm) |
m | J (Hz) |
|---|---|---|---|---|---|
| 16 | C=O | endo 1.75 exo 2.68 |
0.93 | d dd |
18.6 18.6, 3.6 |
| 19 | C=NOH | endo 1.94 exo 3.02 |
1.08 | d dd |
18.4 19, 3.2 |
| 23 | NCO2Me | endoa 2.90 endob 2.97 exoa 3.89 exob 3.98 |
0.99a 1.01b |
d d dd dd |
11 11 11, 2 11, 2 |
| 12 | NH | endo 2.56 exo 3.40 |
0.84 | br d br d |
10.8 10.8 |
| 13 | N | endo 2.28 exo 3.20 |
0.92 | dd d |
12, 1.6 11.6 |
| 25a | CHOH (endo OH) |
endo NDc exo 1.85 |
NDc | NDc ddd |
NDc 14.3, 4.8, 2.6 |
| 25b | CHOH (exo OH) |
endo NDc exo 2.59 |
NDc | NDc ddd |
NDc 14.3, 7.6, 2.4 |
Major rotamer.
Minor rotamer.
ND = not determined.
The proton NMR spectra of the tricyclic and tetracyclic carbamates 20a and 23 show doubling of several peaks (OMe, vinyl protons and, in the case of 23, the C13 bridge head methyl and CH2N) clearly indicating mixtures of rotamers about the N-CO2Me bonds. The rotamer ratios determined from the integrals of the OCH3 singlets were 11:1 for 20a and 1.7:1 for 23. The data for the distinctive peaks for H15exo and H15endo in the spectrum of 23 are presented in Table 1 Noticeably broadened doublets at 1.98 and 2.16 ppm in the spectrum are assigned to the equatorial protons at C12 in the minor and major rotamers on the expectation of a deshielding influence of the proximal carbamate π electrons.
The proton NMR spectrum for the mercurial intermediate (22) in the heterocyclization reaction (Scheme 3) was quite similar to that of the cyclic carbamate product 23, except for the appearance of two additional broad singlets at 3.37 (~0.65H, W1/2 7-8 Hz) and 3.56 ppm (0.35H, W1/2 7-8 Hz). Additional signals confirming a 1.8:1 mixture of rotamers were apparent for H15endo and H15exo, the methoxy singlets, the C13 bridgehead methyls, and other peaks. A chemical shift increment of ΔδH +1.2 to +1.4 ppm for the substitution CH2 → CHHgX was estimated from literature data36 and provided the basis for assignment of the broad singlets to CHHgO2CCF3. The W1/2 values of the rotamer peaks for CHHgO2CCF3 are consistent with overlapping equatorial/axial and axial/axial couplings arising from interaction of equatorial H12α with the adjacent C11 protons. Hence, the HgO2CCF3 group must be axial as would be expected from trans diaxial addition of Hg(O2CCF3)2 across the 12,13 C=C in the mercury-induced cyclization (20a → 22).
X-Ray Crystal Structures
An X-ray diffraction analysis of azatrachylobane picrate salt (24) was carried out to confirm the structure and to obtain bonding parameters for the charged aziridinium ring (Fig. 3 and Table 2). A crystallographic determination was also conducted on methyl ent-trachyloban-19-oate (see 3 and Supporting Information) to ascertain comparative dimensions of the cyclopropane ring in the pentacyclic diterpene. The two C-N bond lengths (1.510 and 1.516 Å) for the aziridine are quite similar, and also of comparable magnitude to those of the cyclopropane ring bonds (1.50, 1.51. and 1.52Å). In contrast, the C12-C13 bond length in the aza analogue salt is considerably shorter (1.472 Å), while the C12-N1-C13 interior bond angle (58.2°) is constricted compared to the two C-C-N bond angles (60.7 and 61.1°), and to the almost equal bond angles within the cyclopropane ring of methyl trachyloban-19-oate (59.5, 59.9, and 60.2°).
Table 2.
Internal bond lengths and bond angles within the aziridinium ring of picrate salt 24
| Bond Lengths (Å) | Bond angles (°) |
|---|---|
| C12-C13 1.472 | C12-C13-N1 60.7 |
| C12-N1 1.510 | C13-C12-N1 61.1 |
| C13-N1 1.516 | C12-N1-C13 58.2 |
In general, literature data for aziridine and its derivatives, if unperturbed by electron-withdrawing substituents on nitrogen, show somewhat longer C-C bonds than their C-N counterparts while both are notably shorter than the analogous bonds in acyclic amines.37-39 Both theoretical calculations40 for the parent aziridine and aziridinium ion and X-ray crystal structures for 8-oxa-3-azatricyclo[3.2.1.02,4]octane derivatives41 indicate shorter C-C ring bonds and longer C-N ring bonds in the protonated forms, in line with the data for 24. These small distortions seem to indicate relatively little carbocation character at the aziridinium carbons, a conclusion similar to that reached for para-substituted aryl aziridinium ions, based on molecular orbital calculations and ESCA results.42
Inhibition of ent-Kaurene Synthase
Genes encoding KS have been cloned from several plant species, and the catalytic activities of full-length proteins, i.e. including the putative N-terminal plastidial targeting sequence, were verified in assays run with recombinant bacterial extracts.4c,14c-14e In the present work, KS from Arabidopsis thaliana (AtKS) was heterologously expressed as a pseudo-mature protein (without the first 41 N-terminal residues) fused to glutathione-S-transferase (GST) in E. coli. The resulting GST-rAtKS fusion protein was purified by affinity chromatography and was immediately utilized in enzymatic assays. Incubation of ent-copalyl PP (5) with GST-rAtKS yielded the expected ent-kaurene (2) product, as verified by GC-MS based comparison to an authentic sample. Kinetic runs were conducted at 10 nM protein concentration according to recently published procedures.11 Reactions were initiated by addition of [15-3H]ent-copalyl PP (5),14e and [14-3H]ent-kaurene was isolated by extraction and analyzed by liquid scintillation counting.11,43 (Table 3)
Table 3.
Pseudo-binding constant (Km) for [15-3H]-ent-copalyl PP (5) with GST-rAtKS and inhibition constants (IC50) for ent, exo-beyeran-16-yl PP, 16-aza-ent-beyerane, and 16-aza-ent-trachylobane (11, 12, and 13).a
| Substrate/Inhibitor | Km or IC50 (M) | IC50 (+ PPi)b (M) |
|---|---|---|
| 5 | 3±1 × 10−7 | – – |
| 11 | 1±0.3 × 10−5 | – – |
| 12 | 1±0.4 × 10−7 | 4±1 × 10−9 |
| 13 | 1±0.2 × 10−6 | 2±0.5 × 10−8 |
Incubation conditions: 10 nM GST-rAtKS + 1.5 μM ent-CPP (5).
Inhibition by PPi alone was negligible at 1 mM concentration.
Studies with native KS partially purified from wild cucumber (Marah macrocarpus) seeds demonstrated that ring rearrangement occurs during the enzyme catalyzed cyclization of ent-copalyl PP.10a Stereo-electronic considerations dictate that the ring D rearrangement cannot be concerted with the preceding cyclization 6 → 7 to the beyeranyl+ intermediate. Thus, there must be a localized secondary beyeranyl carbocation intermediate 7 in the mechanism which undergoes a thermodynamically favorable rearrangement to the tertiary kauranyl carbocation 8. (Scheme 1) One possibility for ‘stabilization’ of the energetically unfavorable secondary carbocation would be covalent bond formation with the PP·Mg anion released in the initiating ionization of ent-copalyl PP (5), producing an exo-beyeranyl PP intermediate (11). Inhibition studies demonstrated that 11 apparently does bind in the GST-rAtKS active site, although it appears to be a relatively weak competitive inhibitor with IC50 = 10 μM, when compared with the pseudo-binding constant of the enzyme for the native substrate (5, Km = 0.3 μM). Incubation of GST-rAtKS with saturating amounts of 11 (50 μM) did not result in the formation of any detectable kaurene, apparently ruling out a role for the tetracyclic PP, at least as a free intermediate, in the KS-catalyzed cyclization reaction.
Inhibition studies with 16-azabeyerane (12), and 16-azatrachylobane (13) were conducted to evaluate the capability of the aza-diterpene analogues to inhibit the normal cyclization reaction and, hence, to estimate the extent to which 12 and 13 mimic secondary carbocation (7) and a bridged non-classical carbocation (not shown), respectively, on the way to the tertiary ent-kauranyl ion 8 (Sch 1, Table 3). The two aza diterpenes are clearly bound in the active site, as indicated by their competitive inhibition effects; 12 with an IC50 = 0.1 μM and 13 with an IC50 = 1 μM, both similar to the 0.3 μM Km for ent-copalyl PP (5). Intriguingly, extended incubation of 13 with led to increased inhibition, suggesting that it may covalently modify GST-rAtKS (i.e. irreversible inhibition), and the IC50 values reported here should be regarded as upper limits on the actual affinity of the binding interaction between 13 and GST-rAtKS. The binding of these heterocyclic analogues was also assayed in the presence of inorganic pyrophosphate (PPi) to mimic the presence of the PPi anion resulting from ionization of 5. The addition of 1 mM PPi, which does not inhibit GST-rAtKS alone, led to a large increase in the ability of 12 to bind to, and to inhibit, GST-rAtKS, with IC50 = 4 nM, corresponding to a 25-fold increase in affinity. Given that the assays contain 10 nM GST-rAtKS, this low IC50 value indicates that the true affinity is sub-nanomolar much as observed previously for abietadiene synthase.18 Similar binding enhancement was also observed with 13, which exhibited an IC50 = 20 nM in the presence of 1 mM PPi. These synergistic effects presumably signify formation of a beyeranyl+ carbocation PP·Mg ion pair intermediate in the KS-catalyzed cyclization reaction mechanism (i.e. as drawn for 7 in Scheme 1).
The less significant synergy exhibited by 13 with PPi might reflect the presumably lower free energy profile for the mimicked trachylobanyl bridged carbocation intermediate (secondary-tertiary) relative to the preceding beyeranyl secondary carbocation. However, particularly given the very similar structures of 12 and 13, it seems more likely that the reduced affinity of 13 relative to 12 (in either the absence or presence of PPi) is a function of the lower basicity of the aziridine nitrogen (parent aziridinium ion pKa ~ 8.0)44 relative to that for the bridged pyrrolidine in 12 (pyrrolidinium ion pKa 11.3).44 Thus azabeyerane would exist exclusively in the ammonium form in the incubation medium at pH 7.4, and it is likely bound to the enzyme in the protonated state, as seems to be the case for azabornane in the active site of bornyl PP synthase.17 By contrast, the proportion of the bridged aziridine in 13 existing in the binding-competent, protonated state would be much lower. Since aziridinium ions are known to act as electrophiles in solution,37,38,45 There is a distinct possibility that the protonated form of 13 reacts slowly with an active site nucleophile, thus inactivating the enzyme and explaining the increased inhibition observed in extended incubations mentioned above.
The relatively weak affinity observed for ent-beyeranyl PP (11) implies that the beyeranyl+ carbocation and the PP·Mg anion leaving group are further separated in the KS active site than a typical covalent C-O bond length (1.42 Å)46. This disparity presumably arises from separation of the PP·Mg anion and the hydrocarbon moieties of 5 following ionization by changes in their relative positions and/or orientations. From previous structural studies it seems most likely that absence of covalent bond formation reflects re-positioning of the hydrocarbon moiety, since the bound PP·Mg counter ions occupy a relatively constant position in various terpene synthase-substrate analogue co-crystals.19
Conclusions
The lack of any detectable conversion of ent-beyeran-16-yl PP (11) to ent-kaurene in incubations with ent-kaurene synthase appears to exclude this secondary PP as a free intermediate in the enzyme-catalyzed bicyclization of ent-copalyl PP (5). The enhanced affinities of 16-aza-ent-beyerane and 16-aza-ent-trachylobane for KS in the presence of inorganic PP, in comparison to the binding of the bicyclic substrate, indicate that the heterocycles behave as transition state inhibitors, and are good candidates for active site probes for this key enzyme associated with gibberellin phytohormone biosynthesis in plants. In addition, the strong synergy exhibited by these compounds with inorganic PP suggests substantial stabilization of the corresponding intermediates, in particular beyeran-16-yl+, by ion pairing with the PP·Mg released in the initial ionization of the ent-copalyl PP substrate. Similar findings have been reported with other terpene synthases,17,18 suggesting that the use of the charged PP·Mg complex as a counter ion to stabilize proximal secondary carbocation intermediates may be a relatively common enzymatic mechanism utilized by these enzymes. However, such counter ion stabilization seems to be limited to carbocations located relatively close to the original PP position, as indicated by the weaker synergy exhibited by distal aza analogues and structural studies.17,19 Indeed, it has been suggested that the PP·Mg anion may drive reactions towards localization of the carbocation intermediates close to the negative charge, particularly in the absence of other stabilizing influences in the active site.11 Finally, the fact that ent-beyeran-16-yl PP does not appear to be a free intermediate further emphasizes the tight control KS must exert over the ionized carbocation intermediates and PP·Mg anion within its active site to prevent recombination of the closely associated ion pair.
Experimental Section (See Supporting Information for procedures and data for other compounds) Methyl 16-ent-Azabeyerane-16-carboxylate (23.)
A literature procedure25a was followed with modification. A solution of commercially available mercuric trifluoroacetate25b (65 mg, 0.15 mmol) and carbamate 20a (44 mg, 0.132 mmol) in 3 mL of THF was purged with nitrogen, covered with aluminum foil, and stirred at room temp for 16 h. The reaction was judged to be complete by TLC analysis (Rf 0.22, 1:4 EtOAc : petroleum ether). A solution of NaBH4 (6 mg, 0.14 mmol) in 0.1 mL of 2.5 M NaOH was added dropwise. After another 8 h, aq. Na2CO3 (1 mL) was added, and stirring was continued for an additional 4 h. The suspension was concentrated to remove THF, and the residue was extracted with ether (2 × 20 mL). The combined ethereal extracts were washed with brine (2 × 10 mL), dried (MgSO4), and concentrated. The resulting solid material (42 mg, 96%) showed the following properties for a 1.7:1 mixture of N-CO2Me rotamers: TLC Rf 0.23 (1:9 EtOAc:hexane); FTIR (CHCl3) νmax 3017, 2950, 1682, 1449, 1387 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.80 (s, 3H, CH3), 0.85 (s, 3H, CH3), 0.93 (s, ca 1.9 H, CH3), 0.94 (s, ca 1.1H CH3), 1.34 (s, ca 1.1H, CH3), 1.20-1.72 (m, 12.5 H), 1.44 (s, 1.9H, CH3), 1.98 ( br d, ~ 0.4 H, J ≅ 8 Hz, H12α), 2.16 (br d, ~0.6 H, J ≅ 8 Hz, H12α), 2.90 (app d, ~ 0.6 H, J ≅10 Hz, H15 endo), 2.97 (app d, ~ 0.4 H, J ≅ 11 Hz, H15 endo), 3.64 (s, ~ 1.9 H, CO2CH3), 3.66 (s, ~1.1 H, CO2CH3), 3.89 (app dd, ~ 0.6 H, J ≅ 11, 2 Hz, H15 exo), 3.98 (app dd, ~0.4H, J ≅ 11, 2 Hz, H15 exo). The crude product was hydrolyzed to azabeyerane (12) without further purification.
16-Aza-ent-beyerane (12)
The carbamate hydrolysis conditions were modeled after a literature procedure.47 A mixture of 23 (44 mg, 0.13 mmol), 1,2-propylene glycol (1 mL), water (0.1 mL), and KOH (336 mg, 6 mmol) was heated at reflux for 18 h, cooled to room temp, diluted with water (5 mL), and extracted with ether (3 × 15 mL). The combined organic extracts were washed with water (1 × 10 mL) and dried (MgSO4). Evaporation of the solvent afforded azabeyerane (12, 33 mg, 91%) as a clear oil: TLC Rf 0.22 (10:89:1 EtOAc : hexane : triethylamine); [α]D25 -10.6 (c = 2.5, CHCl3); FTIR (neat) νmax 3312, 2922, 1699, 1454 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.79 (s, 3H, CH3), 0.78-0.88 (m, 2H ), 0.85 (s, 3H, CH3), 0.93 (s, 3H, CH3), 1.05-1.26 (m, 4H), 1.16 (s, 3H, CH3), 1.33-1.71(m, 12H), 2.56 (d, 1H, J = 10.8 Hz, H15endo), 3.40 (d, 1H, J = 10.8 Hz, H15exo); 13C NMR (125.64 MHz, CDCl3) δ 15.1, 18.6, 20.3, 20.4, 20.6, 22.0, 22.1, 26.9, 33.3, 33.9, 37.9, 39.1, 39.8, 39.9, 42.1, 45.5, 55.3, 56.1, 56.5; HRMS (ESI) m/z calcd for C19H34N (M+H)+ 276.2691. Found 276.2687.
ent-8β-Aminomethyl-13-methyl-12-podocarpene (21).48
The hydrolysis was modeled after a literature procedure.47 A solution of 20a ( 150 mg, 0.45 mmol) and KOH (1 g, 17.85 mmol) in 1.8 mL of methanol and water (0.2 mL) was heated at reflux for 8 h, cooled to room temp, diluted with water (8 mL), and extracted with ether (3 × 20 mL). The combined organic extracts were washed with water (2 × 10 mL) and dried (MgSO4). Evaporation of the solvent afforded primary amine 21 (117 mg, 95%) as a clear oil: [α]D25 -14 (c = 2.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.75-0.98 (m, 2H), 0.81 (s, 3H, CH3), 0.83 (s, 3H, CH3), 0.86 (s, 3H, CH3), 1.08-1.17 (m, 1H), 1.23-1.41 (m, 6H), 1.46-1.61(m, 5H), 1.62(s, 3H, CH3 at C-13), 1.81-2.08 (m, 4H), 2.61 (dd, 1H, J = 13.5, 1.5 Hz, CH2N), 2.70 (d, 1h, J = 13.5 Hz, CH2N), 5.37 (br s, 1H, W1/2 = ~ 7 Hz, =CH); 13C NMR (100.57 MHz, CDCl3) δ 15.9, 18.4, 18.6, 21.6, 22.4, 23.6, 33.4, 36.5, 37.1, 39.7, 40.3, 41.9, 42.6, 53.8, 57.2, 120.9, 131.1; HRMS (ESI) m/z Calcd for C19H34N (M+H)+ 276.2691. Found 276.2688.
16-Aza-ent-trachylobane (13)
The aziridination conditions were modeled after a literature procedure.27 A solution of amine 21 (100 mg, 0.36 mmol) in dry benzene ( 3 mL) was quickly added to a suspension of Pb(OAc)4 (225 mg, 0.51 mmol), anhyd K2CO3(398 mg, 2.88 mmol), and dry benzene ( 2 mL) being stirred and cooled at 0 °C. The mixture was warmed to 15 °C and stirred for 1 h after which 10% aq. NaOH (10 mL, 10%) was added to quench the reaction. The product was extracted with ether (3 × 20 mL). The combined ethereal extracts were washed with water (2 × 15 mL) and brine (1 × 15 mL), and dried (MgSO4). Evaporation of the solvent afforded aziridine 13 (94 mg, 94%) as a clear, unstable, oil that decomposed within 5 h. A solution in hexane stored under N2 at −20 °C remained unchanged for at least several weeks. Properties of 13: [α]D25 -5.5 (c = 2.0, CHCl3); TLC Rf = 0.42 (1:4 EtOAc:hexane); 1H NMR (400 MHz, CDCl3) : δ 0.66-0.85 (m, 2H), 0.77 (s, 3H, CH3), 0.82 (s, 3H, CH3), 0.95 (s, 3H, CH3), 1.03-1.23 (m,3H), 1.23 (s, 3H, CH3 at C-13), 1.27-1.4 (m , ~ 3H), 1.4-1.56 (m, ~ 8H), 1.77 (br s, W1/2 = 8 Hz, ~1 H), 1.78-1.93 (16 line centrosym m, 2H), 2.28 (dd, 1H, J = 12, 1.6 Hz, CH2N), 3.20 (d, 1H, J = 11.6 Hz, CH2N); 13C NMR (100.57 MHz, CDCl3) δ 14.4, 18.0, 18.1, 20.2, 20.6, 21.7, 32.8, 33.4, 35.3, 37.8, 39.1, 40.3, 40.9, 41.9, 43.7, 48.6, 51.6, 55.2, 56.2.
Picrate Derivative of 16-Aza-ent-trachylobane. (24)
A solution of 13 (30 mg, 0.11 mmol) in dry ether (2 mL) was added to a stirred solution of picric acid (0.11 mmol, 26 mg) in 1 mL of methanol at 0 °C. The picrate salt precipitated immediately. After 10 min. the solvents were evaporated. Crystallization from 9:1 benzene and acetone furnished 24 as yellow crystals: yield, 52 mg (94%); mp 232 - 234° C; [α]D25 -28.6 (c = 1.28, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.74-0.85 (m, 2H), 0.79 (s, 3H, CH3), 0.85 (s, 3H, CH3), 1.00 (s, 3H, CH3), 1.08-1.31 (m,2H), 1.34-1.45 (m, 3H), 1.48-1.71 (m, 5H), 1.63 (s, 3H, CH3 at C-13), 1.83 and 1.91 (ABdd, 2H, JAB = 13.6 Hz), 2.15-2.20 (16 line m, 2H), 3.01 (dd, 1H, J = 11.2, 2 Hz, CH2N) 1H), 3.17 (bs, 1H, W1/2 = ca 7 Hz), 3.76 (d, 1H, J = 11.2 Hz, CH2N), 8.85 (s, 2H, ArH), 10.79 (bs, 1H, NH); 13C NMR (100.57 MHz, CDCl3) δ 14.1, 17.06, 17.1, 17.7, 19.7, 21.6, 32.9, 33.2, 34.4, 37.8, 38.7, 40.0, 41.5, 44.7, 45.9, 49.5, 50.9, 53.3, 55.0, 126.5, 128.4, 141.7, 161.8.
Inhibition of ent-Kaurene Synthase
Assays for kinetic analysis were performed with purified GST-rAtKS, the concentration of which was determined using A280 and the calculated extinction co-efficient ε = 172,850 M−1 cm−1, prior to dilution in assay buffer (50 mM Hepes, pH 7.2; 10% (v/v) glycerol; 7.5 mM MgCl2; 0.1 mg/mL α-casein; 5 mM DTT) to 10 nM. Reactions were run at room temp, initiated by the addition of [15-3H]ent-copalyl PP,14e allowed to react for 1 min, and terminated by the addition of KOH to 0.2 M and EDTA to 15 mM, as described.43 The production of ent-kaurene was quantified by extracting the incubation solutions with hexanes and passing the organic extracts through short columns of MgSO4 and silica gel to dry them and to remove any oxygenated diterpenoids resulting from non-specific solvolysis prior to liquid scintillation counting.43 Inhibition studies were carried out with saturating amounts of substrate (1.5 μM [15-3H]ent-copalyl PP) and varying amounts of inhibitor, and in the case of 13 it was necessary to add substrate directly after inhibitor to detect any enzymatic activity. The resulting data were fit to an exponential equation using KaleidaGraph 4.0 (Synergy Software) to determine IC50 values as previously described.18a
Supplementary Material
Acknowledgements
This work was supported by grants from the National Science Foundation (MCB-0416948 to RJP) and the National Institutes of Health (GM076324 to RJP and GM13956 to RMC). We thank Dr. Scott Wilson for assistance with the X-ray crystal structure determinations.
Footnotes
Supporting Information Available General aspects and instrumentation; procedures and characterization data for 15a - 20b, 22, 23, 25a, 25b, and 11; reproductions of NMR spectra; and ORTEP diagrams and tables of X-ray data for 18a, methyl trachyloban-19-oate, and azatrachlobane picrate salt (24). This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.All beyerane, kaurane, and trachylobane derivatives prepared or discussed in this paper belong to the enantiomeric (ent) family of biogenetically related diterpenes. The ent prefix descriptor is attached to the chemical names (eg, ent-beyerane, ent-kaurane, ent-trachylbane) frequently in the text to remind the reader of the absolute configuration. Complete semi-systematic names are given as headings in the Experimental Section and Supporting Information. For IUPAC nomenclature guidelines see: Riguady J, Klesney SP. Nomenclature of Organic Chemistry, Sections A, B, C, D, E, F, and H. Pergamon; Oxford: 1979. , and ref 2b.
- 2 (a).Dev S, Misra R. In: CRC Handbook of Terpenoids: Diterpenoids. Dev S, editor. Vol. IV. CRC Press; Boca Raton, FL: 1986. . Connolly JD, Hill RA. Di-and Higher Terpenoids. Vol. 2. Chapman & Hall; London: 1991. Dictionary of Terpenoids.. Glasby JS. Encyclopedia of the Terpen-oids. Vols. 1 and 2. Wiley; Chichester: 1982. 1982.. Buckingham J, editor. Dictionary of Natural Products. Chapman and Hall; London: 1994. , Hanson JR. Nat. Prod. Rep. 2006;23:875–885. doi: 10.1039/b516326a. and preceding annual reviews in this series. Buckingham J. Dictionary of Natural Products (on-line web edition) Chapman & Hall/CRC Press; London: 2002.
- 3 (a).MacMillan J, Beale MH. Diterpene Biosynthesis. In Isoprenoids Including Carotenoids and Steroids. In: Cane DE, Barton D, Nakanishi K, Meth-Cohn O, editors. Comprehensive Natural Products Chemistry. Vol. 2. Elsevier; Amsterdam: 1999. pp. 217–243. Chap. 8. [Google Scholar]; (b) Mander LN. Nat. Prod. Rep. 2003;20:49–69. doi: 10.1039/b007744p. [DOI] [PubMed] [Google Scholar]
- 4 (a).Kinghorn AD. Medicinal and Aromatic Plants--Industrial Profiles. 2002;19:1–17. (Stevia) [Google Scholar]; (b) Geuns Jan M. C. Phytochemistry. 2003;64:913–921. doi: 10.1016/s0031-9422(03)00426-6. [DOI] [PubMed] [Google Scholar]; (c) Richman AS, Gijzen M, Starratt AN, Yang Z, Brandle JE. Plant J. 1999;19:411–421. doi: 10.1046/j.1365-313x.1999.00531.x. [DOI] [PubMed] [Google Scholar]
- 5.Morris BD, Foster SP, Grugel S, Charlet LD. J. Chem. Ecol. 2005;31:89–102. doi: 10.1007/s10886-005-0976-2. [DOI] [PubMed] [Google Scholar]
- 6 (a).Castrillo A, Heras B. de las, Hortelano S, Rodríguez B, Villar A, Boscá L. J. Biol. Chem. 2001;276:15854–15860. doi: 10.1074/jbc.M100010200. [DOI] [PubMed] [Google Scholar]; (b) Rundle NT, Nelson J, Flory MR, Joseph J, Th’ng J, Aebersold R, Dasso M, Andersen RJ, Roberge M. ACS Chem. Biol. 2006;1:443–450. doi: 10.1021/cb600196w. [DOI] [PubMed] [Google Scholar]; (c) Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ-M, Brandolin G. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 7 (a).Baccelli C, Block S, Van Holle B, Schanck A, Chapon D, Tinant B, Van Meervelt L, Morel N, Quetin-Leclercq J. Planta Medica. 2005;71:1036–1039. doi: 10.1055/s-2005-873123.. (b) For a review on the trachylobane diterpenes, see Fraga BM. Phytochem. Anal. 1994;5:49–56.
- 8 (a).West CA. In: Biosynthesis of Isoprenoid Compounds. Porter JW, Spurgeon SL, editors. Vol 1. Wiley; New York: 1983. Ch. 7. [Google Scholar]; (b) Davis EM, Croteau R. Top. Curr. Chem. 2000;209:53–95. [Google Scholar]
- 9 (a).For a review see, Coates RM. Herz W, Grisebach H, Kirby JW, editors. Progress in the Chemistry of Organic Natural Products. 1976;33:73–230. doi: 10.1007/978-3-7091-3262-3_2.. Coates RM, Kang H-Y. J. Chem. Soc., Chem. Commun. 1987:232–233.
- 10 (a).Coates RM, Cavender PL. J. Am. Chem. Soc. 1980;102:6358–6359. [Google Scholar]; (b) Drengler KA, Coates RM. J. Chem. Soc., Chem. Commun. 1980:856–857. [Google Scholar]; (c) Coates RM, Koch SC, Hegde S. J. Am. Chem. Soc. 1986;108:2762–2764. [Google Scholar]
- 11.Xu M, Wilderman PR, Peters RJ. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7397–7401. doi: 10.1073/pnas.0611454104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12 (a).Duncan JD, West CA. Plant Physiol. 1981;68:1128–1134. doi: 10.1104/pp.68.5.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shen-Miller J, West CA. Phytochemistry. 1985;24:461–464. [Google Scholar]; (c) Railton ID, Fellows B, West CA. Phytochemistry. 1984;23:1261–1267. [Google Scholar]
- 13 (a).Robinson DR, West CA. Biochemistry. 1970;9:70–79. 80–89. doi: 10.1021/bi00803a010. [DOI] [PubMed] [Google Scholar]; (b) Spickett CM, Ponnamperuma A, Abell C. Phytochemistry. 1994;37:971–973. [Google Scholar]; (c) Frost RG, West CA. Plant Physiol. 1977;59:22–29. doi: 10.1104/pp.59.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14 (a).Kawaide H, Sassa T, Kamiya Y. J. Biol. Chem. 2000;275:2276–2280. doi: 10.1074/jbc.275.4.2276., and refs cited. Toyomasu T, Kawaide H, Ishizaki A, Shinoda S, Otsuka M, Mitsuhashi W, Sassa T. Biosci. Biotechnol. Biochem. 2000;64:660–664. doi: 10.1271/bbb.64.660. Yamaguchi S, Sun T, Kawaide H, Kamiya Y. Plant Physiol. 1998;116:1271–1278. doi: 10.1104/pp.116.4.1271. Yamaguchi S, Saito T, Abe H, Yamane H, Murofushi N, Kamiya Y. Plant J. 1996;10:203–213. doi: 10.1046/j.1365-313x.1996.10020203.x. Xu M, Wilderman PR, Morrone D, Xu J, Roy A, Margis-Pinheiro M, Upadhyaya NM, Coates RM, Peters RJ. Phytochemistry. 2007;68:312–326. doi: 10.1016/j.phytochem.2006.10.016. Hayashi K.-i, Kawaide H, Notomi M, Sakigi Y, Matsuo A, Nozaki H. FEBS Lett. 2006;580:6175–6181. doi: 10.1016/j.febslet.2006.10.018.
- 15 (a).Narula AS, Rahier A, Benveniste P, Schuber F. J. Am. Chem. Soc. 1981;103:2408–2409. [Google Scholar]; (b) Rahier A, Taton M, Benveniste P. Biochem. Soc. Trans. 1990:48–52. doi: 10.1042/bst0180048. [DOI] [PubMed] [Google Scholar]; (c) Poulter CD. Acc. Chem. Res. 1990;23:70–77. [Google Scholar]; (d) Abe I, Tomesch JC, Wattanasin S, Prestwich GD. Nat. Prod. Rep. 1994;11:279–302. doi: 10.1039/np9941100279. [DOI] [PubMed] [Google Scholar]
- 16.Burbiel J, Bracher F. Steroids. 2003;68:587–594. doi: 10.1016/s0039-128x(03)00080-1. [DOI] [PubMed] [Google Scholar]
- 17.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau R, Christianson DW. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18 (a).Peters RJ, Ravn MM, Coates RM, Croteau R. J. Am. Chem. Soc. 2001;123:8974–8978. doi: 10.1021/ja010670k. [DOI] [PubMed] [Google Scholar]; (b) Ravn MM, Peters RJ, Coates RM, Croteau R. J. Am. Chem. Soc. 2002;124:6998–7006. doi: 10.1021/ja017734b. [DOI] [PubMed] [Google Scholar]
- 19.Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 20 (a).Ruddat M, Heftmann E, Lang A. Arch. Biochem. Biophys. 1965;110:496–499. doi: 10.1016/0003-9861(65)90441-8. [DOI] [PubMed] [Google Scholar]; (b) Mossetig E, Beglinger U, Dolder F, Lichti H, Quitt P, Waters JA. J. Am. Chem. Soc. 1963;85:2305–2309. [Google Scholar]; (c) Wood HB, Jr., Allerton R, Diehl HW, Fletcher HG., Jr. J. Org. Chem. 1955;20:875–883. [Google Scholar]
- 21 (a).Coates RM, Kang H-Y. J. Org. Chem. 1987;52:2065–2074. [Google Scholar]; (b) Crossley NS, Dowell RJ. Chem. Soc. C. 1971:2496–2498. doi: 10.1039/j39710002496. [DOI] [PubMed] [Google Scholar]
- 22.Mammato DC, Eadon GA. J. Org. Chem. 1975;40:1784–1792. doi: 10.1021/jo00900a024. [DOI] [PubMed] [Google Scholar]
- 23 (a).Shioiri T, Ninomiya K, Yamada S-i. J. Am. Chem. Soc. 94:6203–6205. doi: 10.1021/ja00772a052. 172. [DOI] [PubMed] [Google Scholar]; (b) Yamada S-i., Ninomiya K, Shioiri T. Tetrahedron Lett. 1973;14:2343–2346. [Google Scholar]
- 24.De Kimpe N, Boeykens M, Tehrani KA. J. Org. Chem. 1994;59:8215–8219. [Google Scholar]
- 25 (a).Harding KE, Burks SR. J. Org. Chem. 1981;46:3920–3922. [Google Scholar]; (b) Kocovsky P. In: Encyclopedia of Reagents for Organic Synthesis. Paquette LA, editor. Wiley; Chichester: 1995. pp. 3267–3270. [Google Scholar]
- 26.Nagata W, Hirai S, Kawata K, Aoki T. J. Am Chem. Soc. 1967;89:5045–5046. [Google Scholar]
- 27.Portoghese PS, Sepp DT. Tetrahedron. 1973;29:2253–2256. [Google Scholar]
- 28.Wise ML, Croteau R. In: Comprehensive Natural Product Chemistry. Cane DE, editor. Vol. 2. Elsevier Science; Oxford: 1999. pp. 97–153. [Google Scholar]
- 29 (a).Croteau R, Karp F. Arch. Biochem. Biophys. 1979;198:512–522. doi: 10.1016/0003-9861(79)90526-5. [DOI] [PubMed] [Google Scholar]; (b) Croteau R, Shaskus J. Ibid. 1985;236:535–543. doi: 10.1016/0003-9861(85)90656-3. [DOI] [PubMed] [Google Scholar]
- 30.Cane DE. In: Comprehensive Natural Product Chemistry. Cane DE, editor. Vol. 2. Elsevier Science; Oxford: 1999. pp. 156–200. [Google Scholar]
- 31 (a).Wilcox CF, Sexton M, Jr., Wilcox MF. J. Org. Chem. 1963;28:1079–1082. [Google Scholar]; (b) Coates RM, Bertram EF. J. Org. Chem. 1971;36:2625–2631. [Google Scholar]
- 32 (a).Sobti RR, Dev S. Tetrahedron Lett. 1966;33:3939–3942. [Google Scholar]; (b) Bell RA, Ireland RE, Mander LN. J. Org. Chem. 1966;31:2536–2542. doi: 10.1021/jo01346a025. [DOI] [PubMed] [Google Scholar]
- 33.Assink BK. MS thesis. University of Illinois at Urbana-Champaign; 1999. [Google Scholar]
- 34 (a).Danilov LL, Mal’tsev SD, Shibaev VN. Soviet J. Bioorg. Chem. 1988;14:712–714. 1287–1289. [Google Scholar]; (b) Cramer F, Rittersdorf WR. Tetrahedron. 1967;23:3015–3022. [Google Scholar]
- 35.Pretsch E, Bühlmann P, Affolter C. Structure Determination of Organic Compounds. Springer-Verlag; Berlin: 2000. pp. 177–178. [Google Scholar]
- 36 (a).Kitching W, Atkins AR, Wickham G, Alberta V. J. Org. Chem. 1981;46:563–570. [Google Scholar]; (b) Coxon JM, Steel PJ, Whittington BI, Battiste MA. Ibid. 1989;54:1383–1391. [Google Scholar]; (c) Coxon JM, Steel PJ, Whittington BI. Ibid. 1990;55:4136–4144. [Google Scholar]
- 37 (a).Pearson WH, Lian BW, Bergmeier SC. In: Comprehensive Heterocyclic Chemistry II. Katritsky AR, Rees CW, Scriven EFV, editors. Vol. 1A. Elsevier; Oxford: 1996. pp. 1–60. [Google Scholar]; (b) Rai KM, Hassner A. Ibid. :61–96. [Google Scholar]
- 38 (a).Lwowski W. In: Comprehensive Heterocyclic Chemistry. Katritsky AR, Rees CW, editors. Vol. 7. Pergamon; Oxford: 1984. pp. 1–16. [Google Scholar]; (b) Padwa A, Woolhouse AD. Ibid. :47–93. [Google Scholar]
- 39 (a).Selected X-ray crystal structures of aziridines: Chinnakali K, Fun H-K, Sriraghavan K, Ramakrishnan VT, Razak IA. Acta Cryst. 1998;C54:1299–1301. Dyakonenko VV, Shiskin OV, Zbruev AV, Desenko SM. Acta Cryst. 2005;E61:o667–o668. Cheikh RB, Chaabouni R, Kallel A. Acta Cryst. 1992;C48:283–286. Ko T-M, Olansky L, Moncrief JW. Acta Cryst. 1975;B31:1875–1878.
- 40.Cremer D, Kraka E. J. Am. Chem. Soc. 1985;107:3800–3810. [Google Scholar]
- 41.Pinkerton AA, Gallacher AC, Radil M, Kunze A, Allemann S, Vogel P. Acta Cryst. 1993;B49:328–334. [Google Scholar]
- 42.Crist DR, Turujman SA, Hashmall JA. J. Heterocyc/. Chem. 1991;28:1993–1995. [Google Scholar]
- 43.Peters RJ, Flory JE, Jetter R, Ravn MM, Lee H-J, Coates RM, Croteau RB. Biochemistry. 2000;39:15592–15602. doi: 10.1021/bi001997l. [DOI] [PubMed] [Google Scholar]
- 44.Dean JA. Lange’s Handbook of Chemistry. 15th ed McGraw-Hill; New York: 1999. pp. 8–30. [Google Scholar]
- 45 (a).Chuang T-H, Sharpless KB. Org. Lett. 2000;2:3555–3557. doi: 10.1021/ol000221+. [DOI] [PubMed] [Google Scholar]; (b) Crist DR, Leonard NJ. Angew. Chem. Int. Ed. Engl. 1969;8:962–974. [Google Scholar]
- 46.Carey FA, Sundberg RJ. Advanced Organic Chemistry Part A. 4th ed Kluwer Academic; New York: 2000. p. 13. [Google Scholar]
- 47.Angle SR, Arnaiz DO. Tetrahedron Lett. 1989;30:515–518. [Google Scholar]
- 48.The IUPAC nomenclature was followed according to which the α or β configuration of substituents in ent-diterpenes is that designated in the systematic name for the “normal” antipode. See Rigaudy J, Klesney SP. Nomenclature of Organic Chemistry, Sections A, B, C, D, E, F, and H. Pergamon Press; Oxford: 1979. p. 511.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.