

Abstract
Dyskeratosis congenita (DC) is a multisystem disease caused by genetic mutations that result in defective telomere maintenance. Herein, we describe a 17 year-old patient with severe DC, manifested by bone marrow failure, severe immunodeficiency, and enterocolitis requiring prolonged infliximab therapy, who developed fatal hepatic failure caused by an aggressive, infiltrating hepatic angiosarcoma. While DC patients have known increased risk of developing liver failure and multiple types of malignancy, this report is the first to describe angiosarcoma in a DC patient. Malignancy should thus be considered in the differential diagnosis of progressive liver dysfunction in DC patients.
Keywords: dyskeratosis congenita, bone marrow failure, angiosarcoma, liver failure, pediatric
Introduction
Dyskeratosis congenita (DC) is a multisystem disease characterized by chromosomal instability and caused by a variety of defined genetic mutations leading to dysfunctional telomere maintenance1. The relationship between specific genetic mutations, the resulting degree of telomere dysfunction, and the severity of mutation-specific DC clinical phenotypes is now beginning to be understood. Specifically, mutations in the gene DKC1 are associated with the severe X-linked DC phenotype previously known as the Hoyeraal-Hreidarsson syndrome2. While bone marrow failure, skin and nail dystrophy, and leukoplakia are disease-defining abnormalities in DC, patients with DC are also at risk for developing pulmonary fibrosis; liver failure of unclear etiology; and multiple malignancies including head and neck, skin, and GI cancer 3,4. These predispositions have often been attributed to tissue specific deficits in telomere function; however, because severely affected DC patients with DKC1 mutations often manifest with B and T cell immunodeficiency5, it is possible that poor immune function and the resultant chronic inflammation that occurs in tissues such as the oral mucosa6 and lower GI tract7 in DC patients could contribute to DC cancer predisposition, as chronic inflammation in otherwise healthy individuals is known to increase the risk for GI cancer8. Here, we report a case of hepatic angiosarcoma occurring in an adolescent patient with severe DC, and discuss possible etiologic factors leading to this malignancy.
Case Report
A 17 year-old male with severe dyskeratosis congenita was referred to our Comprehensive Bone Marrow Failure Center for consultative evaluation prior to possible bone marrow transplant. He was formally diagnosed with dyskeratosis congenita at age 8, with genetic evaluation showing a known disease causing mutation in the DKC1 gene (29 C>T, causing an amino acid substitution P10L). Telomere lengths in lymphocytes and granulocytes were considerably less than the first percentile. Prior to this genetic diagnosis, his medical history was significant for in utero growth retardation, short stature, microcephaly, cerebellar dysmetria, developmental delay, nail dystrophy, blunted terminal growth of phalanges, and diffuse reticular rash with skin pigment abnormalities. In early childhood, he developed recurrent sinopulmonary infections, as well as chronic infections with rotavirus and herpes simplex virus. Immunologic evaluation at age 5, using published age adjusted normal values for comparison9, found that he had a significant deficit in CD19+ B lymphocyte number (39 cells/mm3; N 200–2100 cells/mm3), hypogammaglobulinemia (IgG: 87 mg/dL; N 583–1783 mg/dL, IgA 68 mg/dL; N 70–393 mg/dL, IgM 60 mg/dL; N 50–198 mg/dL), and non-protective diphtheria, tetanus, and varicella titers. Further evaluation demonstrated decreased CD4+ T cells (295 cells/mm3; N 500–2400 cells/mm3) and decreased CD16/CD56+ natural killer (NK) cells (58 cells/mm3; N 100–1000 cells/mm3), though with normal total CD3+ (1153 cells/mm3) and CD8+ T cells (669 cells/mm3) and normal mitogen proliferative responses.
Following this evaluation scheduled immune globulin replacement therapy was instituted, and his infectious course improved dramatically. However, he also developed severe biopsy-proven enterocolitis, with symptoms beginning at age 3 that did not improve with subsequent immune globulin replacement. Rather, his enterocolitis required chronic steroid administration by age 5, and institution of tumor necrosis factor-alpha (TNFα) inhibition therapy with infliximab at age 7. His symptoms improved on infliximab, enabling a slow steroid wean over a period of several years. He continued on infliximab until the time of his referral to our center at age 17, with the major clinical events over the preceding decade including esophageal strictures for which he underwent dilatation, chronic malnutrition requiring an enteral feeding tube, and an episode of pneumocystis jiroveci pneumonia at age 12 that fully resolved with treatment.
In the few months prior to initial referral to our center, he had developed progressive peripheral cytopenias, and bone marrow analysis showed a cellularity of about 1%. His absolute neutrophil count remained >1000/μL however, and he had only required red blood cell and platelet transfusions during several recent episodes of acute anemia attributed to bleeding episodes. During this period, he had developed cholestatic liver dysfunction with increased total bilirubin (2.4 mg/dL, N:0.6–1.4 mg/dL), and elevated alkaline phosphatase (492 U/L, N:65–260 U/L), and gamma glutamyl transferase (357 U/L; N 9–29 U/L), with essentially normal liver transaminases. Liver ultrasound showed increased hepatic echogenicity with nodular liver margins and overall small liver appearance concerning for cirrhosis, but with no biliary duct dilation or mass identified. Of note, he had never received androgen therapy. An initial attempt at transjugular liver biopsy prior to his referral to our center was postponed due to uncontrolled mucosal bleeding at the time of attempted pre-procedure intubation. Of note, in relation to this mucosal bleeding, his mild to moderate baseline trismus of unclear etiology had become much more severe beginning 3–4 months prior to this biopsy attempt. Otolaryngologic and oral surgery examinations, as well as imaging studies including MRI and CT revealed no mass lesions and were unable to ascertain the etiology of this trismus, which was accompanied by increased spontaneous bleeding from both nasal and oral cavities.
Over the next several months, despite cessation of infliximab and medications with potential direct hepatotoxicity, and despite negative evaluation for possible infectious etiologies and several normal imaging evaluations of his abdomen, his liver dysfunction continued to worsen. He additionally began to require regular red blood cell and platelet transfusions every 7 to 10 days. Four months after his initial referral, he experienced a sudden dramatic rise in his total bilirubin to 19.1 mg/dL, with elevated conjugated bilirubin of 14.7 mg/dL (N: 0.0–0.3 mg/dL). Liver ultrasound again showed increased echogenicity and heterogeneity without focal mass or large duct dilation. The elevated liver function studies prompted hospital admission and urgent transjugular liver biopsy, which revealed elevated portal venous pressure (22 mmHg) and pathology showing severe canalicular and hepatocellular cholestasis, severe hemosiderosis, and focal sinusoidal dilatation with perisinusoidal fibrosis. No inflammation or extramedullary hematopoiesis was noted though few portal tracts were present in the biopsy, and the only atypical findings were a few large cells immunoreactive to Factor VIII, initially thought to represent megakaryocytes.
His biopsy was complicated by the need for emergent tracheostomy placement due to severe oropharyngeal bleeding following intubation. He was admitted to the pediatric intensive care unit post-operatively for airway management, where over the next two weeks he developed worsening hepatic failure, coagulopathy, hepato-renal syndrome and ultimately respiratory failure. Magnetic resonance (MR) cholangiography exam revealed diffuse hemorrhagic lesions throughout the liver and severe anasarca. During the following days the patient deteriorated rapidly and he subsequently expired due to multi-organ failure.
A limited liver autopsy revealed the diffuse lesions seen on MR imaging to be comprised of multifocal areas of hemorrhage (Figure 1A), with the overall appearance consistent with peliosis hepatis (Figure 1B). Microscopic examination demonstrated multiple foci of dilated hepatic sinusoids accompanied by atrophy of the liver cell plates. In some areas, hepatocytes had entirely disappeared and the resultant spaces were filled with red blood cells, giving a cavernous appearance (Figure 1B). The dilated sinusoids and cavernous areas were lined by scattered malignant cells, which had enlarged, irregular, hyperchromatic nuclei, scanty cytoplasm, and occasional prominent nuclei (Figure 1C). The malignant cells had an infiltrative growth pattern (Figure 1D). Vascular invasion of the portal and hepatic vein branches was observed (Figure 1E). Many of these malignant cells contained Periodic Acid Schiff positive and diastase resistant intracytoplasmic globules. By immunohistochemistry, a subset of these malignant cells were positive for endothelial markers, namely, CD31 (Figure 1F), CD34, and Factor VIII, indicating an endothelial origin. These cells were also positive for vimentin and were negative for pancytokeratin, CD45RO, CD3, PGIIIa, CD163, CD68, D2-40, Glut-1, HepPar, and human herpesvirus 8 (HHV8). Many of the hepatocytes showed evidence of ischemic changes, and a large majority of hepatocytes aberrantly expressed CK7. Hepatocyte atrophy and dropout, diffuse sinusoidal congestion, perisinusoidal and pericellular fibrosis, intrahepatocytic and cannulicular cholestasis, and bile ductular proliferation were present. Excessive iron deposition was noted within the hepatocytic and Kupffer cell cytoplasm. The overall findings were consistent with hepatic angiosarcoma, with diffuse involvement of the liver, invasion of portal and hepatic veins, and resultant peliotic changes.
Figure 1. Hepatic Angiosarcoma in Patient with Dyskeratosis Congenita.
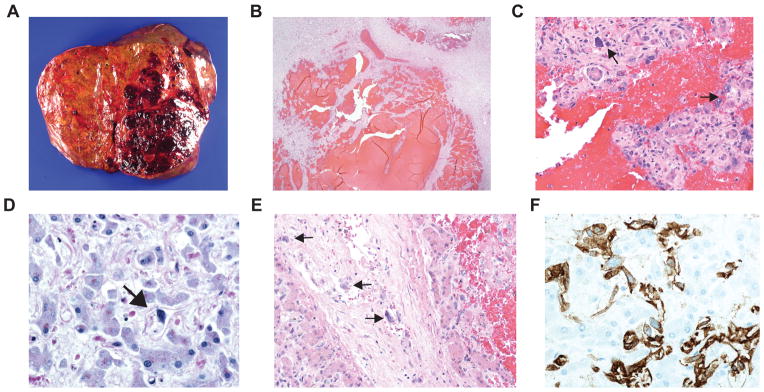
A, macroscopic photograph showing a large hemorrhagic lesion in the left lobe with several smaller lesions in the right lobe of the liver. B, photomicrograph of liver, demonstrating peliotic changes with resultant blood sequestration caused by the tumor (hematoxylin-eosin, original magnification x2). C, photomicrograph of liver, demonstrating enlarged, hyperchromatic tumor cells (black arrows) admixed with normal liver cells (hematoxylin-eosin, original magnification x20). D, photomicrograph of right lobe of the liver (away from macroscopic lesions), demonstrating diffuse infiltration of tumor cells (black arrow) throughout the liver (hematoxylin-eosin, original magnification x40). E, photomicrograph of liver, demonstrating tumor cell (black arrows) infiltration into the wall of an interlobular blood vessel (hematoxylin-eosin, original magnification x20). F, photomicrograph of liver, the tumor cells are immunopositive for CD31, an endothelial marker, consistent with hepatic angiosarcoma (CD31 immunohistochemistry, original magnification x40).
Discussion
Angiosarcoma is extremely uncommon among pediatric soft tissue and visceral tumors, representing <1% of all vascular tumors in children10. Angiosarcomas in children can arise in a number of primary sites, including most frequently the head/neck or heart/mediastinum, but also breast, mesentery, liver and spleen11. Specifically, approximately 40 cases of pediatric primary liver angiosarcomas have been reported12. Because angiosarcoma could arise in the liver or metastasize to the liver from other sites, and because the autopsy in our patient was limited to the liver, we cannot determine whether his liver was the primary site, or whether his severe oropharyngeal bleeding symptoms and trismus of unknown etiology could be secondary to additional tumor involvement in the head and neck region. Unfortunately, prognosis is dismal for patients diagnosed with angiosarcoma involving the liver, even in patients who can tolerate attempts at curative chemotherapy and aggressive surgical approaches12.
Of note, a few patients with pediatric hepatic angiosarcoma described in the literature had a prior diagnosis of infantile hepatic hemangioma, previously known as infantile hepatic hemangioendothelioma. Thus, in these cases, the hepatic angiosarcomas were thought to arise/transform from former infantile hemangiomas12. The negative Glut-1 immunostaining in our case, however, would argue against a pre-existing infantile hepatic hemangioma.
In the workup of this case, a diagnosis of Kaposi’s sarcoma was considered, especially given the severity of our patient’s inherent immunodeficiency caused by his DKC1 mutation, along with his prolonged immunosuppression with infliximab. Kaposi’s sarcoma is a vascular tumor that develops following HHV8 infection in patients with severe immunodeficient states such as HIV infection, prolonged TNFα inhibition with infliximab13, or post-solid organ transplant immunosuppression14. Kaposi’s sarcoma can involve the liver; however, the lack of spindle cell proliferation in fascicular arrangement, degree of cellular pleomorphism, and absence of HHV8 immunoreactivity are incompatible with Kaposi’s sarcoma.
Interestingly, although uncommon, classic angiosarcoma that is histopathologically distinct from Kaposi’s sarcoma has also been reported to arise in immunosuppressed patients. A recent systematic literature review identified 23 cases of angiosarcoma associated with either HIV infection or post-kidney transplant immune suppression15. While not previously linked specifically to classic angiosarcoma, infliximab and other tumor necrosis factor α blocking agents have been linked to a variety of other malignancies including Kaposi sarcoma, hepatocellular carcinoma, and hepatosplenic T cell lymphoma13,16,17, suggesting that prolonged infliximab therapy may have played a role in the development of the hepatic angiosarcoma in our reported case. In another recent report however, hepatic angiosarcoma was described in a patient with the Immunodeficiency, Centromeric region instability, Facial anomalies (ICF) syndrome without exogenous immune suppression18, demonstrating that genetically determined immune deficiency such as that seen in severe DC, may also have been a significant causative factor in the development of angiosarcoma in this case.
In a recent review4, Alter et al. reviewed all literature reports of cancer in DC patients, as well as records of an institution specific cohort, obtaining an incidence rate of DC patients developing cancer of approximately 10–15%. These malignancies were of multiple types, with the most common being head and neck squamous cell carcinoma, skin squamous cell carcinoma, and GI tract carcinomas (esophageal, anorectal, colon, and stomach). Less frequently, lung cancer, lymphoma, and acute myeloid leukemia had also been reported in DC patients. Interestingly, only one previous report of a liver tumor in DC patients is noted, with this tumor consisting of benign hepatic adenoma19. Our review of the current literature revealed no prior reports of malignant vascular tumors in DC patients either with or without liver involvement.
Liver disease is well-known to be associated with DC, though the etiology of liver dysfunction in DC patients remains poorly understood. In patients with less severe DC phenotypes, liver disease often presents in adulthood with a mix of inflammatory and fibrotic disease producing a cholestatic picture and nodular regeneration, with many patients progressing to cirrhosis20. These findings appear to parallel the development of idiopathic pulmonary fibrosis in older DC patients, and may arise from specific effects of telomerase deficiency in the liver itself. However, there are also a number of case reports in which severe liver dysfunction and portal hypertension has arisen in children and young adults with DC in the absence of biopsy proven cirrhosis and in which no clear etiology has been ascertained21–24. Given that we were only able to make the diagnosis of angiosarcoma on autopsy in our patient, and that both liver biopsy and imaging studies were insufficient to make this diagnosis, it is quite possible that a neoplastic process could be the precipitating cause in other cases of severe hepatic dysfunction in young DC patients with severe disease phenotypes.
Peliosis hepatis is a common feature of hepatic angiosarcomas25. While the tumor-associated peliosis described in our patient has not previously been seen in the liver of DC patients, splenic peliosis has been reported in two DC patients, though no association with tumor was found in either patient and the splenic peliosis was instead attributed to cytokine and androgen therapy26. Given that the patient reported herein had never received androgen therapy, the peliotic changes in the liver in this case most likely arose secondary to the angiosarcoma itself.
In conclusion, our case represents the first report of a hepatic angiosarcoma occurring in a patient with severe DC. Given that severe DC patients are known to develop a variety of neoplasms, a neoplastic process should be included in the differential diagnosis for DC patients who develop rapidly progressive organ dysfunction.
Acknowledgments
Source of Funding: T.O. has been supported by the American Society of Hematology Research Training Award for Fellows, and by NHLBI 2 K12 HL087064. M.B. is supported by the Buck Family Endowed Chair in Hematology, and by NIH 2R01 CA105312. We also would like to thank the patient and his family for their participation in our bone marrow failure studies.
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Mason PJ, Bessler M. The genetics of dyskeratosis congenita. Cancer Genet. 2011;204:635–645. doi: 10.1016/j.cancergen.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–486. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- 3.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113:6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jyonouchi S, Forbes L, Ruchelli E, Sullivan KE. Dyskeratosis congenita: a combined immunodeficiency with broad clinical spectrum--a single-center pediatric experience. Pediatr Allergy Immunol. 2011;22:313–319. doi: 10.1111/j.1399-3038.2010.01136.x. [DOI] [PubMed] [Google Scholar]
- 6.Lourenco SV, Boggio PA, Fezzi FA, Sebastiao AL, Nico MM. Dyskeratosis congenita--report of a case with emphasis on gingival aspects. Pediatr Dermatol. 2009;26:176–179. doi: 10.1111/j.1525-1470.2009.00878.x. [DOI] [PubMed] [Google Scholar]
- 7.Sznajer Y, Baumann C, David A, et al. Further delineation of the congenital form of X-linked dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome) Eur J Pediatr. 2003;162:863–867. doi: 10.1007/s00431-003-1317-5. [DOI] [PubMed] [Google Scholar]
- 8.Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143:550–563. doi: 10.1053/j.gastro.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Comans-Bitter WM, de Groot R, van den Beemd R, et al. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr. 1997;130:388–393. doi: 10.1016/s0022-3476(97)70200-2. [DOI] [PubMed] [Google Scholar]
- 10.Coffin CM, Dehner LP. Vascular tumors in children and adolescents: a clinicopathologic study of 228 tumors in 222 patients. Pathol Annu. 1993;28(1):97–120. [PubMed] [Google Scholar]
- 11.Deyrup AT, Miettinen M, North PE, et al. Angiosarcomas arising in the viscera and soft tissue of children and young adults: a clinicopathologic study of 15 cases. Am J Surg Pathol. 2009;33:264–269. doi: 10.1097/PAS.0b013e3181875a5f. [DOI] [PubMed] [Google Scholar]
- 12.Dimashkieh HH, Mo JQ, Wyatt-Ashmead J, Collins MH. Pediatric hepatic angiosarcoma: case report and review of the literature. Pediatr Dev Pathol. 2004;7:527–532. doi: 10.1007/s10024-004-4041-x. [DOI] [PubMed] [Google Scholar]
- 13.Cohen CD, Horster S, Sander CA, Bogner JR. Kaposi’s sarcoma associated with tumour necrosis factor alpha neutralising therapy. Ann Rheum Dis. 2003;62:684. doi: 10.1136/ard.62.7.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokois NU, Perlman EJ, Colombani P, Wise B, Schwarz KB. Kaposi’s sarcoma presenting as a protracted multisystem illness in an adolescent liver transplant recipient. Liver Transpl Surg. 1997;3:541–544. doi: 10.1002/lt.500030511. [DOI] [PubMed] [Google Scholar]
- 15.Bhatia K, Shiels MS, Berg A, Engels EA. Sarcomas other than Kaposi sarcoma occurring in immunodeficiency: interpretations from a systematic literature review. Curr Opin Oncol. 2012;24:537–546. doi: 10.1097/CCO.0b013e328355e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diak P, Siegel J, La Grenade L, Choi L, Lemery S, McMahon A. Tumor necrosis factor alpha blockers and malignancy in children: forty-eight cases reported to the Food and Drug Administration. Arthritis Rheum. 2010;62:2517–2524. doi: 10.1002/art.27511. [DOI] [PubMed] [Google Scholar]
- 17.Seminerio JL, Loftus EV, Jr, Colombel JF, Thapa P, Sandborn WJ. Infliximab for Crohn’s Disease: The First 500 Patients Followed Up Through 2009. Dig Dis Sci. 2012 doi: 10.1007/s10620-012-2405-z. [DOI] [PubMed] [Google Scholar]
- 18.van den Brand M, Flucke UE, Bult P, Weemaes CM, van Deuren M. Angiosarcoma in a patient with immunodeficiency, centromeric region instability, facial anomalies (ICF) syndrome. Am J Med Genet A. 2011;155A:622–625. doi: 10.1002/ajmg.a.33831. [DOI] [PubMed] [Google Scholar]
- 19.Devriendt K, Matthijs G, Legius E, et al. Skewed X-chromosome inactivation in female carriers of dyskeratosis congenita. Am J Hum Genet. 1997;60:581–587. [PMC free article] [PubMed] [Google Scholar]
- 20.Calado RT, Regal JA, Kleiner DE, et al. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS One. 2009;4:e7926. doi: 10.1371/journal.pone.0007926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yazgan Y, Demirturk L, Ozel M, Basekim C. A case of dyskeratosis congenita with portal hypertension associated with jugular venous anomaly. Turk J Gastroenterol. 2006;17:66–69. [PubMed] [Google Scholar]
- 22.Redkar NN, Pandey DB, Jerajani HR, Padhiyar R, Dhokare A. Dyskeratosis congenita with portal hypertension of unknown etiology. J Assoc Physicians India. 2011;59:260–263. [PubMed] [Google Scholar]
- 23.Renoux MC, Mazars N, Tichit R, Counil F. Cyanosis revealing hepatopulmonary syndrome in a child with dyskeratosis congenita. Pediatr Pulmonol. 2010;45:99–102. doi: 10.1002/ppul.21128. [DOI] [PubMed] [Google Scholar]
- 24.Brown KE, Kelly TE, Myers BM. Gastrointestinal involvement in a woman with dyskeratosis congenita. Dig Dis Sci. 1993;38:181–184. doi: 10.1007/BF01296794. [DOI] [PubMed] [Google Scholar]
- 25.Ludwig J, Hoffman HN. Hemangiosarcoma of the liver. Spectrum of morphologic changes and clinical findings. Mayo Clin Proc. 1975;50:255–263. [PubMed] [Google Scholar]
- 26.Giri N, Pitel PA, Green D, Alter BP. Splenic peliosis and rupture in patients with dyskeratosis congenita on androgens and granulocyte colony-stimulating factor. Br J Haematol. 2007;138:815–817. doi: 10.1111/j.1365-2141.2007.06718.x. [DOI] [PubMed] [Google Scholar]