

Abstract
Heterogeneity in systemic sclerosis/SSc confounds clinical trials. We previously identified ‘intrinsic’ gene expression subsets by analysis of SSc skin. Here we test the hypotheses that skin gene expression signatures including intrinsic subset are associated with skin score/MRSS improvement during mycophenolate mofetil (MMF) treatment. Gene expression and intrinsic subset assignment were measured in 12 SSc patients’ biopsies and ten controls at baseline, and from serial biopsies of one cyclophosphamide-treated patient, and nine MMF-treated patients. Gene expression changes during treatment were determined using paired t-tests corrected for multiple hypothesis testing. MRSS improved in four of seven MMF-treated patients classified as the inflammatory intrinsic subset. Three patients without MRSS improvement were classified as normal-like or fibroproliferative intrinsic subsets. 321 genes (FDR <5%) were differentially expressed at baseline between patients with and without MRSS improvement during treatment. Expression of 571 genes (FDR <10%) changed between pre- and post-MMF treatment biopsies for patients demonstrating MRSS improvement. Gene expression changes in skin are only seen in patients with MRSS improvement. Baseline gene expression in skin, including intrinsic subset assignment, may identify SSc patients whose MRSS will improve during MMF treatment, suggesting that gene expression in skin may allow targeted treatment in SSc.
Introduction
Systemic sclerosis (SSc; scleroderma) is a phenotypically diverse disease whose pathological hallmark is fibrosis(Hinchcliff and Varga, 2011). Current classification systems, including autoantibody profiles, cannot reliably predict treatment response or disease course (Merkel et al., 2012). Heterogeneity confounds clinical trials and complicates attempts to elucidate pathogenesis (Merkel et al., 2012).
Genome-wide gene expression analysis of skin is an unbiased approach to quantify SSc heterogeneity(Sargent and Whitfield, 2011). This approach has classified SSc patients into four pathway-centric ‘intrinsic’ gene expression subsets termed fibroproliferative, inflammatory, limited and normal-like(Milano et al., 2008; Pendergrass et al., 2012). SSc intrinsic subsets have been mapped to scleroderma animal models(Greenblatt et al., 2012) and appear stable in patients longitudinally(Pendergrass et al., 2012). This study tests the hypotheses that identification of gene expression signatures in skin including intrinsic subset assignment may identify patients likely to improve during mycophenolate mofetil/MMF (Cellcept®, Roche) treatment, and that identification of changes in gene expression during treatment in improvers may elucidate important deregulated molecular pathways involved in SSc skin disease.
MMF inhibits purine synthesis, reduces lymphocyte proliferation, and attenuates fibrosis in vitro(Ransom, 1995; Roos et al., 2007). Studies demonstrate MRSS improvement in some MMF-treated patients (Derk et al., 2009; Herrick et al., 2010; Le et al., 2011; Vanthuyne et al., 2007). Moreover, a prospective study demonstrated that reduced expression of certain pro-fibrotic proteins in skin accompanied improvement in lung function in some MMF-treated patients (Mendoza et al., 2012) Unfortunately, no biomarkers to predict treatment response have been identified. The present study was conducted to determine whether analyses of gene expression in skin biopsies could identify useful biomarkers to predict response during MMF therapy.
Results
Subject selection and clinical characteristics
Thirty-two subjects (22 SSc patients and ten controls) were included (Table 1). SSc-specific therapies included MMF (n=11), methotrexate (n=2), cyclophosphamide (n=1), minocycline (n=1) (Supplementary Table 1). Of the eleven MMF-treated patients, seven patients met MMF clinical response study entry criteria. Two patients prescribed MMF (SSc04 and SSc07) were ineligible with baseline MRSS <11, and two patients (SScReg 1067 and 1156) were taking MMF at study entry. Of the seven MMF-naïve patients with baseline MRSS ≥11 who were prescribed MMF, four were classified as improvers, and three were classified as non-improvers (Supplementary Fig. 1).
Table 1.
Baseline clinical characteristics
| Control subjects (N = 10) | SSc patients
|
||||
|---|---|---|---|---|---|
| All SSc (N = 22) | Improver during MMF (N = 4) | Non- improver during MMF (N = 3) | Other SSc patients (N = 15) | ||
| Age, Median (range) y | 37 (30–63) | 48 (21–65) | 50 (40–54) | 51 (51–65) | 45 (21–60) |
| Sex, N (%) female | 7 (70%) | 21 (95%) | 4 (100%) | 3 (100%) | 14 (93%) |
| Race, N (%) Caucasian | 6 (60%) | 13 (59%) | 2 (50%) | 2 (67%) | 9 (60%) |
| SSc subtype, N (%) diffuse | NA | 20 (91%) | 4 (100%) | 3 (100%) | 13 (87%) |
| MRSS, Median (range) | NA | 14.5 (4–35) | 18.5 (13–32) | 14 (12–14) | 15 (4–35) |
| Raynaud disease duration, Median (range) mo. | NA | 17.5 (2–152) | 7.5 (4–13) | 14 (5–124) | 27 (2–152) |
| Disease duration from first non- Raynaud, Median (range) mo. | NA | 19 (3–309) | 14 (8–22) | 11 (5–122) | 26 (3–152) |
| ANA primary pattern N (%) patients | |||||
| Homogenous | NA | 7 (32) | 0 (0) | 1 (33) | 6 (40) |
| Nucleolar | NA | 6 (27) | 2 (50) | 2 (67) | 2 (13) |
| Speckled | NA | 9 (41) | 2 (50) | 0 (0) | 7 (47) |
| Centromere | NA | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| SSc-specific antibodies, n (%) | |||||
| Scl-70 | NA | 7 (32) | 0 (0) | 1 (33) | 6 (40) |
| RNA Pol III* | NA | 5/11 (45) | 1/1 (100) | 0/3 (0) | 4/7 (57) |
| Current Therapies, N (%) | |||||
| Methotrexate | NA | 3 (14) | 0 (0) | 1 (33) | 2 (13) |
| Minocycline | NA | 1 (5) | 1 (25) | 0 (0) | 0 (0) |
| Prior Therapies, N (%) | |||||
| Cyclophos-phamide | NA | 2 (9) | 0 (0) | 1 (33) | 1 (7) |
| Imatinib mesylate | NA | 1 (5) | 0 (0) | 0 (0) | 1 (7) |
| Methotrexate | NA | 1 (5) | 0 (0) | 0 (0) | 1 (7) |
| MMF | NA | 4 (18) | 0 (0) | 0 (0) | 4 (27) |
| MMF duration at determination of clinical response, median (range) mo. | 11 (5–17) | 8.5 (6–12) | 14 (5–17) | NA | |
SSc=Systemic sclerosis, MMF-mycophenolate mofetil, MRSS=modified Rodnan skin score, ANA=antinuclear antibodies, N/A=not applicable, Scl-70=anti-topoisomerase I, RNA pol III=RNA polymerase III,
RNA polymerase III results were available for 11/22 SSc subjects.
To validate MRSS response, H&E histology and cartilage oligomeric protein (COMP) immunofluorescence were assessed using pre- and post-treatment arm biopsies from improvers and non-improvers (Farina et al., 2010; Farina et al., 2006; Farina et al., 2009). Patients’ biopsies demonstrated increased fibrosis compared to a representative control. Improvers, and one non-improver whose arm MRSS decreased, showed reduced fibrosis (Fig. 1a). In contrast, two of three non-improvers demonstrated persistent fibrosis. COMP immunofluorescence was significantly reduced in improvers compared to non-improvers (Fig. 1b) (p=0.0016 and p=0.35 respectively, two-sample t-test comparing difference between pre- and post-treatment intensity). These data support the validity of MRSS as an outcome marker.
Figure 1. Changing pathological factors in the skin during treatment.

Hematoxylin and eosin stained skin biopsies. a) Arm biopsy from a healthy control subject (scale bar=20μm), and biopsy pairs (pre- and post-treatment as indicated) from three non-improvers (upper panel) and four improvers (lower panel) during MMF, representative photomicrographs. Total and arm modified Rodnan skin score (MRSS) as well as fibrosis score are listed below. b) COMP immunofluorescence for pre- and post-treatment biopsies for a healthy volunteer (N1000) and four improvers (SSc3, 5, 6 and 10) and three non-improvers (SSc8, 12, 16) with quantification below. Scale bar=50μm.
Table 1 presents clinical characteristics. 95/70% of patients/controls were women respectively. Median SSc disease duration at biopsy was 19/17.5 mo since the first Raynaud/non-Raynaud symptom. 91% of patients had dcSSc, and 100% had positive ANAs. Fourteen (64%) had speckled, 12 (55%) had nucleolar, and 7 (32%) patients had homogenous ANA patterns. Anticentromere antibodies were absent, but seven (32%) patients had positive Scl-70, and 5/11 (45%) had anti-RNA polymerase III autoantibodies. There were no statistically significant differences in age, sex, and ethnicity between patients and controls. There were no statistically significant differences in ANA pattern, SSc-specific serum autoantibodies, prior treatments, baseline MRSS, and disease duration (irrespective of the definition) between patients that were, or were not, prescribed MMF.
No MMF-treated patient had evidence of significant cardiac disease (Supplementary Table 3). 8/9 MMF-treated patients underwent HRCT for suspected interstitial lung disease (ILD). Six had mild-moderate ILD (<50% lung involvement), and two had moderate-severe ILD (≥50% lung involvement; Supplementary Table 3).
To identify factors that may be associated with clinical response regardless of treatment, data from 14 subjects with a baseline MRSS ≥11 and ≥1 follow-up MRSS were examined. Seven patients demonstrated MRSS improvement ≥5 (11mo mean follow-up). There were no statistically significant differences in ANA pattern, SSc-specific autoantibodies, and baseline MRSS or lung parameters between clinical improvers and non-improvers (Supplementary Table 4). These data suggest that the two patient groups (improvers and non-improvers during MMF, and clinical improvers and non-improvers independent of treatment) were similar.
Recapitulation of the SSc intrinsic subsets
To assign patients to the ‘intrinsic’ gene expression subsets defined previously (Milano et al., 2008; Pendergrass et al., 2012), skin biopsies from the cohort were analyzed (Fig. 2). To identify intrinsic genes, we performed intrinsic gene analysis and identified genes with consistent expression among forearm-back pairs from an individual, but with high variation across the cohort. 2775 genes were identified (false discovery rate/FDR 3%) and used for intrinsic subset classification (Fig. 2).
Figure 2. Improvers cluster within the inflammatory intrinsic subset.

We selected 2775 intrinsic genes with a False Discovery Rate of 3%. Genes and microarray samples were clustered hierarchically. The sample dendrogram (a) shows the statistically significant intrinsic groups. Branch points above each * are significant at p ≤ 0.005. The dendrogram branches are colored to reflect the major intrinsic subsets of normal-like (green), inflammatory (purple) and diffuse-proliferation (red). Patient identifiers indicate systemic sclerosis samples (SSc) and normal healthy controls (Norm); those in the MMF study are colored to reflect improvers (blue) and non-improvers (orange). b) Overview of the gene expression profiles. c and d) Inflammatory clusters, e) mitotic fibroproliferative cluster, f) DNA replication proliferation cluster.
We grouped genes and arrays by average linkage hierarchical clustering and identified significant clusters using SigClust with Bonferroni correction for multiple testing(Liu et al., 2008). Four SSc intrinsic subsets were delineated. Several branch points have low corrected p-values (p < 0.005, Fig. 2a) indicating significant differences in gene expression.
These data recapitulate the intrinsic subsets reported previously (Milano et al., 2008; Pendergrass et al., 2012). These groups are normal-like (Fig. 2a, green branches), inflammatory (purple) and fibroproliferative (red). We find consistent intrinsic subset assignment regardless of time point analyzed and treatment (Fig. 2a)(Pendergrass et al., 2012). An overview of expression levels of the 2775 intrinsic genes is shown with specific groups of genes indicated (Fig. 2b). Groups of genes are found that correspond to the normal-like (NL), inflammatory (Fig. 2c and d) and the fibroproliferative subsets (Fig. 2e and f). This provides a third, independent validation of the SSc intrinsic subsets that includes longitudinally collected skin biopsies.
Select genes are shown in Fig. 2c–f. These include CCL2, TNC, CTGF, PAI1 and Granzyme B in the inflammatory groups (Fig. 2c and d). CCL2 stimulates chemotaxis of monocytes and basophils and was identified as a common target deregulated in the inflammatory intrinsic subset, the scleroderma graft versus host disease (sclGVHD) mouse model, and the fibroblasts IL-13 responsive gene signatures(Greenblatt et al., 2012). Two proliferation groups are evident. One proliferation group includes genes involved in mitosis (Fig. 2e) and the second includes genes associated with the process of DNA replication that show peak expression in G1/S phase (Fig. 2f)(Whitfield et al.).
Improvers during MMF therapy map to the inflammatory SSc intrinsic subset
Next, the intrinsic subset of the seven MMF-naive patients who met inclusion criteria was determined (four improvers: SSc03, 05, 06, 10, and three non-improvers: SSc08, 12, 16) (Table 1). It was hypothesized that improvers would map to the inflammatory intrinsic subset because MMF decreases lymphocyte proliferation, and non-improvers would map to one of the other subsets. Median disease duration for improvers during MMF treatment was 7.5 and 14mo defined as the time from the first Raynaud or non-Raynaud symptom to the baseline biopsy date respectively. All improvers demonstrated ANAs (two had isolated speckled, and two demonstrated nucleolar/speckled patterns) and had dcSSc. SSc specific autoantibodies were observed in one treatment improver and one non-improver. 2/3 MMF non-improvers had dcSSc (Table 1).
Patients with MRSS improvement during MMF (Fig. 2a; blue identifiers) were classified in the inflammatory intrinsic subset (p=0.029, Fisher’s exact test). Patients without clinical improvement during MMF were classified in the normal-like (SSc16) or fibroproliferative (SSc08 and SSc12) intrinsic subsets. One treatment naïve patient in the inflammatory subset worsened; one previously receiving MMF for one month showed stable MRSS. These data indicate that a subset of SSc patients who demonstrate an inflammatory gene expression signature improve during MMF while patients with other intrinsic subset signatures are not likely to improve.
Biomarkers of clinical response during MMF therapy
To identify gene expression signatures that may predict MMF response, we examined gene expression at baseline in arm and back biopsies between patients with or without clinical improvement during MMF. There were 393 probes (321 genes) whose expression differed at baseline between improvers and non-improvers (FDR<5%). 113 probes (90 genes) were increased, and 280 probes (231 genes) were decreased in improvers relative to non-improvers during MMF at baseline (Fig. 3a and b).
Figure 3. Comparison of baseline gene expression between improvers and non-improvers.

Baseline gene expression in arm and back samples between improvers (imp) and non-improvers (non-imp) was compared. a) Blue identifiers indicate improvers and gold indicates non-improvers. b) There were 321 genes identified (FDR<5%) with significant differential expression between improvers and non-improvers during MMF.
Analysis of enriched functional annotations in the 90 genes with high expression in improvers showed baseline differences in genes involved in purine metabolism and response to inflammation (PRPS1, NFKB2, CXCL1, FKBP1C). Improvers had higher expression levels of PRPS1 necessary for purine nucleotide biosynthesis, as well as higher levels of NFKB2 family of transcription factors that regulate immunity, stress responses, apoptosis, and differentiation. CXCL1 encodes a secreted growth factor that plays a role in inflammation and as a chemoattractant for neutrophils; FKBP1C is similar to FKBP1A that maintains the inactive conformation of transforming growth factor beta-receptor 1 and blocks the activin signal. Genes with high expression in improvers showed enrichment for genes typically expressed in lymphocytes (p=0.004), monocytes (p=0.035) and cartilage (p=0.028; all Benjamini corrected). Genes with decreased expression in improvers showed enrichment for genes associated with Ras signaling (p=0.018) and regulation of cell communication (p=0.036; both Benjamini corrected). The full list with nominal and corrected p-values are given in Supplementary Tables 5 and 6. Enrichment of NFKB signaling, lymphocyte and chemokine chemoattractants is consistent with assignment of improvers to the inflammatory intrinsic subset, while Ras signaling, which is decreased in improvers, is generally enriched in the fibroproliferative subset (Milano et al., 2008; Pendergrass et al., 2012) (Whitfield, unpublished).
Gene expression changes during MMF in improvers
To identify genes whose expression changed during MMF treatment, we analyzed the gene expression in skin biopsies from MMF-treated patients who met inclusion and response criteria. There were 610 probes (571 genes; FDR <10%) whose expression significantly changed during MMF treatment in improvers exclusively (Fig. 4). Genes with the highest fold change between baseline and post-treatment included PBEF1, CXCL1, HAT1, IL17D, SFRP2, PDGFRL, IL16, COL13A1, THBS2, IGFBP5, WNT3, DKK1/2, and WIF1.
Figure 4. Gene expression changes during MMF treatment between improvers and non-improvers.

571 genes showed changes in expression during MMF treatment (FDR<10%). Patients that were classified as non-improvers show low levels expression of these genes, which either do not change expression or show increased expression.
Genes whose expression increased during MMF treatment in the improvers were enriched in extracellular matrix component (p=0.004, Benjamini corrected). Genes whose expression decreased during MMF treatment in improvers were involved in cell cycle and cell division (e.g., organelle fission, p=6.55E−04, mitotic cell cycle, p=7.05E−04) as well as in NOD-like receptor signaling pathway responsible for NFKB activation, cytokine production and apoptosis (p=0.011). Complete lists with nominal and corrected p-values are provided (Supplemental Tables 7 and 8). There were no significant changes in gene expression between baseline and post-treatment in the non-improvers during MMF when corrected for multiple testing.
Quantitative RT-PCR validation
Expression of genes from the inflammatory intrinsic subset or that changed during MMF treatment was validated. RNA was examined in duplicate by quantitative reverse transcriptase - polymerase chain reactions (qRT-PCR). Figure 5 shows connective tissue growth factor ((CTGF) and interleukin-6 (IL-6); inflammatory intrinsic subset), and thrombospondin-1 (TSP-1; change during MMF treatment) expression values. Mirroring microarray data, improvers compared to non-improvers during MMF demonstrated higher baseline CTGF, IL-6 and TSP-1 expression. CTGF decreased during treatment in improvers and increased or remained stable in non-improvers though the changes were significant in one improver (p=0.006). Pre-treatment IL-6 and TSP-1 levels were higher in improvers than non-improvers (p=0.003 and p=0.10 respectively). IL-6 and TSP-1 expression decreased during treatment in improvers and either remained stable or increased in non-improvers (p=0.25 and p=0.14 respectively).
Figure 5. Validation of biologically relevant microarray findings using quantitative reverse transcription polymerase chain reactions immunofluorescence.
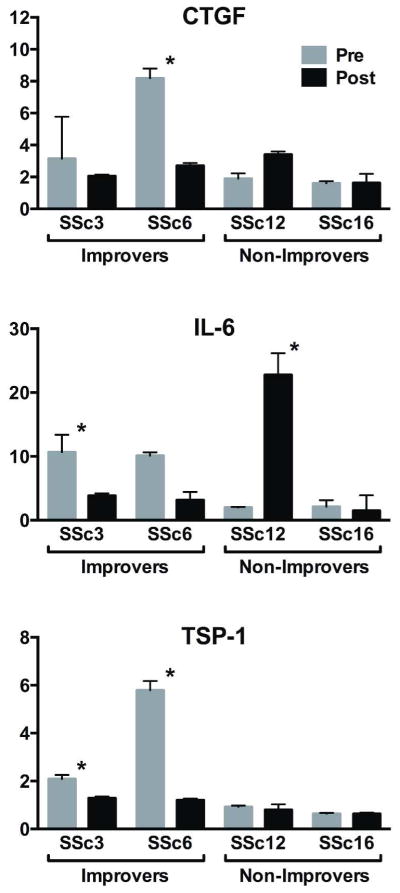
Results are the relative expression values normalized to the mean expression in arm samples of control subjects, *p<0.05.
Discussion
SSc clinical heterogeneity complicates treatment response prediction. Unbiased genome-wide analyses of gene expression in skin biopsies of SSc patients reproducibly separate patients into biologically relevant intrinsic subsets(Milano et al., 2008; Pendergrass et al., 2012), each driven by fundamentally different pathways(Greenblatt et al., 2012; Sargent et al., 2010). These pathway-centric gene expression subsets likely explain SSc clinical heterogeneity. Microarray analyses of skin biopsies from our cohort reproduce the four SSc intrinsic subsets(Milano et al., 2008; Pendergrass et al., 2012). The reproducibility of the SSc intrinsic subsets in the present, as well as two previously recruited cohorts, suggests that intrinsic subset classification will be a useful SSc classification method.
We found that biopsies from improvers during MMF therapy mapped to the inflammatory intrinsic subset while non-improvers were classified as fibroproliferative and normal-like subsets. Additionally, a specific 321-gene baseline expression signature was identified in skin that was associated with MRSS improvement during MMF treatment was absent in non-improvers. Measuring the 321-gene baseline signature and/or intrinsic subset classification may be useful for selection of appropriate patients for MMF therapy to treat SSc skin disease.
There were 571 genes whose expression changed significantly during MMF treatment in improvers but not in non-improvers. Interestingly, many of the genes that are implicated in fibrosis such as COL1A1, COL1A2, TIMP2 and ACTA2 demonstrated statistically significant increases in expression during MMF treatment in improvers. This was an unexpected finding that suggests that dermal repair and tissue remodeling cause transient increased expression of genes classically considered “pro-fibrotic”.
Improvers during MMF had longer disease duration at study entry compared to non-improvers, thus shorter disease duration does not explain response heterogeneity. Baseline clinical characteristics were similar between the seven MMF-treated patients and the entire SSc cohort, and between the clinical improvers and non-improvers independent of treatment (Table 1 and Supplementary Tables 3 and 4). Importantly, clinical response was not associated with autoantibody status. These data suggest that skin gene expression provides additional information that may have clinical relevance.
Results demonstrate that only improvers demonstrate significant changes in gene expression in longitudinally collected skin biopsies. Similar findings were noted in imatinib treated patients as well(Chung et al., 2009). Conversely, in a recent rituximab trial, lack of clinical response coincided with lack of gene expression changes(Lafyatis et al., 2009; Pendergrass et al., 2012). Importantly, gene expression changes can precede MRSS improvement ((e.g. SSc10 demonstrated gene expression response at 6mo (data not shown), and MRSS response at 12mo (i.e. baseline and 6mo MRSS=13, 12mo MRSS=7)).
Study strengths include prospective study design, clinically well–characterized study population, performance of skin scores and biopsies by one investigator. Study limitations include lack of validated definition of active skin disease, randomization and washout procedures, open-label trial design, and small sample size.
The results herein demonstrate that intrinsic subset assignment is a clinically relevant SSc classification method. We provide proof-of-concept that quantitative measurement of genome-wide gene expression in skin using DNA microarray may be useful to identify appropriate patients to receive MMF, and to elucidate genes that are involved in the pathogenesis of SSc skin disease and its resolution during MMF treatment.
Patients and Methods
Inclusion criteria for intrinsic subset analysis
Patients fulfilling American College of Rheumatology SSc criteria (1980) or three out of five criteria for CREST (calcinosis, Raynauds, esophageal dysmotility, sclerodactyly, telangiectasias) were eligible. 22/31 subjects who underwent skin biopsies between November 2008 and September 2010 were included. Nine patients were prescribed MMF (2000 mg/day), and one received oral cyclophosphamide (2mg/kg/day) in divided doses for active SSc skin disease in the treating physician’s opinion. Additionally, two patients were taking MMF (2000 mg/day) at baseline biopsy time. Biopsy pairs (4mm) from the clinically involved (dorsal forearm, 15cm proximal to the ulnar styloid) and clinically uninvolved (back, posterior iliac crest midway between lumbar spinous process and anterior superior iliac spine) skin from the non-dominant side of the body were obtained at baseline. Serial biopsies 3mm proximal (arm) or inferior (back) to previous biopsies were performed at 6 and 12mo for MMF-treated patients. Arm and back biopsies from ten biologically unrelated control subjects recruited from Northwestern University were obtained. One biopsy pair (forearm and back) was placed in RNAlater (Applied Biosystems, Ambion®, Carlsbad, California) and used for DNA microarray analysis; the other biopsy pair was placed in formalin for histology. A single forearm biopsy was obtained for DNA microarray analyses from twelve SSc patients with stable skin disease to have power to detect intrinsic subsets (Supplementary Table 1).
Subjects gave written informed consent in accordance with the Declaration of Helsinki Protocols and Northwestern University Institutional Review Board Guidelines. Control subjects completed demographic and prior medical history questionnaires. Medical histories and physical exams were completed at study visits. One physician blinded to gene expression and clinical data performed MRSS(LeRoy et al., 1988). Serum ANA, anti-topoisomerase I, anticentromere, and anti-RNA polymerase III antibody titers were measured by indirect immunofluorescence at Specialty Laboratories, Valencia, CA.
Patients underwent cardiopulmonary disease screening with Doppler echocardiography, pulmonary function tests and high-resolution computed tomography of the thorax (HRCT) within 3mo of the baseline visit. An echocardiographer, blinded to clinical data, performed quantitative measurements on echocardiograms using a pre-established research protocol. One chest radiologist, also blinded to clinical data, scored HRCT exams(Kazerooni et al., 1997; Strollo and Goldin, 2010). Five lung lobes were scored (0=no, 1=1–5%, 2=6–25%, 3=26–50%, 4=51–75%, and 5=76–100% involvement) for total lung disease degree.
Inclusion and response criteria for response during MMF study
Patients with baseline MRSS ≥11, newly prescribed MMF for active skin disease, willingness to undergo serial skin biopsies, and referral to MH for study participation, were included. Patients were classified as improvers if the MRSS improved ≥5 from baseline (the minimal clinically important difference)(Khanna et al., 2006). A baseline skin score ≥11 was required for inclusion because sclerodactyly contributes 1–6 MRSS points and enrolling patients with MRSS <11 would confound detection of meaningful change.
Skin pathology
Pre- and post-treatment arm biopsies were paraffin-embedded, and 4μm sections were H&E stained. Photomicrographs were taken using an Olympus BX41 microscope and an Olympus DP71 camera at 4X magnification. Two dermatopathologists blinded to clinical data scored dermal fibrosis (0=no fibrosis to 3=severe fibrosis)(Verrecchia et al., 2007).
COMP levels were assessed. Four μm sections were incubated with primary antibodies against COMP (Accurate Chemical & Scientific, Westbury, NY, 1:20 dilution) followed by mouse Alexa-fluor secondary antibodies (Invitrogen, 1:100). Nuclei were identified using DAPI. Immunofluorescence was evaluated in randomly selected fields under a Zeiss UV Meta 510 confocal microscope (Carl Zeiss Inc., Jena, Germany) and staining intensity was quantified with Image J (NIH).
DNA microarray hybridization
Tissue homogenization was performed using Qiagen TissueLyser II. RNA purification was carried out in QIAcube with Qiagen’s RNeasy Fibrous Tissue Mini Kit. The Agilent 2100 Bioanalyzer assessed RNA integrity. Samples had RNA integrity numbers (RIN) > 7. RNA concentration was measured with Thermo Scientific NanoDrop 2000 Spectrophotometer. 200ng total RNA was amplified and labeled with Agilent Quick-Amp Labeling Kits(Milano et al., 2008). Cy3-labeled sample and Cy5-labeled Universal Human Reference RNA (Stratagene), were co-hybridized to Agilent Human Genome (4×44K) Microarrays (G4112F). Data were Log2 Lowess normalized and filtered for probes with intensity ≥2-fold over local background in Cy3 or Cy5 channels. Data were multiplied by −1 to convert to Log2(Cy3/Cy5) ratios. Probes with >20% missing data were excluded.
Intrinsic subset assignment
Intrinsic subsets were determined as previously described(Milano et al., 2008). Genes were rank ordered by “intrinsic score” using a modified F-statistic(Pendergrass et al., 2012). FDR for each intrinsic score was assessed by permuting rows and columns and counting the genes that received ≥ same score in each of 100 data randomizations. At FDR of 3%, 2775 genes were identified and used to assign intrinsic subset.
Data were organized by two-dimensional average linkage hierarchical clustering using Pearson correlation. SigClust was used to determine statistical significance of array clustering(Liu et al., 2008). Bonferroni correction for multiple testing was applied to p-values at branch points using branch point number tested as correction factor(Liu et al., 2008).
Quantitative RT-PCR
RNA was reverse-transcribed to cDNA(Bhattacharyya et al., 2011). Amplicons were analyzed by PCR using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on the Applied Biosystems 7500 Prism Sequence Detection System (Supplementary Table 2). Results are fold-change relative to the mean expression for control arm samples.
Baseline gene expression signature associated with clinical response
Genes differentially expressed at baseline between clinical improvers and non-improvers were identified. Lowess normalized Log2(Cy3/Cy5) gene expression measures for arm and back samples were centered on median expression value across samples. Gene expression differences were detected using two-sample t-tests. Probability of false positives was assessed using positive FDR (pFDR) method (Storey and Tibshirani, 2003) to calculate q-values for each test statistic using Bioconductor package: QVALUE. Genes with FDR<5% were investigated. Functional enrichment was performed with g:GOSt within g:Profiler (Reimand et al., 2011), and DAVID(Dennis et al., 2003). Agilent probe IDs were converted to Ensembl gene IDs via g:Convert. g:GOSt analyses were performed with default options limiting output to significant results (p-value < 0.05 after multiple testing correction). For DAVID, the following annotations were analyzed: Gene Ontology, KEGG and REACTOME pathways, and CGAP SAGE tissue expression. Terms with Benjamini-corrected p-value < 0.05 were evaluated.
Clinical response gene expression signature
A clinical response signature was identified by comparing gene expression in arm and back samples between baseline and post-treatment. Last biopsy obtained was analyzed for non-improvers. Biopsy at time of MRSS improvement was used for improvers. Data were centered, significant changes in gene expression were identified (FDR <10%), and functional enrichment analyses were conducted as previously described.
Statistical analyses
Continuous variables were expressed by median and range. Statistically significant differences were assessed by t-tests or Wilcoxon Rank Sum test. Categorical variables were compared by chi-squared statistic or Fisher’s exact test. For all analyses, a two-sided p-value <0.05 was considered significant. Stata version 10.1 (College Station, TX) was used.
Supplementary Material
Acknowledgments
Funding: This work was supported in part by the NIH K12 HD055884 from the NIH Eunice Kennedy Shriver National Institute of Child Health & Human Development (MH), by a research award from the Arthritis Foundation and the Scleroderma Foundation (MH), by NIH 610-532-800-6001417 (CCH), and by the Scleroderma Research Foundation (MH, MLW), Actelion Entelligence Grant Award (SJS), by NIH U01 AR055063 (MLW and RL) NIH-NCI R25CA134286 (JMM), NIH-NIAMS P60 AR48098 (CCH, JL, RWC) and NIH P50AR060780 (MLW, RL)
We thank Drs. Pedram Gerami, MD and Joan Guitart, MD, Department of Dermatology, Feinberg School of Medicine, for evaluating dermatopathology.
Abbreviations Used
- ANA
antinuclear antibody
- COMP
cartilage oligomeric matrix protein
- CTGF
connective tissue growth factor
- dcSSc
diffuse cutaneous systemic sclerosis
- FDR
false discovery rate
- HRCT
high-resolution computed tomography of the thorax
- H&E
hematoxylin and eosin
- IL-6
interleukin-6
- ILD
interstitial lung disease
- MMF
mycophenolate mofetil
- MRSS
modified Rodnan skin score
- pFDR
positive false discovery rate
- SSc
systemic sclerosis
- TSP-1
thrombospondin-1
- qRT-PCR
quantitative reverse transcriptase-polymerase chain reactions
Footnotes
Conflict of Interest: MLW, MH, RL and CCH have filed patent applications for gene expression biomarkers in scleroderma. MLW is a scientific founder and holds an interest in Celdara Medical, LLC.
References
- 1.Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23:581–90. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharyya S, Sargent JL, Du P, Lin S, Tourtellotte WG, Takehara K, et al. Egr-1 induces a profibrotic injury/repair gene program associated with systemic sclerosis. PloS one. 2011;6:e23082. doi: 10.1371/journal.pone.0023082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung L, Fiorentino DF, Benbarak MJ, Adler AS, Mariano MM, Paniagua RT, et al. Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheum. 2009;60:584–91. doi: 10.1002/art.24221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome biology. 2003;4:P3. [PubMed] [Google Scholar]
- 5.Derk CT, Grace E, Shenin M, Naik M, Schulz S, Xiong W. A prospective open-label study of mycophenolate mofetil for the treatment of diffuse systemic sclerosis. Rheumatology (Oxford) 2009;48:1595–9. doi: 10.1093/rheumatology/kep295. [DOI] [PubMed] [Google Scholar]
- 6.Farina G, Lafyatis D, Lemaire R, Lafyatis R. A four-gene biomarker predicts skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2010;62:580–8. doi: 10.1002/art.27220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farina G, Lemaire R, Korn JH, Widom RL. Cartilage oligomeric matrix protein is overexpressed by scleroderma dermal fibroblasts. Matrix Biol. 2006;25:213–22. doi: 10.1016/j.matbio.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Farina G, Lemaire R, Pancari P, Bayle J, Widom RL, Lafyatis R. Cartilage oligomeric matrix protein expression in systemic sclerosis reveals heterogeneity of dermal fibroblast responses to transforming growth factor beta. Ann Rheum Dis. 2009;68:435–41. doi: 10.1136/ard.2007.086850. [DOI] [PubMed] [Google Scholar]
- 9.Greenblatt MB, Sargent JL, Farina G, Tsang K, Lafyatis R, Glimcher LH, et al. Interspecies comparison of human and murine scleroderma reveals IL-13 and CCL2 as disease subset-specific targets. Am J Pathol. 2012;180:1080–94. doi: 10.1016/j.ajpath.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrick AL, Lunt M, Whidby N, Ennis H, Silman A, McHugh N, et al. Observational study of treatment outcome in early diffuse cutaneous systemic sclerosis. J Rheumatol. 2010;37:116–24. doi: 10.3899/jrheum.090668. [DOI] [PubMed] [Google Scholar]
- 11.Hinchcliff M, Varga J. Managing Systemic Sclerosis and its Complications. The Journal of Musculoskeletal Medicine. 2011;28:380. [Google Scholar]
- 12.Kazerooni EA, Martinez FJ, Flint A, Jamadar DA, Gross BH, Spizarny DL, et al. Thin-section CT obtained at 10-mm increments versus limited three-level thin-section CT for idiopathic pulmonary fibrosis: correlation with pathologic scoring. AJR Am J Roentgenol. 1997;169:977–83. doi: 10.2214/ajr.169.4.9308447. [DOI] [PubMed] [Google Scholar]
- 13.Khanna D, Furst DE, Hays RD, Park GS, Wong WK, Seibold JR, et al. Minimally important difference in diffuse systemic sclerosis: results from the D-penicillamine study. Ann Rheum Dis. 2006;65:1325–9. doi: 10.1136/ard.2005.050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lafyatis R, Kissin E, York M, Farina G, Viger K, Fritzler MJ, et al. B cell depletion with rituximab in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2009;60:578–83. doi: 10.1002/art.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le EN, Wigley FM, Shah AA, Boin F, Hummers LK. Long-term experience of mycophenolate mofetil for treatment of diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2011 doi: 10.1136/ard.2010.142000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- 17.Liu Y, Hayes D, Nobel A, Marron JS. Statistical significance of clustering for high-dimension, low-sample size data. Journal of the American Statistical Association. 2008;103:1281–93. [Google Scholar]
- 18.Mendoza FA, Nagle SJ, Lee JB, Jimenez SA. A prospective observational study of mycophenolate mofetil treatment in progressive diffuse cutaneous systemic sclerosis of recent onset. J Rheumatol. 2012;39:1241–7. doi: 10.3899/jrheum.111229. [DOI] [PubMed] [Google Scholar]
- 19.Merkel PA, Silliman NP, Clements PJ, Denton CP, Furst DE, Mayes MD, et al. Patterns and predictors of change in outcome measures in clinical trials in scleroderma: an individual patient meta-analysis of 629 subjects with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2012;64:3420–9. doi: 10.1002/art.34427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3:e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pendergrass SA, Lemaire R, Francis IP, Mahoney JM, Lafyatis R, Whitfield ML. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol. 2012;132:1363–73. doi: 10.1038/jid.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ransom JT. Mechanism of action of mycophenolate mofetil. Ther Drug Monit. 1995;17:681–4. doi: 10.1097/00007691-199512000-00023. [DOI] [PubMed] [Google Scholar]
- 23.Reimand J, Arak T, Vilo J. g:Profiler--a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–15. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roos N, Poulalhon N, Farge D, Madelaine I, Mauviel A, Verrecchia F. In vitro evidence for a direct antifibrotic role of the immunosuppressive drug mycophenolate mofetil. J Pharmacol Exp Ther. 2007;321:583–9. doi: 10.1124/jpet.106.117051. [DOI] [PubMed] [Google Scholar]
- 25.Sargent JL, Milano A, Bhattacharyya S, Varga J, Connolly MK, Chang HY, et al. A TGFbeta-responsive gene signature is associated with a subset of diffuse scleroderma with increased disease severity. Journal of Investigative Dermatology. 2010;130:694–705. doi: 10.1038/jid.2009.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sargent JL, Whitfield ML. Capturing the heterogeneity in systemic sclerosis with genome-wide expression profiling. Expert review of clinical immunology. 2011;7:463–73. doi: 10.1586/eci.11.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strollo D, Goldin J. Imaging lung disease in systemic sclerosis. Current rheumatology reports. 2010;12:156–61. doi: 10.1007/s11926-010-0095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanthuyne M, Blockmans D, Westhovens R, Roufosse F, Cogan E, Coche E, et al. A pilot study of mycophenolate mofetil combined to intravenous methylprednisolone pulses and oral low-dose glucocorticoids in severe early systemic sclerosis. Clin Exp Rheumatol. 2007;25:287–92. [PubMed] [Google Scholar]
- 30.Verrecchia F, Laboureau J, Verola O, Roos N, Porcher R, Bruneval P, et al. Skin involvement in scleroderma--where histological and clinical scores meet. Rheumatology (Oxford) 2007;46:833–41. doi: 10.1093/rheumatology/kel451. [DOI] [PubMed] [Google Scholar]
- 31.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. MolBiolCell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.