

Abstract
The nuclear translocation and accumulation of IκBα represents an important mechanism regulating transcription of NFκB-dependent pro-inflammatory and anti-apoptotic genes. The nuclear accumulation of IκBα can be induced by post-induction repression in stimulated cells, inhibition of the CRM1-dependent nuclear IκBα export by leptomycin B, and by the inhibition of the 26S proteasome. In addition, IκBα is constitutively localized in the nucleus of human neutrophils, likely contributing to the high rate of spontaneous apoptosis in these cells. In the nucleus, IκBα suppresses transcription of NFκB-dependent pro-inflammatory and anti-apoptotic genes, representing an attractive therapeutic target. However, the inhibition of NFκB-dependent genes by nuclear IκBα is promoter specific, and depends on the subunit composition of NFκB dimers and post-translational modifications of the recruited NFκB proteins. In addition, several recent studies have demonstrated an NFκB-independent role of the nuclear IκBα. In this review, we discuss the mechanisms leading to the nuclear accumulation of IκBα and its nuclear functions as potential targets for anti-inflammatory and anti-cancer therapies.
Keywords: IκBα, NFκB, nuclear protein transport, gene transcription
Introduction
IκBα is a critical regulator of the transcription factor NFκB, which induces expression of a wide range of genes involved in immune and inflammatory responses, cell proliferation and apoptosis [1-5]. Deregulation of IκBα cellular levels and localization results in a variety of diseases, including chronic inflammatory disorders and many types of cancer and leukemia [6-15]. Even though IκBα has been originally discovered as a cytoplasmic inhibitor of NFκB, it is now clear that it has important nuclear functions as well.
NFκB proteins form homodimers or heterodimers consisting of p65 (Rel-A), p50, p52, c-Rel, and Rel-B [16-20]. In the classical model of NFκB activation, IκBα inhibits NFκB activity by masking the nuclear localization signals (NLS) of NFκB dimers and retaining them in an inactive state in the cytoplasm. Following cell stimulation by extracellular stimuli, including inflammatory cytokines, bacterial and viral products, apoptotic signals, and other forms of cellular stress, IκBα is phosphorylated at serine residues 32 and 36 through a cascade of inducible protein kinases that involve IκB kinase (IKK), ubiquitinated, and selectively degraded by the 26S proteasome [21-26]. This results in unmasking of the NLS of the NFκB dimers, which then translocate to the nucleus and stimulate transcription of NFκB-dependent pro-inflammatory and anti-apoptotic genes. Studies have shown that individual NFκB dimers bind various κB sites with differential affinity, which is affected by differences in the affinity of each dimer for the κB site, the ability to interact with associated transcription factors and inhibitors, chromatin environment, and by the post-translational modifications of NFκB proteins [27-32].
One of the first genes induced following NFκB activation is IκBα, since IκBα promoter also contains the NFκB binding region [33-35]. This newly synthesized IκBα can then enter the nucleus, remove NFκB from gene promoters, and transport NFκB proteins back to the cytoplasm [36-39]. This feedback regulation by post-induction repression represents a crucial regulatory mechanism terminating NFκB activation during persistent stimulation, and limiting the NFκB response. Loss of this negative feedback regulation as well as increased degradation of IκBα have been associated with increased NFκB activation in inflammatory diseases as well as in numerous types of cancer and leukemia [6-10].
Regulation of IκBα nuclear transport and accumulation
Sequences determining the nuclear localization of IκBα
IκBα is the most abundant and best-characterized member of the IκB protein family that currently consists of nine IκB proteins: IκBα, IκBβ, IκBε, Bcl-3, IκBz, IκBNS, IκBh, and the precursor proteins p100 and p105. All IκBα proteins are characterized by ankyrin repeat domain (ARD), enabling IκB proteins to form complexes with NFκB dimers and bind other proteins. The IκBα molecule consists of three main regions: N-terminal region where the inducible phosphorylation and ubiquitination occur, the ARD, and an acidic C-terminal sequence that is important for basal degradation of free IκBα [4,5,20,40]. Even though IκBα does not contain the classical nuclear localization sequence (NLS; KK/RXK/R), and its small size (37 kD) would allow a simple diffusion through the nuclear pore complex (NPC), IκBα is transported to the nucleus by an active transport mediated by a nuclear import sequence localized within the ARD of IκBα [41-43].
The nuclear export of IκBα is facilitated by two nuclear export signals (NES) located at the amino terminus (N-NES) [44-47] and carboxyl terminus (C-NES) [37,38]. The nuclear IκBα export is mediated by the NES receptor CRM1, also known as exportin 1, which belongs to the karyopherin β family and shares sequence homology in the Ran-GTP binding domain with members from this family [48,49]. In unstimulated cells, IκBα continuously shuttles between the nucleus and the cytoplasm [38,44]. However, in most cells, the nuclear export of IκBα is dominant over its import, resulting in the cytoplasmic localization of IκBα.
The nuclear translocation and accumulation of IκBα can be induced by three main mechanisms: by the post-induction repression in stimulated cells, by inhibition of the nuclear export of IκBα, and by the proteasome inhibition (Figure 1).
Figure 1.
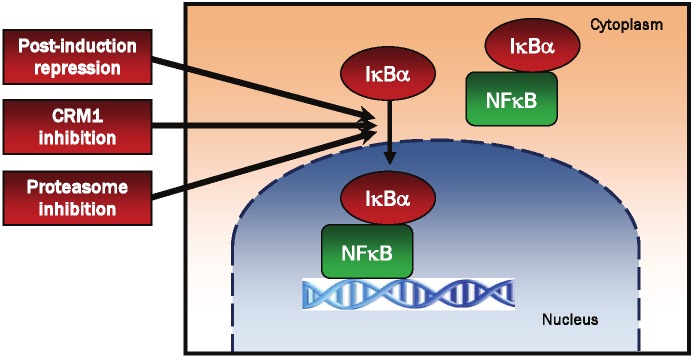
Schematic representation of the main mechanisms inducing the nuclear translocation and accumulation of IκBα. The nuclear translocation and accumulation of IκBα can be induced by the post-induction repression in stimulated cells [36-39], by blocking the nuclear export of IκBα by CRM1 inhibition [54-61], and by the inhibition of the 26S proteasome [69-71].
Induction of nuclear IκBα accumulation by post-induction repression
In continuously stimulated cells, the newly synthesized IκBα translocates to the nucleus, dissociates NFκB dimers from gene promoters and transports them back to the cytoplasm, thus terminating transcription [36-39]. This feedback regulation by post-induction repression represents a crucial mechanism terminating NFκB activation during persistent stimulation. Impaired post-induction repression may result in a persistent activation of NFκB and increased cell survival. Blocking the nuclear export of IκBα by CRM1 inhibitors increases the nuclear IκBα accumulation, suppresses NFκB activity and induces apoptosis, representing an attractive therapeutic target.
Induction of nuclear IκBα accumulation by CRM1 inhibition
Leptomycin B (LMB) is a specific inhibitor of the nuclear protein export that interferes with the interaction between NES and CRM1 by covalently binding to a cysteine residue in the central domain of CRM1 [50-52]. It has been discovered in 1983 as a potent anti-fungal antibiotic produced by Streptomyces [53]. However, since then, LMB has been extensively used to study the nuclear-cytoplasmic shuttling of CRM1-binding proteins, including IκBα [54-57]. Studies from our laboratory have shown that in stimulated human leukocytes, LMB induces nuclear accumulation of IκBα by inhibiting IκBα nuclear export, resulting in the inhibition of NFκB activity, and increased leukocyte apoptosis [57-61]. Even though LMB possesses strong antitumor and anti-inflammatory properties [62,63], its toxicity prevents it from being clinically useful [64,65]. However, using high-content screening technologies and medicinal chemistry approaches based on modifying LMB, several recent studies identified novel selective nuclear export inhibitors (NEI) that maintain the high potency of LMB but are better tolerated [66-68]. These new NEIs have the potential to inhibit the constitutive activity of NFκB in cancer cells and chronic inflammatory disorders by increasing the nuclear levels of IκBα.
Induction of IκBα nuclear accumulation by the proteasome inhibition
We have shown that in addition to the post-induction repression and by blocking the nuclear export of IκBα by the CRM1 inhibition, the nuclear IκBα accumulation can be induced by the proteasome inhibition (Figure 1) [69-71]. Bortezomib (Velcade, PS-341) and other 26S proteasome inhibitors have been developed to inhibit the cytoplasmic degradation of IκBα, thus inhibiting the NFκB signaling in cancer cells [72-75]. However, studies from our laboratory have shown that bortezomib, MG132, MG115 and other proteasome inhibitors inhibit NFκB activity by an additional mechanism that consists of inducing the translocation of IκBα from the cytoplasm to the nucleus in prostate and ovarian cancer cells, HeLa cells, leukemia HL-60 cells, monocytic cells and chronic T cell leukemia Hut-78 cells [69-71]. The proteasome inhibition-induced nuclear IκBα accumulation is dependent on de novo protein synthesis, since cycloheximide (CHX) completely blocks the proteasome-induced nuclear IκBα translocation [69]. This lack of IκBα nuclear translocation in response to the proteasome inhibition in CHX-treated cells could be explained by two mutually non-exclusive mechanisms. In the first model, treatment with CHX might prevent resynthesis of a protein that is otherwise necessary for the proteasome inhibition-induced nuclear translocation of IκBα, but has a short half-life; thus, treatment with CHX would significantly decrease its level. Alternatively, the proteasome inhibition-induced nuclear translocation of IκBα may require that the cellular (cytoplasmic) level of IκBα increases above certain threshold level. When cells are treated with CHX, de-novo synthesis of IκBα is inhibited, and IκBα never reaches this threshold level, even after the degradation of IκBα is blocked by the proteasome inhibition. Similar mechanism has been suggested to account for the proteasome inhibition induced nuclear accumulation of glucocorticoid receptor and the varicella-zoster virus DNA binding protein ORF29p [76-78]. This model is also supported by previous studies that used cells transfected with constructs expressing IκBα and demonstrated that when IκBα is overexpressed, it localizes in the nucleus [79-81]. Since bortezomib has been approved by FDA for the treatment of multiple myeloma and is being tested in clinical trials as a combination therapy to treat other cancers as well [82-85], understanding the mechanism how it induces the nuclear translocation and accumulation of IκBα may lead to the development of more specific and effective therapies in the future.
Induction of IκBα nuclear accumulation by UV light
Interestingly, a recent study by Tsuchiya et al suggested that IκBα translocates into the nucleus and associates with the nuclear IKKβ also in response to UV radiation and other types of oxidative stress [86]. However, in contrast to the proteasome inhibition or to the increased nuclear IκBα accumulation induced by the CRM1 inhibition, this UV light-induced nuclear IκBα translocation does not result in the nuclear IκBα accumulation and inhibition of NFκB activity. On the contrary, the UV light-induced nuclear translocation of IκBα is followed by IκBα degradation and activation of NFκB [86].
Constitutive nuclear localization of IκBα in human neutrophils
In most resting unstimulated cells, IκBα is localized in the cytoplasm and by binding to NFκB dimers, it inhibits their nuclear translocation [1-3,87]. In contrast, in human neutrophils (polymorphonuclear leukocytes), majority (more than 60%) of the total cellular IκBα is localized in the nucleus (Figure 2) [88]. Interestingly, neutrophils are cells that have one of the shortest live spans in the body. They circulate in the blood and in the absence of infection, they undergo apoptosis within 24 hours after the release from bone marrow [89,90]. Even though the NFκB subunits p50 and p65 are present in the nucleus of resting neutrophils as well [88,91], the nuclear IκBα prevents NFκB activation by binding to nuclear p65 NFκB [57]. In response to neutrophil stimulation with lipopolysaccharide (LPS) or pro-inflammatory cytokines, IκBα is phosphorylated by the enzymes of the IKK complex and degraded by the proteasome both in the cytoplasm and in the nucleus [57,92]. However, compared to macrophages and other inflammatory cells, the extent of NFκB activation in human neutrophils is considerably lower [93,94], and this is associated with a decreased production of NFκB-dependent pro-inflammatory cytokines [95,96]. Interestingly, this high nuclear accumulation of IκBα in resting cells is unique to human neutrophils, since in mouse neutrophils, IκBα is localized mainly in the cytoplasm (unpublished data).
Figure 2.
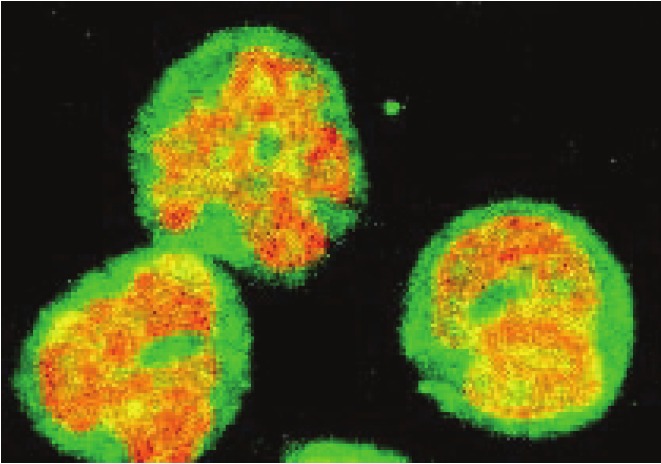
Confocal immunofluorescence microscopy of IκBα in human neutrophils. Resting human neutrophils were fixed and analyzed by confocal laser scanning microscopy using anti-IκBα antibody and FITC-conjugated secondary antibody (green fluorescence) as described [97]. DNA was visualized with propidium iodide (red fluorescence). The figure illustrates the nuclear localization of IκBα in human neutrophils and overlap of the DNA and FITC-IκBα staining (yellow).
The mechanisms responsible for the high nuclear accumulation of IκBα in resting human neutrophils are not understood. There are two possible scenarios. In the first model, the high nuclear IκBα accumulation is the result of an increased nuclear import that is dominant over the nuclear export in human neutrophils. In the second model, the high nuclear IκBα level could be caused by the intranuclear binding of IκBα. This hypothesis is supported by our results indicating that the nuclear IκBα associates with the components of the nuclear matrix in human neutrophils [97]. Our study showed that a further increase of the nuclear accumulation of IκBα in the neutrophils increases caspase-3 activity and accelerates neutrophil apoptosis [57]. Thus, it seems likely that the high nuclear IκBα accumulation in human neutrophils represents one of the underlying mechanisms responsible for the high rate of spontaneous apoptosis in these cells (Figure 3). Since neutrophil apoptosis plays a critical role in the inflammatory response that characterizes sepsis, acute lung injury (ALI), and other inflammatory disorders [98-102], a better understanding of the mechanisms regulating the nuclear accumulation and function of IκBα in the neutrophils will contribute to the development of safer therapies for ALI, sepsis, and other neutrophil-mediated inflammatory disorders.
Figure 3.
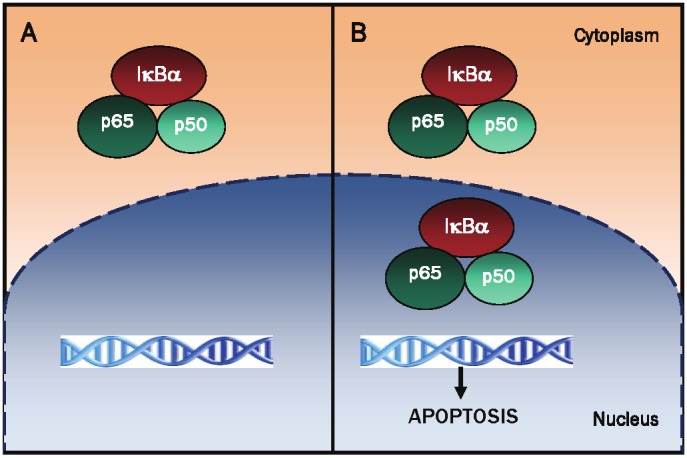
Proposed model of NFκB regulation by nuclear IκBα in human neutrophils. A, In most unstimulated cells, IκBα is localized in the cytoplasm, and by binding to NFκB dimers, it prevents their translocation to the nucleus and NFκB-dependent transcription. B, In human neutrophils, IκBα is localized predominantly in the nucleus [57,88]. However, the nuclear IκBα in the neutrophils associates with NFκB p65 and p50 subunits in the nuclear matrix [97], thus suppressing transcription of NFκB-dependent genes and inducing neutrophil apoptosis.
Nuclear IκBα function
NFκB-dependent function of the nuclear IκBα
The subunit composition of NFκB dimers determines their affinity for IκBα. In vitro, IκBα preferentially binds to p50/65 heterodimer and p65 homodimer, while binding to p50 homodimer is substantially weaker [103-108]. However, even though IκBα can bind to 50 homodimers, it does not inhibit their in vitro DNA-binding activity. The in vitro interaction between IκBα and p65 has a very low dissociation rate resulting in an extremely high affinity and explaining the long half-life observed for the bound IκBα in vivo [109-112]. The precise mechanisms, by which the nuclear IκBα removes NFκB dimers from the target genes in vivo are insufficiently understood. Kinetic studies in living cells indicate a dynamic equilibrium between the promoter-bound and free NFκB dimers [113]. According to this model, the dissociating NFκB dimers may be immediately bound by the free IκBα present in the nucleus. In addition, a most recent study using stopped-flow fluorescence and NMR analysis indicates that the removal of NFκB from promoter DNA is a two-step process [111]. First, IκBα forms a ternary complex with NFκB-DNA, and subsequently, the negatively charged PEST domain of IκBα would displace DNA and dissociate NFκB from the promoter [111]. In vivo, several additional mechanisms are likely to be involved in the termination of NFκB activity. These mechanisms include termination of NFκB activation by p65 phosphorylation/dephosphorylation and acetylation, which regulate affinity for IκBα, nucleosome remodeling and the nuclear degradation of p65 NFκB by the associated proteasome [114-121].
Studies from our laboratory have demonstrated that the recruitment of IκBα to NFκB-dependent promoters is genes specific [60,61,70]. In LPS-stimulated human macrophages, the newly synthesized nuclear IκBα induced by post-induction repression is recruited to TNFα, IL-1β, and IL-6 gene promoters, resulting in the transcriptional suppression of these genes [60]. In contrast, the nuclear IκBα is not recruited to IL-8 promoter and the IL-8 expression is not inhibited by the LMB-induced nuclear IκBα [60]. In vivo, the IL-8 promoter is occupied predominantly by p65 NFκB homodimers phosphorylated on serine 536 [60]. Interestingly, this modification was shown to inhibit p65 binding to IκBα in vitro [119]. These studies indicate that the genes occupied by S536 phosphorylated p65 homodimers are not inhibited by the nuclear IκBα (Figure 4). IKKα, IKKβ and IKKε can phosphorylate p65 on serine 536 [122-127]. However, it is not clear at present whether this phosphorylation occurs before p65 binds to DNA or after, as a part of the preinitiation complex assembly. In this context, both IKKα and IKKβ were shown to be recruited to the promoters of NFκB-dependent as well as NFκB-independent genes [128-132], and could phosphorylate the promoter-bound p65, resulting in a prolonged transcription and decreased binding to the nuclear IκBα. Furthermore, the strength of the in vivo nuclear IκBα-p65 NFκB interaction might be influenced by the DNA sequence of κB response elements in the regulatory regions of NFκB-dependent genes. This would be consistent with studies demonstrating that a single nucleotide can influence the recruitment of specific NFκB dimers and the required cofactors for efficient gene transcription [133,134].
Figure 4.
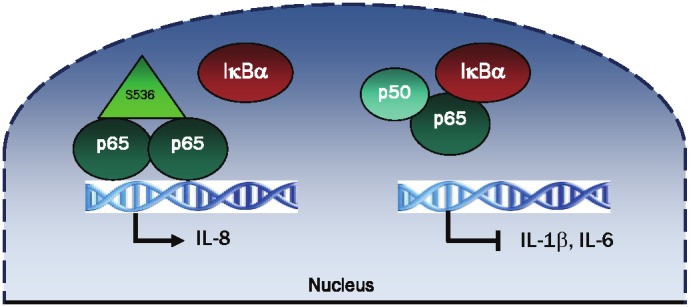
Schematic representation of the regulation of IL-8 transcription by S536 p65 and nuclear IκBα in LPS-stimulated human macrophages. In LPS-stimulated human macrophages, the IL-8 promoter is occupied predominantly by S536 phosphorylated p65 homodimers, which do not bind to IκBα. Consequently, the IL-8 expression is not inhibited by the LMB-induced nuclear IκBα [60]. In contrast, the gene promoters of IL-1β and IL-6 are occupied by p65/p50 heterodimers, and their transcription is repressed by the LMB-induced nuclear IκBα [60].
In addition, the regulation of NFκB-dependent transcription by the nuclear IκBα depends on the subunit composition of NFκB complexes. Our studies indicate that in the chronic T cell leukemia Hut-78 cells, the expression of NFκB-dependent anti-apoptotic genes cIAP1 and cIAP2 is inhibited by the bortezomib-induced nuclear IκBα, while expression of Bcl-2 is not suppressed [70]. Analysis of the in vivo binding of NFκB proteins to cIAP and Bcl-2 promoters by chromatin immunoprecipitation showed that NFκB p65 and p50 subunits are recruited to cIAP1 and cIAP2 promoters, whereas the Bcl-2 promoter is occupied only by NFκB p50. Thus, these data suggest that cIAP1 and cIAP2 promoters associate with NFκB p65/50 heterodimers and this binding and transcription are inhibited by the bortezomib-induced nuclear IκBα. In contrast, Bcl-2 promoter is occupied predominantly by NFκB p50/50 homodimers and its transcription is not inhibited by IκBα.
NFκB-independent function of the nuclear IκBα
The nuclear IκBα not only regulates NFκB binding to NFκB-responsive promoters and NFκB-dependent transcription, but it also physically interacts with different repression elements including nuclear co-repressors, and histone acetyltransferases and deacetylases (HDACs), resulting in transcriptional repression [132,135]. In resting cells, IκBα together with HDACs are recruited to the promoters of Notch target genes correlating with transcriptional repression, whereas in response to NFκB activation, IκBα is released from the chromatin, resulting in Notch-dependent transcriptional activation [136,137]. In addition, IκBα negatively regulates HIV-1 expression by directly binding to the HIV-encoded Tat protein, resulting in the nuclear export and cytoplasmic sequestration of the HIV transactivator [138]. According to this study, IκBα acts as a potent repressor of HIV-1 transcription by inhibiting both NFκB and Tat transacting factors, which are major players in the transcriptional activation and elongation of HIV-1 transcripts [138].
Conclusion
The studies carried out within the last decade clearly demonstrated that in addition to the cytoplasmic retention of NFκB dimers in unstimulated cells, IκBα has important functions in the nucleus as well. Nuclear IκBα is involved in the regulation of numerous pro-inflammatory and anti-apoptotic NFκB-dependent genes as well as NFκB-independent genes through its interactions with HDACs and other transcriptional coregulators. The nuclear translocation and accumulation of IκBα can be induced by the post-induction repression in stimulated cells, by blocking the nuclear export of IκBα by CRM1 inhibitors, and by the proteasome inhibition. A better understanding of the mechanisms regulating the nuclear shuttling of IκBα in stimulated cells, IκBα nuclear translocation and accumulation in response to the proteasome inhibition and the nuclear IκBα accumulation in resting human neutrophils could lead to the development of new therapies aimed at the inhibition of NFκB activity by increased nuclear localization of IκBα. In addition, an important future goal will be to analyze the in vivo NFκB post-translational modifications, and DNA and NFκB subunit preferences of the nuclear IκBα, since they might hold a key to more specific anti-inflammatory and anti-cancer therapies.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (GM079581), National Institute of Allergy and Infectious Diseases (AI085497), and St. John’s University.
References
- 1.Baeuerle PA, Baltimore D. IκB: a specific inhibitor of the NFκB transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh S, Baltimore D. Activation in vitro of NFκB by phosphorylation of its inhibitor IκB. Nature. 1990;344:678–682. doi: 10.1038/344678a0. [DOI] [PubMed] [Google Scholar]
- 3.Beg AA, Baldwin AS Jr. The IκB proteins: multifunctional regulators of Rel/NFκB transcription factors. Genes & Development. 1993;7:2064–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 4.Whiteside ST, Israel A. IκB proteins: structure, function and regulation. Seminars in Cancer Biology. 1997;8:75–82. doi: 10.1006/scbi.1997.0058. [DOI] [PubMed] [Google Scholar]
- 5.Hinz M, Arslan SÇ, Scheidereit C. It takes two to tango: IkBs, the multifunctional partners of NFkB. Immunol Rev. 2012;246:59–76. doi: 10.1111/j.1600-065X.2012.01102.x. [DOI] [PubMed] [Google Scholar]
- 6.Wood KM, Roff M, Hay RT. Defective IκBα in Hodgkin cell lines with constitutively active NFκB. Oncogene. 1998;16:2131–9. doi: 10.1038/sj.onc.1201735. [DOI] [PubMed] [Google Scholar]
- 7.Mozzato-Chamay N, Corbett EL, Bailey RL, Mabey DC, Raynes J, Conway DJ. Polymorphisms in the IκBα promoter region and risk of diseases involving inflammation and fibrosis. Genes Immun. 2001;2:153–5. doi: 10.1038/sj.gene.6363753. [DOI] [PubMed] [Google Scholar]
- 8.Wuerzberger-Davis SM, Chen Y, Yang DT, Kearns JD, Bates PW, Lynch C, Ladell NC, Yu M, Podd A, Zeng H, Huang TT, Wen R, Hoffmann A, Wang D, Miyamoto S. Nuclear export of the NFκB inhibitor IκBα is required for proper B cell and secondary lymphoid tissue formation. Immunity. 2011;34:188–200. doi: 10.1016/j.immuni.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NFκB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135–42. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilmore TD. The Re1/ NFκB /IκB signal transduction pathway and cancer. Cancer Treat Res. 2003;115:241–65. [PubMed] [Google Scholar]
- 11.Aggarwal BB. NFκB: the enemy within. Cancer Cell. 2004;6:203–08. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Li Q, Withoff S, Verma IM. Inflammation-associated cancer: NFκB is the lynchpin. Trends Immunol. 2005;26:318–25. doi: 10.1016/j.it.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh S, Hayden MS. New regulators of NFκB in inflammation. Nature Immunology. 2009;10:158–66. doi: 10.1038/ni.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiDonato JA, Mercurio F, Karin M. NFκB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 15.Perkins ND. The diverse and complex roles of NFκB subunits in cancer. Nat Rev Cancer. 2012;12:121–32. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 16.Verma IM, Stevenson JK, Schwarz EM, van Antwerp D, Miyamoto S. Rel/NFκB/IκB family: intimate tales of association and dissociation. Genes & Development. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 17.Baldwin AS. The NFκB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh S, Karin M. Missing pieces in the NFκB puzzle. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 19.Hayden MS, Ghosh S. Shared principles in NFκB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh G, Wang VY, Huang DB, Fusco A. NFκB regulation: lessons from structures. Immunol Rev. 2012;246:36–58. doi: 10.1111/j.1600-065X.2012.01097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA. Rapid proteolysis of IκBα is necessary for activation of transcription factor NFκB. Nature. 1993;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 22.Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 23.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NFκB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 24.Verma IM, Stevenson J. IκB kinase: beginning, not the end. Proc Natl Acad Sci USA. 1997;94:11758–60. doi: 10.1073/pnas.94.22.11758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NFκB activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 26.Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev. 2012;246:239–53. doi: 10.1111/j.1600-065X.2012.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan F, Lenardo MJ. Specification of DNA binding activity of NFκB proteins. Cold Spring Harb Perspect Biol. 2009;1:1–16. doi: 10.1101/cshperspect.a000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natoli G. Control of NFκB-dependent transcriptional responses by chromatin organization. Cold Spring Harb Perspect Biol. 2009;1:a000224. doi: 10.1101/cshperspect.a000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang B, Yang XD, Lamb A, Chen LF. Posttranslational modifications of NFκB: another layer of regulation for NFκB signaling pathway. Cell Signal. 2010;22:1282–90. doi: 10.1016/j.cellsig.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen S, Smale ST. Selectivity of the NFκB Response. Cold Spring Harb Perspect Biol April. 2010;2:a000257. doi: 10.1101/cshperspect.a000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Natoli G. NFκB and chromatin: ten years on the path from basic mechanisms to candidate drugs. Immunol Rev. 2012;246:183–92. doi: 10.1111/j.1600-065X.2012.01103.x. [DOI] [PubMed] [Google Scholar]
- 32.Smale ST. Dimer-specific regulatory mechanisms within the NFκB family of transcription factors. Immunol Rev. 2012;246:193–204. doi: 10.1111/j.1600-065X.2011.01091.x. [DOI] [PubMed] [Google Scholar]
- 33.Sun SC, Ganchi PA, Ballard DW, Greene WC. NFκB controls expression of inhibitor IκBα: Evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 34.Chiao PJ, Miyamoto S, Verma IM. Autoregulation of IκBα activity. Proc Natl Acad Sci USA. 1994;91:28–32. doi: 10.1073/pnas.91.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Q, Cant CA, Moll T, Hofer-Warbinek R, Wagner E, Birnstiel ML, Bach FH, de Martin R. NFκB subunit-specific regulation of the IκBα promoter. J Biol Chem. 1994;269:13551–7. [PubMed] [Google Scholar]
- 36.Arenzana-Seisdedos F, Thompson J, Rodriguez MS, Bachelerie F, Thomas D, Hay RT. Inducible nuclear expression of newly synthesized IκBα negatively regulates DNA-binding and transcriptional activities of NFκB. Mol Cell Biol. 1995;15:2689–2696. doi: 10.1128/mcb.15.5.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bachelerie F, Rodriguez MS, Dargemont C, Rousset D, Thomas D, Virelizier JL, Arenzana-Seisdedos F. Nuclear export signal of IκBα interferes with the Rev-dependent posttranscriptional regulation of human immunodeficiency virus type I. J Cell Sci. 1997;110:2883–93. doi: 10.1242/jcs.110.22.2883. [DOI] [PubMed] [Google Scholar]
- 38.Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, Dargemont C. Nuclear localization of IκBα promotes active transport of NFκB from the nucleus to the cytoplasm. J Cell Sci. 1997;110:369–378. doi: 10.1242/jcs.110.3.369. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez M, Thompson J, Hay RT, Dargemont C. Nuclear retention of IκBα protects it from signal-induced degradation and inhibits NFκB transcriptional activation. J Biol Chem. 1999;274:9108–9115. doi: 10.1074/jbc.274.13.9108. [DOI] [PubMed] [Google Scholar]
- 40.Jaffray E, Wood KM, Hay RT. Domain organization of IκBα and sites of interaction with NFκB p65. Mol Cell Biol. 1995;15:2166–2172. doi: 10.1128/mcb.15.4.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sachdev S, Hoffmann A, Hannink M. Nuclear localization of IκBα is mediated by the second ankyrin repeat: the IκBα ankyrin repeats define a novel class of cis-acting nuclear import sequences. Mol Cell Biol. 1998;18:2524–34. doi: 10.1128/mcb.18.5.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turpin P, Hay RT, Dargemont C. Characterization of IκBα nuclear import pathway. J Biol Chem. 1999;274:6804–6812. doi: 10.1074/jbc.274.10.6804. [DOI] [PubMed] [Google Scholar]
- 43.Sachdev S, Bagchi S, Zhang DD, Mings AC, Hannink M. Nuclear import of IκBα is accomplished by a Ran-independent transport pathway. Mol Cell Biol. 2000;20:1571–1582. doi: 10.1128/mcb.20.5.1571-1582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson C, Van Antwerp D, Hope TJ. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IκBα. EMBO J. 1999;18:6682–6693. doi: 10.1093/emboj/18.23.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tam WF, Lee LH, Davis L, Sen R. Cytoplasmic sequestration of rel proteins by IκBα requires CRM1-dependent nuclear export. Mol Cell Biol. 2000;20:2269–2284. doi: 10.1128/mcb.20.6.2269-2284.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee SH, Hannink M. The N-terminal nuclear export sequence of IκBα is required for RanGTP-dependent binding to CRM1. J Biol Chem. 2001;276:23599–23606. doi: 10.1074/jbc.M011197200. [DOI] [PubMed] [Google Scholar]
- 47.Huang TT, Miyamoto S. Postrepression activation of NFκB requires the amino-terminal nuclear export signal specific to IκBα. Mol Cell Biol. 2001;21:4737–47. doi: 10.1128/MCB.21.14.4737-4747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–50. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- 49.Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–4. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- 50.Nishi K, Yoshida M, Fujiwara D, Nishikawa M, Horinouchi S, Beppu T. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem. 1994;269:6320–4. [PubMed] [Google Scholar]
- 51.Kudo N, Wolff B, Sekimoto T, Schreiner EP, Yoneda Y, Yanagida M, Horinouchi S, Yoshida M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp Cell Res. 1998;242:540. doi: 10.1006/excr.1998.4136. [DOI] [PubMed] [Google Scholar]
- 52.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA. 1999;96:9112. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamamoto T, Seto H, Beppu T. Leptomycins A and B, new antifungal antibiotics. II. Structure elucidation. J Antibiot (Tokyo) 1983;36:646–50. doi: 10.7164/antibiotics.36.646. [DOI] [PubMed] [Google Scholar]
- 54.Hay RT, Vuillard L, Desterro JM, Rodriguez MS. Control of NFκB transcriptional activation by signal induced proteolysis of IκBα. Philos Trans R Soc Lond B Biol Sci. 1999;354:1601–9. doi: 10.1098/rstb.1999.0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yashiroda Y, Yoshida M. Nucleo-cytoplasmic transport of proteins as a target for therapeutic drugs. Curr Med Chem. 2003;10:741–8. doi: 10.2174/0929867033457791. [DOI] [PubMed] [Google Scholar]
- 56.Ziegler EC, Ghosh S. Regulating inducible transcription through controlled localization. Sci STKE. 2005;284:6. doi: 10.1126/stke.2842005re6. [DOI] [PubMed] [Google Scholar]
- 57.Castro-Alcaraz S, Miskolci V, Kalasapudi B, Davidson D, Vancurova I. NFκB regulation in human neutrophils by nuclear IκBα: correlation to apoptosis. J Immunol. 2002;169:3947–53. doi: 10.4049/jimmunol.169.7.3947. [DOI] [PubMed] [Google Scholar]
- 58.Miskolci V, Ghosh CC, Rollins J, Romero C, Vu HY, Robinson S, Davidson D, Vancurova I. TNFα release from peripheral blood leukocytes depends on a CRM1-mediated nuclear export. Biochem Biophys Res Commun. 2006;351:354–60. doi: 10.1016/j.bbrc.2006.10.045. [DOI] [PubMed] [Google Scholar]
- 59.Ghosh CC, Vu HY, Mujo T, Vancurova I. Analysis of nucleocytoplasmic shuttling of NFκB proteins in human leukocytes. Methods Mol Biol. 2008;457:279–92. doi: 10.1007/978-1-59745-261-8_21. [DOI] [PubMed] [Google Scholar]
- 60.Ghosh CC, Ramaswami S, Juvekar A, Vu HY, Galdieri L, Davidson D, Vancurova I. Gene-specific repression of proinflammatory cytokines in stimulated human macrophages by nuclear IκBα. J Immunol. 2010;185:3685–93. doi: 10.4049/jimmunol.0902230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramaswami S, Manna S, Juvekar A, Kennedy S, Vancura A, Vancurova I. Chromatin immunoprecipitation analysis of NFκB transcriptional regulation by nuclear IκBα in human macrophages. Methods Mol Biol. 2012;809:121–34. doi: 10.1007/978-1-61779-376-9_8. [DOI] [PubMed] [Google Scholar]
- 62.Komiyama K, Okada K, Tomisaka S, Umezawa I, Hamamoto T, Beppu T. Antitumor activity of leptomycin B. J Antibiot (Tokyo) 1985;38:427–9. doi: 10.7164/antibiotics.38.427. [DOI] [PubMed] [Google Scholar]
- 63.Jang BC, Muñoz-Najar U, Paik JH, Claffey K, Yoshida M, Hla T. Leptomycin B, an inhibitor of the nuclear export receptor CRM1, inhibits COX-2 expression. J Biol Chem. 2003;278:2773–6. doi: 10.1074/jbc.C200620200. [DOI] [PubMed] [Google Scholar]
- 64.Kau TR, Silver PA. Nuclear transport as a target for cell growth. Drug Discov Today. 2003;8:78–85. doi: 10.1016/s1359-6446(02)02562-x. [DOI] [PubMed] [Google Scholar]
- 65.Aloisi A, Di Gregorio S, Stagno F, Guglielmo P, Mannino F, Sormani MP, Bruzzi P, Gambacorti-Passerini C, Saglio G, Venuta S, Giustolisi R, Messina A, Vigneri P. BCR-ABL nuclear entrapment kills human CML cells: ex vivo study on 35 patients with the combination of imatinib mesylate and leptomycin B. Blood. 2006;107:1591–8. doi: 10.1182/blood-2005-05-2123. [DOI] [PubMed] [Google Scholar]
- 66.Sakakibara K, Saito N, Sato T, Suzuki A, Hasegawa Y, Friedman JM, Kufe DW, Vonhoff DD, Iwami T, Kawabe T. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood. 2011;118:3922–31. doi: 10.1182/blood-2011-01-333138. [DOI] [PubMed] [Google Scholar]
- 67.Zanella F, Lorens JB, Link W. High content screening: seeing is believing. Trends Biotechnol. 2010;28:237–245. doi: 10.1016/j.tibtech.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 68.Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB, Murli S. Identification of nuclear export inhibitors with potent anticancer activity in vivo . Cancer Res. 2009;69:510–517. doi: 10.1158/0008-5472.CAN-08-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vu HY, Juvekar A, Ghosh C, Ramaswami S, Le DH, Vancurova I. Proteasome inhibitors induce apoptosis of prostate cancer cells by inducing nuclear translocation of IκBα. Arch Biochem Biophys. 2008;475:156–63. doi: 10.1016/j.abb.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Juvekar A, Manna S, Ramaswami S, Chang TP, Vu HY, Ghosh CC, Celiker MY, Vancurova I. Bortezomib induces nuclear translocation of IκBα resulting in gene-specific suppression of NFκB-dependent transcription and induction of apoptosis in CTCL. Mol Cancer Res. 2011;9:183–94. doi: 10.1158/1541-7786.MCR-10-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Juvekar A, Ramaswami S, Manna S, Chang TP, Zubair A, Vancurova I. Electrophoretic mobility shift assay analysis of NFκB transcriptional regulation by nuclear IκBα. Methods Mol Biol. 2012;809:49–62. doi: 10.1007/978-1-61779-376-9_3. [DOI] [PubMed] [Google Scholar]
- 72.Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res. 1999;5:2638–45. [PubMed] [Google Scholar]
- 73.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–6. [PubMed] [Google Scholar]
- 74.Cusack JC Jr, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J, Baldwin AS Jr. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic NFκB inhibition. Cancer Res. 2001;61:3535–40. [PubMed] [Google Scholar]
- 75.Adams J, Elliott PJ. New agents in cancer clinical trials. Oncogene. 2000;19:6687–92. doi: 10.1038/sj.onc.1204088. [DOI] [PubMed] [Google Scholar]
- 76.Santiago-Josefat B, Pozo-Guisado E, Mulero-Navarro S, Fernandez-Salguero PM. Proteasome inhibition induces nuclear translocation and transcriptional activation of the dioxin receptor in mouse embryo primary fibroblasts in the absence of xenobiotics. Mol Cell Biol. 2001;21:1700–9. doi: 10.1128/MCB.21.5.1700-1709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deroo BJ, Rentsch C, Sampath S, Young J, DeFranco DB, Archer TK. Proteasomal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol Cell Biol. 2002;22:4113–23. doi: 10.1128/MCB.22.12.4113-4123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stallings CL, Duigou GJ, Gershon AA, Gershon MD, Silverstein SJ. The cellular localization pattern of Varicella-Zoster virus ORF29p is influenced by proteasome-mediated degradation. J Virol. 2006;80:1497–512. doi: 10.1128/JVI.80.3.1497-1512.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morin PJ, Gilmore TD. The C terminus of the NFκB p50 precursor and an IκB isoform contain transcription activation domains. Nucleic Acids Res. 1992;20:2453–8. doi: 10.1093/nar/20.10.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cressman DE, Taub R. IκBα can localize in the nucleus but shows no direct transactivation potential. Oncogene. 1993;8:2567–73. [PubMed] [Google Scholar]
- 81.Zabel U, Henkel T, Silva MS, Baeuerle PA. Nuclear uptake control of NFκB by MAD-3, an IκB protein present in the nucleus. EMBO J. 1993;12:201–11. doi: 10.1002/j.1460-2075.1993.tb05646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Proteasome inhibition in the treatment of cancer. Cell Cycle. 2005;4:290–6. [PubMed] [Google Scholar]
- 83.Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23:1964–79. doi: 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wright JJ. Combination therapy of bortezomib with novel targeted agents: an emerging treatment strategy. Clin Cancer Res. 2010;16:4094–104. doi: 10.1158/1078-0432.CCR-09-2882. [DOI] [PubMed] [Google Scholar]
- 85.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11:239–53. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsuchiya Y, Asano T, Nakayama K, Kato T Jr, Karin M, Kamata H. Nuclear IKKβ is an adaptor protein for IκBα ubiquitination and degradation in UV-induced NFκB activation. Mol Cell. 2010;39:570–82. doi: 10.1016/j.molcel.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 87.Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS Jr. IκBα interacts with the nuclear localization sequences of the subunits of NFκB: a mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 88.Vancurova I, Miskolci V, Davidson D. NFκB activation in tumor necrosis factor alpha-stimulated neutrophils is mediated by protein kinase C-δ. Correlation to nuclear IκBα. J Biol Chem. 2001;276:19746–52. doi: 10.1074/jbc.M100234200. [DOI] [PubMed] [Google Scholar]
- 89.Haslett C, Savill JS, Whyte MK, Stern M, Dransfield I, Meagher LC. Granulocyte apoptosis and the control of inflammation. Philos Trans R Soc Lond B Biol Sci. 1994;345:327–33. doi: 10.1098/rstb.1994.0113. [DOI] [PubMed] [Google Scholar]
- 90.Whyte M, Renshaw S, Lawson R, Bingle C. Apoptosis and the regulation of neutrophil lifespan. Biochem Soc Trans. 1999;27:802–7. doi: 10.1042/bst0270802. [DOI] [PubMed] [Google Scholar]
- 91.McDonald PP, Bald A, Cassatella MA. Activation of the NFκB pathway by inflammatory stimuli in human neutrophils. Blood. 1997;89:3421–33. [PubMed] [Google Scholar]
- 92.Ear T, Cloutier A, McDonald PP. Constitutive nuclear expression of the IκB kinase complex and its activation in human neutrophils. J Immunol. 2005;175:1834–42. doi: 10.4049/jimmunol.175.3.1834. [DOI] [PubMed] [Google Scholar]
- 93.Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN, Haslett C, Rossi AG. NFκB activation is a critical regulator of human granulocyte apoptosis in vitro . J Biol Chem. 1999;274:4309–18. doi: 10.1074/jbc.274.7.4309. [DOI] [PubMed] [Google Scholar]
- 94.Browning DD, Pan ZK, Prossnitz ER, Ye RD. Cell type- and developmental stage-specific activation of NFκB by fMet-Leu-Phe in myeloid cells. J Biol Chem. 1997;272:7995–8001. doi: 10.1074/jbc.272.12.7995. [DOI] [PubMed] [Google Scholar]
- 95.Cassatella MA. The production of cytokines by polymorphonuclear neutrophils. Immunol Today. 1995;16:21–6. doi: 10.1016/0167-5699(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 96.Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The neutrophil as a cellular source of chemokines. Immunol Rev. 2000;177:195–203. doi: 10.1034/j.1600-065x.2000.17706.x. [DOI] [PubMed] [Google Scholar]
- 97.Miskolci V, Rollins J, Vu HY, Ghosh CC, Davidson D, Vancurova I. NFκB is persistently activated in continuously stimulated human neutrophils. Mol Med. 2007;13:134–42. doi: 10.2119/2006-00072.Miskolci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31:S195–9. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 99.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–7. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 100.Wesche DE, Lomas-Neira JL, Perl M, Chung CS, Ayala A. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol. 2005;78:325–37. doi: 10.1189/jlb.0105017. [DOI] [PubMed] [Google Scholar]
- 101.Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 2010;49:1618–31. doi: 10.1093/rheumatology/keq045. [DOI] [PubMed] [Google Scholar]
- 102.Duffin R, Leitch AE, Fox S, Haslett C, Rossi AG. Targeting granulocyte apoptosis: mechanisms, models, and therapies. Immunol Rev. 2010;236:28–40. doi: 10.1111/j.1600-065X.2010.00922.x. [DOI] [PubMed] [Google Scholar]
- 103.Urban MB, Schreck R, Baeuerle PA. NFκB contacts DNA by a heterodimer of the p50 and p65 subunit. EMBO J. 1991;10:1817–1825. doi: 10.1002/j.1460-2075.1991.tb07707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ganchi PA, Sun SC, Greene WC, Ballard DW. A novel NFκB complex containing p65 homodimers: implications for transcriptional control at the level of subunit dimerization. Mol Cell Biol. 1993;13:7826–7835. doi: 10.1128/mcb.13.12.7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bell S, Matthews JR, Jaffray E, Hay RT. IκBα inhibits DNA binding of NFκB p50 homodimers by interacting with residues that contact DNA. Mol Cell Biol. 1996;16:6477–6485. doi: 10.1128/mcb.16.11.6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Malek S, Huxford T, Ghosh G. IκBα functions through direct contact with the nuclear localization signals and the DNA binding sequences of NFκB. J Biol Chem. 1998;273:25427–25435. doi: 10.1074/jbc.273.39.25427. [DOI] [PubMed] [Google Scholar]
- 107.Simeonidis S, Stauber D, Chen G, Hendrickson WA, Thanos D. Mechanisms by which IκB proteins control NFκB activity. Proc Natl Acad Sci USA. 1999;96:49–54. doi: 10.1073/pnas.96.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Phelps CB, Sengchanthalangsy LL, Huxford T, Ghosh G. Mechanism of IκBα binding to NFκB dimers. J Biol Chem. 2000;275:29840–29846. doi: 10.1074/jbc.M004899200. [DOI] [PubMed] [Google Scholar]
- 109.Bergqvist S, Croy CH, Kjaergaard M, Huxford T, Ghosh G, Komives EA. Thermodynamics reveal that helix four in the NLS of NFκB p65 anchors IκBα, forming a very stable complex. J Mol Biol. 2006;360:421–34. doi: 10.1016/j.jmb.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NFκBxDNA dissociation by IκBα. Proc Natl Acad Sci USA. 2009;106:19328–33. doi: 10.1073/pnas.0908797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sue SC, Alverdi V, Komives EA, Dyson HJ. Detection of a ternary complex of NFκB and IκBα with DNA provides insights into how IκBα removes NFκB from transcription sites. Proc Natl Acad Sci USA. 2011;108:1367–72. doi: 10.1073/pnas.1014323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Komives EA. Consequences of fuzziness in the NFκB/IκBα interaction. Adv Exp Med Biol. 2012;725:74–85. doi: 10.1007/978-1-4614-0659-4_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bosisio D, Marazzi I, Agresti A, Shimizu N, Bianchi ME, Natoli G. A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NFκB-dependent gene activity. EMBO J. 2006;25:798–810. doi: 10.1038/sj.emboj.7600977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the NFκB response. J Exp Med. 2004;200:107–113. doi: 10.1084/jem.20040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen LF, Fischle W, Verdin E, Greene WC. Duration of nuclear NFκB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 116.Ashburner BP, Westerheide SD, Baldwin AS Jr. The p65 (RelA) subunit of NFκB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Greene WC, Chen LF. Regulation of NFκB action by reversible acetylation. Novartis Found Symp. 2004;259:208–217. [PubMed] [Google Scholar]
- 118.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NFκB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 119.Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NFκB pathway. J Biol Chem. 2005;280:34538–34547. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- 120.Dong J, Jimi E, Zhong H, Hayden MS, Ghosh S. Repression of gene expression by unphosphorylated NFκB p65 through epigenetic mechanisms. Genes Dev. 2008;22:1159–1173. doi: 10.1101/gad.1657408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Natoli G, Chiocca S. Nuclear ubiquitin ligases, NFκB degradation, and the control of inflammation. Sci Signal. 2008;1:1. doi: 10.1126/stke.11pe1. [DOI] [PubMed] [Google Scholar]
- 122.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IκB kinases phosphorylate NFκB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274:30353–6. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 123.Yang F, Tang E, Guan K, Wang CY. IKKβ plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol. 2003;170:5630–5. doi: 10.4049/jimmunol.170.11.5630. [DOI] [PubMed] [Google Scholar]
- 124.Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, Doi T, Saiki I. TNFα-induced IKK phosphorylation of NFκB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–23. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- 125.O'Mahony AM, Montano M, Van Beneden K, Chen LF, Greene WC. Human T-cell lymphotropic virus type 1 tax induction of biologically Active NFκB requires IκB kinase-1-mediated phosphorylation of RelA/p65. J Biol Chem. 2004;279:18137–45. doi: 10.1074/jbc.M401397200. [DOI] [PubMed] [Google Scholar]
- 126.Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and IL-1-inducible phosphorylation of p65 NFκB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)- α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated IL-8 transcription. J Biol Chem. 2004;279:55633–43. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- 127.Adli M, Baldwin AS. IKK-i/IKKε controls constitutive, cancer cell-associated NFκB activity via regulation of Ser-536 p65/RelA phosphorylation. J Biol Chem. 2006;281:26976–84. doi: 10.1074/jbc.M603133200. [DOI] [PubMed] [Google Scholar]
- 128.Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IKKα in NFκB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 129.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKKα is critical for cytokine-induced gene expression. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 130.Anest V, Cogswell PC, Baldwin AS Jr. IKKα and p65/RelA contribute to optimal epidermal growth factor-induced c-fos gene expression independent of IκBα degradation. J Biol Chem. 2004;279:31183–31189. doi: 10.1074/jbc.M404380200. [DOI] [PubMed] [Google Scholar]
- 131.Park KJ, Krishnan V, O'Malley BW, Yamamoto Y, Gaynor RB. Formation of an IKKα dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell. 2005;18:71–82. doi: 10.1016/j.molcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 132.Espinosa L, Bigas A, Mulero MC. Alternative nuclear functions for NFκB family members. Am J Cancer Res. 2011;1:446–59. [PMC free article] [PubMed] [Google Scholar]
- 133.Natoli G. Tuning up inflammation: how DNA sequence and chromatin organization control the induction of inflammatory genes by NFκB. FEBS Lett. 2006;580:2843–2849. doi: 10.1016/j.febslet.2006.02.072. [DOI] [PubMed] [Google Scholar]
- 134.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a κB site can determine cofactor specificity for NFκB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 135.Viatour P, Legrand-Poels S, van Lint C, Warnier M, Merville MP, Gielen J, Piette J, Bours V, Chariot A. Cytoplasmic IκBα increases NFκB-independent transcription through binding to histone deacetylase (HDAC) 1 and HDAC3. J Biol Chem. 2003;278:46541–46548. doi: 10.1074/jbc.M306381200. [DOI] [PubMed] [Google Scholar]
- 136.Espinosa L, Ingles-Esteve J, Robert-Moreno A, Bigas A. IκBα and p65 regulate the cytoplasmic shuttling of nuclear corepressors: cross-talk between Notch and NFκB pathways. Mol Biol Cell. 2003;14:491–502. doi: 10.1091/mbc.E02-07-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aguilera C, Hoya-Arias R, Haegeman G, Espinosa L, Bigas A. Recruitment of IκBα to the hes1 promoter is associated with transcriptional repression. Proc Natl Acad Sci USA. 2004;101:16537–16542. doi: 10.1073/pnas.0404429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Puca A, Fiume G, Palmieri C, Trimboli F, Olimpico F, Scala G, Quinto I. IκBα represses the transcriptional activity of the HIV-1 Tat transactivator by promoting its nuclear export. J Biol Chem. 2007;282:37146–57. doi: 10.1074/jbc.M705815200. [DOI] [PubMed] [Google Scholar]