

Abstract
Naive CD4+ cells differentiate into T helper (Th1, Th2, Th9, Th17) and regulatory T (Treg) cells to execute their immunologic function. Whereas TGF-β suppresses Th1 and Th2 cell differentiation, this cytokine promotes Th9, Th17 and Foxp3+ regulatory T cells depending upon the presence of other cytokines. IL-6 promotes Th17, but suppresses regulatory T cell differentiation. Moreover, natural but not TGF-β-induced regulatory T cells convert into Th17 cells in the inflammatory milieu. Here an update of T cell differentiation and conversion, as well as underlying mechanisms are given.
Keywords: T helper cells, Th1, Th2, Th9, Th17, Tfh, Foxp3+ regulatory T cells, cytokine, differentiation, autoimmunity
Naive CD4+ cells differentiate into T helper (Th1, Th2, Th9, Th17) and regulatory T (Treg) cells to execute their functional activities. Th1, Th2 and Th17 cells play an important role in the protective immune response against intracellular pathogens and extracellular parasites, nonetheless, excessive immune responses exerted by these T helper cells also cause autoimmune and inflammatory diseases. Foxp3+ Treg cells are essential for the immune tolerance and play a crucial role in the limitation of the excessive immune and inflammatory response executed by these T helper cells. Although Treg and Th17 cells have a completely different function in the immune responses, the differentiation of both cell subsets does need TGF-β. In this review, I will discuss the differentiation and relation of Tregs and Th17 cells, in particular Treg cell conversion to Th17 cells. I have also discussed the underlying mechanisms and functional significance of these differentiations and conversions.
Functional characteristics and categories of regulatory T cells
It is now well accepted that CD4+CD25+Foxp3+ regulatory or suppressor cells are critically involved in immune tolerance and homeostasis. In the early 1970s, Gershon and colleagues initially reported that thymocytes from his experimental animal model contained a such cell population they called “suppressor T cells” and assumed they belong to CD8+ cell subset [1]. This suggestion was not appreciated until Sakaguchi et al found that a population of CD4+CD25+ cells rather than CD8+ T cells in the thymus did indeed possess immunosuppressive activity that is now referred to as “regulatory T cells or natural regulatory T cells, nTregs [2].
CD4+CD25+ cell population also exists in humans, although only the CD4+CD25bright cell population appears to display an immune suppressive activity. A better approach for the identification of human Treg cells is to target the CD4+CD25+CD127-/low population [3]. More recently, CD4+CD25+CD127-CD45RO+Foxp3+ cells are identified as real human suppressor cells [4].
CD25 is also an activation marker for lymphocytes. Thus, the utility of CD25 expression as a Treg marker is limited since it does not discriminate between activated T effector cells and Tregs. Fortunately, the nuclear transcription factor Foxp3 has been identified as a more specific marker for Treg cells. Foxp3 is critically involved in the development and function of Treg cells [5]. In mice, the lack of functional Foxp3 expression results in a fatal lymphoproliferative disorder known as scurfy and mutations of the human FOXP3 gene results in a human syndrome known as IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked), which is characterized by autoimmune disease expression in multiple endocrine organs [6].
Despite the fact that Foxp3-GFP “knock-in” studies clearly demonstrate that there is a very broad spectrum of CD25 expression on Treg cells and that the intranuclear location of Foxp3 makes it difficult to use this protein for immunoaffinity-based purification methods although recent study has identified a new technique to improve the isolation of the live Treg cells [7], CD4+CD25+ cells are still widely used in the field of the biology of Treg cells without using genetically modified tissues, particular in human studies. Although Foxp3 is considered as a specific marker for Tregs in mouse, this may not be the case for human Tregs. Recent data demonstrate that FOXP3 (FOXP3 for human cells and Foxp3 for mouse cells) may be upregulated in rapidly proliferating human T cells and might be viewed as an activation marker for human T cells [8]. More studies are needed to determine how FOXP3 might also be expressed on rapidly proliferating human T effector cells and more specific molecular markers to identify human Tregs are also desirable.
Many studies have revealed that the numbers of CD4+CD25+ cells and CD4+FOXP3+ cells in patients with various autoimmune diseases are diminished and that this Treg deficit is associated with disease severity and activity [9]. The peripheral Treg deficit in patients with autoimmune diseases is not resultant from their redistribution to different organs [10]. However, diminishment of Tregs in the autoimmune diseases is not a universal finding. Other groups have actually observed the converse; that the numbers of human CD4+CD25+ cells can be increased under these circumstances [11]. Since CD25 and FOXP3 can also be classified as activated makers, this aspect may reflect the disparity between these findings. Miyara et al have further classified human FOXP3+ cells into three cell subsets: CD45RA+FOXP3low, CD45RA-FOXP3hi and CD45RA-FOXP3low. Functional assay demonstrated that the CD45RA-FOXP3low subset contains non-suppressor cells, that the CD45RA+FOXP3low subset contains resting Tregs and that active Tregs are found in the CD45RA-FOXP3hi subset. Using these criteria, they found that Treg cell numbers were indeed diminished in patients with active autoimmune disease [4].
In addition to Treg frequency, others have also reported that the functional activity of Tregs has been altered in some autoimmune diseases. For example, the suppressive activity of CD4+CD25+ cells isolated from patients with active rheumatoid arthritis was significantly reduced [12]. It is possible that some intrinsic defect in CD4+CD25+ cells in these patients accounts for their reduced functional activity. Similarly, the frequency of CD4+CD25+ cells in patients with multiple sclerosis (MS) is unaltered, however, the functional activity of these cells to suppress T cell immune responses including antigen-specific or non-specific stimulation is diminished [13-15]. These results suggest that the manipulation of nTregs to restore their numbers and function may be therapeutic.
Although most reports claim that CD4+CD25+ in peripheral blood mononuclear cell (PBMC) belong to natural Treg cells, we and others would suggest that CD4+CD25+ cells in PBMCs consist of a mixture of both thymic-derived nTregs and those induced in the periphery (induced Tregs, iTregs) [16-18]. Until today, there are no specific markers that can distinguish nTregs from iTregs, although Shevach’s group recently reported that Helios, an Ikaros family transcription factor, may be helpful for distinguishing these cell population [19], while others reported that Helios is also highly expressed on Th2 and T follicular helper cells and may be associated with the differentiation of these cells [20]. We recently observed activated Foxp3- T cells also express Helios (unpublished data). NrP-1 expression provides another biological marker to distinguish nTregs from iTregs [21,22], however, it specificity needs to be further validated since others reported that NrP1 is not a marker for human Treg cells [23]. it is, therefore, necessary to identify more reliable molecular marker(s) to distinguish various subsets of Treg cells.
It has been well known that the adoptive transfer of nTregs can prevent the appearance and development of autoimmune diseases in many animal models. Nonetheless, there are also considerable numbers of studies demonstrating that the therapeutic effect of nTregs on established autoimmune diseases is fairly unsatisfactory. For example, the efficacy of adoptive transfer of nTregs to established collagen-induced arthritis (CIA) is poor for controlling the disease progression [24]. Injection of nTregs to established lupus had mild protective effects and it was unable to suppress lupus glomerulonephritis and sialoadenitis [25,26]. Moreover, adoptive transfer of nTregs failed to control Th17-mediated autoimmune gastritis [27].
There are several possibilities that could explain the inability of nTregs to treat CIA and other autoimmune diseases. First, pro-inflammatory cytokines may hamper their suppressive activity. Pasare et al have reported that Treg suppressive activity can be abolished by IL-6 [28]. Valencia et al also revealed that elevated TNF-α may interfere with the suppressive capacity of nTregs in patients with rheumatoid arthritis (RA) [12]. There is no question that these pro-inflammatory cytokines are elevated in patients with RA and other autoimmune diseases [29]. Secondly, Th17 cells may be resistant to the suppressive effects exerted by nTregs. This could explain how nTregs are able to prevent development of disease before Th17 cells become established, while demonstrating ineffective suppression after disease expression is evident where Th17 cells have been developed. Third, nTregs are inherently unstable and can be converted to Th1, Th2, Th17 and Tfh effector cells when they encounter an inflammatory milieu [24,30-34].
There are still other reasons that could hamper the utilization of nTregs in clinical therapy. First, the intranuclear location of Foxp3 makes it difficult to purify human nTregs for functional study. Second, nTregs constitute only 1-2% of human CD4+ T cells and this is also difficult to gain the sufficient numbers for therapeutic requirement. Although several groups have claimed that expansion of human nTregs in vitro can overcome this problem [35], other laboratories have reported that repeated expansion alters Treg phenotype and function [36]. Third, the expansion of nTregs from patients with RA and MS for therapeutic purposes may be problematic due to potential other intrinsic defects in RA and MS nTregs. nTreg instability, Teff cell resistance and the influence of an inflammatory milieu may individually or collectively account for the inability of nTregs to control established autoimmune diseases.
Of great interest, the plasticity or instability of nTregs under inflammatory conditions could be overcome with cytokines or other compounds. Our group recently reported that while nTregs become Th17 cells in the presence of IL-6, these cells also lost their suppressive role in the progression of the lupus-like syndromes and CIA. We also determined that pretreatment of nTregs with IL-2 combined with TGF-β, or all-trans retinoic acid (atRA), a vitamin A metabolite, can render these nTregs resistant to Teff cell conversion and allow them to begin to suppress lupus and CIA progression [34,37]. Recently, we found that atRA also maintains the stability of nTregs in human (unpublished data). This indicates that the manipulation of nTregs still holds a potential promise in the treatment of autoimmune diseases.
Like nTregs, iTregs generated ex vivo with IL-2 and TGF-β also share similar phenotypes related to Treg cells, and suppress immune responses and immune cell-mediated diseases. Importantly, adoptive transfer of iTregs not only prevents autoimmune diseases in many animal models, but also attenuates the disease syndromes when iTregs were infused when or after diseases are established [38-47]. It is very likely that iTregs are stable and sustain their immune suppressive activity in the inflammatory condition [34]. Because sufficient numbers of iTregs can be easily gained and antigen-specific iTregs can be easily developed in the certain environment, it implicates that the manipulation of iTregs has a great potential to treat autoimmune and inflammatory diseases.
iTregs and their differentiation and development
CD4+ Treg subsets can be further classified into three main populations, thymus-derived, naturally occurring CD4+CD25+Foxp3+ cells (nTregs) described as above, endogenous induced Tregs in vivo and those that can be induced ex vivo from CD25- precursors in peripheral lymphoid organs (iTregs) [48]. Although IL-10-induced Tr1 cells represent another cell population of iTregs, they do not express Foxp3 and produce considerable levels of IL-10 [49]. As IL-10 may promote autoimmune response through stimulating B cell activation and its level is highly increased in patients with active systemic lupus erythematosus (SLE) [50], Tr1 may not be suitable for the treatment of SLE and other autoimmune diseases. TGF-β-induced Tregs will be defined as iTregs in this review.
While Yamagiwa et al reported that TGF-β promotes endogenous CD4+CD25+ nTreg cell expansion [51], our group first reported that TGF-β does have an ability to induce CD4+CD25- cells to become CD4+CD25+ Treg cells in vitro [48]. When Foxp3 was identified as Treg marker, several groups immediately found that TGF-β can induce Foxp3 expression in iTregs [38,39,52]. Additionally, other studies have also clearly demonstrated the development of Foxp3+ Tregs in vivo is also through TGF-β-dependent mechanism [53].
Phenotypically, both nTregs and iTregs express similar molecules such as CD25, CD122, CTLA-4, GITR, CCR4, CD62L, PD1 and Foxp3, and express CD45RBlow in mice and CD45RO in humans. CD4+CD25+Foxp3+ cells in the periphery have been considered as a mixed population comprised of nTregs and iTregs. Although Helios and NrP-1 might possibly help to distinguish nTregs from iTregs [19,21], more specific molecular markers are needed to distinguish both Treg cell populations.
Although both nTreg and iTreg subsets share similar phenotypes and display comparable suppressive activity, several factors distinctly affect their development, stability and function. First, nTregs develop in the thymus through recognition of self-antigens. A high and medium affinity cognate interaction between self-peptide: MHC complex and T cell receptor is required for this process. They also require CD28 co-stimulation because they do not develop in CD28 deficient mice [54]. Although IL-2 and TGF-β play an important role in the maintenance of the pool size of nTregs, both cytokines are redundant for their development since both IL-2 and TGF-β knock-out mice contain CD4+CD25+Foxp3+ regulatory T cells in the thymus [55]. Although one group recent reported that TGF-β is essential for the nTreg cell development [56], most researchers believe that TGF-β is redundant in nTreg development although this cytokines is important for the maintaining pool size of the nTreg cells [57].
By contrast, the generation of iTregs is dependent upon the presence of both TGF-β and TGF-β receptor signals since the absence of TGF-β or TGF-β receptors or blocking the TGF-β receptor signal prevents the induction of Foxp3 expression and the subsequent functional suppressive capacity [33,58]. Similarly, IL-2 plays an essential role in the differentiation of Foxp3+ iTregs. TGF-β fails to induce Foxp3+ iTregs from naïve CD4+CD25- precursor cells in IL-2 deficient mice [59]. The conversion of CD4+CD25- cells in the periphery to CD25+ iTregs requires a suboptimal TCR stimulation and thus environmental antigens may sufficiently trigger iTreg development. The absence of CD28 co-stimulatory molecules does not affect the differentiation of iTregs (Lan Q and Zheng SG, unpublished data), but inhibitory CTLA-4 co-stimulation and CTLA-4/B7.1 signaling is crucially required for the generation of iTregs [60]. This conclusion is further documented by an observation that the blocking of CTLA-4/B7.1 signal abolished the capacity of TGF-β to induce iTregs in wild type mice [61]. OX40/OX40L, an alternate CD28/B7-independent co-stimulatory pathway, also negatively regulates the development and function of both nTregs and iTregs. While stimulation of mature nTregs by OX40 results in the loss of suppression of T cell proliferation and cytokine production, the generation of iTregs is completely abolished by OX40 although OX40 does not affect the generation of nTregs [62].
Recently, Housley et al reported that while the TNF-R2 expression is essential for nTregs-mediated suppression of colitis, its expression is not required for iTreg-mediated suppression [63]. Differing IL-2 and co-stimulatory molecule requirements for Treg development, and TNFRII expression requirements for the suppressive function of both nTregs and iTregs suggests that nTregs and iTregs are possibly heterogeneous populations and that integration of both Treg subsets is required for the maintenance of normal immune homeostasis. It is also likely that both nTreg and iTreg subsets can either act in concert or separately on different targets. In addition, as anti-TNF-α therapy has been widely used in treating patient with rheumatoid arthritis, further studies are required to understand whether this therapy differentially regulates nTregs and/or iTregs development in individual diseases.
Function and differentiation of Th17 cells
Recent studies have provided a line of new evidence demonstrating the existence of a third subset of effector CD4+ cells in addition to the classic Th1 and Th2 cells, the differentiation and growth of which is directed by a combination of TGF-β1 and IL-6 or IL-21 [64-67]. These T cells have been designated Th17 cells based on their production of IL-17A and F. Despite the evidence for the role of Th1 cells in autoimmune disease, recent incontrovertible findings have revealed that pathologies previously attributed to Th1 cells may in fact be mainly mediated by Th17 cells in some autoimmune diseases. The best evidence for this comes from studies using anti-IFN-γ-treated mice, IFN-γ- or IFNR-deficient mice, mice deficient in IL-12p35, IL-12 receptor β2 (IL-12Rβ2), or STAT1, which are critical molecules in IL-12/IFN-γ–Th1-mediated responses, are capable of developing even severe collagen-induced arthritis (CIA) or experimental autoimmune encephalomyelitis (EAE) despite interference of the proper functioning of the prototypic Th1 cytokine IFN-γ [68-73]. Moreover, these findings concur with reports demonstrating that CIA is suppressed in IL-17-deficient mice and that administration of neutralizing anti-IL-17 antibodies at preclinical and advanced stages significantly reduces disease severity [74]. Similarly, IL-17R-deficient mice or IL-17R IgG1 fusion protein significantly attenuates colonic inflammation in acute trinitrobenzenesulfonic acid (TNBS)-induced colitis although this protection occurred in the presence of equivalent induction of local IL-23 and higher levels of IL-12p70 and interferon-gamma in IL-17R knockout mice compared with wild-type mice [75].
Interleukin-17 (IL-17A) is one of the prototypic IL-17 family members that are predominately produced by CD4+ memory cells [76]. IL-17 receptor is ubiquitously expressed and ligand binding causes the secretion of a range of other factors known to drive inflammatory responses such as rheumatoid arthritis (RA), multiple sclerosis (MS) and colitis [71,72,75]. IL-17 stimulates epithelial, endothelial, and fibroblastic cells to secrete pro-inflammatory factors such as IL-6, IL-8, GM-CSF, CXCL1, CCL20 as well as prostaglandin E2 [77]. Secreted CXCL1 and CCL20 result in the recruitment of neutrophils and/or macrophages to the area of inflammation and enable cell movement and tissue damage. Thus, research directed at controlling autoimmune inflammatory diseases will require a better characterization of the developmental and functional properties of Th17 in these diseases.
Naive CD4+ T cells can be induced to differentiate towards Th1, Th2, Th9, Th17 and Treg phenotypes according to the local cytokine milieu. While IL-12 favors the differentiation towards Th1 cells through transcription factor T-bet [78], IL-4 towards Th2 via GATA-3 [79], IL-2 and TGF-β towards iTregs via Foxp3 [38,39,48], the combination of TGF-β and IL-6 induces Th17 cells through transcription factor orphan nuclear receptor RORγt [80]. IL-21 could substitute for IL-6 to promote Th17 differentiation in this condition [67]. IL-23 was initially considered as a key cytokine to induce Th17 cell production [81], however, recent studies revealed it is dispensable for the differentiation of Th17 cells but critical for Th17 expansion and survival [82]. IL-1β also promotes the allergic asthma by enhancing Th17 cell differentiation [83]. TGF-β signal is crucial for Th17 cell differentiation although one group recently reported that Th17 cells can be differentiated in the lack of TGF-β signal [84].
It has been demonstrated that the skewing of naïve T cells towards Th17 and iTreg is mutually exclusive. While we and others have recently revealed IL-2 critically drives TGF-β-treated CD4+ cells to differentiate into Foxp3+ iTreg cells [59,85], IL-6 or IL-21, however, exclusively drives TGF-β-treated T cells to become Th17 cells [64-67]. It seems IL-6 has a dominate effect on promoting Th17 cells compared to IL-2 promoting Treg cell differentiation. Our data demonstrated that naive CD4+ cells preferentially differentiate into Th17 rather than Tregs in the presence of IL-6, IL-2 and TGF-β no matter whether low or high doses of IL-2 are included in the cultures although others have reported that IL-2 constrains Th17 cell differentiation [86].
Foxp3+ Tregs conversion to Th17 and other T effector cells
While Foxp3+ nTregs suppress Th1 and Th2 cell differentiation and function, it is less defined whether these cells similarly suppress Th17 differentiation and function. Yang et al have reported that Foxp3 inhibited Th17 differentiation by antagonizing the function of the transcription factors RORγt and ROR. However, IL-6 overcame this suppressive effect of Foxp3 on Th17 differentiation [87]. It has been known that adoptive transfer of nTregs to Th17-mediated diseases was less therapeutic [24,27], suggesting that nTregs may be less effective on suppressing Th17 cell differentiation, function and Th17-mediated diseases.
In fact, nTregs can convert into Th17 cells and other T effector cells in the certain environments. As TGF-β either promotes Foxp3+ iTregs, Th9 or 17 cells depending upon other cytokines involved (Figure 1), and as nTreg cells express a membrane-bound form of TGF-β and this TGF-β has functional activities, it is reasonable to assume that IL-6 can convert nTregs to become Th17 and other T helper cells [30]. To demonstrate this, Xu et al used the purified nTregs from Foxp3 GFP knock in-mice to exclude the possibility that CD4+CD25+Foxp3- non-Tregs made this conversion. We used both wild type and Foxp3 GFP knock-in mice to confirm this observation [34]. Endogenous TGF-β produced by nTregs is critically required for this conversion since blocking TGF-β receptor I signal or using nTregs from TGF-β receptor II dominant mice resulted in the failure of Th17 conversion [33,34]. Moreover, activation of nTregs with IL-6 resulted in decreased Foxp3 expression and suppressive activity both in vitro and in vivo. Furthermore, adoptive transfer experiments revealed that nTregs treated with IL-6 ex vivo lost their ability to protect mice from a lupus-like disease [34]. Moreover, it has recently been demonstrated that nTregs can be converted into Th17 cells in an in vivo model [88]. Others have also reported that Th17 cells derived from Tregs share common features with Th17 cells generated from naive precursors, including expression of the chemokine receptor CCR6 [89]. This conversion is not the result of outgrowth of a contaminating Th17 precommitted population because it is indicated by the demonstration of double-positive cells for the Treg transcription factor Foxp3 and IL-17 [34,90]. Human nTregs also can convert into Th17 cells when stimulated with IL-1 and IL-6 although the role of TGF-β in this conversion is less clear. IL-17 production from Treg cells also occurs in vivo [91]. Thus, in an IL-1 and/or IL-6 rich inflammatory milieu, nTregs may be unstable and lose the functional activity.
Figure 1.
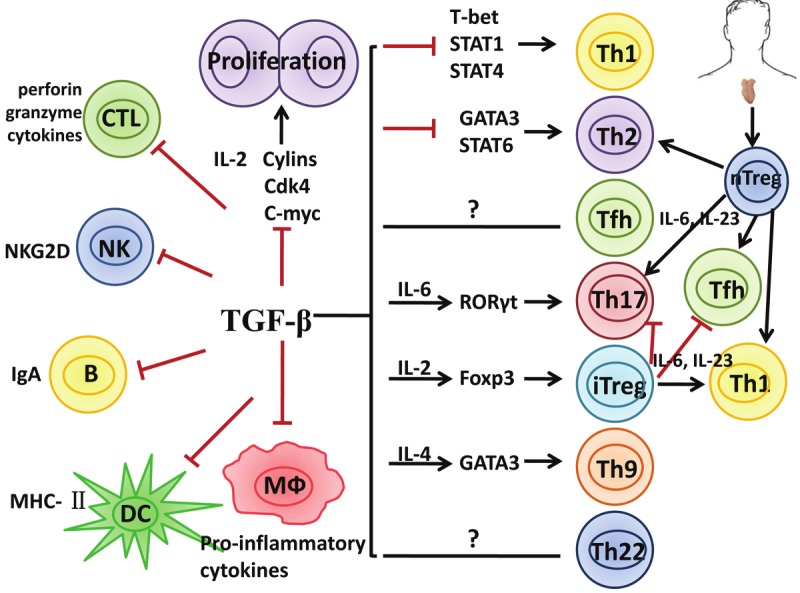
Multi effects of TGF-β on regulatory and effector T cells and interrelation between Treg and T effector cells. TGF-β inhibits the differentiation, proliferation and function of various immune cells including Th1, Th2 and Tfh cells. TGF-β also promotes iTreg, Th9 and Th17 cell differentiation depending upon the cytokine environment. Additionally, TGF-β inhibits maturation and function of other immune cells such as CD8+ CTL, NK cell, DC and macrophages. Both nTregs and iTregs suppress Th1 and Th2, only iTregs suppress Th17 cells. nTregs convert into Th1, Th2, Th17 and Tfh cells and lost suppressive activity, iTregs are resistant to T effector cell conversion except Th1 but maintaining suppressive activity.
In addition to Th17 cell conversion, nTregs can be converted into other subsets of T effector cells. Wan and Flavell found that Foxp3+ nTregs can convert to Th2 cells when endogenous Foxp3 gene expression is attenuated in these cells [31]. Interestingly, T cells expressing decreased Foxp3 still preferentially became Th2 effectors even in a Th1-polarizing environment. It is therefore likely that these cells instructed Th2 differentiation of conventional T cells, which contributed to the immune diseases observed in these mice [31]. Moon et al also reported that acetyl salicylic acid also changes Th17-type into Th2-type inflammation cells in mice model with asthma [92]. When these cells were strongly stimulated with antigen or anti-CD3/CD28 antibodies, nTregs also lost Foxp3 and became Th1 cells [33]. IL-2 also promotes Treg cells to become Th1-like cells although Foxp3+INF-γ+ cells are still suppressive [93]. In human, nTregs can also be converted into Th1 cells and most lost suppressive activity (Lu L and Zheng SG, manuscript submitted). Moreover, these cells can become T follicular helper (Tfh) cells [32]. Tsuji et al have demonstrated that adoptive transfer of nTregs to immune deficient mice, Foxp3+ CD4+ cells can differentiate into Tfh cells in mouse Peyer’s patches. The conversion of Foxp3+ T cells into Tfh cells requires the loss of Foxp3 expression and subsequent interaction with B cells [32].
Previous studies have clearly demonstrated that naive rather than memory CD4+ cells preferentially differentiate into Foxp3+ Tregs cells in the presence of exogenous TGF-β [59]. It is no surprise that Th1, Th2 and Th17 cells are unable to differentiate into Foxp3+ Treg cells even they have been primed with exogenous TGF-β (Zheng SG et al, unpublished data). However, Th17 cells can be converted into Th1 and Th2 cells, suggesting that Th17 cells are not stable phenotypes [94,95]. The functional significance of Th17 to Th1 and Th2 cell conversion is unclear so far. In addition, the epigenetic modifications were remarkably stable during these cells conversion [96].
In sharp contrast, TGF-β-induced iTregs were found to be completely resistant to the Th17 conversion by IL-6. This difference cannot be explained by insufficient production of TGF-β by iTregs since both nTregs and iTregs expressed similar levels of membrane-bound TGF-β (20-25%) and secreted similar levels of active TGF-β (about 40 ng/ml). Furthermore, the resistance of iTregs to Th17 conversion also cannot be explained by alterations in TCR stimulation since anti-CD3/CD28 activated nTregs can still differentiate into Th17 cells upon IL-6 stimulation. To account for this difference between nTregs and iTregs, we found that the combination of IL-2 and TGF-β down-regulated IL-6 receptor expression and function in activated T cells. We have observed that both cytokines markedly decreased IL-6 receptor alpha-chain (CD126) and beta-chain (CD132) expression on CD4+ cells and these cells expressed significantly lower level of phosphorylated STAT3 when stimulated by IL-6 [34]. Unexpectedly, Yang et al had a conversed report showing iTregs can convert into Th17 cells in the presence of pro-inflammatory cytokines [87]. To solve this contradiction, O’Connor et al reinvestigated the fates of both nTregs and iTregs in the pro-inflammatory condition. They did find that iTregs are completely resistant to Th17 conversion although nTregs do in the presence of IL-6, IL-23 and TGF-β [97]. Interestingly, they also demonstrated that iTregs rather than nTregs became Th1-like cells. Although these Th1-like cells began to express T-bet and produce INF-γ, they are less pathogenic compared to conventional Th1 cells, conversely, they still suppressed naive T cell clonal expansion and protected against the development of EAE [97].
We further observed the differences of stability of both Treg cell subsets in vivo. About 50% of nTregs converted to Th17 cells in draining LNs ten days after cells transfer to established collagen-induced arthritis. Conversely, iTregs were completely resistant to Th1, Th2, and Th17 cell conversion. When these cells were sorted for in vitro analysis, nTregs mostly lost suppressive activity, whereas the functional activity of iTregs was mostly intact [88]. These results indicate iTregs are stable and functional in the inflammatory condition and may have a greater advantage to treat autoimmune and inflammatory diseases compared with nTregs.
Nonetheless, others have also reported that TGF-β-induced iTregs were unstable in vitro [98] and in vivo following antigen-stimulation [99], and lack protective activity to prevent lethal graft versus host disease (GVHD) [98,100]. It has been claimed that the Foxp3 promoter on TGF-β-induced iTregs but not nTregs is methylated and accounts for their instability [98]. However, we have recently observed that the methylation status in Foxp3 gene loci does not affect Foxp3 stability. Moreover, addition of atRA to TGF-β promoted iTreg stability and maintenance in vitro and in vivo and this effect is unrelated to CpG methylation in Foxp3 promoter but related to acetylation of Foxp3 histone [101]. Others have also observed protective human TGF-β-induced Tregs that exhibit methylated Foxp3 [35]. To explain these controversial results, we consider the technical reasons are possibly responsible for the generation of unstable, ineffective TGF-β-induced iTregs in these groups. They have used high concentrations of plate-bound anti-CD3 with TGF-β, whereas our group has used suboptimal concentrations of anti-CD3 and anti-CD28 coated beads with IL-2 and TGF-β. It has been known that strong, sustained TCR stimulation activates the mTOR/Akt signaling pathway which facilitates Teff cell differentiation and inhibits Foxp3 expression and Treg differentiation [102]. Treg generation is best established with suboptimal TCR stimulation that facilitates Foxp3expression [16].
These studies also raise the possibility that nTregs and iTregs may have distinct roles in the adaptive immune response. In response to microbial infections nTregs could possibly serve as a first line of host defense by differentiation to IL-17 producing cells, which contribute to neutrophil mobilization and have other pro-inflammatory effects. After eradication of invading pathogens, the late appearance of TGF-β-induced iTregs would not only terminate the antigen-specific response, but also prevent the emergence of non-specifically stimulated or cross-reactive self-reactive T cells. Accordingly, failure of this mechanism could result in an immune-mediated disease.
Acknowledgements
This work was in part supported by grants from the NIH AR 059103, NIH AR 084359, Arthritis Foundation; Wright Foundation; the Arthritis National Research Foundation; the Clinical Research Feasibility Fund; the James H. Zumberge Faculty Research and Innovation Fund; the Outstanding Youth Scientist Investigator Award from National Nature Science Foundation of China (30728007), Science Foundation of Shanghai Pudong new area (PKJ2009-Y41), and the American College of Rheumatology Research and Education’s Within Our Reach: Finding a Cure for Rheumatoid Arthritis campaign.
References
- 1.Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970;18:723–737. [PMC free article] [PubMed] [Google Scholar]
- 2.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 3.Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, Solomon M, Selby W, Alexander SI, Nanan R, Kelleher A, Fazekas de St Groth B. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203:1693–1700. doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, Mathian A, Nakahata T, Yamaguchi T, Nomura T, Ono M, Amoura Z, Gorochov G, Sakaguchi S. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 5.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 6.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Wang J, Shi W, Brand DD, Liu Z, Fan H, Zheng SG. Isolation of purified and live Foxp3+ regulatory T cells using FACS sorting on scatter plot. J Mol Cell Biol. 2010;2:164–169. doi: 10.1093/jmcb/mjq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, Roncarolo MG, Levings MK. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19:345–354. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- 9.Tritt M, Sgouroudis E, d’Hennezel E, Albanese A, Piccirillo CA. Functional waning of naturally occurring CD4+ regulatory T-cells contributes to the onset of autoimmune diabetes. Diabetes. 2008;57:113–123. doi: 10.2337/db06-1700. [DOI] [PubMed] [Google Scholar]
- 10.Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, Nochy D, Debre P, Piette JC, Gorochov G. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol. 2005;175:8392–8400. doi: 10.4049/jimmunol.175.12.8392. [DOI] [PubMed] [Google Scholar]
- 11.Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S. Dysfunctional CD4+,CD25+ regulatory T cells in untreated active systemic lupus erythematosus secondary to interferon-alpha-producing antigen-presenting cells. Arthritis Rheum. 2008;58:801–812. doi: 10.1002/art.23268. [DOI] [PubMed] [Google Scholar]
- 12.Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–261. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haas J, Hug A, Viehover A, Fritzsching B, Falk CS, Filser A, Vetter T, Milkova L, Korporal M, Fritz B, Storch-Hagenlocher B, Krammer PH, Suri-Payer E, Wildemann B. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35:3343–3352. doi: 10.1002/eji.200526065. [DOI] [PubMed] [Google Scholar]
- 14.Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D, Mueller N, Hardt C, Kreuzfelder E, Grosse-Wilde H. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol. 2006;180:178–184. doi: 10.1016/j.jneuroim.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. 2008;29:429–435. doi: 10.1016/j.it.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Zhou X, Kong N, Zou H, Brand D, Li X, Liu Z, Zheng SG. Therapeutic potential of TGF-beta-induced CD4(+) Foxp3(+) regulatory T cells in autoimmune diseases. Autoimmunity. 2011;44:43–50. doi: 10.3109/08916931003782163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan Q, Fan H, Quesniaux V, Ryffel B, Liu Z, Zheng SG. Induced Foxp3+ regulatory T cells: a potential new weapon to treat autoimmune and inflammatory diseases? J Mol Cell Biol. 2012;4:22–8. doi: 10.1093/jmcb/mjr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serre K, Benezech C, Desanti G, Bobat S, Toellner KM, Bird R, Chan S, Kastner P, Cunningham AF, Maclennan IC, Mohr E. Helios is associated with CD4 T cells differentiating to T helper 2 and follicular helper T cells in vivo independently of Foxp3 expression. PLoS One. 2011;6:e20731. doi: 10.1371/journal.pone.0020731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, Ruminski P, Weiss D, Von Schack D, Bluestone JA. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209:1713–1722. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, Albert J, Sparwasser T, Sakaguchi S, Westendorf AM, Schadendorf D, Buer J, Helfrich I. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med. 2012;209:2001–2016. doi: 10.1084/jem.20111497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milpied P, Renand A, Bruneau J, Mendes-da-Cruz DA, Jacquelin S, Asnafi V, Rubio MT, MacIntyre E, Lepelletier Y, Hermine O. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol. 2009;39:1466–1471. doi: 10.1002/eji.200839040. [DOI] [PubMed] [Google Scholar]
- 24.Ding ZC, Blazar BR, Mellor AL, Munn DH, Zhou G. Chemotherapy rescues tumor-driven aberrant CD4+ T-cell differentiation and restores an activated polyfunctional helper phenotype. Blood. 2010;115:2397–2406. doi: 10.1182/blood-2009-11-253336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol. 2006;177:1451–1459. doi: 10.4049/jimmunol.177.3.1451. [DOI] [PubMed] [Google Scholar]
- 26.Bagavant H, Tung KS. Failure of CD25+ T cells from lupus-prone mice to suppress lupus glomerulonephritis and sialoadenitis. J Immunol. 2005;175:944–950. doi: 10.4049/jimmunol.175.2.944. [DOI] [PubMed] [Google Scholar]
- 27.Huter EN, Stummvoll GH, DiPaolo RJ, Glass DD, Shevach EM. Cutting edge: antigen-specific TGF beta-induced regulatory T cells suppress Th17-mediated autoimmune disease. J Immunol. 2008;181:8209–8213. doi: 10.4049/jimmunol.181.12.8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 29.Wakkach A, Fournier N, Brun V, Breittmayer JP, Cottrez F, Groux H. Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity. 2003;18:605–617. doi: 10.1016/s1074-7613(03)00113-4. [DOI] [PubMed] [Google Scholar]
- 30.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25- Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 31.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 32.Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science. 2009;323:1488–1492. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- 33.Lu L, Wang J, Zhang F, Chai Y, Brand D, Wang X, Horwitz DA, Shi W, Zheng SG. Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells. J Immunol. 2010;184:4295–4306. doi: 10.4049/jimmunol.0903418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J Immunol. 2008;180:7112–7116. doi: 10.4049/jimmunol.180.11.7112. [DOI] [PubMed] [Google Scholar]
- 35.Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, Riley JL, June CH, Miller JS, Wagner JE, Blazar BR. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am J Transplant. 2011;11:1148–1157. doi: 10.1111/j.1600-6143.2011.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, Olek S, Dietmaier W, Andreesen R, Edinger M. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol. 2009;39:1088–1097. doi: 10.1002/eji.200838904. [DOI] [PubMed] [Google Scholar]
- 37.Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, Brand D, Liu Z, Zheng SG. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol. 2010;185:2675–2679. doi: 10.4049/jimmunol.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. 2004;172:5213–5221. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 40.Weber SE, Harbertson J, Godebu E, Mros GA, Padrick RC, Carson BD, Ziegler SF, Bradley LM. Adaptive islet-specific regulatory CD4 T cells control autoimmune diabetes and mediate the disappearance of pathogenic Th1 cells in vivo. J Immunol. 2006;176:4730–4739. doi: 10.4049/jimmunol.176.8.4730. [DOI] [PubMed] [Google Scholar]
- 41.DiPaolo RJ, Brinster C, Davidson TS, Andersson J, Glass D, Shevach EM. Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J Immunol. 2007;179:4685–4693. doi: 10.4049/jimmunol.179.7.4685. [DOI] [PubMed] [Google Scholar]
- 42.Fantini MC, Becker C, Tubbe I, Nikolaev A, Lehr HA, Galle P, Neurath MF. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut. 2006;55:671–680. doi: 10.1136/gut.2005.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Selvaraj RK, Geiger TL. Mitigation of experimental allergic encephalomyelitis by TGF-beta induced Foxp3+ regulatory T lymphocytes through the induction of anergy and infectious tolerance. J Immunol. 2008;180:2830–2838. doi: 10.4049/jimmunol.180.5.2830. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen TL, Sullivan NL, Ebel M, Teague RM, Dipaolo RJ. Antigen-Specific TGF-{beta}-Induced Regulatory T Cells Secrete Chemokines, Regulate T Cell Trafficking, and Suppress Ongoing Autoimmunity. J Immunol. 2011;187:1745–1753. doi: 10.4049/jimmunol.1004112. [DOI] [PubMed] [Google Scholar]
- 45.Zheng SG, Wang JH, Koss MN, Quismorio F Jr, Gray JD, Horwitz DA. CD4+ and CD8+ regulatory T cells generated ex vivo with IL-2 and TGF-beta suppress a stimulatory graft-versus-host disease with a lupus-like syndrome. J Immunol. 2004;172:1531–1539. doi: 10.4049/jimmunol.172.3.1531. [DOI] [PubMed] [Google Scholar]
- 46.Xu W, Lan Q, Chen M, Chen H, Zhu N, Zhou X, Wang J, Fan H, Yan CS, Kuang JL, Warburton D, Togbe D, Ryffel B, Zheng SG, Shi W. Adoptive transfer of induced-Treg cells effectively attenuates murine airway allergic inflammation. PLoS One. 2012;7:e40314. doi: 10.1371/journal.pone.0040314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lan Q, Zhou X, Fan H, Chen M, Wang J, Ryffel B, Brand D, Ramalingam R, Kiela PR, Horwitz DA, Liu Z, Zheng SG. Polyclonal CD4+Foxp3+ Treg cells induce TGFbeta-dependent tolerogenic dendritic cells that suppress the murine lupus-like syndrome. J Mol Cell Biol. 2012;4:409–419. doi: 10.1093/jmcb/mjs040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng SG, Gray JD, Ohtsuka K, Yamagiwa S, Horwitz DA. Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25- precursors. J Immunol. 2002;169:4183–4189. doi: 10.4049/jimmunol.169.8.4183. [DOI] [PubMed] [Google Scholar]
- 49.Pot C, Apetoh L, Kuchroo VK. Type 1 regulatory T cells (Tr1) in autoimmunity. Semin Immunol. 2011;23:202–8. doi: 10.1016/j.smim.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu SL, Wong CK, Wong PT, Chen DP, Szeto CC, Li EK, Tam LS. Down-Regulated NOD2 by Immunosuppressants in Peripheral Blood Cells in Patients with SLE Reduces the Muramyl Dipeptide-Induced IL-10 Production. PLoS One. 2011;6:e23855. doi: 10.1371/journal.pone.0023855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282–7289. doi: 10.4049/jimmunol.166.12.7282. [DOI] [PubMed] [Google Scholar]
- 52.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 53.Liang S, Alard P, Zhao Y, Parnell S, Clark SL, Kosiewicz MM. Conversion of CD4+ CD25- cells into CD4+ CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J Exp Med. 2005;201:127–137. doi: 10.1084/jem.20041201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 55.Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, Shevach EM. CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. J Exp Med. 2002;196:237–246. doi: 10.1084/jem.20020590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 57.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu L, Ma J, Wang X, Wang J, Zhang F, Yu J, He G, Xu B, Brand DD, Horwitz DA, Shi W, Zheng SG. Synergistic effect of TGF-beta superfamily members on the induction of Foxp3+ Treg. Eur J Immunol. 2010;40:142–152. doi: 10.1002/eji.200939618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–2027. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- 60.Zheng SG, Wang JH, Stohl W, Kim KS, Gray JD, Horwitz DA. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J Immunol. 2006;176:3321–3329. doi: 10.4049/jimmunol.176.6.3321. [DOI] [PubMed] [Google Scholar]
- 61.Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol. 2006;177:4376–4383. doi: 10.4049/jimmunol.177.7.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J Immunol. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 63.Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, Rajan TV, Clark RB. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol. 2011;186:6779–6787. doi: 10.4049/jimmunol.1003868. [DOI] [PubMed] [Google Scholar]
- 64.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 65.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 66.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 67.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, Huang S, Boissier MC, Fournier C. High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol. 1997;158:5501–5506. [PubMed] [Google Scholar]
- 69.Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P. Accelerated procollagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol. 1997;158:5507–5513. [PubMed] [Google Scholar]
- 70.Kageyama Y, Koide Y, Yoshida A, Uchijima M, Arai T, Miyamoto S, Ozeki T, Hiyoshi M, Kushida K, Inoue T. Reduced susceptibility to collagen-induced arthritis in mice deficient in IFN-gamma receptor. J Immunol. 1998;161:1542–1548. [PubMed] [Google Scholar]
- 71.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-gamma. Eur J Immunol. 1996;26:2393–2398. doi: 10.1002/eji.1830261019. [DOI] [PubMed] [Google Scholar]
- 73.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 74.Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:382–388. doi: 10.1097/01.MIB.0000218764.06959.91. [DOI] [PubMed] [Google Scholar]
- 76.Voo KS, Wang YH, Santori FR, Boggiano C, Arima K, Bover L, Hanabuchi S, Khalili J, Marinova E, Zheng B, Littman DR, Liu YJ. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A. 2009;106:4793–4798. doi: 10.1073/pnas.0900408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 79.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 80.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 81.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 83.Besnard AG, Togbe D, Couillin I, Tan Z, Zheng SG, Erard F, Le Bert M, Quesniaux V, Ryffel B. Inflammasome - IL-1 - Th17 response in allergic lung inflammation. J Mol Cell Biol. 2012;4:3–10. doi: 10.1093/jmcb/mjr042. [DOI] [PubMed] [Google Scholar]
- 84.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O’Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 86.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Shea JJ. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 87.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng XH, Jetten AM, Dong C. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kong N, Lan Q, Chen M, Wang J, Shi W, Horwitz DA, Quesniaux V, Ryffel B, Liu Z, Brand D, Zou H, Zheng SG. Antigen-specific TGF-beta-induced regulatory T cells but not natural Tregs ameliorate autoimmune arthritis by shifting the balance of Th17 toward Treg cells. Arthritis Rheum. 2012 doi: 10.1002/art.34513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh SP, Zhang HH, Foley JF, Hedrick MN, Farber JM. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J Immunol. 2008;180:214–221. doi: 10.4049/jimmunol.180.1.214. [DOI] [PubMed] [Google Scholar]
- 90.Afzali B, Mitchell P, Lechler RI, John S, Lombardi G. Translational mini-review series on Th17 cells: induction of interleukin-17 production by regulatory T cells. Clin Exp Immunol. 2010;159:120–130. doi: 10.1111/j.1365-2249.2009.04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ayyoub M, Deknuydt F, Raimbaud I, Dousset C, Leveque L, Bioley G, Valmori D. Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc Natl Acad Sci U S A. 2009;106:8635–8640. doi: 10.1073/pnas.0900621106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moon HG, Tae YM, Kim YS, Gyu Jeon S, Oh SY, Song Gho Y, Zhu Z, Kim YK. Conversion of Th17-type into Th2-type inflammation by acetyl salicylic acid via the adenosine and uric acid pathway in the lung. Allergy. 2010;65:1093–1103. doi: 10.1111/j.1398-9995.2010.02352.x. [DOI] [PubMed] [Google Scholar]
- 93.Feng T, Cao AT, Weaver CT, Elson CO, Cong Y. Interleukin-12 converts Foxp3+ regulatory T cells to interferon-gamma-producing Foxp3+ T cells that inhibit colitis. Gastroenterology. 2011;140:2031–2043. doi: 10.1053/j.gastro.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Panzer M, Sitte S, Wirth S, Drexler I, Sparwasser T, Voehringer D. Rapid In Vivo Conversion of Effector T Cells into Th2 Cells during Helminth Infection. J Immunol. 2012;188:615–23. doi: 10.4049/jimmunol.1101164. [DOI] [PubMed] [Google Scholar]
- 95.Mathur AN, Chang HC, Zisoulis DG, Kapur R, Belladonna ML, Kansas GS, Kaplan MH. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood. 2006;108:1595–1601. doi: 10.1182/blood-2006-04-015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cohen CJ, Crome SQ, Macdonald KG, Dai EL, Mager DL, Levings MK. Human Th1 and th17 cells exhibit epigenetic stability at signature cytokine and transcription factor Loci. J Immunol. 2011;187:5615–5626. doi: 10.4049/jimmunol.1101058. [DOI] [PubMed] [Google Scholar]
- 97.O’Connor RA, Leech MD, Suffner J, Hammerling GJ, Anderton SM. Myelin-reactive, TGF-beta-induced regulatory T cells can be programmed to develop Th1-like effector function but remain less proinflammatory than myelin-reactive Th1 effectors and can suppress pathogenic T cell clonal expansion in vivo. J Immunol. 2010;185:7235–7243. doi: 10.4049/jimmunol.1001551. [DOI] [PubMed] [Google Scholar]
- 98.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J Immunol. 2011;186:6329–6337. doi: 10.4049/jimmunol.1100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, Huehn J, Ganser A, Forster R, Prinz I. Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur J Immunol. 2009;39:3091–3096. doi: 10.1002/eji.200939432. [DOI] [PubMed] [Google Scholar]
- 101.Lu L, Ma J, Li Z, Lan Q, Chen M, Liu Y, Xia Z, Wang J, Han Y, Shi W, Quesniaux V, Ryffel B, Brand D, Li B, Liu Z, Zheng SG. All-trans retinoic acid promotes TGF-beta-induced Tregs via histone modification but not DNA demethylation on Foxp3 gene locus. PLoS One. 2011;6:e24590. doi: 10.1371/journal.pone.0024590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, Shokat KM, Fisher AG, Merkenschlager M. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]