

Abstract
The preparation of triethylphosphine adducts of cyclic disilylated or digermylated germylenes was achieved by reaction of 1,4-dipotassio-1,1,4,4-tetrakis(trimethylsilyl)tetramethyltetrasilane with GeBr2·(dioxane) and PEt3. Phosphine abstraction with B(C6F5)3 allowed formation of the base-free germylenes, which undergo 1,2-trimethylsilyl shifts to the germylene atom to form the respective silagermene or digermene, which further dimerize in [2 + 2] cycloadditions to tricyclic compounds. The reasons responsible for the germylenes’ completely different reactivities in comparison to the previously studied analogous stannylenes and plumbylenes were elucidated in a theoretical study.
1. Introduction
Over the last few years the advent of stable N-heterocyclic carbenes (NHCs) has revolutionized several branches of chemistry.1−3 However, even before the first stable carbenes were reported already examples of stable germylenes, stannylenes, and plumbylenes were known.4 Having a close relationship to NHCs, most of these compounds derive their stability from the π-donation of attached nitrogen substituents, while compounds with more electropositive substituents were found to be much more reactive and thus more difficult to prepare and isolate. Seminal work by Klinkhammer and colleagues has nevertheless shown that stable examples of silylated stannylenes and plumbylenes can be obtained in a straightforward way.5−7 More recently Escudié, Castel and coworkers have reported that silylated and germylated stannylenes can be stabilized using coordinating NHCs.8 The same strategy was shown to stabilize also the silylated chlorogermylene (Me3Si)3SiGeCl.8 Previous attempts by Stalke and Heine9 and by Klinkhammer10 showed that the reaction of [tris(trimethylsilyl)silyl]lithium with GeBr2 did not give bis[tris(trimethylsilyl)silyl]germanium as a stable compound but its isomer hexakis(trimethylsilyl)disilagermirane as the product of a rearrangement reaction. Using (Me3Si)3GeLi as the nucleophile, Mallela et al. reported similar chemistry leading to hexakis(trimethylsilyl)germirane.11
Recently, we have reported on the synthesis of examples of cyclic disilylated stannylenes12 and plumbylenes.13 The difference between the aforementioned work by Klinkhammer5 and our attempt was that we utilized a 1,4-tetrakis(trimethylsilyl)tetramethyltetrasilanylene14,15 unit, which can be considered as a linked variation of two tris(trimethylsilyl)silyl groups. While we initially expected these compounds to exhibit a chemical behavior similar to that of the acyclic analogues, it turned out that the cyclic nature renders the tetrylene atom more accessible so that dimerization becomes a more facile process.12,13
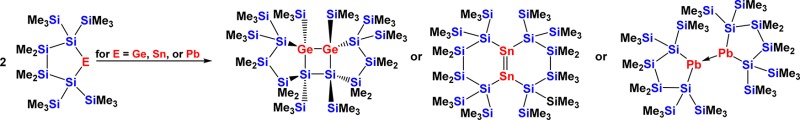
With the elements Pb, Sn, and Ge exhibiting decreasing stability of the divalent state in this order, the respective disilylated tetrylenes reflect this by showing different mechanisms of stabilization. While bis[tris(trimethylsilyl)silyl]lead5 was found to be monomeric even in the solid state, for the cyclic plumbylene13 a dimerization in the crystal was observed (Figure 1). The higher congener bis[tris(trimethylsilyl)silyl]tin was found to be monomeric in solution and dimeric in the solid state,5 while for the cyclic stannylene12 dimerization to a distannene in solution and the solid state was observed (Figure 1). The synthesis of adducts of a disilylated germylene in the current study, together with the findings of Stalke9 and Klinkhammer,10 show that the decreased stability of disilylated germylenes leads to a different mechanism of stabilization in comparison to analogous stannylenes12 and plumbylenes.13 These results are consistent with older work by Klinkhammer10,16 and a very recent account by Lai, Li, and co-workers.17
Figure 1.

Dimers of cyclic disilylated stannylene and plumbylene.
2. Results and Discussion
Synthesis
Reaction of 1,4-dipotassiotetrasilane 1(14,15) or the respective 1,4-digermanium analogue 1a(18) with GeBr2·(dioxane) and PEt3 (Scheme 1) led to the clean formation of the germylene·PEt3 adducts 2 and 2a. With these precursors in hand, it was possible to release the free germylenes 3 and 3a by abstracting the phosphane with the strong Lewis acid B(C6F5)3 (Scheme 1). As expected, germylenes 3 and 3a are not stable but undergo a facile dimerization process. The interesting result is, however, that the dimerization process is not related to that observed for the analogous stannylene.12 The latter was found to dimerize to a distannene (Figure 1),12 whereas germylenes 3 and 3a dimerize to the tricyclic compounds 5 and 5a (Scheme 1).
Scheme 1. Germylene Adduct Formation, Followed by Abstraction of Base, 1,2-Silyl Shift, and Dimerization.

For the stannylene dimerization process the first step was found to be the formation of an exocyclic distannene, which then rearranges to its endocyclic isomer.12 In contrast to this, the first step of the germylene dimerization is a 1,2-trimethylsilyl shift from the α-position to the germanium atom. The thus formed silagermene (4) or digermene (4a) undergoes a [2 + 2] cycloaddition to form 5 and 5a. The first step of this reaction sequence is likely analogous to what happened in the reactions described by Stalke,9 Klinkhammer,10 and Mallela.11 However, the acyclic examples can react by another 1,2-trimethylsilyl shift to give an isomeric silylene, which eventually inserts into the Si–Si bond, thus forming a three-membered ring. While the cyclic silagermene 4 and digermene 4a could also undergo an additional 1,2-trimethylsilyl shift, final insertion of the thus formed silylene or germylene into a Si–Si or Si–Ge bond is not likely, due to the cyclic nature of the molecule. Lacking opportunities for further intramolecular stabilization, 4 and 4a therefore react in an intermolecular [2 + 2] cycloaddition.
Reactions of 2 and 2a with 2,3,4,5-tetramethylimidazol-2-ylidene (NHCMe)19 proceeded smoothly to exchange PEt3 against the carbene ligand and afforded the carbene-stabilized germylenes 6 and 6a (Scheme 1).
NMR Spectroscopy
The NMR spectra of 2 and 2a do not exhibit very unusual chemical shifts. As the tricoordinated germanium atoms have configurational stability, the respective 1H, 13C, and 29Si spectra display different symmetry (vide infra). The 31P resonances at 14.8 ppm for 2 and 15.0 ppm for 2a are almost identical. They are interesting in comparison with the analogous stannylene and plumbylene PEt3 adducts, for which resonances at −1.0 and −60 ppm, respectively, were observed.12,1329Si resonances for the trimethylsilyl groups were found at −7.9 ppm for 2 and −2.0 and −4.1 ppm for 2a, which corresponds to a typical chemical shift difference between SiMe3 groups attached to either Si or Ge. In addition, the SiMe2 shifts of −22.7 ppm for 2 and −16.9 ppm for 2a and the resonance of the quaternary Si atom of 2 at −127.1 ppm are perfectly reasonable and do not differ much from the analogous compound with a dimethylgermylene unit in the ring.20 The 29Si NMR spectra of 6 and 6a are fairly similar to those of 2 and 2a, with all resonances shifted slightly to lower field. The 13C NMR shifts of the carbene carbon atoms of 6 and 6a were found at 174.0 and 175.6 ppm, which in comparison to the signal for the free carbene (213.7 ppm)21 also indicates the substantial Lewis acidity of the germylenes 3 and 3a.
When PEt3 was abstracted from 2 at low temperature, direct NMR spectroscopic observation of silagermene 4 was possible. Especially the sp2-hybridized silicon atom with a chemical shift of δ 149.6 ppm is quite diagnostic for the detection of this molecule. This resonance is very close to that observed for the related stable silagermene (tBuMe2Si)2Ge=Si(SitBuMe2)2 (δ(29Si) 144.0 ppm).22 In addition, 29Si NMR chemical shift calculations at the MP2/GIAO level predict for the silagermene (Me3Si)2Ge=Si(SiMe3)2 a silicon NMR chemical shift of δ(29Si) 158 ppm, which is very close to the experimentally observed values of persilylated silagermenes.22,23 The tricyclic compound 5 also exhibits an interesting 29Si NMR spectrum with two resonances at −69.7 and −94.1 ppm. While 4-fold silylated silicon atoms typically resonate around −130 ppm, the incorporation of such units into cyclotetrasilanes is accompanied by a downfield shift of some 40 ppm.998,999 The presence of strongly branched substituents26 causes further downfield shift behavior, so that we assign the Si(SiMe3) group in the central ring to the resonance at −69.7 ppm, whereas the Si(SiMe3)2 unit is associated with the resonance at −94.1 ppm.
As mentioned above, 1H, 13C, and 29Si NMR spectroscopic analysis of phosphane adducts 2 and 2a at ambient temperature revealed configurational stability of the germanium atom for 2a. This is clearly indicated by the presence of well-resolved signal sets for the different sides of the five-membered ring. Under the same conditions compound 2 exhibits broad signals for the trimethylsilyl and methyl groups. The same behavior was observed also for compounds 6 and 6a. While for the digermylated 6a configurational stability of the Ge atom at ambient temperature was observed, the same conditions corresponded to the coalescence temperature of 6. Activation parameters of 2 and 6 were determined by VT-NMR spectroscopic analysis/Eyring plot. For 2a and 6a values for ΔG⧧ are based on the determination of the coalescence temperature (Table 1).
Table 1. Experimentally Determined and Computed (in Italics) Activation Parameters of Germylene Donor Complexes 2, 6, 2a, and 6a.
| 2 | 6 | 2a | 6a | |
|---|---|---|---|---|
| Tcoalescence (K) | 269 | 293 | 348 | 391 |
| ΔH⧧ (kJ mol–1) | 23.7 ± 3.2 | 50.2 ± 3.1 | ||
| ΔS⧧ (J mol–1 K–1) | –121 ± 10 | –34.3 ± 11.6 | ||
| ΔG⧧ (kJ mol–1) | 59.8 ± 6.3a | 60.3 ± 6.5a | 73.9 | 78.4 |
| (55.6 ± 6.0)b | (60.2 ± 6.5)b | |||
| Ea (kJ mol–1) | 26.2 ± 3.2a | 52.6 ± 3.1a | ||
| (25.9 ± 3.2)b | (52.6 ± 3.1)b | |||
| ΔG⧧(calcd) (kJ mol-1)c | 56 | 61 | 76 | 81 |
| BDE(kJ mol-1)d | 130 | 173 | 137 | 196 |
For T = 298 K.
For T = Tc.
Calculated by M06-2X/def2tzvp (Ge), 6-311+G(d,p) (P), 6-31G(d) (Si, C, H), and PCM methods using the specific parameter for toluene. Free Gibbs energies G were calculated at the indicated temperature T and at p = 23.71 MPa (234 atm); see the Supporting Information for further details.
BDE denotes the bond dissociation energy of the Ge–P (2, 2a) or Ge–C bond (6, 6a) at 0 K and 0.101 MPa (1 atm).
The results of a computational study indicate that the reversible process observed by variable-temperature (VT) NMR spectroscopy for compounds 2, 2a, 6, and 6a occurs in each case intramolecularly. In this process the configuration at the pyramidalized germanium atom is inverted via a transition state in which the germanium atom adopts a trigonal-planar coordination environment (see Figure 2 for an example). Therefore, this process clearly parallels the well-known inversion processes of isolobal amines and phosphanes. The computed barriers for the intramolecular inversion process are in all four cases very close to the experimentally determined values. The largest deviation, ΔΔG⧧, was found for the inversion process of compound 2 (ΔΔG⧧ = 4 kJ mol–1). Moreover, all experimentally determined barriers are significantly smaller than the calculated bond dissociation energy, BDE, of the germanium–donor bond (see Table 1), which discards a dissociation–recomplexation process as a possible alternative.
Figure 2.
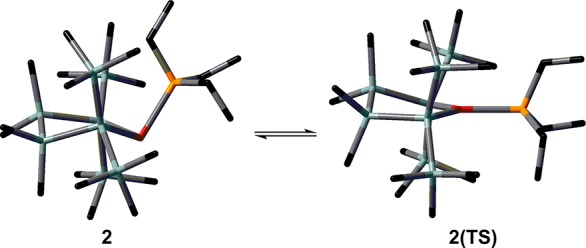
Tube representation of the germylene–phosphane complex 2 and its transition state, 2(TS), for inversion. (M06-2X/def2tzvp (Ge), 6-311+G(d,p) (P), 6-31G(d) (Si, C, H); hydrogen atoms omitted for clarity; color code Ge (red), Si (gray), P (orange), C (black)).
Computational Study
In view of the previous reports by Stalke,9 Klinkhammer,10 and Mallela,11 and in light of our recent and actual experimental results,12,13 a computational study24,25 which gives insights into the reactivity of germylene 3 and into the exclusive formation of the head-to-head dimer 5 ought to address possible isomerization reactions of germylene 3 to silagermene 4, hausane 7, and silylene 8. Furthermore, the relative thermodynamic stabilities of different dimers of silagermene 4 and germylene 3, such as the head-to-tail dimer 9 and the digermenes 10 and 11 need to be evaluated.

The results of our computations suggest that the 1,2 silyl shift which transforms the germylene 3 into the silagermane 4 is slightly exergonic (ΔG298 = −11 kJ mol–1) and is connected with a barrier of ΔG298 ⧧ = 69 kJ mol–1 (Figure 3). Although the isomeric hausane 7 is also close in energy (G298rel = −3 kJ mol–1), it is separated from the silagermene 4 by a prohibitively high barrier of ΔG298 ⧧ = 185 kJ mol–1 (calculated for the forward reaction 4 → 7) which practically excludes its formation from silagermene 4 under the applied reaction conditions. Similarly, silylene 8 is markedly destabilized in comparison to other investigated isomers (G298rel(8) = 66 kJ mol–1), most probably due to the occurrence of sterically unfavorable vicinal 1,1,2,2-tetrakis(trimethylsilyl) substitution in this compound. This result suggests that silylene 8 is not of further relevance for the discussion.
Figure 3.
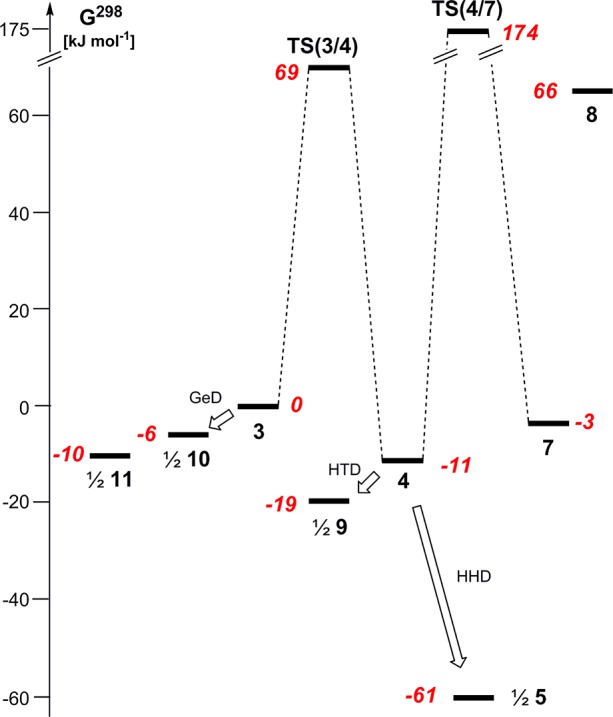
Reaction coordinate for the transformation of germylene 3 into silagermene 4 and hausane 7 and their free Gibbs energies at 298.15 K, G298, relative to their dimers 5 and 9–11 (G298 calculated at M06-2X/def2tzvp (Ge), 6-31G(d) (Si, C, H), are given in red italics): GeD, germylene dimerization; HTD, head to tail dimerization; HHD, head-to-head dimerization.
From a thermodynamic point of view, the dimerization of germylene 3 to give digermene 10 and finally, after skeletal rearrangement,12 the endocyclic digermene 11 (Figure 3) is practically thermoneutral in comparison to the formation of the silagermene 4. In contrast, the head-to-tail (HTD) and in particular the head-to-head (HHD, Figure 3) dimerizations of silagermene 4 to the tricyclic compounds 5 (HHD) and 9 (HTD) are strongly exergonic. These computational results suggest that, in agreement with the experimental results, the transient germylene 3 is converted under thermodynamic control preferentially to the head-to-head dimer 5. The large difference in Gibbs free energies between the HHD isomer 5 and the HTD isomer 9 of ΔG298(5/9) = 84 kJ mol–1 is a result of the greater steric congestion in the HTD 9, as for smaller model systems this marked energy difference is not reflected by the results of the calculations. For example, the calculations for the two different cyclic dimers of H2Si=Ge(H)SiH3 predict an energy difference of merely 6 kJ mol–1 in favor of the head-to-head dimer.
Quite similar results were obtained for the transformation of the germanium-substituted germylene 3a. In this case the 1,2-silyl shift proceeds with a slightly smaller barrier (ΔG298 ⧧ = 52 kJ mol–1) to give the more stable digermene 4a (Grel298(4a) = −13 kJ mol–1). Again the dimerization to give the head-to-head dimer 5a is favored over the formation of the corresponding head-to-tail dimer 9a (ΔG298(5a/9a) = 83 kJ mol–1).
X-ray Crystallography
Crystal structure analysis provided molecular structures of compounds 2, 2a, 5, 5a, 6, and 6a in the solid state (Table S1, Supporting Information). As expected, compound pairs 2 and 2a, 5 and 5a, and 6 and 6a display isotypic behavior. The structures of 2 (Figure S-1, Supporting Information) and 2a (Figure 4) feature the five-membered ring in an envelope conformation with one of the E(SiMe3)2 units out of plane. The base ligands coordinate in an approximately orthogonal way to the plane which is spanned by the divalent tetrel atom and its two neighboring atoms of 2, 2a, 6, and 6a. For the NHC adducts 6 (Figure S-3, Supporting Information) and 6a (Figure 5) the angle between the EGe(II)E plane and the base–Ge bond base is more acute (6, 104.9°; 6a, 104.2°) in comparison to the PEt3 adducts (2, 113.6°). For 6 and 6a thus almost planar five-membered rings were observed.
Figure 4.
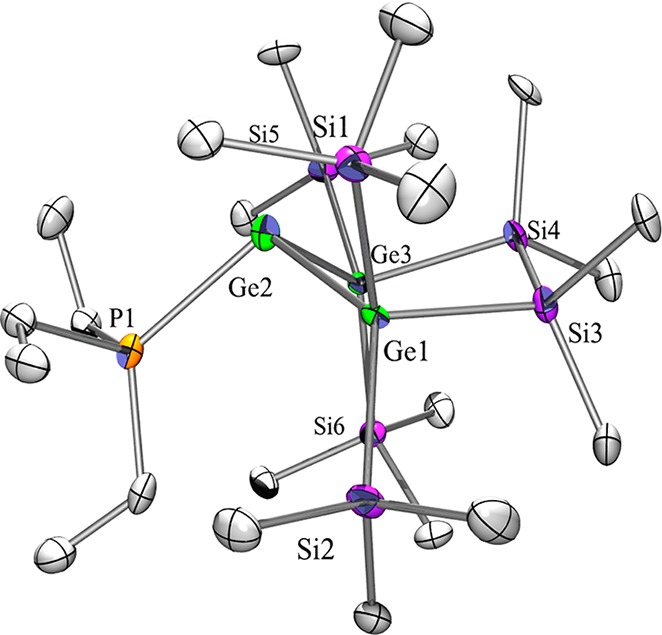
Molecular structure of 2a in the solid state.
Figure 5.
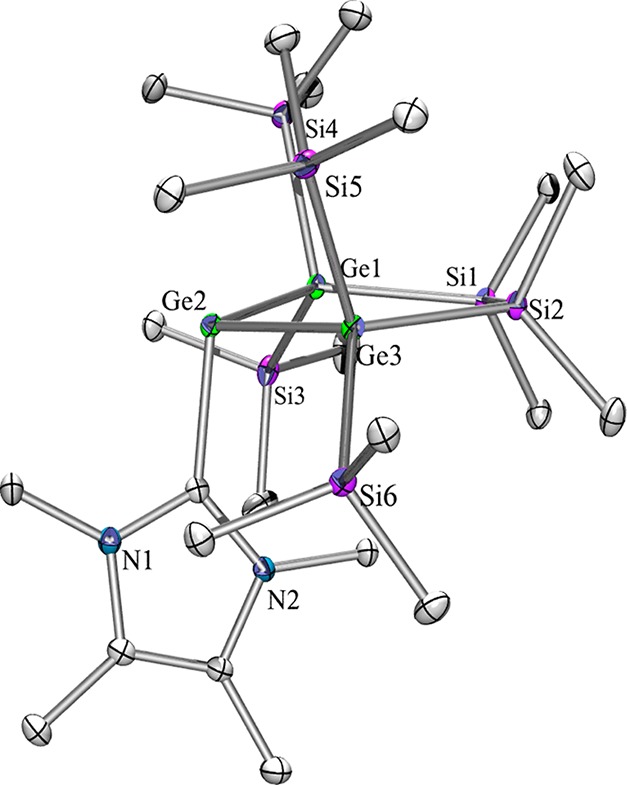
Molecular structure of 6a in the solid state.
The tricyclic compounds 5 (Figure S-2, Supporting Information) and 5a (Figure 6) are interesting in that sense that they are highly branched. The fact that the [2 + 2] cycloaddition of the respective silgermene or digermene occurs in a head-to-head fashion leads to a dimer with a Ge–Ge bond with maximum branched atoms in the α-positions. The steric bulk of this arrangement leads to a very long Ge–Ge bond of 2.575(1) Å for 5a. The direct influence of sterics can be estimated when this bonds is compared to the other Ge–Ge bond of the four-membered ring of 5a, which are with 2.4832(9) and 2.4617(9) Å much shorter. Related structures containing digermane units with silyl or germyl substituents feature Ge–Ge bond lengths ranging from 2.457 to 2.491 Å.
Figure 6.
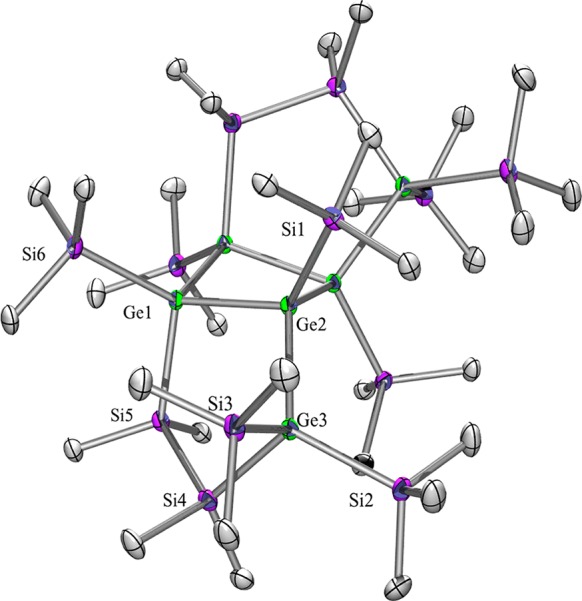
Molecular structure of 5a in the solid state.
3. Conclusion
Although the chemistry of germylenes has received much attention in recent times, most of the studied compounds are either N-heterocyclic compounds or at least carry π-donating substituents such as amino or alkoxy groups. Tetrylenes with more electropositive substituents are much more reactive, and so far no stable examples of silylated germylenes have been reported. Reports dealing with these compounds so far either describe rearrangement reactions of the formed germylenes9,10 or utilize adduct formation of the compounds.8
In the current account the preparation of cyclic disilylated and digermylated germylene PEt3 adducts was accomplished by reaction of either a 1,4-dipotassiotetrasilane or an analogous digermanide with GeBr2·PEt3. Subsequent reaction with an N-heterocyclic carbene led to substitution of the phosphine by the carbene. By treatment of the adducts with B(C6F5)3 the respective germylenes could be released. In comparison to analogous stannylenes and plumbylenes, the thus formed free germylenes are less stable and achieve stabilization by a different mode of action. For the germylenes studied in the current investigation an intermolecular way of stabilization by means of dimerization as digermene is unfavorable. Instead, a 1,2-trimethylsilyl shift from the germylene’s α-position to the dicoordinated germanium atom leads to a cyclic silagermene or digermene. These compounds, though still not stable, can be detected by NMR spectroscopy before undergoing a dimerization process via a formal [2 + 2] cycloaddition reaction.
4. Experimental Section
General Remarks
All reactions involving air-sensitive compounds were carried out under an atmosphere of dry nitrogen or argon using either Schlenk techniques or a glovebox. All solvents were dried using a column-based solvent purification system.27 GeBr2·(dioxane),34 1,3,4,5-tetramethylimidazol-2-ylidene,191,14,15 and 1a(18) were prepared according to literature procedures. Potassium tert-butanolate was purchased from Merck. All other chemicals were obtained from different suppliers and used without further purification.
1H (300 MHz), 13C (75.4 MHz), 31P (124.4 MHz), and 29Si NMR spectra (59.3 MHz) were recorded on a Varian INOVA 300 spectrometer. If not noted otherwise, C6D6 was used as the solvent for all samples. To compensate for the low isotopic abundance of 29Si, the INEPT pulse sequence28,29 was used for the amplification of the signal.
X-ray Structure Determination
For X-ray structure analyses the crystals were mounted onto the tip of glass fibers, and data collection was performed with a Bruker-AXS SMART APEX CCD diffractometer using graphite-monochromated Mo Kα radiation (0.71073 Å). The data were reduced to Fo2 and corrected for absorption effects with SAINT30 and SADABS,31,32 respectively. The structures were solved by direct methods and refined by full-matrix least-squares methods (SHELXL97).33 If not noted otherwise, all non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were located in calculated positions to correspond to standard bond lengths and angles. All diagrams were drawn with 30% probability thermal ellipsoids, and all hydrogen atoms were omitted for clarity. Unfortunately the obtained crystal quality of some substances was poor. This fact is reflected by quite high R and low θ values.
Crystallographic data (excluding structure factors) for the structures of compounds 2, 2a, 5, 5a, 6, and 6a reported in this paper have been deposited with the Cambridge Crystallographic Data Center as supplementary publication nos. CCDC 866887 (2), 866886 (2a), 866885 (5), 866890 (5a), 866889 (6), and 866888 (6a). Copies of the data can be obtained free of charge at http://www.ccdc.cam.ac.uk/products/csd/request/.
2-Germa-1,1,3,3-tetrakis(trimethylsilyl)-2,2,3,3-tetramethylcyclopentasilan-2-ylidene–Triethylphosphane Adduct (2)
Compound 1 (freshly prepared from (Me3Si)3Si(SiMe2)2Si(SiMe3)3 (1.85 g, 3.03 mmol) and KOtBu (714 mg, 6.36 mmol) in 10 mL of DME) dissolved in 40 mL of THF/DME (3/1) was slowly added to a mixture of GeBr2·(dioxane) (1.07 g, 3.33 mmol) and PEt3 (393 mg, 3.33 mmol) in 20 mL of THF at −80 °C. The mixture turned orange, and a white precipitate was formed. The mixture was stirred for 12 h and slowly reached ambient temperature. The solvent was removed under reduced pressure, and the dark orange residue was extracted with pentane (4 × 10 mL). After concentration to 10 mL and storage at −20 °C yellow crystals of 2 (1.42 g, 71%) were obtained. Mp: 139–144 °C dec. 1H NMR (δ in ppm): 0.43 (s, 36H, SiMe3), 0.51 (s, 12H, SiMe2), 0.82 (m, 9H, CH3CH2), 1.61 (m, 6H, CH2P). 13C NMR (δ in ppm): −1.2 (SiMe2), 3.8 (broad, SiMe3), 8.3 (d, 2JC,P = 5.7 Hz, CH3CH2), 19.9 (d, 1JC,P = 19.7 Hz, CH2P). 29Si NMR (δ in ppm): −7.9 (broad, s, SiMe3), −22.7 (d, 3JSi,P = 9.7 Hz, SiMe2), −127.1 (d, 2JSi,P = 15.2 Hz, SiSiMe3). 31P NMR (δ in ppm): 14.8. Anal. Calcd for C22H63GePSi8 (656.03): C. 40.28, H. 9.68. Found: C, 39.36; H, 9.48. UV absorption: λ1 261 nm (shoulder) (ε1 = 2.2 × 104 M–1 cm–1), λ2 = 361 nm (ε2 = 3.7 × 103 M–1 cm–1), λ3 = 414 nm (ε3 = 1.9 × 103 M–1 cm–1).
1,2,3-Trigerma-1,1,3,3-tetrakis(trimethylsilyl)-2,2,3,3-tetramethycyclopentasilan-2-ylidene–Triethylphosphane Adduct (2a)
The procedure for 2 was carried out using 1a (freshly prepared from (Me3Si)3Ge(SiMe2)2Ge(SiMe3)3 (1.40 g, 2.00 mmol), KOtBu (471 mg, 4.20 mmol), GeBr2·(dioxane) (705 mg, 2.20 mmol), and PEt3 (307 mg, 2.60 mmol). Yellow crystalline 2a was isolated (710 mg, 48%). Mp: 149–153 °C dec. 1H NMR (δ in ppm): 0.42 (s, 18H, SiMe3), 0.51 (s, 18H, SiMe3), 0.53 (s, 6H, SiMe2), 0.54 (s, 6H, SiMe2), 0.85 (m, 9H, CH3CH2), 1.61 (m, 6H, CH2P). 13C NMR (δ in ppm): −0.5 (SiMe2), −0.1 (SiMe2), 4.0 (SiMe3), 5.4 (SiMe3), 8.7 (d, 2JC,P = 5.3 Hz, CH3CH2), 20.1 (d, 1JC,P = 18.9 Hz, CH2P). 29Si NMR (δ in ppm): −2.0 (d, 3JSi,P = 13.6 Hz, SiMe3), −4.1 (d, 3JSi,P = 7.9 Hz, SiMe3), −16.9 (d, 3JSi,P = 7.8 Hz, SiMe2). 31P NMR (δ in ppm): 15.0. Anal. Calcd for C22H63Ge3PSi6 (745.14): C. 35.46; H. 8.52. Found: C. 35.30; H. 8.36. UV absorption: λ1 259 nm (shoulder) (ε1 = 2.1 × 104 M–1 cm–1), λ2 340 nm (ε2 = 3.9 × 103 M–1 cm–1), λ3 421 nm (ε3 = 2.4 × 103 M–1 cm–1).
1,2-Digerma-1,2,3,3,6,7,10,10-octakis(trimethylsilyl)octamethyltricyclo[5.3.0.02,6]decasilane (5)
A solution of 2 (200 mg, 0.305 mmol) was dissolved in 5 mL of pentane and slowly added to a stirred solution of B(C6F6)3 (156 mg, 0.305 mmol) in 5 mL of pentane. The mixture turned orange during the addition, and a colorless precipitate was formed. After 2 h the mixture was hydrolyzed with dilute H2SO4 (0.5 M), the organic layer was separated, the aqueous phase was extracted with Et2O (3 × 30 mL), and the extract was dried over Na2SO4. The solvent was removed under reduced pressure, and the yellowish residue crystallized from hexane, giving colorless crystals (105 mg, 64%) of 5. Mp: 180–186 °C dec. 1H NMR (δ in ppm): 0.37 (s, 18H, SiMe3), 0.41 (s, 18H, SiMe3), 0.42 (s, 18H, SiMe3), 0.50 (s, 6H, SiMe), 0.57 (s, 6H, SiMe), 0.58 (s, 18H, SiMe3), 0.62 (s, 6H, SiMe), 0.74 (s, 6H, SiMe). 13C NMR (δ in ppm): 1.0 (SiMe), 1.6 (SiMe), 2.3 (SiMe), 4.6 (SiMe), 5.1 (SiMe3), 5.3 (SiMe3), 6.1 (SiMe3), 7.1 (SiMe3). 29Si NMR (δ in ppm): −1.5 (SiMe3), −6.7 (SiMe3), −6.9 (SiMe3), −8.7 (SiMe3), −17.2 (SiMe2), −25.1 (SiMe2), −69.7 (SiSiMe3), −94.1 (SiSiMe3). Anal. Calcd for C32H96Ge2Si16 (1075.75): C, 35.73;, H, 8.99. Found: C, 35.72; H, 8.82. UV absorption: λ1 344 nm (ε1 = 5.2 × 103 M–1 cm–1).
1,2,3,6,7,10-Hexagerma-1,2,3,3,6,7,10,10-octakis(trimethylsilyl)octamethyltricyclo[5.3.0.02,6]decasilane (5a)
The procedure for 5 was carried out using 2a (224 mg, 0.300 mmol) and tris(pentafluorophenyl)borane (154 mg, 0.300 mmol). Light yellow crystalline 5a was isolated (85 mg, 45%). Mp: 198–201 °C dec. 1H NMR (δ in ppm): 0.40 (s, 18H, SiMe3), 0.44 (s, 18H, SiMe3), 0.45 (s, 18H, SiMe3), 0.52 (s, 6H, SiMe), 0.58 (s, 24H, SiMe3/SiMe), 0.66 (s, 6H, SiMe), 0.82 (s, 6H, SiMe). 13C NMR (δ in ppm): 1.2 (SiMe), 1.8 (SiMe), 2.6 (SiMe), 5.2 (SiMe), 5.2 (SiMe3), 5.6 (SiMe3), 6.4 (SiMe3), 6.8 (SiMe3). 29Si NMR (δ in ppm): −0.7 (SiMe3), −1.0 (SiMe3), −1.3 (SiMe3), −1.8 (SiMe3), −11.6 (SiMe2) −21.0 (SiMe2). Anal. Calcd for C32H96Ge6Si12 (1253.97): C, 30.65; H, 7.72. Found: C, 30.61; H, 7.82. UV absorption: λ1 262 nm (ε1 = 2.5 × 104 M–1 cm–1), λ2 338 nm (ε2 = 4.4 × 103 M–1 cm–1).
2-Germa-1,1,3,3-tetrakis(trimethylsilyl)-2,2,3,3-tetramethycyclopentasilan-2-ylidene–NHC Adduct (6)
Compound 2 (66 mg, 0.10 mmol) and 1,3,4,5-tetramethylimidazol-2-ylidene (13 mg, 0.10 mmol) were dissolved in pentane (4 mL) and stirred for 10 min. The orange solution was filtered to remove residual K2S precipitate from the NHC synthesis. The filtrate was evaporated under reduced pressure. The yellow residue was dissolved in pentane (3 mL) and stored at −35 °C for 3 days, affording yellow prismatic crystals of 6 (60 mg, 90%). Mp: 147–153 °C dec. 1H NMR (δ in ppm, toluene-d8 at 25 °C): 0.38 (s, very broad, 36H, SiMe3), 0.59 (s, 12H, SiMe2), 1.38 (s, 6H, CH3C), 3.56 (s, 6H, CH3N). 13C NMR (δ in ppm, toluene-d8 at 25 °C): −0.6 (broad, SiMe2), 3.7 (broad, SiMe3), 8.0 (MeC), 36.4 (MeN), 105.0 (NCC), 174.0 (NCN). 29Si NMR (δ in ppm, toluene-d8 at 25 °C): −18.8 (SiMe2), −120.0 (SiSiMe3). Anal. Calcd for C23H60GeN2Si8 (662.06): C, 41.73; H, 9.13; N, 4.23. Found: C, 41.41; H, 8.88; N, 4.24. UV absorption: λ1 287 nm (shoulder) (ε1 = 2.2 × 104 M–1 cm–1), λ2 377 nm (ε2 = 4.0 × 103 M–1 cm–1).
1,2,3-Trigerma-1,1,3,3-tetrakis(trimethylsilyl)-2,2,3,3-tetramethycyclopentasilan-2-ylidene–NHC Adduct (6a)
The procedure for 6 was carried out using 2a (75 mg, 0.10 mmol) and 1,3,4,5-tetramethylimidazol-2-ylidene (12 mg, 0.10 mmol). Yellow crystals of 6a (66 mg, 88%) were obtained. Mp: 180–184 °C dec. 1H NMR (δ in ppm): 0.19 (s, 18H, SiMe3), 0.56 (s, 6H, SiMe2), 0.60 (s, 18H, SiMe3), 0.62 (s, 6H, SiMe2), 1.27 (s, 6H, CH3C), 3.46 (s, 6H, CH3N). 13C NMR (δ in ppm): 0.2 (SiMeSiMe), 0.8 (SiMeSiMe), 4.4 (SiMe3), 4.8 (SiMe3), 8.3 (MeC), 36.5 (MeN), 105.3 (MeC), 175.6 (NCN). 29Si NMR (δ in ppm): −2.8 (SiMe3), −3.9 (SiMe3), −13.2 (SiMe2). Anal. Calcd for C23H60GeN2Si8 (751.17): C, 36.78; H, 8.05; N, 3.75. Found: C, 36.90; H, 7.85; N, 7.46. UV absorption: λ1 278 nm (shoulder) (ε1 = 2.4 × 104 M–1 cm–1), λ2 371 nm (ε2 = 3.9 × 103 M–1 cm–1).
Acknowledgments
Support of the study was provided by the Austrian Fonds zur Förderung der wissenschaftlichen Forschung (FWF) via projects P-22678 and P-25124 and by the Deutsche Forschungsgemeinschaft (DFG) via project Mu1440/8-1. The High End Computing Resource Oldenburg (HERO) at the CvO University is thanked for computer time.
Supporting Information Available
CIF files, tables, and figures giving crystallographic data for compounds 2, 2a, 5, 5a, 6, and 6a as well as details of the theoretical study. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools; Díez-González S., Ed.; Royal Society of Chemistry: Cambridge, U.K., 2010. [Google Scholar]
- N-Heterocyclic Carbenes in Transition Metal Catalysis; Glorius F., Ed.; Springer: Berlin, Heidelberg, 2007; Topics in Organometallic Chemistry 21. [Google Scholar]
- Hahn F. E.; Jahnke M. C. Angew. Chem., Int. Ed. 2008, 47, 3122–3172. [DOI] [PubMed] [Google Scholar]
- Mizuhata Y.; Sasamori T.; Tokitoh N. Chem. Rev. 2009, 109, 3479–3511. [DOI] [PubMed] [Google Scholar]
- Klinkhammer K. W.; Schwarz W. Angew. Chem., Int. Ed. Engl. 1995, 34, 1334–1336. [Google Scholar]
- Klinkhammer K. Polyhedron 2002, 21, 587–598. [Google Scholar]
- Stürmann M.; Saak W.; Klinkhammer K. W.; Weidenbruch M. Z. Anorg. Allg. Chem. 1999, 625, 1955–1956. [Google Scholar]
- Katir N.; Matioszek D.; Ladeira S.; Escudié J.; Castel A. Angew. Chem., Int. Ed. 2011, 50, 5352–5355. [DOI] [PubMed] [Google Scholar]
- Heine A.; Stalke D. Angew. Chem. 1994, 106, 121–123. [Google Scholar]
- Klinkhammer K. W. In Organosilicon Chemistry III; Auner N., Weis J., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp 82–85. [Google Scholar]
- Mallela S. P.; Hill S.; Geanangel R. A. Inorg. Chem. 1997, 36, 6247–6250. [DOI] [PubMed] [Google Scholar]
- Arp H.; Baumgartner J.; Marschner C.; Müller T. J. Am. Chem. Soc. 2011, 133, 5632–5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arp H.; Baumgartner J.; Marschner C.; Zark P.; Müller T. J. Am. Chem. Soc. 2012, 134, 6409–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser C.; Kickelbick G.; Marschner C. Angew. Chem., Int. Ed. 2002, 41, 989–992. [DOI] [PubMed] [Google Scholar]
- Fischer R.; Frank D.; Gaderbauer W.; Kayser C.; Mechtler C.; Baumgartner J.; Marschner C. Organometallics 2003, 22, 3723–3731. [Google Scholar]
- Klinkhammer K. W. Chem. Eur. J. 1997, 3, 1418–1431. [Google Scholar]
- Xiao X.-Q.; Zhao H.; Xu Z.; Lai G.; He X.-L.; Li Z.. Chem. Commun. 2013, 49, 2706–2708. [DOI] [PubMed] [Google Scholar]
- Fischer J.; Baumgartner J.; Marschner C. Organometallics 2005, 24, 1263–1268. [Google Scholar]
- Kuhn N.; Kratz T. Synthesis 1993, 561–562. [Google Scholar]
- Fischer J.; Gaderbauer W.; Baumgartner J.; Marschner C. Heterocycles 2006, 67, 507–510. [Google Scholar]
- Bourissou D.; Guerret O.; Gabbaï F. P.; Bertrand G. Chem. Rev. 2000, 100, 39–92. [DOI] [PubMed] [Google Scholar]
- Iwamoto T.; Okita J.; Yoshida N.; Kira M. Silicon 2010, 2, 209–216. [Google Scholar]
- Computed at MP2/GIAO/6-311G(d,p) (Si, Ge) and 6-31G(d) levels (C, H) using a structure of C2v molecular symmetry, optimized at M06-2X/def2tzvp.
- Chen Y. S.; Gaspar P. P. Organometallics 1982, 1, 1410–1412. [Google Scholar]
- Wagner H.; Wallner A.; Fischer J.; Flock M.; Baumgartner J.; Marschner C. Organometallics 2007, 26, 6704–6717. [Google Scholar]
- All calculations were done at the M06-2X/def2tzvp (Ge) and 6-31G(d) (Si, C, H) levels of theory. This model chemistry was calibrated using smaller systems for energies against the results of higher level computations up to the CCSD level and, for structures, against the experimental solid-state structures; see the Supporting Information for details.
- All calculations were done using the G09 program, Version B.01, 2010.
- Krempner C. Polymers 2012, 4, 408–447. [Google Scholar]
- Pangborn A. B.; Giardello M. A.; Grubbs R. H.; Rosen R. K.; Timmers F. J. Organometallics 1996, 15, 1518–1520. [Google Scholar]
- Morris G. A.; Freeman R. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar]
- Helmer B. J.; West R. Organometallics 1982, 1, 877–879. [Google Scholar]
- SAINTPLUS: Software Reference Manual, Version 6.45; Bruker-AXS: Madison, WI, 1997–2003. [Google Scholar]
- Sheldrick G. M.SADABS Version 2.10; Bruker AXS Inc., Madison, WI, 2003.
- Blessing R. H. Acta Crystallogr., Sect. A 1995, 51, 33–38. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. A 2007, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- Leigh W. J.; Harrington C. R.; Vargas-Baca I. J. Am. Chem. Soc. 2004, 126, 16105–16116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.