

Abstract
Membranous nephropathy (MN) is a major cause of idiopathic nephrotic syndrome in adults, often progressing to end-stage kidney disease. The disease is mediated by IgG antibodies that form subepithelial immune complexes upon binding to antigens expressed by podocytes or planted in the subepithelial space. Subsequent activation of the complement cascade, podocyte injury by the membrane attack complex and the expansion of the glomerular basement membrane cause proteinuria and nephrotic syndrome. The blueprint for our current understanding of the pathogenic mechanisms of MN has largely been provided by studies in rat Heymann nephritis, an excellent animal model that closely replicates human disease. However, further progress in this area has been hindered by the lack of robust mouse models of MN that can leverage the power of genetic approaches for mechanistic studies. This critical barrier has recently been overcome by the development of new mouse models that faithfully recapitulate the clinical and morphologic hallmarks of human MN. In these mouse models, subepithelial ICs mediating proteinuria and nephrotic syndrome are induced by injection of cationized bovine serum albumin, by passive transfer of heterologous anti-podocyte antibodies, or by active immunization with the NC1 domain of α3(IV) collagen. These mouse models of MN will be instrumental for addressing unsolved questions about the basic pathomechanisms of MN and also for preclinical studies of novel therapeutics. We anticipate that the new knowledge to be gained from these studies will eventually translate into much needed novel mechanism-based therapies for MN, more effective, more specific, and less toxic.
Keywords: Membranous nephropathy, mouse models, immune complexes, nephrotic syndrome, anti-podocyte antibodies, glomerular basement membrane, type IV collagen
Human membranous nephropathy
Membranous nephropathy (MN) is one of the most common causes of nephrotic syndrome in adults. The clinical course in majority of patients appears to be indolent and slowly progressive. However, over years, approximately up to 40% of patients with MN develop progressive renal insufficiency leading to end stage kidney disease [1]. Another third of patients suffer from complications arising from the long term consequences of nephrotic syndrome. In most cases (75-80%), MN manifests as a primary (idiopathic) glomerular disease, although MN can also be secondary to systemic lupus erythematosus, infections such as hepatitis B, exposure to toxins, use of certain drugs, and malignancy. Morphologic hallmarks of MN on the renal biopsy are subepithelial immune deposits containing antigen, IgG, and complement as well as glomerular basement membrane (GBM) thickening and podocyte foot process effacement.
The pathogenesis of MN has largely been established in the rat model of Heymann nephritis (see below), in which the target antigen responsible for in situ deposition of immune complexes (ICs) was ultimately identified as megalin. As megalin is not expressed by the human podocyte, the search for the major human antigen continued for decades. The first human antigen identified in a rare form of antenatal feto-maternal alloimmune MN was neutral endopeptidase [2]. It was not until recently that the M-type phospholipase A2 receptor (PLA2R1), a 180 kDa member of the mannose receptor family and a transmembrane glycoprotein expressed by human podocytes, was identified as the major antigenic target in human primary MN [3]. Circulating anti-PLA2R antibodies correlate very well with clinical disease activity [4], although they have not been formally proved to be pathogenic in animal models (see below). Other reports have identified antibodies to several intracellular antigens, which may be secondarily induced by cell damage and expressed at the cell surface, in human MN. These include superoxide dismutase, aldose reductase, alpha-enolase, and probably other antigens [5,6]. An intriguing case of a dietary antigen implicated in MN, cationic bovine serum albumin (BSA), was recently described in several young infants [7].
The subepithelial immune deposits pathognomonic for MN may form in several ways [8]. The paradigm established with megalin in Heymann nephritis, which appears to hold true for the human cases of fetomateral alloimmune (anti-NEP) as well as anti-PLA2R-associated idiopathic MN, involves the binding of circulating antibodies in situ to an intrinsic podocyte protein [9]. Once ICs form, they activate the complement cascade, leading to the assembly of the membrane attack complex (C5b-9) within the podocyte cell membrane, which causes sub-lethal podocyte injury and proteinuria (Figure 1). The ICs and C5b-9 are shed, due to membrane turnover and other adaptive cellular mechanisms, into the GBM in a subepithelial position. The injured podocyte also increase the production and secretion of basement membrane components, which are laid down around and over the immune deposits, leading to an apparent thickening of the GBM (this is why the disease was first described as “membranous”). In addition to in situ IC formation, the target antigen can also be extrinsic to the glomerulus. In some cases, a cationic antigen such as cationic BSA or the hepatitis B e antigen can first become ‘planted’ in the subepithelial space due to its charge, and later incite the formation of immune deposits. In other cases, entire circulating ICs, for example in lupus, may ultimately end up in a subepithelial position.
Figure 1.
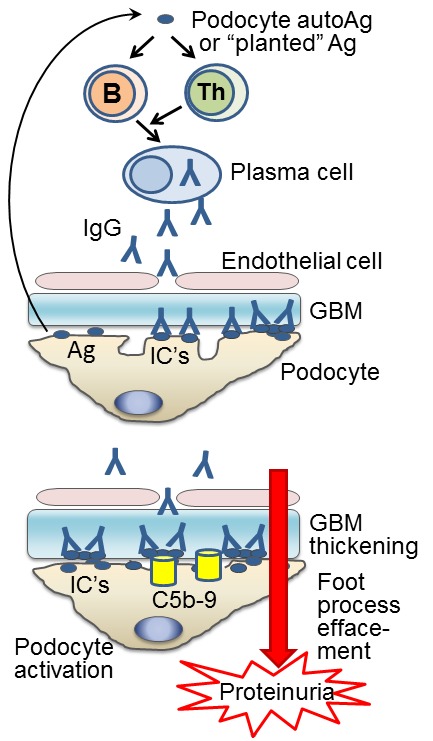
Pathogenesis of MN. Top: MN is initiated by maladaptive immune responses against autoantigens expressed by podocytes (e.g. human PLA2R, rat megalin) or antigens that can be “planted” in the subepithelial space, such as cationic proteins (cBSA, α3NC1). B cells, with assistance from cognate helper T(Th) cells, differentiate into plasma cells that produce pathogenic autoantibodies, mostly of IgG4 subclass. Upon binding to target antigens, the antibodies form ICs that deposit subepithelially (beneath podocytes), on the outer side of the GBM. Bottom: Subepithelial ICs induce complement activation and podocyte injury by C5b-9, leading to podocyte activation, effacement of foot processes, increased production of matrix components and thickening of the GBM, ultimately resulting in massive leakage of plasma proteins in urine (proteinuria).
Current treatments for MN are non-specific, toxic, and too often ineffective [10,11]. A recent Cochrane systematic review and meta-analysis concluded that, despite the 36 randomized controlled trials evaluated for the study, there is still insufficient data to strongly recommend a given treatment in any one particular case [12]. As recently highlighted at the NIDDK Workshop on Glomerular Disease (Bethesda, April 2012), the development of novel targeted therapies for MN remains a key priority in the field. Achieving this goal requires a better understanding of the pathogenic mechanisms. Because experiments in humans are not possible for obvious ethical reasons, good animal models faithfully recapitulating the pathogenesis of human MN are essential for investigating the disease mechanisms and for preclinical studies of drug candidates [13]. Although animal models of MN have been described in many species (including dogs, cats, rabbits and pigs), the experimental model of rat Heymann nephritisis by far the most commonly used, providing a de facto “gold standard” against which other models are compared [14].
Rat models of membranous nephropathy: Heymann nephritis
Rat Heymann nephritis has emerged as the best model for studying the disease mechanisms because it faithfully recapitulates human MN in terms of histopathology and its major clinical aspects [15]. In active Heymann nephritis, pathogenic antibodies are produced by the animal’s own immune system, thus recapitulating human MN. The disease is induced by immunization with Fx1A, an extract of rat kidneys containing proximal tubular brush border antigens, in complete Freund adjuvant [16-18]. Immunized rats develop anti-Fx1A antibodies within ~2 weeks, granular capillary loop staining for IgG and C3 and subepithelial IC’s by 3-4 weeks, proteinuria by 6-8 weeks, and nephrotic syndrome after ~12 weeks. The glomerular antigen involved in the formation of subepithelial deposits in Heymann nephritis was eventually identified as a complex of megalin and receptor-associated protein, which is expressed in the tubular brush border and on rat podocytes, but not on human podocytes [19]. Some disadvantages of this active model---including much greater variability in outcome andanimal discomfort because of repeated footpad immunizations---restricted its usage. Passive Heymann nephritis (PHN) is induced in various strains of rats by passive transfer of heterologous sheep or rabbit antibodies against rat tubular brush border or Fx1A antigens [20,21]. Intravenous or intraperitoneal injection of anti-Fx1A antibodies (as isolated IgG or antiserum) induces GBM thickening, podocyte abnormalities, subepithelial deposits of IgG, C3 and C5b-9, and heavy proteinuria after 3 to 7 days. In this heterologous phase, kidney pathology is exclusively mediated by ICs formed by heterologous IgG. The autologous phase is initiated at 2-4 weeks, when rats develop an immune response against the foreign IgG, and is characterized by glomerular deposition of rat IgG which exacerbates proteinuria.
Mouse models of membranous nephropathy: the road less travelled by
Although Heymann nephritis has been tremendously useful in defining the paradigm by which complement-mediated cell injury occurs in MN, this rat model is limited by the lack of genetically-engineered rat strains, as well as an antigen that is now known to be irrelevant in human disease. Murine models are highly desirable because of the broad availability of genetically engineered strains and mouse-specific reagents, convenient manipulation, low costs, and short gestation period. The development of a robust murine model of MN has been a work in progress for decades. However, achieving this goal has provenelusive despite many efforts by several groups [14]. Most mouse models of MN are unsatisfactory because of the minimal or transient or complement-independent proteinuria. Below, we review the progress in this field, with particular emphasis on three recently developed mouse models of MN characterized by subepithelial ICs, heavy proteinuria, and nephrotic syndrome. Despite significant differences in how disease is induced, these three models closely recapitulate the clinic and morphologic hallmarks of human MN (Figure 2) and compare favorably with rat Heymann nephritis.
Figure 2.
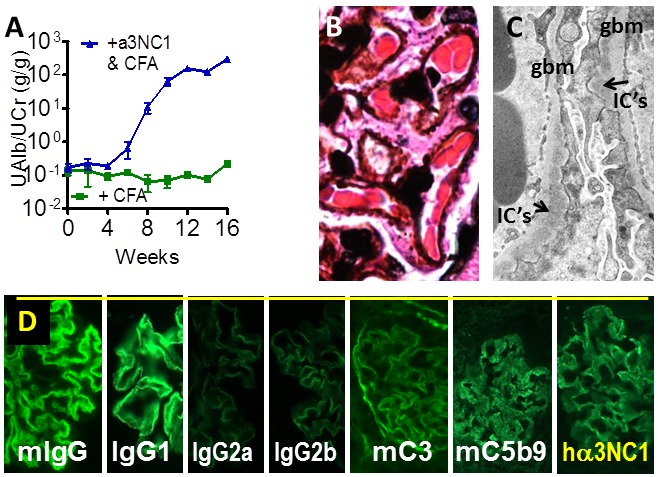
Recapitulation of clinical and histologic hallmarks of MN in mouse models. A. DBA/1 mice immunized with α3NC1 develop massive albuminuria and nephrotic syndrome. B. Light microscopy shows the absence of glomerular inflammation and formation of GBM “spikes” (silver stain). C. Electron microscopy shows expansion of the GBM to surround electron-dense subepithelial IC’s (arrows) and podocyte foot process effacement. D. Immunofluorescence staining shows granular capillary loop staining for mouse IgG (predominantly IgG1 with small amounts of IgG2a and IgG2b), mouse complement C3 and C5b-9, and exogenous antigen (hα3NC1).
Early efforts to develop a murine model of MN predated the identification of the major antigens in human MN and sought to recapitulate the induction of Heymann nephritis in rats, as described above. Subepithelial ICs in the absence of proliferative changes were induced in C57Bl10 mice by a single injection of rabbit IgG raised against homologous (mouse) pronase-digested renal tubular antigens [22]. The heterologous phase was characterized by the fixation of rabbit IgG to the mouse capillary walls and brush borders of the proximal tubules in a linear or fine-granular pattern, with a transient increase of glomerular permeability. In the autologous phase, an immune response to heterologous IgG led to the development of subepithelial ICs containing rabbit IgG, mouse IgG1, and small amounts of mouse C3. Histologic changes were mild, differing very little from control mice, and neither GBM “spikes” nor effacement of podocyte foot processes were observed. Rabbit IgG was found to fix directly to an antigen in the cell membrane of the glomerular visceral epithelium, implying that subepithelial IC’s formed in situ. However, these mice developedonly transient albuminuria, and not nephrotic syndrome. This has been attributed to the predominance in the epimembranous ICs deposits of mouse IgG1, an IgG subclass that does not activate complement. Nevertheless, the predominant subclass of serum and kidney-bound autoAbs in human idiopathic MN is human IgG4, which is functionally equivalent to mouse IgG1 [23,24]. Also, proteinuria and glomerular C3 deposition do in fact occur in other mouse models of MN in which mouse IgG1 is prevalent in serum and in subepithelial IC deposits (see below).
In a search for antibodies against intrinsic podocytes antigens capable of inducing more severe and persistent proteinuria, a rat IgG1 mAb against mouse aminopeptidase A, named ASD-4, was developed [25]. Mice injected with mAb ASD-4 immediately developed massive, dose-dependent albuminuria that lasted for at least 16 days. The binding of rat IgG to the glomerular capillary wall was initially homogenous but changed into a granular pattern after one day. Glomerular deposition of mouse C3 was absent. By light microscopy, no glomerular lesions could be observed, and no influx of polymorphonuclear leukocytes, mononuclear cells or platelets was seen. Electron microscopy showed fusion of the podocytes foot processes and widely scattered electron-dense immune deposits in the slit pores but no GBM thickening (unlike human MN). Neither complement activation nor glomerular inflammation was required for the development of albuminuriain this model. Most likely, albuminuria is the result of a direct disturbance of the glomerular filtration barrier by the deposited ASD4.
Murine membranous nephropathy induced by a planted antigen, cationized BSA
Exogenous cationic proteins, such as cationized bovine serum albumin (cBSA), bind to anionic sites in the glomerular filtration barrier and form subepithelial ICs by a “planted antigen” mechanism. This leads to development of proteinuria and glomerular lesions recapitulating MN in rabbits and in several other species [26]. In 2004, the first mouse model exhibiting subepithelial ICs, severe proteinuria and nephrotic syndrome was established by immunizing mice with cBSA [27]. Induction of disease was dose-dependent and strain specific. BALB/c and ICR strains were susceptible, but C57BL/6 mice were resistant. After repeated injections with cBSA (every other day for 6 weeks), mice developed severe proteinuria, hypoalbuminemia, and hypercholesterolemia--clinical symptoms characteristic of nephrotic syndrome. Overt proteinuria appeared in week 4 and reached a plateau at week 8. Serum albumin concentration declined markedly after week 6, while serum cholesterol concentration increased at week 6 and was maximal at week 8. The blood urea nitrogen and serum creatinine concentrations did not change. Histological findings revealed diffuse thickening of the GBM with spike formation but no hypercellularity. Immunofluorescence staining showed IgG and C3 deposition along the GBM in a granular pattern with a discrete beaded appearance. Ultrastructural analysis identified severely irregular thickening of the GBM and subepithelial deposits. In situ formation of subepithelial ICs was suggested in this model because the serum concentration or circulating ICs did not increase significantly before strong positive IF staining indicative of ICs deposition was observed. The levels of mouse IgG1 anti-cBSA antibodies were significantly higher than those of mouse IgG2a, implying a Th2 bias, because mouse IgG1 is associated with Th2 and mouse IgG2a with Th1 polarization. A subsequent study using T1/T2 double transgenic mice expressing human Thy1 under the control of interferon gamma and mouse Thy1.1 under the control of IL-4 found that both peripheral and renal immune reactions are strongly polarized toward Th2-type immune responses during the course of MN in this model, and Th2 cells but not Th1 cells were significantly correlated with hypercholesterolemia and proteinuria [28]. The pattern of injury and outcomes in glomerulonephritis are influenced by the Th subsets, with Th1 predominance implicated in proliferative and crescentic glomerulonephritis, and Th2 predominance associated with membranous nephropathy [29]. In idiopathic MN, Th2 cytokines increase and stimulate B cells to produce IgG4 [30,31]. Hence, in mice and man, the development of MN appears to depend on host factors that favor a Th2 polarization.
The importance of adaptive immune responses in this model has been demonstrated using severe combined immuno-deficient (SCID) mice, which are resistant to murine MN induced by cBSA [32]. Granular immune complex deposits and pathologic proteinuria were induced in SCID mice by passive transfer of serum from MN mice, but not of serum from normal control mice, nor by adoptive transfer of splenocytes. Overall, cBSA-induced murine MN may recapitulate the pathogenesis of some forms of early childhood MN in which dietary cationic BSA has been implicated as an antigen [7]. However, this model has not gained widespread adoption, perhaps because disease induction is cumbersome, requiring numerous injections of cBSA in the tail vein.
Passive murine membranous nephropathy induced by anti-podocyte antibodies
Since studies of Heymann nephritis (and subsequently in human MN) established the importance of antibodies against specific podocyte antigens in the pathogenesis of MN, it has been reasoned that the administration of anti-podocyte antibodies could induce MN in mice. In 2011, one of us (C.M.S. et al.) has induced characteristic features of MN in mice injected with sheep anti-podocyte antiserum [33]. Sheep antiserum used in this study was raised against a murine podocyte cell line from Endlich et al [34]. C57BL/6 mice injected with this antiserum developed nephrotic syndrome with severe edema, proteinuria, hypoalbuminemia, elevated cholesterol and triglycerides commencing by day 9 and peaking day 14, with baseline serum creatinine and urea nitrogen values. The histologic changes showed swollen podocytes, podocyte loss and glomerular basement membrane thickening. Immune staining demonstrated glomerular deposition of mouse IgG of all subclasses in a linear and granular pattern along the GBM and in the subepithelial space. Electron microscopy revealed extensive podocyte foot process effacement and subepithelial IC deposits. Immunoelectron microscopy localized sheep antibodies to podocyte foot processes and to the GBM. Mice exhibited an inflammatory response characterized by a periglomerular accumulation of F4/80+ macrophages and a glomerular infiltration with CD3+ T-cells, Ly6G+ neutrophils and MAC-2+ macrophages, suggesting an inflammatory mediation of disease. Interestingly, C3 deficient mice (C57BL/6) were slightly but not significantly protected from podocyte damage, despite linear deposition of C3 along the GBM, indicating a partly complement-independent mechanism. Twenty proteins were identified as possible antigens to the sheep anti-podocyte serum by mass spectrometry suggesting multiple antigens rather than a single antigen acting to initiate the disease. Injection of the heterologous sheep anti-podocyte serum into Balb/c mice resulted in an enhanced glomerular IC deposition with a decreased inflammatory reaction compared to C57BL/6 mice, more closely resembling the pathologic features of human MN.
Notably, murine MN was not observed in similar studies using heterologous antibodies raised against a murine podocyte cell lin developed by Mundel et al [35]. A mouse model of immune-mediated podocyte damage was induced in mice sensitized with rabbit IgG in complete Freund’s adjuvant by injection of polyclonal rabbit anti-mouse podocytes antibodies [36]. Mice developed albuminuria 10 days later. P athologic changes were restricted to podocyte swelling and loss, linear glomerular IgG and C3 deposition, loss of linear immunostaining of slit membrane protein and segmental foot process effacement with rare subepithelial immune deposits at mesangial hinge areas. However, extensive subepithelial ICs deposits, characteristic of human MN, were not found. The course and extent of disease were not attenuated by complement depletion. The inflammatory response was characterized by slight glomerular infiltration with T-cells. Another group has found that sheep antibodies raised against the same mouse podocyte cell line cause little glomerular damage in wild type mice, despite in vivo IgG binding to glomerular capillary wall [37]. Thus, the presence of heterologous antibodies targeting specific mouse podocyte antigens may be critical for the induction of murine MN by passive immunization.
Active murine membranous nephropathy induced by immunization with α3NC1
While studying the role of activating Fcγ receptors in murine anti-GBM antibody-mediated glomerulonephritis, several of us (J.J.Z., W.L., D.B.B. et al) unexpectedly found that DBA/1 mice immunized with human NC1 monomers of α3(IV) collagen (α3NC1) in complete Freund‘s adjuvant and boosted once with the same antigen in incomplete Freund‘s adjuvant develop severe albuminuria (Figure 2A), followed by hypoalbuminemia, hyperlipidemia, and often edema--features typical of the nephrotic syndrome [38]. Pathologic changes in the kidneys recapitulated hallmark features of human MN, rather than anti-GBM disease, as had initially been anticipated. Similar to human MN, light microscopy showed minimal glomerular inflammation, very few crescents (<5%) and characteristic GBM “spikes” by silver stain (Figure 2B). Electron microscopy showed subepithelial IC’s deposits, GBM thickening and podocyte foot process effacement (Figure 2C). Immunofluorescence microscopy showed linear-granular capillary loop staining for of mouse IgG, C3c, C5b-9, and exogenous human α3NC1 antigen (Figure 2D). Serum and kidney-bound antibodies were mostly non-complement fixing mouse IgG1, with small amounts of mouse IgG2a and IgG2b, thus recapitulating the predominance of human IgG4 in human MN. Proteinuric mice excreted C5b-9 in urine, as reported in human MN [39].
Other groups have also noted the development of subepithelial ICs in DBA/1 mice repeatedly immunized with human α3NC1, but clinical features of MN were not assessed [40]. When DBA/1 mice are subjected to a more aggressive immunization protocol (four α3NC1 immuni-zations instead of two), the kidney disease initially presents as MN until 8 weeks post-immunization, but later crescentic glomerulonephritis and tubulointerstitial inflammationalso develop [41]. In this model, C57Bl/6 mice develop milder glomerular injury than DBA/1 mice, while AKR and NOD mice are relatively resistant, despite mIgG deposition in the kidneys [40]. Immunization with rat α3NC1 monomers (highly homologous to autologous murine α3NC1) also induced subepithelial ICs in C57Bl/6 mice, whereas B6. FcγRIIB-/- mice developed both subepithelial and subendothelial ICs, the latter possibly due to spontaneously produced lupus-like autoantibodies [42].
An important question is how subepithelial ICs form in α3NC1-induced murine MN. Co-localization of exogenous α3NC1 antigen with mouse IgG and C3 in the GBM (but not in kidney tubular basement membranes) is consistent with a planted antigen mechanism or deposition of small circulating IC’s. Glomerular deposition of extrinsic antigens from the circulation occurs in other active models of MN, such as rats immunized with human brush border antigens and rabbits immunized with rat tubular antigens [43,44]. Human α3NC1 has a high isoelectric point (pI ~ 9), comparable to cBSA or other cationic antigens that form subepithelial ICs by a planted antigen mechanism. In addition, α3NC1 has been shown to bind to several receptors expressed on the podocyte cell surface, including α3β1 and αvβ3 integrins [45,46]. Both human α3NC1 and ICs composed of human α3NC1 and a mouse IgG1 anti-α3NC1 mAb bind to mouse podocytes (Corina Borza, personal communication).
Development of kidney pathology and the MN phenotype in α3NC1-immunized mice is strictly dependent on the formation of subepithelial IC’s. Even though anti-α3NC1 autoAbs that bind to mouse GBM in vivo are also produced, the absence of typical features of anti-GBM antibody disease (such as the influx of inflammatory cells in glomeruli and formation of crescents) implies that anti-GBM antibodies formed in this model are non-pathogenic. Other NC1 antigens that elicit murine anti-GBM autoantibodies but not form subepithelial ICs (e.g. NC1 hexamers from bovine GBM) induce neither proteinuria nor any glomerular pathology in wild type DBA/1 or C57Bl/6 mice (W.L., D.B.B. and others, manuscript in preparation). The inability of murine anti-GBM autoAbs to mediate severe glomerulonephritis (in contrast to Goodpasture autoAbs in human anti-GBM disease) may be due to prevalence of mouse IgG1 autoAbs, which do not activate complement and have lower affinity for activating Fcγ receptors than murine IgG2a and IgG2b, hence a reduced ability to elicit inflammation.
All currently available evidence points to a key role of complement activation in the development of proteinuria and pathogenesis of MN in α3NC1-immunized mice. Proteinuric mice have glomerular deposition of murine C3 and C5b-9 and urinary excretion of C5b-9. Ablation of activating IgG Fc receptors in congenic D1. FcRγ-/- does not influence the severity of α3NC1-induced murine MN, implying that subepithelial IC’s mediate pathology via other effector pathways. AKR and NOD mice, which are naturally deficient in complement C5 [47] are resistant to α3NC1-induced g lomerular pathology [40]. In preliminary studies presented in abstract form at the Renal Week 2012 meeting, several of us (W.L., D.B.B. and others) found that the genetic ablation or pharmacologic inhibition of factor B, a key component of the alternative complement pathway, prevents glomerular C3 deposition and proteinuria in α3NC1-immunized mice [48].
A possible role of α3(IV) collagen in the pathogenesis of some forms of human MN is suggested by the association between anti-GBM/Goodpasture disease and MN. At least 25 cases have been reported, reviewed in [49]. Anti-GBM/Goodpasture disease can either precede or is followed by MN, though simultaneous presentation is more common. In these patients, the linear GBM IgG staining found on the renal biopsy is likely due to Goodpasture autoantibodies binding to the NC1 domains of α345(IV) collagen in the GBM. Subepithelial IC’s may form due to Goodpasture autoantibodies binding to podocyte-associated or nascent α3(IV) collagen secreted by podocytes but not yet incorporated in the GBM, or to α3(IV) collagen fragments released from the GBM by proteases.
Notably, other GBM antigens have also been implicated in various animal models of MN. Pre-sensitized rats injected with heterologous antibodies to rat GBM heparan sulfate proteoglycans develop subepithelial IC deposits with clinical and histologic features of MN [50]. Anti-laminin antibodies play a central role in the formation of subepithelial IC deposits and heavy proteinuria in HgCl2-induced nephropathy in Brown-Norway rats [51]. Thickening of the GBM due to periodic expansions of lamina rara externa forming a beaded pattern has been observed in Swiss-Webster mice immunized with a crude preparation of human GBM. Linear GBM staining was observed for mouse IgG1, but capillary loop staining for mouse IgG2a, IgG2b and IgG3 changed from a linear GBM to a granular pattern. Electron dense subepithelial IC deposits were not observed and proteinuria was modest, perhaps due to the absence of C3 deposition in the GBM [52]. The antigen(s) implicated in this model have not been identified. How GBM antigens are implicated the development of MN is not currently understood.
Murine membranous lupus nephritisin MRL/lpr mice deficient for IL-27 receptor (WSX-1)
MN can be secondary to other causes, including autoimmune diseases such as systemic lupus erythematosus (SLE). SLE is an autoimmune disease characterized by the production of autoantibodies to DNA and other nuclear antigens and multi-organ inflammation. Lupus nephritis (LN) is a major complication in SLE, caused by the formation and deposition in the glomerular capillary wall of IC’s that induce glomerular inflammation and injury. Based on the morphology on the renal biopsy, about 10-20% of cases feature continuous granular subepithelial immune deposits, often with concomitant mesangial immune deposits and variable mesangial cell proliferation, and are classified as class V lupus nephritis, also known as membranous lupus nephritis [53]. Membranous lupus nephritis is the most common form of secondary MN in the United States. MRL/lpr mice, a model for SLE, develop spontaneous glomerulonephritis recapitulating diffuse proliferative glomerulonephritis in human lupus nephritis class IV, an autoimmune response driven by Th1 cells producing IFN-γ. Ablation of the WSX-1 gene, which encodes a subunit of the IL-27 receptor, impairs proper Th1 differentiationin mice, skewing immune responses to Th2 [54]. Ablation of WSX-1 gene in MRL/lpr micedramatically altersthe kidney pathology from diffuse proliferative glomerulonephritis to membranous lupus, with a predominance of mouse IgG1 in glomerular IC deposits, reduced IFN-γ production, and elevated IL-4 expression [55]. This indicates that the Th1/Th2 balance has a major influence on the phenotype of glomerulonephritis, and the Th2 predominance promotes the development of membranous lupus nephritis.
Transgenic mouse models of membranous nephropathy in development
Given the recent identification of PLA2R as the major autoantigen in human MN, it is imperative to generate a model in which the pathogenesis of anti-PLA2R antibodies can be tested. Initial collaborative attempts to generate a transgenic mouse model by expressing human PLA2R1 in the CD1 (ICR) strain were unsuccessful (L. Beck, D. Salant, G. Lambeau, S. Quaggin, V. Eremina; unpublished observations). Despite generating three founder lines that expressed human PLA2R under the murine nephrin promoter, and despite detection of human PLA2R expression by glomeruli via PCR, there was no detectable expression of the human protein in the kidneys or glomeruli of any of the founder lines. Injection of normal or anti-PLA2R-positive human IgG did not lead to proteinuria in either control or the transgenic mice. A recent abstract by Zahner et al. presented at the Renal Week 2012 annual meeting describes the generation of a tetracycline-inducible human PLA2R1 transgenic mouse line with podocyte expression of the tetracycline transactivator directed by the podocin promoter [56]. While these investigators were able to show glomerular expression of human PLA2R, there was not yet any data as to whether or not the animals became proteinuric upon injection of anti-PLA2R. Of note, it has been difficult to generate large quantities of recombinant PLA2R protein in culture, possibly related to the finding that PLA2R is able to induce cellular senescence [57] and therefore similar issues may be hampering the establishment of a murine model with this particular antigen. Cultured cells and potentially murine podocytes in vivo may lack the appropriate machinery (chaperonins or other binding proteins) need for optimal export from the endoplasmic reticulum. We are hopeful that as investigators discover more about the PLA2R molecule and other human podocyte antigens, increasingly more sophisticated murine models of human MN can be generated.
Future directions and concluding remarks
The recent development of mouse models that recapitulate the hallmarks of human MN as faithfully as rat Heymann nephritis models has overcome a critical barrier that has long hindered progress in the field. The murine models described herein, as well as other models that may be developed in the future, will be essential for answering unresolved questions about the patho-mechanisms of MN that could not be effectively addressed using rat models. Whereas studies in Heymann nephritis models demonstrated that the activation of the terminal complement cascade and subsequent assembly of C5b-9 on podocytes arecritical mediators of podocyte injury and proteinuria, our understanding of the mechanisms of complement activation by subepithelial ICs remains rudimentary. The specific roles of the classic, lectin and alternative pathways of complement activation and the importance of various complement regulatory proteins in the pathogenesis of MN remain to be elucidated. Addressing these basic questions requires mouse models in which subepithelial ICs and proteinuria can be induced reliably and reproducibly, and proteinuria is complement dependent--regardless of the specific antigens implicated. Models exhibiting these features, as has been shown so far for α3NC1-induced murine MN, will not only help define the basic mechanisms of complement activation by subepithelial ICs, but also are ideally suited for preclinical studies of new therapeutics aiming toarrest pathogenic complement activation and podocyte damage. We anticipate that the new knowledge gained from these studies will translate into much needed novel mechanism-based therapies for MN: more effective, more specific and less toxic. To paraphrase the immortal words of Robert Frost, taking the road less travelled can make all the difference. CMS is supported by the Deutsche Forschungsgesellschaft (KFO228 “Immunoathogenesis and Therapy of Glomerulonephritis”).
Acknowledgements
DBB is supported by NIH grant R01 DK080799 and by a Normal S. Coplon extramural research grant from Satellite Healthcare. JJZ is supported by The Youth Innovation Fund of the First Affiliated Hospital of Zhengzhou University. LHB is supported by NIH grant RO1 DK097053. CMS is supported by the Deutsche Forschungsgesellschaft (KFO228 “Immunoathogenesis and Therapy of Glomerulonephritis”).
Disclosure of conflict of interest
The authors declare they have no conflict of interest.
References
- 1.Fervenza FC, Sethi S, Specks U. Idiopathic membranous nephropathy: diagnosis and treatment. Clin J Am Soc Nephrol. 2008;3:905–919. doi: 10.2215/CJN.04321007. [DOI] [PubMed] [Google Scholar]
- 2.Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, Deschenes G, Ronco PM. Antenatal membranous glomerulo-nephritis due to anti-neutral endopeptidase antibodies. N Engl J Med. 2002;346:2053–2060. doi: 10.1056/NEJMoa012895. [DOI] [PubMed] [Google Scholar]
- 3.Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hofstra JM, Beck LH Jr, Beck DM, Wetzels JF, Salant DJ. Anti-phospholipase A receptor antibodies correlate with clinical status in idiopathic membranous nephropathy. Clin J Am Soc Nephrol. 2011;6:1286–1291. doi: 10.2215/CJN.07210810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, Trivelli A, Magnasco A, Petretto A, Santucci L, Mattei S, Gatti R, Scolari F, Kador P, Allegri L, Ghiggeri GM. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol. 2010;21:507–519. doi: 10.1681/ASN.2008121259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murtas C, Bruschi M, Candiano G, Moroni G, Magistroni R, Magnano A, Bruno F, Radice A, Furci L, Argentiero L, Carnevali ML, Messa P, Scolari F, Sinico RA, Gesualdo L, Fervenza FC, Allegri L, Ravani P, Ghiggeri GM. Coexistence of different circulating anti-podocyte antibodies in membranous nephropathy. Clin J Am Soc Nephrol. 2012;7:1394–1400. doi: 10.2215/CJN.02170312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Debiec H, Lefeu F, Kemper MJ, Niaudet P, Deschenes G, Remuzzi G, Ulinski T, Ronco P. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med. 2011;364:2101–2110. doi: 10.1056/NEJMoa1013792. [DOI] [PubMed] [Google Scholar]
- 8.Nangaku M, Couser WG. Mechanisms of immune-deposit formation and the mediation of immune renal injury. Clin Exp Nephrol. 2005;9:183–191. doi: 10.1007/s10157-005-0357-8. [DOI] [PubMed] [Google Scholar]
- 9.Couser WG, Steinmuller DR, Stilmant MM, Salant DJ, Lowenstein LM. Experimental glomerulonephritis in the isolated perfused rat kidney. J Clin Invest. 1978;62:1275–1287. doi: 10.1172/JCI109248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glassock RJ. The treatment of idiopathic membranous nephropathy: a dilemma or a conundrum? Am J Kidney Dis. 2004;44:562–566. [PubMed] [Google Scholar]
- 11.Waldman M, Austin HA 3rd. Controversies in the treatment of idiopathic membranous nephropathy. Nat Rev Nephrol. 2009;5:469–479. doi: 10.1038/nrneph.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Schieppati A, Cai G, Chen X, Zamora J, Giuliano GA, Braun N, Perna A. Immunosuppression for Membranous Nephropathy: A Systematic Review and Meta-Analysis of 36 Clinical Trials. Clin J Am Soc Nephrol. 2013;8:787–96. doi: 10.2215/CJN.07570712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quigg RJ. Why study membranous nephropathy in rats? Kidney Int. 2003;64:2318–2319. doi: 10.1046/j.1523-1755.2003.00382.x. [DOI] [PubMed] [Google Scholar]
- 14.Jefferson JA, Pippin JW, Shankland SJ. Experimental Models of Membranous Nephropathy. Drug Discov Today Dis Models. 2010;7:27–33. doi: 10.1016/j.ddmod.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cybulsky AV, Quigg RJ, Salant DJ. Experimental membranous nephropathy redux. Am J Physiol Renal Physiol. 2005;289:F660–671. doi: 10.1152/ajprenal.00437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heymann W, Hackel DB, Harwood S, Wilson SG, Hunter JL. Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med. 1959;100:660–664. doi: 10.3181/00379727-100-24736. [DOI] [PubMed] [Google Scholar]
- 17.Noble B, Van Liew JB, Andres GA, Brentjens JR. Factors influencing susceptibility of LEW rats to Heymann nephritis. Clin Immunol Immunopathol. 1984;30:241–254. doi: 10.1016/0090-1229(84)90059-x. [DOI] [PubMed] [Google Scholar]
- 18.Allison ME, Wilson CB, Gottschalk CW. Pathophysiology of experimental glomerulonephritis in rats. J Clin Invest. 1974;53:1402–1423. doi: 10.1172/JCI107689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol. 1995;6:35–47. doi: 10.1681/ASN.V6135. [DOI] [PubMed] [Google Scholar]
- 20.Edgington TS, Glassock RJ, Dixon FJ. Autologous immune complex nephritis induced with renal tubular antigen. I. Identification and isolation of the pathogenetic antigen. J Exp Med. 1968;127:555–572. doi: 10.1084/jem.127.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgington TS, Glassock RJ, Watson JI, Dixon FJ. Characterization and isolation of specific renal tubular epithelial antigens. J Immunol. 1967;99:1199–1210. [PubMed] [Google Scholar]
- 22.Assmann KJ, Tangelder MM, Lange WP, Tadema TM, Koene RA. Membranous glomerulonephritis in the mouse. Kidney Int. 1983;24:303–312. doi: 10.1038/ki.1983.159. [DOI] [PubMed] [Google Scholar]
- 23.Oliveira DB. Membranous nephropathy: an IgG4-mediated disease. Lancet. 1998;351:670–671. doi: 10.1016/S0140-6736(97)04122-6. [DOI] [PubMed] [Google Scholar]
- 24.Doi T, Mayumi M, Kanatsu K, Suehiro F, Hamashima Y. Distribution of IgG subclasses in membranous nephropathy. Clin Exp Immunol. 1984;58:57–62. [PMC free article] [PubMed] [Google Scholar]
- 25.Assmann KJ, van Son JP, Dijkman HB, Koene RA. A nephritogenic rat monoclonal antibody to mouse aminopeptidase A. Induction of massive albuminuria after a single intravenous injection. J Exp Med. 1992;175:623–635. doi: 10.1084/jem.175.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Border WA, Ward HJ, Kamil ES, Cohen AH. Induction of membranous nephropathy in rabbits by administration of an exogenous cationic antigen. J Clin Invest. 1982;69:451–461. doi: 10.1172/JCI110469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen JS, Chen A, Chang LC, Chang WS, Lee HS, Lin SH, Lin YF. Mouse model of membranous nephropathy induced by cationic bovine serum albumin: antigen dose-response relations and strain differences. Nephrol Dial Transplant. 2004;19:2721–2728. doi: 10.1093/ndt/gfh419. [DOI] [PubMed] [Google Scholar]
- 28.Wu CC, Chen JS, Chen SJ, Lin SH, Chen A, Chang LC, Sytwu HK, Lin YF. Kinetics of adaptive immunity to cationic bovine serum albumin-induced membranous nephropathy. Kidney Int. 2007;72:831–840. doi: 10.1038/sj.ki.5002426. [DOI] [PubMed] [Google Scholar]
- 29.Holdsworth SR, Kitching AR, Tipping PG. Th1 and Th2 T helper cell subsets affect patterns of injury and outcomes in glomerulonephritis. Kidney Int. 1999;55:1198–1216. doi: 10.1046/j.1523-1755.1999.00369.x. [DOI] [PubMed] [Google Scholar]
- 30.Kuroki A, Iyoda M, Shibata T, Sugisaki T. Th2 cytokines increase and stimulate B cells to produce IgG4 in idiopathic membranous nephropathy. Kidney Int. 2005;68:302–310. doi: 10.1111/j.1523-1755.2005.00415.x. [DOI] [PubMed] [Google Scholar]
- 31.Hirayama K, Ebihara I, Yamamoto S, Kai H, Muro K, Yamagata K, Kobayashi M, Koyama A. Predominance of type-2 immune response in idiopathic membranous nephropathy. Cytoplasmic cytokine analysis. Nephron. 2002;91:255–261. doi: 10.1159/000058401. [DOI] [PubMed] [Google Scholar]
- 32.Wu CC, Lu KC, Lin YF, Chen JS, Huang CF, Chen CC, Lin SH, Chu P, Sytwu HK. Pathogenic role of effector cells and immunoglobulins in cationic bovine serum albumin-induced membranous nephropathy. J Clin Immunol. 2012;32:138–149. doi: 10.1007/s10875-011-9614-7. [DOI] [PubMed] [Google Scholar]
- 33.Meyer-Schwesinger C, Dehde S, Klug P, Becker JU, Mathey S, Arefi K, Balabanov S, Venz S, Endlich KH, Pekna M, Gessner JE, Thaiss F, Meyer TN. Nephrotic syndrome and subepithelial deposits in a mouse model of immune-mediated anti-podocyte glomerulonephritis. J Immunol. 2011;187:3218–3229. doi: 10.4049/jimmunol.1003451. [DOI] [PubMed] [Google Scholar]
- 34.Schiwek D, Endlich N, Holzman L, Holthofer H, Kriz W, Endlich K. Stable expression of nephrin and localization to cell-cell contacts in novel murine podocyte cell lines. Kidney Int. 2004;66:91–101. doi: 10.1111/j.1523-1755.2004.00711.x. [DOI] [PubMed] [Google Scholar]
- 35.Mundel P, Reiser J, Zuniga Mejia Borja A, Pavenstadt H, Davidson GR, Kriz W, Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 36.Meyer TN, Schwesinger C, Wahlefeld J, Dehde S, Kerjaschki D, Becker JU, Stahl RA, Thaiss F. A new mouse model of immune-mediated podocyte injury. Kidney Int. 2007;72:841–852. doi: 10.1038/sj.ki.5002450. [DOI] [PubMed] [Google Scholar]
- 37.Bao L, Haas M, Pippin J, Wang Y, Miwa T, Chang A, Minto AW, Petkova M, Qiao G, Song WC, Alpers CE, Zhang J, Shankland SJ, Quigg RJ. Focal and segmental glomerulosclerosis induced in mice lacking decay-accelerating factor in T cells. J Clin Invest. 2009;119:1264–1274. doi: 10.1172/JCI36000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang JJ, Malekpour M, Luo W, Ge L, Olaru F, Wang XP, Bah M, Sado Y, Heidet L, Kleinau S, Fogo AB, Borza DB. Murine membranous nephropathy: immunization with alpha3(IV) collagen fragment induces subepithelial immune complexes and FcgammaR-independent nephrotic syndrome. J Immunol. 2012;188:3268–3277. doi: 10.4049/jimmunol.1103368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brenchley PE, Coupes B, Short CD, O’Donoghue DJ, Ballardie FW, Mallick NP. Urinary C3dg and C5b-9 indicate active immune disease in human membranous nephropathy. Kidney Int. 1992;41:933–937. doi: 10.1038/ki.1992.143. [DOI] [PubMed] [Google Scholar]
- 40.Hopfer H, Maron R, Butzmann U, Helmchen U, Weiner HL, Kalluri R. The importance of cell-mediated immunity in the course and severity of autoimmune anti-glomerular basement membrane disease in mice. Faseb J. 2003;17:860–868. doi: 10.1096/fj.02-0746com. [DOI] [PubMed] [Google Scholar]
- 41.Hopfer H, Holzer J, Hunemorder S, Paust HJ, Sachs M, Meyer-Schwesinger C, Turner JE, Panzer U, Mittrucker HW. Characterization of the renal CD4(+) T-cell response in experimental autoimmune glomerulonephritis. Kidney Int. 2012;82:60–71. doi: 10.1038/ki.2012.73. [DOI] [PubMed] [Google Scholar]
- 42.Sharp PE, Martin-Ramirez J, Boross P, Mangsbo SM, Reynolds J, Moss J, Pusey CD, Cook HT, Tarzi RM, Verbeek JS. Increased incidence of anti-GBM disease in Fcgamma receptor 2b deficient mice, but not mice with conditional deletion of Fcgr2b on either B cells or myeloid cells alone. Mol Immunol. 2012;50:49–56. doi: 10.1016/j.molimm.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Edgington TS, Glassock RJ, Dixon FJ. Autologous immune-complex pathogenesis of experimental allergic glomerulonephritis. Science. 1967;155:1432–1434. doi: 10.1126/science.155.3768.1432. [DOI] [PubMed] [Google Scholar]
- 44.Barabas AZ, Lannigan R. Immune-complex nephritis in the rabbit produced by injections of rat renal tubular fraction 3 antigen. Br J Exp Pathol. 1981;62:94–102. [PMC free article] [PubMed] [Google Scholar]
- 45.Borza CM, Borza DB, Pedchenko V, Saleem MA, Mathieson PW, Sado Y, Hudson HM, Pozzi A, Saus J, Abrahamson DR, Zent R, Hudson BG. Human podocytes adhere to the KRGDS motif of the alpha3alpha4alpha5 collagen IV network. J Am Soc Nephrol. 2008;19:677–684. doi: 10.1681/ASN.2007070793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borza CM, Pozzi A, Borza DB, Pedchenko V, Hellmark T, Hudson BG, Zent R. Integrin {alpha}3beta1, a Novel Receptor for {alpha}3(IV) Noncollagenous Domain and a Trans-dominant Inhibitor for Integrin {alpha}vbeta3. J Biol Chem. 2006;281:20932–20939. doi: 10.1074/jbc.M601147200. [DOI] [PubMed] [Google Scholar]
- 47.Nilsson UR, Muller-Eberhard HJ. Deficiency of the fifth component of complement in mice with an inherited complement defect. J Exp Med. 1967;125:1–16. doi: 10.1084/jem.125.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo W, Olaru F, Ge L, Rops AL, Van der Vlag J, Holers VM, Thurman JM, Borza DB. Alternative Complement Pathway Is Essential for Glomerular C3 Deposition and Progressive Proteinuria in Murine Membranous Nephropathy [Abstract FR-OR140] . J Am Soc Nephrol. 2012;23:62A. [Google Scholar]
- 49.Basford AW, Lewis J, Dwyer JP, Fogo AB. Membranous nephropathy with crescents. J Am Soc Nephrol. 2011;22:1804–1808. doi: 10.1681/ASN.2010090923. [DOI] [PubMed] [Google Scholar]
- 50.Makino H, Lelongt B, Kanwar YS. Nephritogenicity of proteoglycans. II. A model of immune complex nephritis. Kidney Int. 1988;34:195–208. doi: 10.1038/ki.1988.165. [DOI] [PubMed] [Google Scholar]
- 51.Icard P, Pelletier L, Vial MC, Mandet C, Pasquier R, Michel A, Druet P. Evidence for a role of antilaminin-producing B cell clones that escape tolerance in the pathogenesis of HgCl2-induced membranous glomerulopathy. Nephrol Dial Transplant. 1993;8:122–127. [PubMed] [Google Scholar]
- 52.Bolton WK, Benton FR, Sturgill BC. Autoimmune glomerulotubular nephropathy in mice. Clin Exp Immunol. 1978;33:463–473. [PMC free article] [PubMed] [Google Scholar]
- 53.Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, Balow JE, Bruijn JA, Cook T, Ferrario F, Fogo AB, Ginzler EM, Hebert L, Hill G, Hill P, Jennette JC, Kong NC, Lesavre P, Lockshin M, Looi LM, Makino H, Moura LA, Nagata M. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65:521–530. doi: 10.1111/j.1523-1755.2004.00443.x. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida H, Hamano S, Senaldi G, Covey T, Faggioni R, Mu S, Xia M, Wakeham AC, Nishina H, Potter J, Saris CJ, Mak TW. WSX-1 is required for the initiation of Th1 responses and resistance to L. major infection. Immunity. 2001;15:569–578. doi: 10.1016/s1074-7613(01)00206-0. [DOI] [PubMed] [Google Scholar]
- 55.Shimizu S, Sugiyama N, Masutani K, Sadanaga A, Miyazaki Y, Inoue Y, Akahoshi M, Katafuchi R, Hirakata H, Harada M, Hamano S, Nakashima H, Yoshida H. Membranous glomerulonephritis development with Th2-type immune deviations in MRL/lpr mice deficient for IL-27 receptor (WSX-1) J Immunol. 2005;175:7185–7192. doi: 10.4049/jimmunol.175.11.7185. [DOI] [PubMed] [Google Scholar]
- 56.Zahner G, Hoxha E, Helmchen U, Stahl RA. The Generation of Inducible Podocyte Specific Human Phospholipase A2-Receptor-1 Transgenic Mice [Abstract FR-PO874] . J Am Soc Nephrol. 2012;23:568A. [Google Scholar]
- 57.Augert A, Payre C, de Launoit Y, Gil J, Lambeau G, Bernard D. The M-type receptor PLA2R regulates senescence through the p53 pathway. EMBO Rep. 2009;10:271–277. doi: 10.1038/embor.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]