

SUMMARY
Emerging evidence suggests that dysregulation stress hormones, such as glucocorticoids, in aged persons put them at a higher risk to develop Alzheimer’s disease (AD). However, the mechanisms underlying such vulnerability remain to be unraveled. Pharmacologic inhibition of 5-lipoxygenase (5LO), an active player in AD pathogenesis whose protein level increases with aging in the human, has been shown to blunt glucocorticoid-mediated amyloid β (Ab) formation in vitro. In the current paper we investigated the role of this pathway in modulating the development of the corticosteroid-dependent AD-like phenotype in the triple transgenic mice (3xTg). Dexamethasone was administered for one week to 3xTg or 3xTg genetically deficient for 5LO (3xTg/5LO−/−) mice and its effect on memory, amyloid-β and tau levels, and metabolism assessed. At the end of the treatment, we observed that dexamethasone did not induce changes in behavior. Compared with controls, treated mice did not show significant alterations in brain soluble Aβ levels. While total tau protein levels were unmodified in all groups, we found that dexamethasone significantly increased tau phosphorylation at S396, as recognized by the antibody PHF-13, which was specifically associated with an increase in the GSK3β activity. Additionally, dexamethasone-treated mice had a significant increase of the tau insoluble fraction, and reduction of the post-synaptic protein PDS-95. By contrast, these modifications were blunted in the 3xTg/5LO−/− mice. Our findings highlight the functional role that 5LO plays in stress-induced AD tau pathology and support the hypothesis that pharmacologic inhibition of this enzyme could be a useful tool for individuals with this risk factor.
Keywords: Alzheimer’s disease, transgenic mouse models, glucocorticoid, 5-lipoxygenase, amyloid, tau protein
INTRODUCTION
While Alzheimer’s disease (AD) is a disease of late life, non-heritable environmental factors are thought to accelerate accumulation of AD pathology, Aβ plaques and tau tangles, well before cognitive decline is detected (Villemagne et al., 2011). Recent evidence has suggested that high circulating levels of glucocorticoids could be one such environmental factor. AD patients display higher levels of circulating glucocorticoids than age-matched controls and higher levels of glucocorticoids confer greater risk of developing dementia in cognitively intact aged persons (Wilson et al., 2006; Csernansky et al., 2006, Huang et al. 2009). These observations have been recapitulated in animal models of the disease: stress worsens the AD phenotype, and glucocorticoid antagonists such as mifepristone reduce Aβ and tau pathology (Lee, et al. 2009; Baglietto-Vargas, et al. 2013).
We have previously shown that pharmacologic inhibition of the enzyme 5-lipoxygenase (5LO), a central nervous system protein whose expression increases with age, blunts dexamethasone-mediated amyloid β (Aβ) formation in vitro (Puccio et al., 2011). Additionally, we have shown that knockout of 5LO reduces AD-like amyloidosis and improves memory in an APP transgenic mouse model of AD (Chu and Pratico, 2011). Therefore, while glucocorticoids accelerate AD neuropathology and inhibition or knockout of 5LO reduces it, it is still an unanswered question whether 5LO contributes to the in vivo glucocorticoid-mediated exacerbation of the full spectrum AD-like phenotype, which includes amyloid and tau pathology as well as behavioral deficits. In the current work, we investigated how knockout of 5LO modulated the effect of dexamethasone treatment in the triple transgenic (3xTg) mouse model of AD, a system in which both the signature pathologies of AD, Aβ plaques and tau tangles, are expressed. At the end of the treatment, we found that dexamethasone treatment that did not result in obvious memory deficit was nonetheless able to induce hyperphosphorylation of tau, increase in its insoluble fraction as well as disrupt synaptic integrity in 3xTg animals. However, the 3xTg mice that lacked 5LO resisted both tau pathology and synaptic deficits. These results suggest that targeting 5LO could be attractive for AD prophylaxis in individuals with elevated stress hormones.
RESULTS
Cognitive deficits are not evident in 3xTg animals treated with a 7d course of dexamethasone
We administered dexamethasone intraperitoneally (5mg/kg) or saline (PBS) daily to 3xTg and 3xTg/5LO−/− mice aged 3.5 months for 7 days. At the end of this treatment, we observed that neither dexamethasone nor knockout of 5LO produced differences in Y-maze behavior in total number of entries or alternation percentage (Fig 1C, D). We also found no differences in any of the animal groups with regard to behavior in the fear conditioning paradigm, with all groups displaying similar contextual as well as cued recall freezing responses (Fig 1A, B).
Figure 1.

Cognitive deficits are not evident in 3xTg or 3xTg/5LO−/− animals treated with a 7d course of dexamethasone. A) Contextual recall freezing responses in the fear conditioning behavioral paradigm of 3xTg mice treated with PBS/saline (n=5), 3xTg mice treated with dexamethasone 5mg/kg for 7d (n=7), 3xTg/5LO−/− mice (n=4) treated with PBS/saline and 3xTg/5LO−/− mice (n=5) treated with dexamethasone. B) Cued recall in the same animals. C) Spontaneous alternating behavior and D) total entries in the Y-maze.
5LO knockout prevents dexamethasone-induced PHF13 tau phosphorylation
Because we and others have reported that 30 day administration of a similar dose of dexamethasone increases soluble Aβ species in the Tg2576 mouse model of AD-like amyloidosis, we next assayed the brains of our animals for soluble Aβ40 and Aβ42. We found that 7d treatment of dexamethasone was not sufficient to significantly alter Aβ 1–40 and 1–42 levels (data not shown). We next investigated the effect of dexamethasone on tau levels and metabolism. As shown in Fig. 2A, B, we found that dexamethasone significantly elevated levels of phosphorylated tau at S396, as recognized by the PHF13 antibody, but not at S396/S404, as recognized by the PHF1 antibody. By contrast, the observed changes in PHF13 levels were absent in the 3xTg mice genetically deficient for 5LO. Brain immunohistochemistry analyses further supported our biochemical results, as we found increased immunostaining for PHF13 but not for PHF-1 in the 3xTg/5LO but not in the 3xTg/5LO−/− mice (Fig. 2D). Neither dexamethasone treatment in both groups nor the 5LO knockout altered levels of RIPA-soluble total tau, as recognized by the HT7 antibody. By contrast, dexamethasone increased insoluble total tau in 3xTg mice as shown in Fig 2C, but neither control nor treated 3xTg/5LO−/− animals showed significant levels of insoluble tau.
Figure 2.

5LO knockout prevents dexamethasone-induced PHF13 tau phosphorylation. A) Representative western blots for total tau, HT7, PHF13 and PHF1 tau in cortical brain homogenates of 3xTg and 3xTg/5LO−/− animals treated with PBS or dexamethasone. B) Densitometric analysis of PHF13 tau immunoreactivity. C) Representative western blot for insoluble brain HT7. D) Representative immunohistochemical staining for PHF13 and PHF1 tau. *p<0.05
Dexamethasone-mediated increase in the activity of GSK3β is absent in animals that lack 5LO
To investigate the mechanism whereby 5LO modulates dexamethasone-mediated tau phosphorylation, we assayed for two tau kinases that have been shown to be altered in the brains of AD patients: glycogen synthase kinase 3 (GSK3) and cyclin-dependent kinase 5 (cdk5). We found no changes in the steady state levels of either total GSK3α and GSK3β, or phosphorylated ser21 GSK3α and ser9 GSK3β protein subunits as shown in Fig. 3A, B. Likewise, we found no changes in cdk5 nor its coactivators p35 or p25. However, we observed that the ex vivo activity of GSK3β was significantly elevated in the 3xTg mice after dexamethasone treatment which was not present in the brains of 3xTg/5LO−/− animals (Fig 3C). By contrast, we found that neither dexamethasone nor knockout of 5LO altered the activity of cdk5 (Fig. 3D).
Figure 3.

Dexamethasone-mediated increase in the activity of GSK3β is absent in animals that lack 5LO. A) Representative western blots for GSK3α, GSK3β, pSer 21 GSK3α, pSer9 GSK3β, cdk5, p35, and p25 in cortical brain homogenates of 3xTg and 3xTg/5LO−/− animals treated with PBS or dexamethasone. B) Densitometric analyses of immunoreactivities to the antibodies shown in panel A. Kinase activity of C) GSK3β and D) cdk5 in brains of animals.
Dexamethasone dysregulates synaptic protein in 3xTg animals but not in 3xTg/5LO−/−
Recent evidence has shown that synaptic dysfunction can occur in AD before presentation of obvious behavioral symptoms (Chakroborty et al., 2012). We assayed levels of two synaptic proteins, the postsynaptic density protein 95 (PSD95) and the presynaptic protein synaptophysin (SYP), to see whether dexamethasone altered their steady state levels. As shown in Fig. 4A, B, we found that dexamethasone significantly reduced levels of PSD95 but not synaptophysin in the 3xTg mice. Interestingly, we found that not only did animals lacking 5LO resist dexamethasone-mediated PSD95 reduction, but they also displayed greater overall levels of PSD95 than 3xTg animals. Finally, neither dexamethasone treatment not knockout of 5LO altered steady-state levels of the glucocorticoid receptor.
Figure 4.
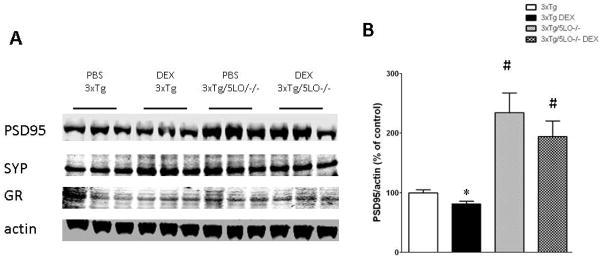
Dexamethasone alters synaptic protein levels in 3xTg animals but not in 3xTg/5LO−/−. A) Representative western blots for PSD95, synaptophysin and glucocorticoid receptor in the cortical brain homogenates of PBS and dexamethasone treated 3xTg and 3xTg/5LO−/− animals. B) Densitometric analysis of PSD95 shown in panel A. *p<0.05
DISCUSSION
While the cause of sporadic AD remains unknown, a combination of environmental and genetic factors has been implicated in its pathogenesis. Psychosocial stress leading to elevated levels of stress hormones, i.e., glucocorticoids, is considered one important environmental risk factor that can influence AD onset (Wilson et al., 2006; Csernansky et al., 2006, Huang et al. 2009). However, the mechanisms(s) by which corticosteroids accelerate the AD neuropathology and influence the development of its phenotype remains to be fully elucidated.
5LO has recently emerged as an important molecular target in neuropsychiatric disease, though relatively little is known about its role downstream of glucocorticoid receptor activation and signaling. Chronic treatment with corticosterone increases message and protein of 5LO in the rat cerebellum, and blockade of the glucocorticoid receptor reduces dexamethasone-induced message and expression of 5LO in cultured primary rat cerebellar granular neurons (Uz et al., 1999; Uz et al. 2001), suggesting 5LO expression is directly affected by stress hormones in the brain.
In the current study, we show for the first time that the 5LO enzymatic pathway is involved in the dexamethasone-dependent increase of tau phopshorylation by modulating the activity of the GSK3β kinase in a rodent model of AD with plaques and tangle, the 3xTg mouse.
Multiple glucocorticoid and stress paradigms have been used to interrogate the role and mechanism of stress in rodent models of AD, with impairment of cognition coincident with elevation of the classical neuropathology (Jeong, et al., 2006; Kang, et al., 2007 Lee, et al., 2009; Joshi et al., 2012). However, because glucocorticoids are known to independently disrupt memory processes, studies in which stress hormone treatments cause frank cognitive insults are unable to separate between glucocorticoid effects AD pathology and direct effects on memory. Here we report for the first time a short-term dexamethasone treatment paradigm that causes changes in AD pathology before any memory impairment can be detected.
Our observation that dexamethasone did not alter Aβ levels are at odds with previous work by Green et al. 2006 in this animal model, reporting a modest increase using a similar dexamethasone paradigm, which we believe is attributable to differences in animal gender, Aβ ELISA, extraction buffer and the fact that whole brain homogenization was used for assay. Since there is evidence that there exists a gender-based dimorphism in stress response in these animals, this finding is not unexpected (Clinton et al., 2007). Nevertheless, in their work, Green and colleagues showed somatodendritic mislocalization of tau following dexamethasone treatment, which mirrors our finding of elevated levels of S396 tau, a phosphorylation event on tau that has been proposed to initiate the aggregation of fibrillar and paired-helical fragments. Since at this early age we found that 5LO knockout resisted ser396 phosphorylation of tau caused by dexamethasone, this suggests that 5LO is required for progression of tauopathy in the corticosteroid-dependent AD context. Further supporting this observation, insoluble HT7 tau immunoreactivity was elevated in the brains of dexamethasone treated animals but barely detectable in the brains of 5LO knockout animals.
Associated with the significant increase in tau phosphorylation and its insoluble fraction, we found that dexamethasone significantly increased the activity but not the levels of GSK3β. This protein is a major kinase that phosphorylates tau and has been shown to be dysregulated in the brains of AD patients (Pei et al., 1999). Our observation is in accordance with previous work showing that glucocorticoids activate GSK3β by increasing its activity (Smith et al., 2002). Interestingly, while we observed that this kinase activity was significantly altered in the dexamethasone treated animals, we observed no changes in protein levels or activity for another kinase involved in tau metabolism, cdk5. This finding could explain the selective phosphorylative effect of dexamethasone on specific tau epitopes in our model. While the specific pathways linking glucocorticoid receptor activation to tau phosphorylation in AD still remain poorly defined, others have shown that leukotrienes, downstream metabolites of 5LO, can modulate GSK3β in an Akt-dependent manner (Kim et al., 2018). We hypothesize that pathway may be relevant in glucocorticoid-induced tau pathology in this AD model.
Pathologic tau phosphorylation and fibrillary neurofibrillary tangle formation have been shown to compromise neuronal ultrastructure, leading to destabilization of synapses (Busciglio et al., 1995). In our paper we observed that the increase in tau phosphorylation was indeed associated with reduction in the levels of PSD95, which suggests synaptic impairment. Our finding that in 3xTg animals lacking 5LO, PSD95 levels are not only unaffected by dexamethasone but are significantly greater than 3xTg controls suggests that these animals tolerate better AD-induced insults to synaptic functioning.
Since stress and stressful experiences are unavoidable aspects of the environment, a therapeutic strategy targeting stress-mediated pathological changes in AD before symptom onset could be beneficial, especially considering the failure of recent clinical trials evaluating AD-targeted therapeutics (i.e., γ-secretase inhibitors, immunization against Aβ, etc), which suggest that intervening after cognitive decline occurs is a poor strategy. Considering that validated biomarkers for tracking early AD disease progression are in their infancy, targeting environmental factors that are known to exacerbate the AD phenotype is perhaps a more prudent approach.
Our work by showing that 5LO plays a functional role in the glucocorticoid-mediated induction of tau pathology and synaptic disruption support the novel idea that this protein could be an attractive target for pharmacologic intervention in individuals that are particularly vulnerable to stress as an environmental risk factor.
EXPERIMENTAL PROCEDURES
Animals
All animal procedures were approved by the Institutional Animal Care and Usage Committee, in accordance with the U.S. National Institutes of Health guidelines and approved by the Temple University’s Animal Care and Use Committee. The 3xTg mice harboring a mutant amyloid precursor protein (APP; KM670/671NL), a human mutant PS1 (M146V) knockin, and tau (P301L) transgenes; 3xTg wild type (WT), and mice genetically deficient for 5LO (5LOKO) used in the study were reported previously (Oddo et al. 2003; Goulet et al. 1994). All the animals were backcrossed 10 times on the same genetic background. The 5LOKO mice were crossbred several times with 3xTg mice to obtain founder animals (3xTg/5LOKO), which were then crossed with each others and the animals from these crosses used for the studies. Naïve female 3xTg and 3xTg/5LO−/− mice aged 3.5 months were used for this study. All mice were housed on a 12 hours light/dark cycle in the Medical Research Building at the Temple University Health Sciences Campus, which is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International. Standard mouse chow and water were provided ad libitum. Animals were intraperitoneally either given phosphate-buffered saline (PBS) as control, or dexamethasone (DEX; Sigma-Aldrich) at a dose of 5mg/kg for 7 d (n=5 3xTg (PBS), n = 7 3xTg (DEX), n = 4 3xTg/5LO−/− (PBS), n = 5 3xTg/5LO−/− (DEX)). Mice were sacrificed and brains were removed 24h after behavioral assessment as previous described (Joshi et al., 2012). Cortices were dissected from one hemibrain and stored −80° C while the other hemisection was fixed in paraformaldehyde overnight, processed and paraffin-embedded, and used for immunohistochemistry analyses.
Behavioral paradigms
Following 7d treatment with DEX or PBS, all animals were assayed in the Y-maze and fear conditioning paradigms. All apparatuses were cleaned with 70% ethanol between animal trials and allowed to dry completely. The Y-maze and fear-conditioning paradigms were carried out as previously described (Joshi and Praticò, 2013). The Y-maze apparatus consisted of three arms 32 cm (long) ×10 cm (wide) with 26-cm walls (San Diego Instruments). For fear conditioning, tests were conducted in a chamber equipped with black methacrylate walls, a transparent front door, a speaker and grid floor (Start Fear System; Harvard Apparatus). Testing was always performed in the same room and at the same time to ensure environmental consistency.
Biochemical analyses
Mouse brain cortical homogenates were sequentially extracted first in radio-immunoprecipitation assay buffer (RIPA) containing EDTA-free protease inhibitor (Roche) and phosphatase inhibitor (Thermo Fisher) for the soluble fractions and then in formic acid (FA) for insoluble fractions as previously described (Chu et al. 2012). Aβ1–40 and Aβ1–42 levels were assayed by a sensitive sandwich enzyme-linked immunosorbent assay (ELISA) kit (Wako Chemicals, USA) in accordance to the manufacturer’s protocols.
Immunoblotting
Brain homogenate RIPA or pH-neutralized FA samples were electrophoretically separated using 10% Bis-Tris gels or 3% to 8% Tris-acetate gel (Bio-Rad), according to the molecular weight of the target molecule, and then transferred onto nitrocellulose membranes (Bio-Rad). Membranes were blocked with Odyssey blocking buffer and incubated with primary antibodies overnight at 4° C. After 3 washing cycles in TBS-T, membranes were incubated with IRDye secondary antibodies (LI-COR) at 22° C for 1 hour. Signals were developed with Odyssey Infrared Imaging Systems (LI-COR). Actin was always used as an internal loading control. Antibodies and dilutions were as follows: HT7 (1:200, Cell Signal), PHF13 (1:200; Pierce), PHF1 (1:200; a generous gift from Dr. Peter Davies), GSK3α/β (1:200; Santa Cruz), pGSK3α/β (1:200; Santa Cruz), cdk5 (1:200; Santa Cruz), p35/p25 (1:200; Santa Cruz), synaptophysin (SYP; 1:200; Santa Cruz), PSD95 (1:200; Cell Signal), β-actin (1:1000; Santa Cruz)
Immunohistochemistry
Immunohistochemistry analysis was performed as previously described (Chu et al. 2012). Briefly, 6 μm brain sections were deparaffinized, hydrated, and after blocking with 2% serum with citric acid used to retrieve antigen. Sections were incubated with primary antibody against PHF13 (1:20) or PHF1 (1:200) overnight at 4° C. Sections were washed, incubated with appropriate secondary antibody and finally developed using the avidin-biotin complex method (Vector Laboratories) with 30,30-diaminobenzidine as chromogen.
Kinase activity
GSK and cdk5 kinase activities were carried out as previously described (Joshi et al. 2013; Chu et al. 2012). Briefly, GSK3β and cdk5 were immunoprecipitated from 75 ug of brain lysate using 1.5μg of GSK3β and cdk5 antibody respectively (Santa Cruz). The immunoprecipitates were washed with lysis buffer three times followed once by HBS (10mM HEPES, pH 7.4, 150mM NaCl). The kinase activity of the immunoprecipitated Cdk5 was determined by using histone H1 (Santa Cruz Biotechnology) as substrate, while activity of GSK3β was determined by using phospho-glycogen synthase peptide 2 (Millipore). In both experiments, immunoprecipitates were incubated in HBS containing 15mM MgCl2, 50mm adenosine triphosphate (ATP), 1mM dithiothreitol, and 1μCi of [32P]ATP. After 30 minutes of incubation at 37°C, the reaction products were determined by a liquid scintillation counter.
Statistical analysis
Data are presented as the mean ± standard error of the mean. One-way analysis of variance (ANOVA), with Bonferroni multiple comparison tests were performed using Prism 5.0 (GraphPad Software, La Jolla, CA). All data are presented as mean +/− standard error of the mean. Significance was set at p <0.05.
Acknowledgments
This work was supported in part by grants from the National Institute of Health (AG33568) and the Alzheimer Art Quilt Initiative.
Footnotes
AUTHOR CONTRIBUTIONS
Y.B.J and D.P conceived and designed the study, and wrote the manuscript. Y.B.J and J.C. carried out experiments. All authors reviewed the manuscript.
References
- Busciglio J, Lorenzo A, Yeh J, Yanker BA. beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:878–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- Baglietto-Vargas D, Medeiros R, Martinez-Coria H, LaFerla FM, Green KN. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and tau pathology. Biol Psych. 2013 doi: 10.1016/j.biopsych.2012.12.003. pii: S0006-3223(12)01097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Kim J, Schneider C, Jacobson C, Molgo J, Stuzmann GE. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer’s disease mice. J Neurosci. 2012;32:8341–53. doi: 10.1523/JNEUROSCI.0936-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Giannopoulos PF, Ceballos-Diaz C, Golde TE, Pratico D. 5-lipoxygenase gene transfer worsens memory, amyloid, and tau brain pathologies in a mouse model of Alzheimer’s disease. Ann Neurol. 2012;72:442–54. doi: 10.1002/ana.23642. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chu J, Praticò D. 5-lipoygenase as an endogenous modulator of amyloid beta formation in vivo. Ann Neurol. 2011;69:34–46. doi: 10.1002/ana.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton LK, Billings LM, Green KN, Caccamo A, Ngo J, Oddo S, McGaugh JL, LaFerla FM. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis. 2007;28:76–82. doi: 10.1016/j.nbd.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, Morris JC. Plasma cortisol and pregression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry. 2006;163:2164–2169. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firuzi O, Zhuo J, Cinnici CM, Wisniewski T, Pratico D. 5-lipoxygenase gene disruption reduces amyloid-beta pathology in a mouse model of Alzheimer’s disease. FASEB J. 2008;22:1169–1178. doi: 10.1096/fj.07-9131.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulet JL, Snouwaert JN, Latourt AM, Coffman TM, Koller BH. Altered inflammatory responses in leukotriene-deficient mice. Proc Natl Acad Sci. 1994;91:12852–12856. doi: 10.1073/pnas.91.26.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CW, Lui CC, Chang WN, Lu CH, Wang YL, Chang CC. Elevated basal cortisol level predicts lower hippocampal volume and cognitive decline in Alzheimer’s disease. J Clin Neurosci. 2009;16:1283–1286. doi: 10.1016/j.jocn.2008.12.026. [DOI] [PubMed] [Google Scholar]
- Kim MH, Lee YJ, Kim MO, Kim JS, Han HJ. Effect of leukotriene D4 on mouse embryonic stem cell migration and proliferation: involvement of PI3K/Akt as well as GSK-3β/β-catenin signaling pathways. J Cell Biochem. 2010;111:686–98. doi: 10.1002/jcb.22755. [DOI] [PubMed] [Google Scholar]
- Jeong YH, Park CH, Yoo J, Shin KY, Ahn SM, Kim HS, Lee SH, Emson PC, Suh YH. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer’s disease model. FASEB J. 2006;20:729–731. doi: 10.1096/fj.05-4265fje. [DOI] [PubMed] [Google Scholar]
- Joshi YB, Pratico D. Knockout of 5-lipoxygenase results in age-dependent anxiety-like behavior in female mice. PLoS One. 2011;6:e29448. doi: 10.1371/journal.pone.0029448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi YB, Pratico D. The involvement of 5-lipoxygenase activating protein in anxiety-like behavior. J Psychiatr Res. 2013 doi: 10.1016/j.jpsychires.2012.12.011. pii: S0022-3956(12)00381-0. 10.1016. [Epub before print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi YB, Chu J, Pratico D. Stress hormone leads to memory deficits and altered tau phosphorylation in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2012;31:167–8. doi: 10.3233/JAD-2012-120328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc Natl Acad Sci U S A. 2007;104:10673–10678. doi: 10.1073/pnas.0700148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS, Kim KS, Lee JK, Han PL. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem. 2009;108:165–175. doi: 10.1111/j.1471-4159.2008.05769.x. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- Puccio S, Chu J, Pratico D. Involvement of 5-lipoxygenase in the corticosteroid-dependent amyloid-beta formation: in vitro and in vivo evidence. PLoS One. 2011;6:e15163. doi: 10.1371/journal.pone.0015163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Coetzee GA, Frenkel B. Glucocorticoids inhibit cell cycle progression in differentiating osteoblasts via glycogen synthase kinase-3beta. Journal of Biological Chemistry. 2002;277:18191–18197. doi: 10.1074/jbc.M109708200. [DOI] [PubMed] [Google Scholar]
- Uz T, Dwivedi Y, Savani PD, Impagnatello F, Pandey G, Manev H. Glucocorticoids stimulate inflammatory 5-lipoxygenase gene expression and protein translocation in the brain. J Neurochem. 1999;73:693–9. doi: 10.1046/j.1471-4159.1999.0730693.x. [DOI] [PubMed] [Google Scholar]
- Uz T, Dwivedi T, Qeli A, Peters-Golden M, Pandey G, Manev H. Glucocorticoid receptors are required for up-regulation of neuronal 5-lipoxygenase (5LOX) expression by dexamethasone. FASEB J. 2001;15:1792–4. doi: 10.1096/fj.00-0836fje. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bougeat P, Ackermann U, Jones G, Szoeke C, Salvado O, Martins R, O’Keeke G, Mathis CA, Klunk WE, Ames D, Masters CL, Rowe CC. Longitudinal assessment of Aβ and cognition in aging and Alzheimer’s disease. Ann Neurology. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27:143–153. doi: 10.1159/000095761. [DOI] [PubMed] [Google Scholar]