

Abstract
Structural characterization of the phosphate complex with a thiophene-based macrocycle suggests that two dihydrogen phosphates in a dimeric form are encapsulated in the cavity via several hydrogen bonds from NH···O and CH···O interactions. In the lattice framework, the two dimers are linearly hydrogen-bonded to form a tetramer. 1H NMR titrations suggest that the host forms a 1:1 complex with phosphate, showing an association constant of 120 M−1 in D2O at pH = 5.5. The host guest complexation was further confirmed by ESI-MS in a gas phase.
Keywords: Azamacrocycle, Ditopic complex, Anion binding, Phosphate complex
Phosphate is ubiquitous in Nature and is widely used in the production of fertilizers [1]. It is also used as preservatives in foods and as additives to household detergents. Furthermore, phosphate is a key component of nucleic acids (DNA and RNA) and is known to play an important role in many enzymatic reactions [2]. Furthermore, crystallographic findings can aid in identifying accurate bonding patterns involved in a host guest complex. Therefore, there is an increasing interest in understanding interactions of phosphate anions with synthetic receptors particularly in aqueous solution [3]. However, the high free energy of hydration of phosphate significantly reduces its ability to complex with a synthetic molecule in water [4]. Phosphate binding has been reported by several classes of neutral receptors including amides [5], thioamides [6], ureas [7], thioureas [8], pyrroles [9] and indoles [10]; however, most of these binding studies have been performed in organic solvents. On the other hand, polyamines tend to be soluble in a polar solvent and can effectively be used in binding phosphate anions in water over a wide range of pH [11]. For examples, Martell and coworkers reported a m-xylyl-based hexaazamacrocycle forming an inclusion complex with a pyrophosphate, where the anion is hydrogen bonded to four protonated amines through two oxygen atoms [12]. Bianchi, García-España, Paoletti and coworkers studied a smaller macrocycle[18]aneN6 which in its tetraprotonated form was found to interact with two pyrophosphate anions via NH···O and CH···O bonds [13]. Bowman-James and coworkers isolated crystals of [26]aneN6C6 with mixed phosphoric acid/dihydrogen phosphate anion grown at low pH, showing a ditopic complex in which two anions were held above and below the macrocycle [14]. Increasing the dimension from monocycle to bicycle, Lu and coworkers obtained crystals of phosphate complexes with both hexa- and octa-protonated forms of p-xylyl-based cryptands [15]. However, only the octaprotonated cryptand provided an inclusion complex with one phosphate anion, suggesting that electrostatic interactions play a key role in the complex formation.
In Nature phosphate was structurally identified in the phosphate binding protein (PBP) in which the anion was held with a total of 12-hydrogen bonds [16]. Katayev, Sessler and coworkers synthesized an oligopyrrolic macrocycle which was found to complex a dianioic phosphate showing hydrogen-bond networks similar to those present at the active sites of PBP [17]. During the course of our study, we isolated crystals of phosphate complex of L, and characterized a dimeric phosphate species in the form of (H2PO4)22− within the macrocyclic cavity. Herein, we report the binding aspects of L with phosphate in water and structural characterization of the phosphate complex.
The compound L was previously reported by Dancey et al.[18] and prepared as described before [19]. Phosphate salt was obtained by adding a few drop of phosphoric acid in a solution of the free amine L (50 mg) in CH3OH (2 mL). The white precipitate formed immediately was filtered and washed with diethyl ether. Crystals suitable for X-ray analysis were obtained from slow evaporation of the salt solution in water-methanol system.
The structure of the phosphate complex of L was determined by X-ray diffraction analysis [20]. As shown in Figure 1, all the secondary amines are protonated forming a charged cavity. The macrocyle adopts a rectangular shape and two aromatic rings are almost parallel with an Ar···Ar distance (centroid to centroid) of 9.394 Å. The distance between the central nitrogens (N1···N14 is 6.741 Å which is larger than 6.296 Å observed in the perchlorate complex of L [19]. The macrocyle is found to host two dihydrogen phosphates via several hydrogen bonds from NH···O and CH···O interactions (see Table 1). One phosphate (labeled A) is bonded to the macrocycle via two strong NH···O interactions (N1···O2A = 2.69(2) and N14···O1A = 2.723(16) Å) with the central nitrogens (N1 and N14) and one weak NH···O interaction (N4···O2A = 3.21(3) Å) with one protonated secondary amine (N4) connected to an aromatic group. While other phosphate (labeled B) is bonded to the macrocycle via one strong NH···O (N4H···O4B = 2.70(2) Å) and two CH···O (3.162 and 3.276 Å) interactions. Therefore, each anion is connected to the macrocycle with three hydrogen bonds, suggesting that the charged macrocycle effectively interacts with the negatively charge phosphate.
Fig 1.
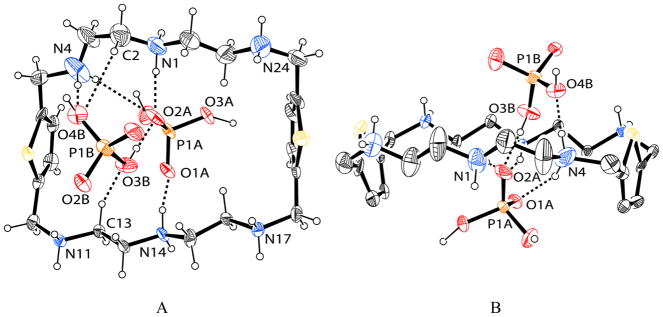
ORTEP drawing of [H6L(H2PO4)2]4+ motif, with thermal ellipsoids at the 50% probability level: (A) side view and (B) view along the tertiary N-N axis.
Table 1.
Hydrogen bonding parameters (Å, °) for H2PO4− binding in L
| D–H···A | D···A | < D–H–A |
|---|---|---|
| N1–H1B···O2A | 2.69(2) | 172.0 |
| N4–H4B···O2A | 3.21(3) | 130.6 |
| N4–H4B···O4B | 2.70(2) | 178.0 |
| N14–H14B···O1A | 2.723(16) | 157.0 |
| C2–H2A···O4B | 3.276 | 133.69 |
| C13–H13A···O3B | 3.162 | 117.78 |
| O3B–H3BO···O2A | 2.531(16) | 179.4 |
The observed oxygen-nitrogen bond distances for the phosphate binding interactions with an average of 2.83 Å are comparable to those reported for the phosphate complex of m-xylyl-based macrocycle ranging from 2.60 to 2.75 Å [12] and [18]aneN6 ranging from 2.71(6) to 3.037 (5) Å [13]. The anions remain on the both side of the macrocycle, as observed in the phosphate structure of [26]aneN6C6 reported previously [14]. However, in the present case the two phosphate anions are connected via one strong hydrogen bond (OH···O = 2.533 Å) to form a dimer. As shown in Figure 1B, the dimer sits in a perpendicular fashion to the “macrocyclic plane” rather than coplanar with it. Such arrangement was observed for the pyrophosphate structure with m-xylyl-based macrocycle reported by Martell [12]. Interestingly, each phosphate group is further connected linearly with another phosphate through two hydrogen bonds to form a tetramer as (H2PO4)44− (Figure 2A), as viewed in an extended structure along the c axis. The linear tetrameric phosphate is encircled by two macrocycles which are anti-parallel to each other (Figure 2B). The structural evidence of dimeric phosphate (H2PO4)22− formed by synthetic hosts was previously reported [21]. In our case the dimer is further H-bonded to two additional macrocycle-bound H2PO4− to form a linear tetramer.
Fig 2.

(A) Tetrameric (H2PO4)44− and (B) Space filling view of the tetrameric phosphate encircled by two macrocycles.
Solution binding affinity of the host for phosphate was carried out by 1H NMR titrations in D2O at two different pH (pH = 7.0 and 5.5). The solution pH was adjusted by the concentrated solution of TsOH and NaOD dissolved in D2O. The addition of NaH2PO4 (50 mM in D2O) to the host solution (5 mM in D2O) resulted into a downfield shift of ligand’s protons. The change in the 1H NMR signals with an increasing amount of the anion solution was analyzed by the non-linear regression method [22], giving the best fit to a 1:1 binding model (Figure 3).The calculated association constant (Kas) of L for phosphate was 120 M−1 at pH 5.5. However, the host was found to interact weakly (Kas<20 M−1) under neutral condition (pH = 7.0), suggesting that the binding is primarily influenced by electrostatic interactions.
Fig 3.

1H NMR titration curves for phosphate binding with the host (5 mM) in D2O at pH = 5.5. Changes in the chemical shifts of different protons (a = NHCH2CH2 and b = NHCH2CH2 are shown against an increasing amount of NaH2PO4 (50 mM) at room temperature.
The formation of the host-guest complex was further supported by ESI-MS experiments in a positive mode. For this purpose, a solution of phosphate complex of L was prepared in the mixture of MeOH and H2O (50:50, v/v). As shown in Figure 4, there is an intense peak at m/z 520.8 which is assigned for the singly charged species, [H4LPO4]+. The peak at m/z 260.6 is due to the formation of doubly charged [H5LPO4]2+ formed during the experiment. The peak at m/z 423.2 and 212.2 correspond to the free ligand [HL]+and [H2L]2+, respectively. The results obtained from this experiment confirm the formation of a 1:1 complex between the charged macrocycle and the phosphate anion in a gas phase, supporting the stoichiometry observed in solution.
Figure 4.

ESI-MS (positive ion mode) spectrum of the phosphate complex. The solution was prepared from the phosphate salt of L (1.0×10−5 M) in MeOH/H2O (50:50, v/v).
In summary, we have structurally characterized that a simple macrocycle encapsulates a dimeric form of dihydrogen phosphate by strong hydrogen bonding interactions. The two dimers are further connected in a linear fashion to form a phosphate tetramer. Such assembly of anions is assisted by two macrocycles which are encircled around the tetramer with multiple hydrogen bonds.
Supplementary Material
Highlight.
A thiophene-based macrocycle encapsulates two dihydrogen phosphates.
The two phosphate dimers are linearly hydrogen-bonded to form a tetramer.
The host binds a phosphate with an association constant of 120 M−1 in D2O at pH = 5.5.
The host guest complexation is confirmed by ESI-MS in gas phase.
Acknowledgments
The National Science Foundation is acknowledged for a CAREER award (CHE-1056927) to MAH. This work was supported by the National Institutes of Health (G12RR013459). The 500 NMR instrument used for this work was funded by the National Science Foundation (CHE-0821357). The assistance of Dr. Douglas R. Powell at the University of Oklahoma is acknowledged for the crystal structure analysis.
Appendix A. Supplementary material
CCDC 808448 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Supplementary data associated with this article can be found, in the online version, at doi: ???.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Bianchi A, Garcia-España E, Bowman-James K, editors. Supramolecular Chemistry of Anions. Wiley-VCH; New York: 1997. [Google Scholar]; (b) Steed JW, Atwood JL. Supramolecular Chemistry. 2. John Wiley & Sons, Ltd; Hoboken, NJ: 2009. [Google Scholar]
- 2.Bazzicalupi C, Bencini A, Biagini S, Faggi E, Meini S, Giorgi C, Spepi A, Valtancoli B. J Org Chem. 2009;74:7349–7363. doi: 10.1021/jo901423m. [DOI] [PubMed] [Google Scholar]
- 3.Amendola V, Boiocchi M, Esteban-Gómez D, Fabbrizzi L, Monzani E. Org Biomol Chem. 2005;3:2632–2639. doi: 10.1039/b504931h. [DOI] [PubMed] [Google Scholar]
- 4.Bruce JI, Dickins RS, Govenlock LJ, Gunnlaugsson T, Lopinski S, Lowe MP, Parker D, Peacock RD, Perry JJB, Aime S, Botta M. J Am Chem Soc. 2000;122:9674–9684. [Google Scholar]
- 5.Bondy CR, Loeb SJ. Coord Chem Rev. 2003;240:77–99. [Google Scholar]
- 6.(a) Hossain MA, Kang SK, Llinares LM, Powell D, Bowman-James K. Inorg Chem. 2003;42:5043–5045. doi: 10.1021/ic034735r. [DOI] [PubMed] [Google Scholar]; (b) Inoue Y, Kanbara T, Yamamoto T. Tetrahedron Lett. 2003;44:5167–5169. [Google Scholar]
- 7.(a) Brooks SJ, Caltagirone C, Cossins AJ, Gale PA, Light M. Supramol Chem. 2008;20:349–355. [Google Scholar]; (b) Lorenzo A, Aller E, Molina P. Tetrahedron. 2009;65:1397–1401. [Google Scholar]
- 8.Sessler JL, Cho DG, Stepien M, Lynch V, Waluk J, Yoon ZS, Kim D. J Am Chem Soc. 2006;128:12640–12641. doi: 10.1021/ja064675z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Jiang QQ, Darhkijav B, Liu H, Wang F, Li Z, Jiang YB. Chem - An Asian J. 2010;5:543–549. doi: 10.1002/asia.200900519. [DOI] [PubMed] [Google Scholar]; (b) Liu WX, Yang R, Li AF, Li Z, Gao YF, Luo XX, Ruan YB, Jiang YB. Organic and Biomol Chem. 2009;7:4021–4028. doi: 10.1039/b910255h. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kim J, Juwarker H, Liu X, Lah MS, Jeong KS. Chem Commun. 2010;46:764–766. doi: 10.1039/b919519j. [DOI] [PubMed] [Google Scholar]; (b) Ju J, Park M, Suk JM, Lah MS, Jeong KS. Chem Commun. 2008:3546–3548. doi: 10.1039/b804284e. [DOI] [PubMed] [Google Scholar]
- 11.García-Españ E, Díaz P, Llinares JM, Bianchi A. Coord Chem Rev. 2006;250:2952–2988. [Google Scholar]
- 12.Nation DA, Reibenspies JH, Martell AE. Inorg Chem. 1996;35:4597–4603. [Google Scholar]
- 13.Bianchi A, Escuder B, Fusi V, Garcia-España E, Giorgi G, Marcelino V, Paoletti P, Valtancoli B. J Am Chem Soc. 1999;121:6807–6815. [Google Scholar]
- 14.Gerasimchuk OA, Mason S, Llinares JM, Song MP, Alcock NW, Bowman-James K. Inorg Chem. 2000;39:1371–1375. doi: 10.1021/ic9911116. [DOI] [PubMed] [Google Scholar]
- 15.Yang Li-Zi, Li Yu, Jiang Long, Feng Xiao-Long, Lu Tong-Bu. CrystEngComm. 2009;11:2375–2380. [Google Scholar]
- 16.(a) Luecke H, Quiocho FA. Nature. 1990;347:402–406. doi: 10.1038/347402a0. [DOI] [PubMed] [Google Scholar]; (b) Yao N, Ledvina PS, Choudhary A, Quiocho FA. Biochemisrty. 1996;35:2079–2085. doi: 10.1021/bi952686r. [DOI] [PubMed] [Google Scholar]
- 17.Katayev EA, Sessler JL, Khrustalev VN, Ustynyuk YA. J Org Chem. 2007;72:7244–7252. doi: 10.1021/jo071106g. [DOI] [PubMed] [Google Scholar]
- 18.Dancey KF, Fenton ED, Moss S, Jones G, Price R. Synthetic Communications. 1986;16:795–801. [Google Scholar]
- 19.Saeed MA, Thompson JJ, Fronczek FR, Hossain MA. CrystEngComm. 2010;12:674–676. doi: 10.1039/b918152k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crystal data for 10010: C20H58.51N6O26P6S2, M = 1049.10, monoclinic, a = 15.984(9) Å, b = 20.991(12) Å, c = 26.596(16) Å, α = 90.00°, β = 101.21(2)°, γ = 90.00°, V = 8753(9) Å3, T = 100(2) K, space group C2/c, Z = 8, 22421 reflections measured, 3647 independent reflections (Rint = 0.0905). The final R1 value was 0.1513 (I > 2σ(I)).
- 21.Evans IR, Howard JAK, Evans JSO. Cryst Growth Des. 2008;8:1635–1639. [Google Scholar]
- 22.Schneider HJ, Kramer R, Simova S, Schneider U. J Am Chem Soc. 1988;110:6442–6448. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.