

Abstract
Plasmacytoid dendritic cells (pDCs) and neutrophils are detected in psoriatic skin lesions and implicated in the pathogenesis of psoriasis. pDCs specialize in the production of type I interferon (IFNI), a cytokine that plays an important role in chronic autoimmune-like inflammation, including psoriasis. Here, we demonstrate that IFNI production in pDCs is stimulated by DNA structures containing the neutrophil serine protease cathepsin G (CatG) and the secretory leukocyte protease inhibitor (SLPI), which is a controlling inhibitor of serine proteases. We also demonstrate the presence of neutrophil-derived DNA structures containing CatG and SLPI in lesional skin samples from psoriasis patients. These findings suggest a previously unappreciated role for CatG in psoriasis by linking CatG and its inhibitor SLPI to the IFNI-dependent regulation of immune responses by pDCs in psoriatic skin.
Keywords: Serine protease, neutrophil extracellular traps, psoriasis, autoimmunity
Introduction
Plasmacytoid dendritic cells (pDCs) constitute a distinct subset of dendritic cells that specializes in the rapid and robust production of type I interferons (IFNI) such as IFN-α and IFN-β in response to the activation of the Toll-like receptors TLR7 and TLR9 [1]. These endosome-localized TLR receptors allow pDCs to uniquely respond to pathogen-derived nucleic acids and, through the production of IFNI, to inhibit viral replication and activate immune responses [2]. In addition to their contribution to antiviral defenses, pDCs have emerged as a pivotal cell type in autoimmune and chronic autoinflammatory conditions such as systemic lupus erythematosus (SLE) and psoriasis [3-6]. The contribution of pDCs to these disorders is largely attributed to the production of IFNI by these cells following the activation of TLR9 by endogenous DNA. Although pDCs normally do not respond to host DNA, self-DNA can also trigger the production of INFI by pDCs if it reaches the DNA sensors inside the cell. The delivery of self-DNA to endosomal TLR9 was first reported to require the coupling of the DNA to an antimicrobial peptide of the cathelicidin family of proteins, LL-37 [5]. Several other proteins/peptides, primarily of neutrophil origin, have subsequently been identified along with DNA as potent triggers of IFNI in pDCs. These include the human neutrophil peptides of the α-defensin family [4], the high mobility group box 1 protein (HMGB) [3] and human neutrophil elastase (HNE) together with the secretory leukocyte proteinase inhibitor (SLPI) [7]. A candidate source of these molecules is the neutrophil extracellular trap (NET), which is a weblike structure that is composed of DNA as well as nuclear, granular and cytoplasmic proteins and is released by neutrophils into the extracellular environment [8].
The mechanisms that regulate NET formation are still largely unknown but are thought to involve several steps that are triggered by neutrophil activation. These steps include the production of reactive oxygen species (ROS), the the translocation of HNE to the nucleus, followed by chromatin decondensation, which is mediated by PAD4-dependent histone citrulination and/or HNE-dependent histone processing. During the later phases of NET generation, the entire cell is filled with loose karyoplasm that mixes with the cytoplasm. Finally, the decondensed chromatin containing granular and cytoplasmic proteins is deposited extracellularly [9-12].
Although it can be envisaged that the various proteins exposed on the NET are equipped with pDC-activating functions, we recently demonstrated that this process may be protein-specific. Whereas HNE together with SLPI and DNA induced marked production of IFNI by pDCs, another protein closely related to HNE, azurocidin, did not stimulate IFNI synthesis under the same experimental conditions [7]. Azurocidin, similarly to HNE, was identified as a component of the NET by a proteomic analysis [8]. Therefore, these data suggest that NET proteins differ in their ability to trigger IFNI production by pDCs. Because pDCs play a key role in psoriasis, it is important to identify the NET components that stimulate these cells.
HNE and cathepsin G (CatG) are serine proteases that are stored in the primary (azurophil) granules of neutrophils [13]. They share many functions that are primarily related to their proteolytic activity, including the processing/degradation of cell surface and extracellular matrix molecules [13,14]. Both proteases are inhibited by SLPI, which is an inhibitor expressed by keratinocytes and immune cells such as granulocytes in psoriatic skin [7,15,16]. Because CatG, similarly to HNE, is deposited by neutrophils into the extracellular milieu either through extracellular discharge of the granules’ contents or through NET expulsion, it may participate in the pathogenesis of psoriasis as a soluble molecule and/or a NET constituent.
In this study we report that CatG, similarly to HNE, co-localizes with SLPI and DNA deposits in psoriatic skin and, together with DNA and SLPI, strongly stimulates pDCs to secrete IFNI. These data place CatG and SLPI in a pathway that regulates pDC activation in psoriasis.
Materials and methods
Materials
Recombinant SLPI was purchased from R&D Systems. HNE and CatG were isolated using fresh whole human blood from healthy donors as a starting material, as previously described [17]. Pig pancreatic trypsin and bovine α-chymotrypsin were active-site-titrated with p-nitrophenyl guanidobenzoate [18] or p-nitrophenyl acetate [19] and used to standardize a solution of human α1-PI and α1-antichymotrypsin (ACH), respectively. The final inhibitor preparations were more than 95% pure and 80% active. These inhibitors were then used as a secondary standard to determine the activity of HNE and CatG. Activity was measured in 0.1 M Tris-HCl and 0.5 M NaCl, pH 7.4, using 0.5 mM MeO-Suc-Ala-Ala-Pro-Val-pNA or Suc-Ala-Ala-Pro-Phe-pNA as a substrate, respectively. The final preparations of HNE and CatG were more than 95% pure and 90% active. The protein content in the HNE and CatG preparations was estimated using an A280 (1%) of 9.85 or 6.64, respectively. Human plasma ACH was isolated as previously described [20]. Human genomic DNA was isolated from peripheral blood neutrophils of healthy donors using a method based on guanidine thiocyanate (GTC). GTC lysates were extracted using phenol: chloroform. DNA was precipitated from the water phase using isopropanol and resuspended in TE buffer, as previously described [21]. The DNA purity, as measured as the A260/A280 ratio, was between 1.8 and 2.0. Bafilomycin A1 was obtained from Sigma-Aldrich.
Patients
All human studies were performed in accordance with the guidelines established by the Jagiellonian University Institutional Bioethics Committee, under approved protocols. The Declaration of Helsinki protocols were followed. In total, six psoriasis patients (age 28.7 ± 6.6 years; F:M 2:4) and 20 healthy individuals (age 28.7 ± 5.1 years; F:M 7:13) were enrolled in these studies. The severity of the psoriatic skin lesions was assessed according to the Psoriasis Area Severity Index score (PASI) (minimum, 0 points; maximum, 72 points) and ranged from 25.3 to 39.6 (mean ± SD 32.5 ± 5.4). Patients on UV therapy or systemic or local corticosteroid treatments were excluded from the studies. Healthy control subjects had no clinical signs of dermatologic diseases.
Isolation of pDCs and neutrophils
Human blood was collected, and the peripheral blood mononuclear cells (PBMC) and granulocyte-enriched fractions were harvested following LSM1077 (PAA Laboratories) gradient separation, as described by the manufacturer. pDCs were enriched from PBMCs using negative selection with biotinylated mAbs directed against CD3, CD14, CD16, CD19, CD20 and CD56 along with anti-biotin MACS Microbeads (Miltenyi Biotech), according to the manufacturer’s recommendations. The cells were separated by magnetic sorting using an AutoMACS. The pDCs purity was ~95%, as determined by CD123 and BDCA-2 immunoreactivity and flow cytometry analysis.
Neutrophils were further purified from the granulocyte-enriched fractions. The high-density fraction, containing neutrophils and erythrocytes, was mixed with a 1% solution of polyvinyl alcohol (Merck) in PBS and incubated for 20 min at room temperature. Neutrophils were harvested from the upper phase and subjected to hypotonic lysis to remove contaminating red blood cells. The purity of the isolated cells was examined by flow cytometry based on FSC and SSC parameters and/or HNE immunoreactivity. Granulocytes were routinely > 98% pure.
pDC treatment
Purified pDCs (5x104-5x105 cells) were seeded into round-bottom 96-well plates in 50 µl of RPMI. The factors were either added directly into pDC cultures, or first premixed with DNA or with each other in 10 µl RPMI, incubated for 15 min at room temperature, diluted five times and added to the pDC cultures. The following factors were used at the indicated final concentrations: CatG, HNE, SLPI and ACH (all at 1 µM), and/or human neutrophil-derived DNA at 2 µg/ml. In the indicated experiments, bafilomycin was added 30 min before the addition of the premixed stimuli to the pDC cultures. The pDCs were then cultured at 37°C under 5% CO2 for 24h in RPMI supplemented with 10% FBS and human recombinant IL-3 (50 ng/ml) (Peprotech), which improves pDC survival in vitro. The conditioned media were collected and centrifuged at 500 g for 5 min to remove the cells and tested by ELISA. Cytotoxicity was evaluated based on colorimetric measurement of the activity of lactate dehydrogenase (LDH) released from damaged cells and by staining pDCs with propidium iodide and annexin V followed by flow cytometry. The viability pDCs after 24 h of culture in vitro was approximately 80-85%. Cell treatment with the indicated factors, including bafilomycin, did not affect cell viability.
RT-qPCR
Total RNA was extracted as previously described [22] and converted to cDNA using NxGen™ M-MulV reverse transcriptase (Lucigen) with oligo-dT primers (Oligo.pl). Real time PCR was performed on the 7500 Fast system (Applied Biosystems) using SYBR Green I-containing universal PCR master mix (A&A Biotechnology) and primers specific for human IFN alpha (5’-ACCCATTTCAACCAGTCTAGCAG-3’, 5’-GTGGGTTTGAGGCAGATCACA) and GAPDH (5’-CCGAGCCACATCGCTCAGAC-3’, 5’-GTTGAGGTCAATGAAGGGGTC-3’). The relative gene expression normalized to GAPDH was calculated using the 2-ΔΔCT method [23].
ELISA
IFNI levels were quantified by ELISA using human IFN-α-specific matched antibody pairs (eBioscience) according to the manufacturer’s recommendations. The reaction was developed using TMB substrate (BD Science).
Immunohistochemistry
Frozen 6 µm sections were prepared from skin biopsies. Sections were fixed in acetone, blocked with mouse IgG and stained with the following antibodies: first, biotin-mouse anti-human SLPI (Abcam) and rabbit-anti human CatG (Athens Research and Technology), followed by PE-streptavidin (BD Pharmingen) and APC-goat anti-rabbit IgG F(ab)2 (Jackson ImmunoResearch). The sections were counterstained with Hoechst 33258 (Invitrogen). Images were captured with a fully motorized fluorescence microscope (NIKON, Eclipse) and analyzed by NIS elements software (Nikon).
Results
To determine whether CatG co-localizes with DNA and SLPI in areas of skin inflammation, we immunohistochemically analyzed involved psoriatic skin, which is known to be abundantly infiltrated by neutrophils. As demonstrated in Figure 1, the skin contained CatG+ SLPI+ clusters of neutrophils or numerous single neutrophils at various stages of activation. Although double-positive (CatG+, SLPI+) neutrophils were the predominant infiltrating cells, we also observed occasional immune cells that stained for SLPI but not for CatG, which suggests a smaller contribution from other SLPI+ cells. Several different patterns of CatG staining were observed; CatG was found bound to the extracellular matrix scaffold, localized to the cell surface, or associated with decondensed chromatin in the extracellular milieu. Notably, SLPI often co-localized with CatG either at the cell surface or in diffuse chromatin material characterized as NETs. However, in several cases we observed separate staining for SLPI and CatG (Figure 1).
Figure 1.
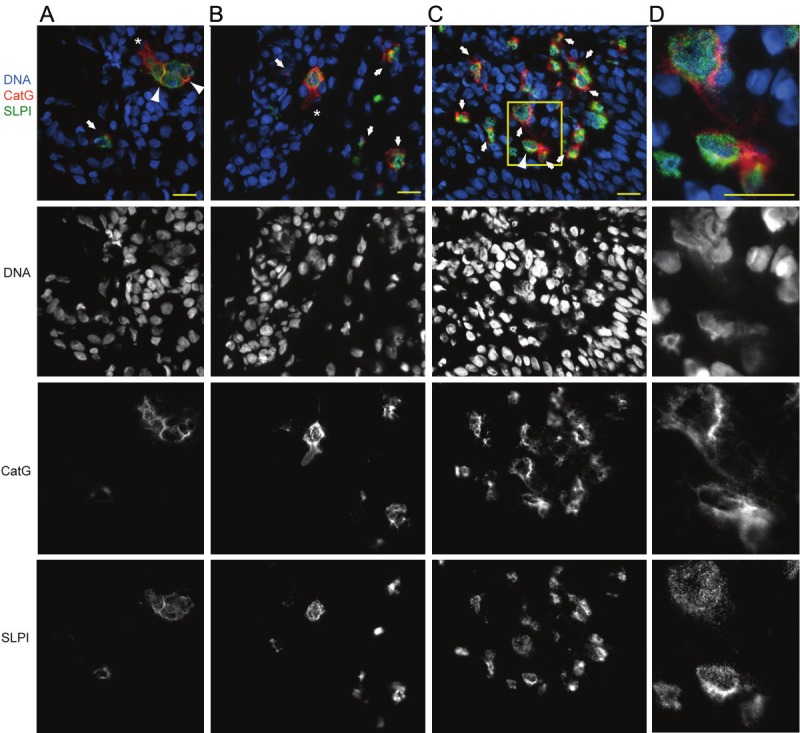
CatG- and SLPIcontaining NETs are present in psoriatic skin. Fluorescence microscopy images of lesional skin stained for CatG (red), SLPI (green) and DNA (blue). Netting cells (A-C), enlarged in (D), were identified by diffuse nuclei (demonstrating a lower-intensity Hoechst staining pattern), intracellular chromatin dispersion or extracellular fibrous DNA deposits. Cells with dispersed chromatin that co-stained with CatG and SLPI are shown by arrows. Neutrophils undergoing CatG release that is not associated with DNA and/or SLPI are shown by asterisks. Arrowheads highlight CatGSLPI-DNA co-localization at the cell surface. Data are from one donor and are representative of at least three donors. Scale bar = 10 µm.
Our previous immunohistochemical data demonstrated that neutrophils are often found in close association with pDCs in psoriatic skin [7]. The presence of CatG- and SLPI-bearing DNA deposits in psoriatic skin suggests that the induction of IFNI synthesis by skin-infiltrating pDCs may depend on the presence of self-DNA/CatG/SLPI complexes. To test whether such complexes can trigger the production of IFNI by pDCs, we stimulated pDCs isolated from human blood of healthy donors with CatG and/or SLPI coupled to neutrophil-derived DNA. As shown in Figure 2A, CatG/SLPI/DNA complexes significantly stimulated the production of IFNI by pDCs. CatG, SLPI or DNA alone was each insufficient to activate pDCs to secrete IFNI. However, the dual complex containing CatG and DNA showed an increased tendency to trigger the production of IFNI in pDCs (Figure 2A), and the stimulatory effect was much stronger for the dual complex containing CatG and SLPI. Because the effect of the dual complexes was much less apparent that that of the triple complex (Figure 2A), in subsequent experiments we focused on the complex of DNA with CatG and SLPI.
Figure 2.
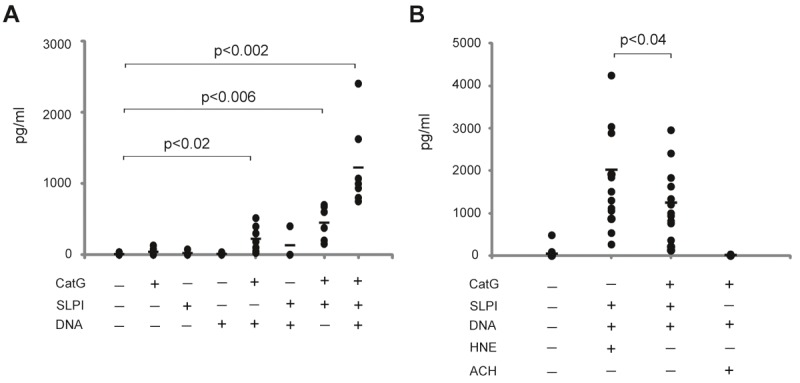
IFNI production is stimulated by CatG- and SLPI-binding DNA structures. Purified pDCs were stimulated with the indicated factors, which were added directly into the pDC culture or were first premixed with DNA or each other. The final concentrations of the factors were as follows: DNA, 2 μg/ml; CatG, HNE, SLPI and ACH, 1µM. Supernatants of stimulated pDCs were collected and the IFNI levels were determined by ELISA. The amount of secreted IFNI is displayed as pg/ml. Each data point represents one experiment, and the mean value in each group, i.e., n = 4-7 (A) or n = 5-14 (B), is indicated by a horizontal line. Statistically significant differences between untreated cells and treated cells (A) or CatG/SLPI/DNA and HNE/SLPI/DNA (B) are shown with the p value from a paired, two-tailed Student’s t test. (Data with p < 0.05 were considered statistically significant).
We previously reported that a mixture of neutrophil DNA with HNE and SLPI was a very potent stimulator of pDCs [7]. We next evaluated whether HNE and CatG are equally effective in triggering IFNI production in pDCs by analyzing IFNI levels in conditioned media of pDCs stimulated with CatG/SLPI/DNA or HNE/SLPI/DNA for 24 h. The replacement of CatG with HNE resulted in a stronger response, which suggests that in comparison with HNE, CatG has a more limited effect on IFNI production (Figure 2B). To determine whether other endogenous CatG inhibitors are capable of inducing IFNI in pDCs, we replaced SLPI with α1antichymotrypsin (ACH). ACH is the dominant inhibitor of CatG and other chymotrypsinlike enzymes, and it is abundantly produced by the liver and to a lesser extent by epithelial cells [24,25]. Therefore, ACH may be present in psoriatic skin because of diffusion from blood plasma or local production by the skin epithelium. As demonstrated in Figure 2B, in contrast to SLPI, ACH in complex with CatG and DNA did not stimulate IFNI production in pDCs. Given that replacement of SLPI with ACH resulted in a lack of IFNI production by pDCs, these data suggest a selective role for SLPI in controlling the serine-protease-mediated activation of DNA sensors in pDCs.
To determine whether self-DNA-dependent IFNI production by pDCs is induced by endosome-localized TLRs, pDCs were pretreated with bafilomycin, which is a specific inhibitor of vacuolar-type H+ ATPase and thus inhibits the acidification of vacuolar compartments. Because endosomal acidification is required for endosomal TLR signaling, bafilomycin is used as a specific inhibitor of endosomal TLRs activation [26]. As demonstrated in Figure 3, bafilomycin markedly inhibited DNA/CatG/SLPI-mediated IFNI production by pDCs at the mRNA and protein levels. These findings support the involvement of TLR9 in the induction of IFNI by CatG- and SLPI-bearing DNA.
Figure 3.
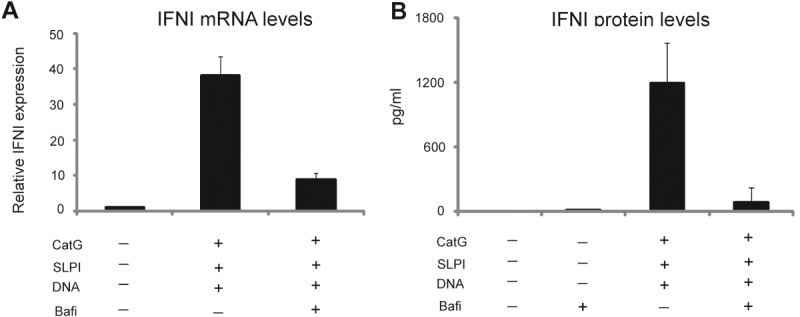
CatG/SLPI/DNA complexes stimulate IFNI production via endosome-localized receptors. Purified pDCs were stimulated for 24 h with the indicated factors: DNA, 2 μg/ml; CatG and SLPI, 1 microM. Bafilomycin (Bafi) was pre-incubated with the pDCs at 25 nM for 30 min, followed by the addition of the CatG/SLPI/DNA mixture. Cells and supernatants were collected and subjected to RT-qPCR (A) or ELISA (B). A. The IFNI expression data are normalized to GAPDH and presented relative to unstimulated cells. The mean ± SD of three independent experiments is shown. B. IFNI levels in conditioned media are displayed as pg/ml. The mean ± SD of three independent experiments is shown.
Discussion
In this study, we identified the NET component CatG as an effective trigger of IFNI production in pDCs. Our data suggest that CatG is likely to play a role in the activation of pDCs by allowing TLR9 to sense extracellular self-DNA. However, to efficiently deliver endogenous DNA to TLR9, CatG appears to require SLPI because in the absence of SLPI, the synthesis of IFNI was negligible or much less apparent. In this regard, we also demonstrated the presence of CatG- and SLPI-enriched DNA deposits in the lesional skin of psoriasis patients, which suggests that these structures may contribute to psoriasis by inducing IFNI production by pDCs that have been recruited to the skin.
Based on in vitro studies, human CatG has long been proposed to be involved in inflammatory processes through the proteolytic modification or cleavage of many proteins, including bacterial virulence factors [14,27]. In addition, this protease was reported to have antibacterial activity that is independent of its enzymatic function [28], which suggests that it contributes on multiple levels to antimicrobial defenses. Human and mouse CatG differ in substrate specificity, and this difference is mainly attributed to peptide cleavage at basic (tryptic) and aromatic (chymotryptic) sites by human enzymes, with a lack of tryptic activity but higher chymotryptic activity for mouse CatG [29]. However, mice deficient in CatG are more susceptible to fungal and bacterial infections, for example in models of lung infection with Streptococcus pneumoniae or intravenous injection of S. aureus or Aspergillus fumigatus [30-32]. Together, these observations support a protective role for CatG in immunity against pathogens. In contrast, in a non-infectious mouse model of renal ischemia-reperfusion injury or acute arthritis induced by passive transfer of monoclonal antibodies against type II collagen, an absence of CatG resulted in decreased inflammation and tissue damage [33,34], which indicates that CatG is also an important component of tissue injury. The defensive and/or pro-inflammatory functions of CatG may be mediated by intracellular protein hydrolysis within lysosomes and/or cleavage of extracellular substrates following the release of CatG from activated neutrophils. In addition, CatG may localize to the cell surface, where it remains catalytically active and possibly resistant to inhibition by naturally occurring proteinase inhibitors [35]. In a cell surface-bound state, CatG regulates neutrophil effector functions such as integrin clustering, which is required for cell spreading and ROS production [36]. In agreement with the observation of CatG plasma membrane expression by activated neutrophils, our data indicate that high levels of CatG are associated with the cell surface in neutrophils that are infiltrating psoriatic skin. Although cell-surface-bound CatG has been proposed to be a secreted protease loaded onto binding sites on the cell membrane [35], in view of recent findings, it is also likely that neutrophils undergoing NET formation expose CatG at some point at the cell surface. If so, CatG in combination with SLPI and DNA at the cell membrane of skin-recruited neutrophils may be well-suited to activate pDCs, which, as we have demonstrated previously, are in contact with neutrophils in psoriatic skin [7]. Alternatively, the association of CatG with SLPI at the cell surface may represent a control mechanism to limit excessive proteolysis or other CatG-mediated functions.
In addition to the DNA decorated with CatG and SLPI that is found at the neutrophil surface, we also noted CatG- and SLPI-enriched DNA deposits in the extracellular environment of psoriatic skin. These deposits resembled, to some degree, the HNE- and SLPI-containing NETs that we reported previously in the lesional skin of patients suffering from psoriasis [7]. Although it remains to be determined whether CatG and HNE co-localize on NETs in lesional skin, it is likely that these enzymes have, at least to some degree, diverse distributions on DNA deposits. Although they have similar biochemical and physiological features, CatG and HNE differ in several properties, most notably in their ability to promote the nuclear decondensation that precedes NET release, which is attributed to HNE but not to CatG [11]. Therefore, the coupling of HNE and CatG to DNA may be separated in time and/or in space, thus resulting in a distinct pattern of HNE and CatG distribution on NETs.
Another intriguing aspect of our studies is the dependence of HNE and CatG on SLPI for effectively controlling the immunogenicity of extracellular self-DNA ([7] and present studies). Several of the most potent known inhibitors of HNE and CatG, such as α1-proteinase inhibitor, ACH and SLPI, may be present in psoriatic skin, either delivered by plasma or serum influx to the afflicted tissue or produced locally by keratinocytes or immune cells. However, in contrast to SLPI, neither α1-proteinase inhibitor nor ACH was effective in stimulating IFNI production by pDCs when combined with self-DNA and HNE or CatG, respectively ([7] and present studies). These data suggest that regulation of the catalytic activity of serine proteases may not be a unifying or sufficient property for an inhibitor to support IFNI production by pDCs in response to self-DNA. In future work, it would be interesting to determine what structural or functional assets of SLPI are responsible for its unique cooperation with neutrophil proteases for the delivery of self-DNA to TLR9.
The IFNI that is produced by pDCs following stimulation by NETs has been reported to promote autoinflammatory conditions such as SLE [3,4]. In contrast, lupus-prone mice that are unable to generate NETs because of a deficiency in NADPH oxidase (Nox2) demonstrate more severe lupus than their Nox2-sufficient counterparts [37], which supports the importance of NETs in homeostasis. Our findings that DNA/CatG/SLPI complexes in the skin can stimulate IFNI production in pDCs raise the question of whether these structures promote the proinflammatory responses of pDCs that support the initiation and/or augmentation of psoriasis, and/or contribute to the resolution of inflammation in psoriatic skin through IFNI. Notably, pDC-derived IFNI also has been proposed to promote wound healing following skin injury [38]. Therefore, although aberrant production of IFNI by activated pDCs may exacerbate the symptoms of psoriasis, IFNI secretion by these cells following stimulation with self-DNA in a complex with CatG or HNE and SLPI is also likely to play an anti-inflammatory role in psoriasis by supporting a healing response in the damaged skin. Our discovery of CatG/SLPI/DNA-mediated IFNI production by pDCs may contribute to a better understanding of the growing complexity of NETs in auto-inflammatory diseases.
Acknowledgments
This work was supported in part by the Team Award and Polish National Science Center grant 2011/02/A/NZ5/00337 (to JC). The Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University is a beneficiary of the structural funds from the European Union (grant No: POIG.02.01.00-12-064/08).
References
- 1.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 2.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, Zal T, Mellman I, Schroder JM, Liu YJ, Gilliet M. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 6.Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, Burg G, Liu YJ, Gilliet M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. 2005;202:135–143. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skrzeczynska-Moncznik J, Wlodarczyk A, Zabieglo K, Kapinska-Mrowiecka M, Marewicz E, Dubin A, Potempa J, Cichy J. Secretory leukocyte proteinase inhibitor-competent DNA deposits are potent stimulators of plasmacytoid dendritic cells: implication for psoriasis. J Immunol. 2012;189:1611–1617. doi: 10.4049/jimmunol.1103293. [DOI] [PubMed] [Google Scholar]
- 8.Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639. doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinkmann V, Goosmann C, Kuhn LI, Zychlinsky A. Automatic quantification of in vitro NET formation. Front Immunol. 2013;3:413. doi: 10.3389/fimmu.2012.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neeli I, Radic M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol. 2013;4:38. doi: 10.3389/fimmu.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5:1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Cichy J, Bals R, Potempa J, Mani A, Pure E. Proteinase-mediated release of epithelial cellassociated CD44. Extracellular CD44 complexes with components of cellular matrices. J Biol Chem. 2002;277:44440–44447. doi: 10.1074/jbc.M207437200. [DOI] [PubMed] [Google Scholar]
- 15.Doumas S, Kolokotronis A, Stefanopoulos P. Anti-inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect Immun. 2005;73:1271–1274. doi: 10.1128/IAI.73.3.1271-1274.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wingens M, van Bergen BH, Hiemstra PS, Meis JF, van Vlijmen-Willems IM, Zeeuwen PL, Mulder J, Kramps HA, van Ruissen F, Schalkwijk J. Induction of SLPI (ALP/HUSI-I) in epidermal keratinocytes. J Invest Dermatol. 1998;111:996–1002. doi: 10.1046/j.1523-1747.1998.00425.x. [DOI] [PubMed] [Google Scholar]
- 17.Baugh RJ, Travis J. Human leukocyte granule elastase: rapid isolation and characterization. Biochemistry. 1976;15:836–841. doi: 10.1021/bi00649a017. [DOI] [PubMed] [Google Scholar]
- 18.Theodore Chase Jr, Elliott S. Titration of trypsin, plasmin and trombin with p-nitrophenyl p'-guanidinobenzoate HCl. Methods in Enzymology. 1970;19:20–27. [Google Scholar]
- 19.Bender ML, Begue-Canton ML, Blakeley RL, Brubacher LJ, Feder J, Gunter CR, Kezdy FJ, Killheffer JV Jr, Marshall TH, Miller CG, Roeske RW, Stoops JK. The determination of the concentration of hydrolytic enzyme solutions: alpha-chymotrypsin, trypsin, papain, elastase, subtilisin, and acetylcholinesterase. J Am Chem Soc. 1966;88:5890–5913. doi: 10.1021/ja00976a034. [DOI] [PubMed] [Google Scholar]
- 20.Travis J, Garner D, Bowen J. Human alpha1-antichymotrypsin: purification and properties. Biochemistry. 1978;17:5647–5651. doi: 10.1021/bi00619a010. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1982. [Google Scholar]
- 22.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Cichy J, Potempa J, Chawla RK, Travis J. Stimulatory effect of inflammatory cytokines on alpha 1-antichymotrypsin expression in human lung-derived epithelial cells. J Clin Invest. 1995;95:2729–2733. doi: 10.1172/JCI117975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cichy J, Potempa J, Chawla RK, Travis J. Regulation of alpha 1-antichymotrypsin synthesis in cells of epithelial origin. FEBS Lett. 1995;359:262–266. doi: 10.1016/0014-5793(95)00064-g. [DOI] [PubMed] [Google Scholar]
- 26.Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol. 2011;186:4794–4804. doi: 10.4049/jimmunol.1000702. [DOI] [PubMed] [Google Scholar]
- 27.Lopez-Boado YS, Espinola M, Bahr S, Belaaouaj A. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J Immunol. 2004;172:509–515. doi: 10.4049/jimmunol.172.1.509. [DOI] [PubMed] [Google Scholar]
- 28.Bangalore N, Travis J, Onunka VC, Pohl J, Shafer WM. Identification of the primary antimicrobial domains in human neutrophil cathepsin G. J Biol Chem. 1990;265:13584–13588. [PubMed] [Google Scholar]
- 29.Raymond WW, Trivedi NN, Makarova A, Ray M, Craik CS, Caughey GH. How immune peptidases change specificity: cathepsin G gained tryptic function but lost efficiency during primate evolution. J Immunol. 2010;185:5360–5368. doi: 10.4049/jimmunol.1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hahn I, Klaus A, Janze AK, Steinwede K, Ding N, Bohling J, Brumshagen C, Serrano H, Gauthier F, Paton JC, Welte T, Maus UA. Cathepsin G and neutrophil elastase play critical and nonredundant roles in lung-protective immunity against Streptococcus pneumoniae in mice. Infect Immun. 2011;79:4893–4901. doi: 10.1128/IAI.05593-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 32.Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 2000;12:201–210. doi: 10.1016/s1074-7613(00)80173-9. [DOI] [PubMed] [Google Scholar]
- 33.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophilderived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimoda N, Fukazawa N, Nonomura K, Fairchild RL. Cathepsin g is required for sustained inflammation and tissue injury after reperfusion of ischemic kidneys. Am J Pathol. 2007;170:930–940. doi: 10.2353/ajpath.2007.060486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol. 1995;131:775–789. doi: 10.1083/jcb.131.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raptis SZ, Shapiro SD, Simmons PM, Cheng AM, Pham CT. Serine protease cathepsin G regulates adhesion-dependent neutrophil effector functions by modulating integrin clustering. Immunity. 2005;22:679–691. doi: 10.1016/j.immuni.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. 2012;4:157ra141. doi: 10.1126/scitranslmed.3004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, Arai N, Gallo RL, Digiovanni J, Gilliet M. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med. 2010;207:2921–2930. doi: 10.1084/jem.20101102. [DOI] [PMC free article] [PubMed] [Google Scholar]