

Summary
Background
Inorganic polyphosphates (polyP), which are secreted by activated platelets (short chain polyP) and accumulate in some bacteria (long chain polyP), support the contact activation of factor XII (FXII), and accelerate the activation of factor XI (FXI).
Objectives
The aim of the present study was to evaluate the role of FXI in polyP-mediated coagulation activation and experimental thrombus formation
Methods and Results
Pretreatment of plasma with antibodies that selectively inhibit FXI activation by activated FXII (FXIIa) or factor IX (FIX) activation by activated FXI (FXIa) were not able to inhibit the procoagulant effect of long or short polyP in plasma. In contrast, the FXIIa inhibitor, corn trypsin inhibitor (CTI), blocked the procoagulant effect of long and short polyP in plasma. In a purified system, long polyP significantly enhanced the rate of FXII and prekallikrein (PK) activation, and the activation of FXI by thrombin, but not by FXIIa. In FXI-deficient plasma, long polyP promoted clotting of plasma in a FIX-dependent manner. In a purified system, the activation of FXII and PK by long polyP promoted FIX activation and prothombin activation. In an ex vivo model of occlusive thrombus formation, inhibition of FXIIa with CTI but not of FXI with a neutralizing antibodies abolished the prothrombotic effect of long polyP.
Conclusions
We propose that long polyP promotes FXII-mediated blood coagulation bypassing FXI. Accordingly, some polyP containing pathogens may have evolved strategies to exploit polyP-initiated FXII activation for virulence, and selective inhibition of FXII may improve the host response to pathogens.
Keywords: factor XI, factor XII, polyphosphate, thrombin generation, thrombosis
Introduction
It has recently been shown that inorganic polyphosphates (polyP) accumulate in a variety of microorganisms [1], are secreted by activated platelets [2] and, when administered to mice, can induce pathologic blood coagulation in a factor XII (FXII)-dependent manner [3,4]. Depending on polymer length, the ability of polyP to trigger the contact pathway exhibits differential effects on blood clotting. For instance, long polymers of polyP (~1000 phosphate units, such as those present in microorganisms) have been shown to be more efficient at triggering the contact activation pathway as compared to shorter polyP (~60–100 phosphate units, secreted by human platelets) [5]. Short polyP has been shown to accelerate both factor V (FV) activation by either activated factor X (FXa) or thrombin and factor XI (FXI) activation by either activated FXI (FXIa) or thrombin [6,7], providing a mechanism by which platelets could accelerate the activation of the coagulation cascade during thrombus formation.
FXII is activated in a process involving high molecular weight kininogen (HK) and plasma prekallikrein (PK)[8]. Activated FXII (FXIIa) catalyzes the activation of FXI, which in turn activates factor IX (FIX) to activated FIX (FIXa), leading to thrombin generation and fibrin formation. This series of reactions forms the basis of the activated partial thromboplastin time (aPTT) measurement in plasma or blood. Individuals with severe FXII deficiency have a prolonged aPTT but do not suffer from abnormal bleeding [9]. However, some FXI-deficient patients exhibit postoperative or posttraumatic bleeding, especially in tissues with increased profibrinolytic activity [10]. In contrast, FIX deficiency causes the severe bleeding disorder hemophilia B in all subjects, indicating that FIX is required for normal hemostasis. Current models of thrombin generation provide a rationale for these phenotypic differences as follows: FIX is activated by both the FVIIa/tissue factor complex and FXIa [11], FXI is activated by FXIIa, thrombin and autoactivation by FXIa [12], while FXII autoactivates when blood comes into contact with foreign material and surfaces [8].
Consistent with the observed role of FXI in experimental thrombosis, humans with severe FXI deficiency are largely protected from deep vein thrombosis and ischemic stroke [13,14], suggesting that FXI activation contributes to the pathogenesis of human thromboembolic disease. Experimentally, inhibition of FXI interrupts experimental thrombosis without bleeding side effects in baboons and mice [15,16], and asymptomatic mice lacking FXII or FXI have been shown to be resistant to arterial thrombotic occlusion [17,18], providing a rationale for the targeted inhibition of the FXII/FXI pathway as a safe antithrombotic strategy. However, it is not clear whether FXIIa mediates its prothrombotic effect through FXI alone. The observation that the aPTT of FXII-deficient plasma is longer than that of FXI-deficient plasma suggests that there are procoagulant targets for FXIIa other than FXI. It has been shown that FXII-deficient mice are resistant to FeCl3-induced arterial occlusion to a larger extent than FIX and FXI deficient mice [15], suggesting that FXII also contributes to thrombosis in a FXI-independent manner. Platelet polyP injection has been shown to induce coagulopathy in mice in a FXII-dependent manner [3], but the role of FXI in the pathogenesis remains unclear. The goal of our study was to determine whether FXII activation in the presence of long polyP promotes blood coagulation and experimental occlusive thrombus formation in a FXI-dependent manner.
Materials and methods
Reagents
Short (~70–85 phosphate units in length) and long (>1000 phosphate units in length) polyP were prepared as described previously [5]. PolyP concentrations are reported in terms of phosphate monomer concentration (monomer formula, NaPO3). Anti-factor XI antibodies 1A6 and 14E11 were generated as described [12,15]. Anti-human FIX antibody, α-FXIIa, PK, HK, kallikrein and corn trypsin inhibitor (CTI) were from Enzyme Research Laboratories, Inc. (South, IN, USA). Plasma-derived FXI, FXIa, FIX, FX, FXa, prothrombin, α-thrombin and FIX were from Haematologic Technologies, Inc (Essex Junction, VT, USA). Congenital FXI-deficient plasma was from George King Bio-Medical (KS, USA). Polybrene, soybean trypsin inhibitor and hirudin were from Sigma-Aldrich (St Louis, MO, USA). S-2366 was from Diapharma Group, Inc (West Chester, OH, USA). Spectrozyme TH, Spectrozyme FXIIa and Spectrozyme FXa were from American Diagnostica, Inc (Stamford, CT, USA). Rivaroxaban, a direct FXa inhibitor, was from Bayer AG (Wuppertal, Germany). Enoxaparin (Lovenox,®), a low molecular weight heparin that preferentially inhibits FXa, was from Sanofi-Aventis (Canada). Dextran sulfate (average Mr, 500 000) was from Fisher (Pittsburgh, PA, USA). All other reagents were from previously described commercial sources [19].
Plasma clotting assay
Human venous blood was drawn in accordance with an IRB-approved protocol. Platelet-poor plasma (PPP) was prepared by centrifugation of citrated whole blood (in 0.32% w/v sodium citrate) from three separate donors at 2150×g for 10 min. Pooled PRP was further centrifuged at 2150×g for 10 min. Clotting times of human PPP were measured with a KC4 Coagulation Analyzer (Trinity Biotech, Ireland). Samples were pretreated at room temperature (RT) for 10 min with blocking FXI antibodies, 1A6 (20 μg/ml) or 14E11 (20 μg/ml), 50 μg/ml CTI, 1 μg/ml enoxaparin, or 2 μg/ml rivaroxaban. Long or short polyP in 25 mM Hepes, pH 7.4 and 1%BSA was mixed in coagulometer cuvettes with prewarmed plasma. In selected experiments, 10 pM α-thrombin was added and incubated for 1 minute at 37°C. Clotting was initiated with addition of 25 mM CaCl2. In selected experiments, FIX- or FX-depleted or FXI-deficient plasmas were used.
FXII activation
FXII (200nM) was incubated at 37°C with PK (50nM) and HK (50nM) in the presence or absence of long or short polyP (10 μM) in 25 mM Hepes, pH 7.4, 150 mM NaCl, and 0.1% BSA (HBS). Samples were removed and quenched with polybrene (6 μg/ml) to neutralize polyP and soybean trypsin inhibitor (50 μg/ml) to inactivate kallikrein, after which the generation of FXIIa was quantified by measuring rates of Spectrozyme FXIIa hydrolysis at 405 nm. Rates of Spectrozyme FXIIa hydrolysis were converted to FXIIa concentrations using a standard curve. In some experiments, FXII (200nM) was incubated in the presence or absence of long polyP (10 μM) or dextran sulfate (2 μg/ml) in the absence of PK and HK.
PK activation
PK (50 nM) was incubated at 37°C with HK (50 nM) and α-FXIIa (100 pM) in the presence or absence of long or short polyP (10 μM) in HBS. Samples were removed and quenched with polybrene (6 μg/ml) to neutralize polyP and CTI (50 μg/ml) to inactivate α-FXIIa, after which the generation of kallikrein was quantified by measuring rates of Spectrozyme FXIIa hydrolysis at 405nm. Rates of Spectrozyme FXIIa hydrolysis were converted to kallikrein concentrations using a standard curve. In some experiments, PK (200 nM) was incubated at 37°C with HK (200 nM) in presence or absence of long or short polyP (10 μM).
FXI activation
FXI (30 nM) was incubated with 5 nM α-thrombin or 5 nM α-FXIIa in the absence or presence of short and long polyP (15 μM) at 37°C in HBS. Samples were removed and quenched with polybrene (6 μg/ml) to neutralize polyP and hirudin (10 U/ml) to inactivate thrombin or CTI (50ug/ml) to inactivate FXIIa, after which the generation of FXIa was quantified by measuring rates of S-2366 hydrolysis at 405nm. Rates of S-2366 hydrolysis were converted to FXIa concentrations using a standard curve. Selected experiments were performed in the presence of FXII (200 nM), PK (50 nM) and HK (50 nM).
FX activation
FX (150 nM) was incubated with 50 nM FXIa or 50 nM FXIIa at 37°C in HBS. Samples were serially removed and quenched with 10 μM aprotinin to inactivate FXIa or CTI (50 μg/ml) to inactivate FXIIa, after which FXa generation was quantified by measuring rates of Spectrozyme FXa hydrolysis at 405 nm. Rates of Spectrozyme FXa hydrolysis were converted to FXa concentrations using a standard curve.
FIX activation
FIX (100 nM) was incubated with 5 nM FXIa, 50 nM kallikrein or 50 nM FXIIa at 37°C in 25 mM Hepes, pH 7.4, 150 mM NaCl, CaCl2 5 mM and 0.1% BSA (HBS-Ca) for 30 min. 10 μl sample were mixed with 10 μl of Laemmli sample buffer (Bio-Rad) with 0.5M dithiothreitol (100°C, 5 minutes), separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and immunoblotted with an anti-FIX antibody and HRP-conjugated secondary antibodies. Protein was detected using ECL (Amersham Biosciences).
FIX activation in plasma
FXI-deficient plasma was incubated with 50 μM long polyP in the absence of calcium at 37°C, followed by immunoprecipitation of FIX. Plasma was precleared with 50 μl Protein A/G PLUS-Agarose (Santa Cruz) prior to incubation with 3 μg of a goat anti FIX antibody for 1 hour, followed by the addition of 20 μl Protein A/G agarose. Captured protein complexes were washed with PBS buffer and eluted into 50 μl Laemmli sample buffer with 0.5M dithiothreitol (100°C, 5 minutes), separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and immunoblotted with an anti-FIX antibody and HRP-conjugated secondary antibodies. Protein was detected using ECL.
Prothrombin activation
Prothrombin (1 μM) was incubated with 50 nM α-FXIIa, 50 nM FXIa or 50 nM FXa at 37°C in HBS-Ca. Samples were serially removed and quenched with 10 μM aprotinin to inactivate FXIa, CTI (50 μg/ml) to inactivate α-FXIIa, and 10 μg/ml rivaroxaban to inactivate FXa, after which thrombin generation was quantified by measuring rates of Spectrozyme TH hydrolysis at 405 nm. Rates of Spectrozyme TH hydrolysis were converted to thrombin concentrations using a standard curve. Prothrombin activation in presence of FXIIa and FXIa were performed in presence of rivaroxaban (20 μg/ml), in order to eliminate any possibility of FX contamination in the α-FXIIa or FXIa preparation.
Capillary occlusion assay
Glass capillary tubes (0.2 × 2mm) were incubated for 1 hr at RT with a solution of collagen (100 μg/ml), followed by washing with PBS and incubation for 1 hr at RT with 0.1 nM tissue factor (TF). Surface was blocked with 5 mg/ml denatured BSA for 1 hr. Sodium citrate (0.38 % w/v) anticoagulated whole blood was sequentially supplemented with 7.5 mM Ca2+ and 3.75 mM Mg2+ in 1 ml aliquots. Flow through the capillary was driven by the force of gravity, and the height of the sample reservoir was regulated in order to produce an initial shear rate of 300 s−1 [20,21].
Analysis of data
Data are shown as means ± SEM. Statistical analysis was performed using paired Student’s t test on Microsoft Excel. Probability values of P < 0.05 were selected to be statistically significant.
Results
Short and long polyP promotes clotting of plasma independent of FXI
Previous studies have shown that polyP was able to initiate clotting of plasma in a FXII-dependent manner. In our system, polyP of the size secreted by human platelets (~70–85 phosphate units long, short polyP) decreased the clotting of plasma from ~600 sec down to ~350 sec in a concentration-dependent manner, with maximum effect observed above 5 μM short polyP (Fig. 1A). PolyP of the size present in bacteria (>1000 phosphate units long, long polyP) decreased clotting time to ~150 seconds in a concentration-dependent manner (Fig. 1B). Pretreatment of plasma with CTI, which inhibits FXIIa, abrogated the ability of long polyP to promote clotting. Pretreatment of plasma with the blocking FXI antibody, 1A6, which blocks FIX activation by FXIa [12] and FXI activation by FXIIa (Fig. S1), failed to block the procoagulant effect of either short or long polyP (Fig. 1A, 1B), suggesting that polyP shortens the clotting time of plasma in a manner dependent on FXII yet independent of FXI.
Figure 1. Short and long polyP promotes clotting of plasma independent of FXI.

(A and B)Recalcified plasma in the presence of vehicle (◇), 50 μg/ml CTI (□) or 20 μg/ml 1A6 (▲) was incubated for 1 minute at 37°C with short (A) or long (B) polyP. (C and D) Citrated plasma in the presence of vehicle (◇), 50 μg/ml CTI (■), 20 μg/ml 1A6 (○), 20 μg/ml 14E11 (△), or CTI plus 1A6 (●), was incubated for 1 minute at 37°C with 10 pM α-thrombin and short (C) or long (D) polyP. Data are mean ± SE (n of at least 3).
Previous studies have shown that polyP of varying polymer lengths was able to potentiate the activation of FXI by thrombin and thrombin-initiated clotting of plasma in a FXI-dependent manner [7]. As shown in figure 1C and 1D, short and long polyP decreased α-thrombin-initiated clotting of plasma. Under these conditions, pretreatment of plasma with CTI, which inhibits FXIIa, or the blocking FXI antibody, 14E11, which inhibits FXI activation by FXIIa [12], failed to block the procoagulant effect of either short or long polyP (Fig. 1C, 1D). In the absence of polyP, pretreatment of plasma with 1A6 prolonged the clotting time from 407 ± 96 to 1135 ± 34 seconds (Fig. 1C, 1D), in accord with the notion that feedback activation of FXI plays a key role in promoting α-thrombin-induced coagulation [12]. Our data show that the ability of either long or short polyP to promote coagulation was unaffected by the pretreatment of plasma with 1A6 (Fig. 1C, D). These results show that short and long polyP shortened the α-thrombin induced clotting times of plasma in a FXI-independent manner.
Activation of FXII by long polyP promotes α-thrombin-initiated clotting of plasma independent of FXI
Previous studies have shown that both long and short polyP were able to accelerate FV activation by thrombin and FXa [6]. Therefore, plasma was supplemented with 30 nM FVa in order to eliminate the contribution of polyP-mediated FV activation in α-thrombin-initiated clotting of plasma. As we show in figure 1C, short polyP promoted α-thrombin-induced coagulation in a concentration-dependent manner. We found that the addition of short polyP failed to further promote the clotting of plasma supplemented with FVa (Fig. 2A). In contrast, our data show that long polyP decreased the clotting time of FVa-supplemented plasma in a concentration-dependent manner (~300 sec vs. ~150 sec in the presence of vehicle or 50 μM long polyP, respectively; Fig. 2B). The ability of long polyP to promote clotting of FVa-supplemented plasma was unaffected by the addition of CTI or 1A6 alone (Fig. 2B). Strikingly, the combination of CTI with 1A6, but not 14E11, abrogated the ability of long polyP to promote clotting of FVa-supplemented plasma (Fig. 2B, S2). Our data suggests that the ability of long polyP to shorten α-thrombin-induced clotting times is dependent on feedback activation of FXI, yet independent of FXI activation by FXIIa.
Figure 2. Activation of FXII by long polyP promotes thrombin-initiated clotting of plasma independent of FXI.
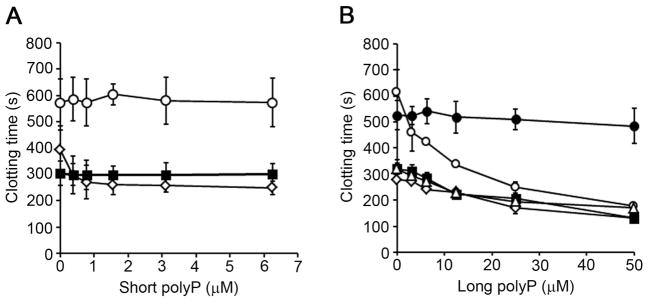
(A) Short polyP was added to citrated plasma in the presence of vehicle (◇) or 30 nM FVa (■) or 20 μg/ml 1A6 plus FVa (○). Clotting was initiated with 10 pM α-thrombin. (B) Long polyP was added to citrated plasma supplemented with 30 nM FVa in the presence of vehicle (◇), 50 μg/ml CTI (■), 20 μg/ml 1A6 (○), or CTI plus 1A6 (●). Clotting was initiated with 10 pM α-thrombin. Data are mean ± SE (n = 3).
It is known that FXIIa is able to activate FVII, while kallikrein is able to activate FIX and prothrombin [22,23]. The ability of FXIIa to activate FX, FIX or prothrombin has not been studied. In a purified system, we found that FXIa, but not FXIIa, activated FX (Fig. S3A). As shown in figure S3B, FXIIa did not activate FIX. While our data show that FXIIa, but not FXIa, induced conversion of prothrombin to thrombin in a calcium-dependent manner (Fig. S3C–D), the kinetics of the activation of prothrombin by FXIIa were very slow (Km = 0.82 ± 0.04 μM, kcat of 0.0012 ± 0.0001 min−1; Fig. S3E). We validated the finding that kallikrein was able to promote FIX and prothrombin generation (Fig. S3A and S3D, respectively). Taken together, these data provide a mechanism by which FXII and PK activation by long polyP may promote FIX and prothrombin activation in a FXI-independent manner.
Activation of FXII by long polyP promotes α-thrombin-initiated clotting of plasma independent of FXI in presence of FXa inhibitors
We aimed to determine the role of FX in promoting α-thrombin-initiated clotting of plasma by long or short polyP. Accordingly, plasma was supplemented with FXa inhibitors, enoxaparin or ribaroxaban. We observed that α-thrombin-initiated clotting of plasma was prolonged to ~1200 seconds in the presence of FXa inhibitors (Fig 3A). The presence of short polyP failed to potentiate clotting of plasma in the presence of FXa inhibitors (Fig. 3A). In contrast, long polyP decreased the clotting time of plasma in a concentration-dependent manner, even in the presence of FXa inhibitors (Fig. 3A). As shown in figure 3B, plasma pretreated with 1A6 failed to clot after 2000 seconds in presence of rivaroxaban. In contrast, the pretreatment of plasma with 14E11 did not affect the α-thrombin-initiated clotting time of plasma in presence of rivaroxaban. The ability of long polyP to promote clotting of plasma pretreated with rivaroxaban was unaffected by the addition of either 1A6 or 14E11 (Fig. 3B). Interestingly, plasma pretreated with CTI failed to clot after 2000 seconds in presence of rivaroxaban. The procoagulant effect of long polyP was inhibited when plasma was pretreated with CTI (Fig. 3B). These results show that the activation of FXII by long polyP shortens the α-thrombin clotting time of plasma in a FXI-independent manner.
Figure 3. Activation of FXII by long polyP promotes thrombin-initiated clotting of plasma independent of FXI in presence of FXa inhibitors.
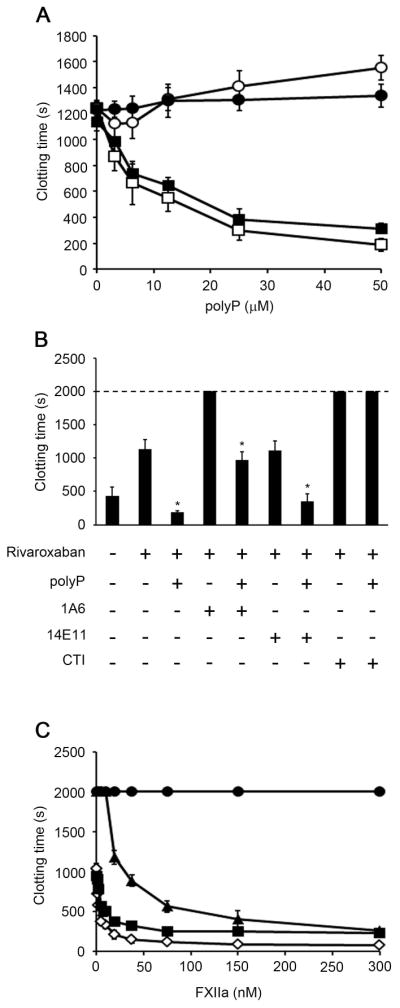
(A) Plasma clotting time was measured in citrated plasma pretreated with 2 μg/ml rivaroxaban (■, ●) or 1 μg/ml enoxaparin (○, □). Clotting was initiated by the addition of 10 pM α-thrombin and either short (●, ○) or long (■, □) polyP. (B) Plasma clotting time was measured in citrated plasma pretreated with 2 μg/ml rivaroxaban incubated for 1 minute at 37°C with 10 pM α-thrombin in presence of vehicle, 50 μM long polyP, 20 μg/ml 1A6, 20 μg/ml 14E11 or 50 μg/ml CTI. (C) Plasma clotting time was measured in citrated plasma pretreated with 2 μg/ml rivaroxaban incubated for 1 minute at 37°C with 10 pM α-thrombin and FXIIa in the presence of vehicle (◇), 20 μg/ml 1A6 (▲), 20 μg/ml 14E11 (■), or 50 μg/ml CTI (●). The clotting time was recorded, with a maximum observation period of 2000 seconds. Data are mean ± SE (n = 3). (*P < 0.05)
As shown in figure 3C, addition of FXIIa to plasma in the presence of FXa inhibitors accelerated clotting from ~1000 sec to ~90 sec in a concentration-dependent manner. We observed that the pretreatment of plasma with 14E11 or 1A6 did not inhibit the procoagulant effect of α-FXIIa. However, the ability of α-FXIIa to promote the plasma clotting was abrogated by the presence of CTI (Fig. 3C).
PolyP promotes FXII and PK activation and the activation of FXI by thrombin, but not by FXIIa
We next designed experiments to determine whether polyP promotes FXII activation in a purified system. We found that long, but not short polyP increased the rate of FXII activation in presence of PK and HK. (Fig. 4A), confirming the observation that long polyP of the size accumulated in bacteria are more potent at activating the contact pathway than polyP of the size present in platelets [5]. We found that long and short polyP did not increase FXII autoactivation (Fig. S4A). We next investigated whether long polyP could directly promote PK activation. As shown in figure 4B, long and short polyP enhanced the rate of PK autoactivation. Furthermore, our data show that activation of PK by FXIIa was significantly increased by long, but not short polyP (fig. 4C).
Figure 4. PolyP promotes FXII and PK activation and the activation of FXI by thrombin, but not by FXIIa.

(A) FXII activation was measured in a purified system following the addition of 200 nM FXII, 50 nM PK and 50 nM HK in the presence of vehicle (◇) or 5 μM long (▲) or short (■) polyP. FXIIa generation was measured with a chromogenic substrate. (B) PK autoactivation following addition of 200 nM PK and 200 nM HK in the presence of vehicle (◇) or 5 μM long (▲) or short (■) polyP. Kallikrein generation was measured with a chromogenic substrate. (C) PK activation following addition of 50 nM PK, 50 nM HK and 100 pM FXIIa in the presence of vehicle (◇) or 5 μM long (▲) or short (■) polyP. Kallikrein generation was measured with a chromogenic substrate. (D) FXI activation was measured following addition of 5 μM long (□) or short (△) polyP to 30 nM FXI. In separate experiments, 5 nM α-thrombin was added to 30 nM FXI in the presence of vehicle (◇) or 5 μM long (▲) or short (■) polyP. FXIa generation was measured with a chromogenic substrate. (E) FXI activation following addition of 30 nM FXI and 5 nM α-FXIIa in the presence of vehicle (◇) or 5 μM long (▲) or short (■) polyP. FXIa generation was measured with a chromogenic substrate. (F) FXI activation following addition of 50 nM FXI, 200 nM FXII, 50 nM PK and 50 nM HK in the presence of vehicle (◇) or 5 μM long (▲) or short (■) polyP. FXIa generation was measured with a chromogenic substrate. Data are mean ± SE (n = 3).
We next designed experiments to determine whether polyP promotes FXI activation in a purified system. As shown in figure 4D, long, but not short polyP enhanced the rate of FXI activation by α-thrombin. We found that dextran sulfate increased the activation of FXI by FXIIa (Fig. S4B, C). In contrast, neither long nor short polyP increased FXI activation by FXIIa (Fig. 4E). Interestingly, in the presence of FXII, PK and HK, long and short polyP blocked the generation of FXIa (Fig. 4F). These data suggest that long polyP of the size accumulated in bacteria may play an important role in FXI activation by α-thrombin, but not by FXIIa.
Activation of FXII by long polyP promotes FIX and prothrombin activation in the absence of FXI
Our results suggested that the activation of FXII by long polyP could promote coagulation largely independent of FXIa generation by FXIIa or FIXa generation by FXIa. We therefore designed experiments to validate whether long polyP and FXIIa were able to promote coagulation in the absence of FXI. Our data show that long polyP decreased the clotting time of FXI-deficient plasma (Fig. 5A). The procoagulant activity of long polyP was inhibited by a blocking FIX antibody, and eliminated by the presence of CTI or rivaroxaban (Fig. 5A). Our data show that 150 nM FXIIa was able to decrease the clotting time of FXI-deficient plasma (Fig. 5A). The presence of blocking FXI antibodies, 1A6 or 14E11, did not inhibit the procoagulant effect of FXIIa, confirming the absence of FXI in FXI-deficient plasmas. Moreover, the addition of CTI or rivaroxaban abrogated the ability of FXIIa to induce clotting in FXI-deficient plasma (Fig. 5A). Addition of a blocking FIX antibody inhibited the ability of FXIIa to promote clotting in FXI-deficient plasma (Fig. 5A). These results suggest that the activation of FXII by long polyP shortens the clotting time of plasma by a mechanism that is dependent on FIX yet independent of FXI.
Figure 5. Activation of FXII by long polyP promotes FIX activation in absence of FXI.

(A) Plasma clotting time was measured in recalcified FXI-deficient plasma in the presence of vehicle (−), 150 nM α-FXIIa or 50 μM long polyP. In selected experiments, FXI-depleted plasma was pretreated with 20 μg/ml 1A6, 20 μg/ml 14E11, 20 μg/ml anti-FIX mAb, 50 μg/ml CTI or 20 μg/ml rivaroxaban for 10 minutes. The clotting time was recorded, with a maximum observation period of 1000 seconds. (B) Plasma clotting time was measured in recalcified FIX-depleted plasma in the presence of vehicle (−) or 150 nM FXIIa, 50 μM long polyP, or 10 nM FXIa. In selected experiments, FIX-depleted plasma was pretreated with 20 μg/ml 1A6, 20 μg/ml 14E11, 20 μg/ml anti-FIX mAb, 50 μg/ml CTI or 20 μg/ml rivaroxaban for 10 minutes. The clotting time was recorded, with a maximum observation period of 2500 seconds. Data are mean ± SE (n = 3). (C) FIX activation in a purified system following the addition of 100 nM FIX, 100 nM FXII, 50 nM PK and 50 nM HK in the presence of vehicle or 10 μM long polyP. FXIa generation was measured by western blot. (D) Prothrombin activation was measured in a purified system following the addition of 100 nM FXII, 50 nM PK and 50 nM HK to 1 μM prothrombin in the presence of vehicle (◇, ◆) or 10 μM long polyP (△, ▲), in the presence (◆, ▲, ●) or absence of Ca2+ (◇, △, ○). Long polyP plus FXII and PK in the absence of prothrombin resulted in no hydrolysis of the thrombin substrate (●, ○). Thrombin generation was measured with a chromogenic substrate. (E) Long polyP induced FIX activation in FXI-deficient plasma. Plasma was incubated with 50 μM long polyP in the absence of calcium in the presence or absence of CTI (50 μg/ml), then subjected to SDS-PAGE and western blotting with an antibody against FIX.
As shown in figure 5B, long polyP had only a minimal effect on clotting in FIX-depleted plasma. In contrast, we found that 150 nM FXIIa was able to decrease the clotting time of FIX-depleted plasma to ~700 sec (Fig. 5B). Addition of a blocking FIX antibody had a negligible effect on the clotting time of FIX-depleted plasma initiated by FXIIa. Interestingly, 14E11 and 1A6 partially blocked the ability of FXIIa to clot plasma (Fig. 5B), indicating that FXIa may promote coagulation in a FIX-independent manner. We found that 10 nM FXIa was able to clot FIX-depleted plasma (Fig. 5B). Addition of aprotinin, which inhibits FXIa, abrogated the ability of FXIa to clot FIX-depleted plasma. In contrast, addition of CTI did not affect the ability of FXIa to clot FIX-depleted plasma, indicating that the effect of FXIa is not explained by its capacity to activate FXII [24] in our system (Fig. 5B). As shown in figure S4B, FXIa was able to activate FX, and the antibody 1A6 did not block the activity of FXIa towards FX, suggesting that the effect of FXIa in FIX-depleted plasma could be explained by its capacity to promote FXa generation.
As shown in figure S4B, kallikrein, but not FXIIa was able to activate FIX. We next designed experiments to determine whether the activation of prekallikrein by long polyP was able to activate FIX. In a purified system, in the presence of FXII, PK and HK, long polyP promoted FIX activation (Fig. 5C), suggesting that the activation of kallikrein by long polyP may be able to promote FIX activation. As kallikrein and FXIIa were able to activate prothrombin (Fig. S4D), we next designed experiments to characterize whether the activation of FXII and prekallikrein by long polyP was able to generate thrombin. In a purified system in the presence of prothrombin, FXII, PK and HK, long polyP significantly increased the generation of thrombin in a calcium-independent manner (Fig. 5D). We did not detect any hydrolysis of the thrombin substrate in the absence of prothrombin.
We next designed experiments to measure the generation of FIXa by long polyP in plasma. We incubated FXI-deficient plasma with 50 μM long polyP in the absence of calcium, followed by western blotting to measure the generation of FIXa. Our results demonstrate that long polyP triggered the conversion of FIX to FIXa. The presence of CTI blocked the ability of long polyP to activate FIX (Fig. 5E)
Activation of FXII by long polyP promotes occlusive thrombus formation in a FXI-independent manner
Experiments were designed to determine whether long polyP or FXIIa were able to promote occlusive thrombus formation in whole blood. Citrated-whole blood was recalcified immediately before being driven under constant pressure through capillary tubes that had been coated with collagen and TF. Under these conditions, an occlusive thrombus formed after ~20 min. Pretreatment of blood with 1A6 prolonged the time to occlusion to ~28 min (Fig. 6); in contrast, the pretreatment of blood with 14E11 failed to prolong the time to occlusion, suggesting that the feedback activation of FXI plays a role in promoting thrombus formation in our system. Pretreatment of blood with CTI prolonged the time to occlusion to ~28 min, indicating a role for FXIIa in the promotion of occlusive thrombus formation. The pretreatment of blood with long polyP decreased the time to occlusion to ~12 min. Neither 1A6 nor 14E11 was able to inhibit the ability of long polyP to promote occlusive thrombus formation; in contrast, CTI blocked the ability of long polyP to reduce the time to occlusion, suggesting that long polyP promotes occlusive thrombus formation in a FXII-dependent, yet FXI-independent, manner.
Figure 6. Activation of FXII by long polyP promotes experimental thrombus formation in a FXI-independent manner.
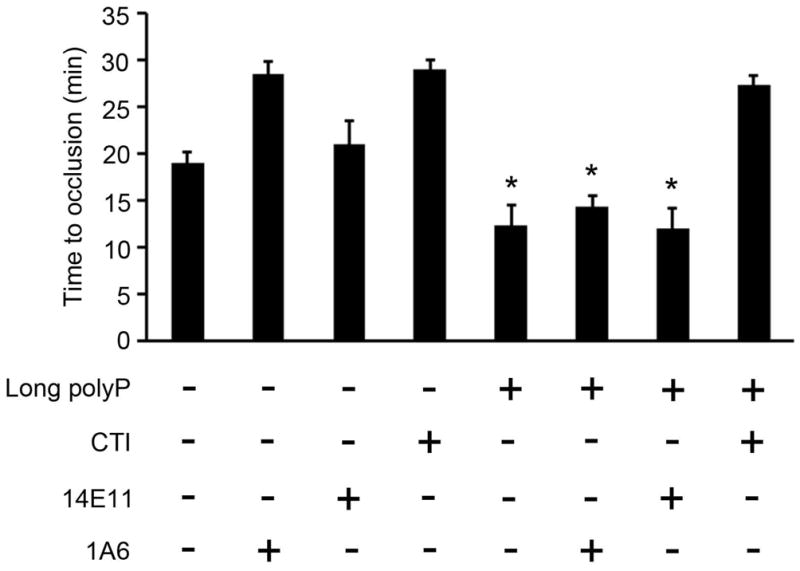
Citrate anticoagulated human blood was recalcified and perfused through a collagen/TF-coated glass capillary tube. Blood flow was driven by a constant pressure gradient and the time to occlusion of the tube was recorded. Blood was pretreated with 100 μM long polyP in presence of vehicle, 20 μg/ml 1A6, 20 μg/ml 14E11 or 50 μg/ml CTI. Data are mean ± SE (n = 3). (*P < 0.05)
Discussion
A role for the contact system in thrombosis has been postulated based upon the fact that FXII-deficient mice are protected in models of arterial thrombosis, pulmonary embolism and stroke [17, 25]. Moreover, we demonstrated that the FXI-blocking antibody, 14E11, which inhibits FXI activation by FXIIa, was protective in a non-human primate model of thrombosis and murine models of thrombosis [15], sepsis [27], and stroke [28], supporting the hypothesis that FXIIa-mediated activation of FXI contributes to pathological thrombus formation in vivo. However, the mechanism by which FXII contributes to thrombus formation has remained ill-defined.
A number of negatively charged compounds have been identified that activate FXII in vivo: extracellular RNA [28], collagen [27, 28], neutrophil extracellular traps (NETs) and platelet polyP have been shown to be prothrombotic in a FXII-dependent manner in mouse models of thrombosis [3,31]. The polymer length of polyP is known to vary; for instance, very long chains of polyP accumulate in microorganisms (~1000 phosphate units) [1], while short chains of polyP are secreted by platelets (~60–100 phosphates units) [2]. PolyP has been shown to trigger the contact pathway and accelerate FXI activation by either FXIa or thrombin [6,7]. The data from our ex vivo model of occlusive thrombus formation demonstrated that the prothrombotic effect of long polyP was dependent of FXIIa generation, yet largely independent of FXIa generation. Recent studies have shown that FXII-deficient mice are resistant to deep vein thrombosis, and that disintegration of NETs confers protection against venous thrombosis [31]. Interestingly, in this model, FXI-deficiency only had a minor impact on venous thrombogenesis. As NETs promote FXII activation, these findings raise the possibility that the role of FXII in thrombosis is not restricted to the activation of FXI. For instance, FXIIa has been shown to activate FVII [22] and modulate thrombus formation by regulating fibrin structure [32].
In a purified system, we found that FXIIa failed to activate FXI in the presence of either long or short polyP and PK and HK. Previous studies have demonstrated that polyP derived from E. coli (very long chain polymers [33]) was able to efficiently activate the kallikrein-kinin system in plasma [3]. Moreover, polysaccharide dextran sulfate, misfolded protein aggregates and mast cell-released heparin have been shown to initiate FXII-dependent kallikrein and bradykinin generation, which, unexpectedly, is not associated with FXIa generation [34–36]. In contrast to these polyanionic surfaces, E. coli polyP has been shown to be able to generate FXIa in plasma [3]. Based upon the fact that long polyP is a potent cofactor for the activation of factor FXI by thrombin and FXIa [7], perhaps the generation of FXIa by long polyP is largely dependent upon thrombin rather than FXIIa. Thus far, two forms of activated FXII have been identified, termed α-FXIIa and β-FXIIa. In plasma, α-FXIIa, which can activate FXI, is formed by a single cleavage of FXII. β-FXIIa is formed when α-FXIIa is cleaved a second time by kallikrein. This form of FXII has lost its capacity to induce FXIa formation, yet β-FXIIa retains the ability to activate PK [37]. We hypothesize that the generation of different forms of FXIIa by long polyP may explain why the activation of FXIIa promotes kallikrein generation but not FXIa.
Our study supports the notion that the ability of long polyP to promote coagulation is in part dependent upon FXIIa and FIXa generation, which can by-pass FXI to generate thrombin. Using purified proteins, we found that the long polyP in the presence of FXII, PK and HK promoted FIX and prothrombin activation. In FXI-deficient plasma, we observed that polyP was able to generate FIXa in a FXII-dependent manner. In a purified system, we observed that FXIIa was able to activate prothrombin directly, but not FIX, albeit the prohibitively slow rate of thrombin generation by FXIIa may diminish the physiological relevance of this reaction. Alternatively, kallikrein has been reported to directly activate not only prothrombin [23,39] but also FIX in the absence of calcium in vitro [22,38]. The fact that long polyP is able to increase the generation of kallikrein suggests that long polyP may be able to generate FIXa and thrombin in the absence of FXI due to its capacity to generate kallikrein (Fig. 7).
Figure 7. Summary of the role of long polyP in the promotion of coagulation bypassing FXI.
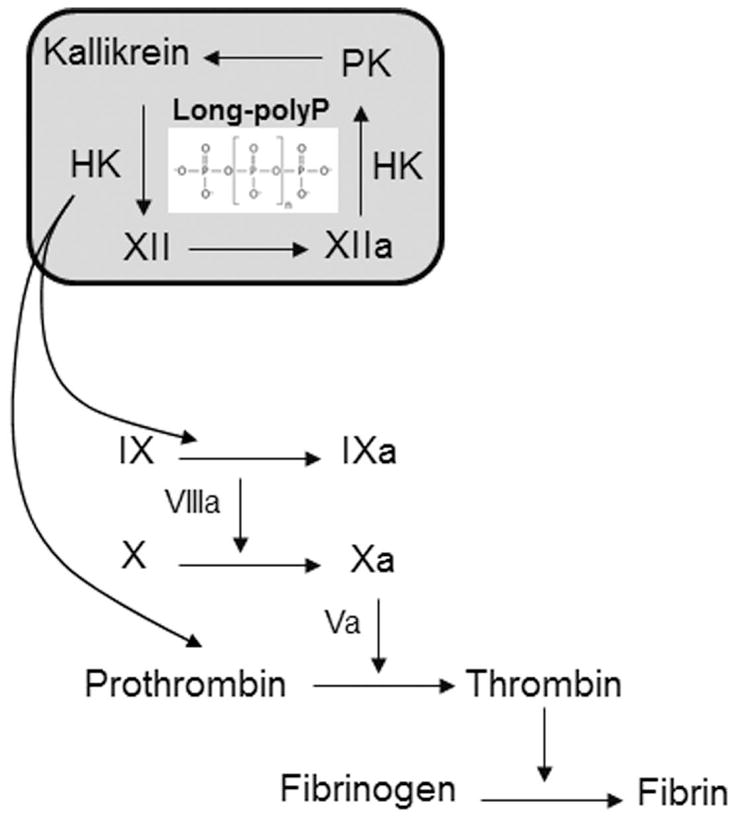
The activation of FXII and PK by long polyP promotes the activation of FIX and prothrombin in a manner independent of FXI activation.
FXII is believed to contribute to thrombus formation in sepsis-associated disseminated intravascular coagulation [40]. Along these lines, bacteria have been shown to assemble contact pathway coagulation factors on their surface, leading to the activation of FXII [41]. In this study, we showed that long polyP of the size present in bacteria was able to promote thrombus formation in a FXII-dependent manner. Damaged microorganisms have been shown to release intracellular polyP [42], while some microorganisms express large amounts of long polyP on the exterior of their cells [43]. Our data, in accord with other study [5], suggest that bacterial polyP can trigger blood coagulation and thrombus formation via the contact pathway as part of the host response to pathogens. Like bacteria, brain contains very long-chain polyP (~ 800 phosphate units) [44]. Interestingly, FXII-deficient mice are protected from pathological thrombosis in a model of stroke [25]. This observation supports the notion that release of long polyP by damaged brain cells due to cerebral ischemia may play a role in contact activation and subsequent thrombus formation in a FXII-dependent manner.
Clinical studies have shown a direct relationship between high levels of FXI and thrombosis in humans [13,14,45,46]; however, the direct association between FXII and thrombosis is not clear. There are several reports suggesting that FXII deficiency may predispose patients to thrombosis [47,48]. In contrast, other studies have shown that in most of the cases of thrombosis associated with FXII deficiency, there were other prothrombotic risk factors present [49]. Indeed, the risk of cardiovascular death in patients with severe FXII-deficiency has been reported to be similar to that of individuals having 100% FXII plasma levels, indicating that any potential thromboprotection may be counterbalanced by other mechanisms in patients with severe congenital FXII-deficiency [50]. Clinical studies have shown an association between elevated plasma FXIIa levels with an increased prevalence of coronary heart disease and a risk factor for current coronary events [51–53], supporting the notion that FXII contributes to thrombosis formation in humans. Taken together, our data suggest that the activation of FXII by long chain polyP can promote FIX and prothrombin activation bypassing FXI to promote thrombus formation (Fig. 7).
Supplementary Material
Acknowledgments
We thank Jiaqing Pang and Dr. David Farrell for technical support. This work was supported by the National Institutes of Health (NIH) grants R01 HL47014 (to J.H.M.), R01HL101972 (A.G. and O.J.T.M.), R44HL106919 (E.I.T), R44AI088937 (E.I.T. and A.G.), and the Oregon Clinical and Translational Research Institute (OCTRI), grant number (UL1TR000128) from the National Center for Advancing Translational Sciences (NCATS). O.J.T.M. is an American Heart Association (AHA) Established Investigator (13EIA12630000).
Footnotes
Disclosure of Conflicts of Interest: A.G., E.I.T., and Oregon Health & Science University have a significant financial interest in Aronora Inc, a company that may have a commercial interest in the result of this research. This potential conflict of interest has been reviewed and managed by the Oregon Health & Science University Conflict of Interest in Research Committee. The remaining authors declare no competing financial interests.
References
- 1.Kornberg A, Rao NN, Ault-Riché D. Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem. 1999;68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- 2.Ruiz FA, Lea CR, Oldfield E, Docampo R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem. 2004;279:44250–7. doi: 10.1074/jbc.M406261200. [DOI] [PubMed] [Google Scholar]
- 3.Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renné T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–56. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackman N, Gruber A. Platelet polyphosphate: an endogenous activator of coagulation factor XII. J Thromb Haemost. 2010;8:865–7. doi: 10.1111/j.1538-7836.2010.03832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith SA, Choi SH, Davis-Harrison R, Huyck J, Boettcher J, Rienstra CM, Morrissey JH. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood. 2010;116:4353–9. doi: 10.1182/blood-2010-01-266791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci USA. 2006;103:903–8. doi: 10.1073/pnas.0507195103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. 2011;118:6963–70. doi: 10.1182/blood-2011-07-368811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colman RW, Schmaier AH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–43. [PubMed] [Google Scholar]
- 9.Ratnoff OD, Colopy JE. A familial hemorrhagic trait associated with a deficiency of a clot-promoting fraction of plasma. J Clin Invest. 1955;34:602–13. doi: 10.1172/JCI103109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost. 2009;7 (Suppl 1):84–7. doi: 10.1111/j.1538-7836.2009.03395.x. [DOI] [PubMed] [Google Scholar]
- 11.Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–93. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 12.Kravtsov DV, Matafonov A, Tucker EI, Sun M-F, Walsh PN, Gruber A, Gailani D. Factor XI contributes to thrombin generation in the absence of factor XII. Blood. 2009;114:452–8. doi: 10.1182/blood-2009-02-203604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- 14.Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113–7. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- 15.Cheng Q, Tucker EI, Pine MS, Sisler I, Matafonov A, Sun M-F, White-Adams TC, Smith SA, Hanson SR, McCarty OJT, Renné T, Gruber A, Gailani D. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981–9. doi: 10.1182/blood-2010-02-270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tucker EI, Marzec UM, White TC, Hurst S, Rugonyi S, McCarty OJT, Gailani D, Gruber A, Hanson SR. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–44. doi: 10.1182/blood-2008-06-163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Renné T, Pozgajová M, Grüner S, Schuh K, Pauer H-U, Burfeind P, Gailani D, Nieswandt B. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–81. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Cheng Q, Xu L, Feuerstein GZ, Hsu M-Y, Smith PL, Seiffert DA, Schumacher WA, Ogletree ML, Gailani D. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3:695–702. doi: 10.1111/j.1538-7836.2005.01236.x. [DOI] [PubMed] [Google Scholar]
- 19.Itakura A, Aslan JE, Sinha S, White-Adams TC, Patel IA, Meza-Romero R, Vandenbark AA, Burrows GG, Offner H, McCarty OJ. Characterization of human platelet binding of recombinant T cell receptor ligand. J Neuroinflammation. 2010;7:75. doi: 10.1186/1742-2094-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berny MA, Patel IA, White-Adams TC, Simonson P, Gruber A, Rugonyi S, McCarty OJT. Rational design of an ex vivo model of thrombosis. Cell Mol Bioeng. 2010;3:187–9. [Google Scholar]
- 21.White-Adams TC, Berny MA, Patel IA, Tucker EI, Gailani D, Gruber A, McCarty OJT. Laminin promotes coagulation and thrombus formation in a factor XII-dependent manner. J Thromb Haemost. 2010;8:1295–301. doi: 10.1111/j.1538-7836.2010.03850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seligsohn U, Osterud B, Brown SF, Griffin JH, Rapaport SI. Activation of human factor VII in plasma and in purified systems: roles of activated factor IX, kallikrein, and activated factor XII. J Clin Invest. 1979;64:1056–65. doi: 10.1172/JCI109543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stief TW. Kallikrein activates prothrombin. Clin Appl Thromb Hemost. 2008;14:97–8. doi: 10.1177/1076029607308036. [DOI] [PubMed] [Google Scholar]
- 24.Matafonov A, Sarilla S, Sun M, Sheehan JP, Serebrov V, Verhamme IM, Gailani D. Activation of factor XI by products of prothrombin activation. Blood. 2011;118:437–45. doi: 10.1182/blood-2010-10-312983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer H-U, Burfeind P, Renné C, Gailani D, Nieswandt B, Renné T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203:513–8. doi: 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJT, Gailani D, Gruber A. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood. 2012;119:4762–8. doi: 10.1182/blood-2011-10-386185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leung PY, Hurst S, Berny-Lang MA, Verbout NG, Gailani D, Tucker EI, Wang RK, McCarty OJT, Gruber A. Inhibition of Factor XII-Mediated Activation of Factor XI Provides Protection Against Experimental Acute Ischemic Stroke in Mice. Transl Stroke Res. 2012;3:381–9. doi: 10.1007/s12975-012-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, Von Bruehl M-L, Sedding D, Massberg S, Günther A, Engelmann B, Preissner KT. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA. 2007;104:6388–93. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilner GD, Nossel HL, LeRoy EC. Activation of Hageman factor by collagen. J Clin Invest. 1968;47:2608–15. doi: 10.1172/JCI105943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van der Meijden PEJ, Munnix ICA, Auger JM, Govers-Riemslag JWP, Cosemans JMEM, Kuijpers MJE, Spronk HM, Watson SP, Renné T, Heemskerk JWM. Dual role of collagen in factor XII-dependent thrombus formation. Blood. 2009;114:881–90. doi: 10.1182/blood-2008-07-171066. [DOI] [PubMed] [Google Scholar]
- 31.Von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–35. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konings J, Govers-Riemslag JWP, Philippou H, Mutch NJ, Borissoff JI, Allan P, Mohan S, Tans G, Ten Cate H, Ariëns RAS. Factor XIIa regulates the structure of the fibrin clot independently of thrombin generation through direct interaction with fibrin. Blood. 2011;118:3942–51. doi: 10.1182/blood-2011-03-339572. [DOI] [PubMed] [Google Scholar]
- 33.Rao NN, Roberts MF, Torriani A. Amount and chain length of polyphosphates in Escherichia coli depend on cell growth conditions. J bacteriol. 1985;162:242–7. doi: 10.1128/jb.162.1.242-247.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johne J, Blume C, Benz PM, Pozgajová M, Ullrich M, Schuh K, Nieswandt B, Walter U, Renné T. Platelets promote coagulation factor XII-mediated proteolytic cascade systems in plasma. Biol Chem. 2006;387:173–8. doi: 10.1515/BC.2006.023. [DOI] [PubMed] [Google Scholar]
- 35.Oschatz C, Maas C, Lecher B, Jansen T, Björkqvist J, Tradler T, Sedlmeier R, Burfeind P, Cichon S, Hammerschmidt S, Müller-Esterl W, Wuillemin WA, Nilsson G, Renné T. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity. 2011;34:258–68. doi: 10.1016/j.immuni.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 36.Maas C, Govers-Riemslag JWP, Bouma B, Schiks B, Hazenberg BPC, Lokhorst HM, Hammarström P, Ten Cate H, De Groot PG, Bouma BN, Gebbink MFBG. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118:3208–18. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Revak SD, Cochrane CG, Bouma BN, Griffin JH. Surface and fluid phase activities of two forms of activated Hageman factor produced during contact activation of plasma. J Exp Med. 1978;147:719–29. doi: 10.1084/jem.147.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osterud B, Bouma BN, Griffin JH. Human blood coagulation factor IX. Purification, properties, and mechanism of activation by activated factor XI. J Biol Chem. 1978;253:5946–51. [PubMed] [Google Scholar]
- 39.Qian Y, Pan J, Zhou X, Weiser P, Lu H, Zhang L. Molecular mechanism underlines heparin-induced thrombocytopenia and thrombosis. Prog Mol Biol Transi Sci. 2010;93:395–421. doi: 10.1016/S1877-1173(10)93017-2. [DOI] [PubMed] [Google Scholar]
- 40.Colman RW. The role of plasma proteases in septic shock. N Engl J Med. 1989;320:1207–9. doi: 10.1056/NEJM198905043201809. [DOI] [PubMed] [Google Scholar]
- 41.Herwald H, Mörgelin M, Olsén A, Rhen M, Dahlbäck B, Müller-Esterl W, Björck L. Activation of the contact-phase system on bacterial surfaces--a clue to serious complications in infectious diseases. Nat Med. 1998;4:298–302. doi: 10.1038/nm0398-298. [DOI] [PubMed] [Google Scholar]
- 42.Noegel A, Gotschlich EC. Isolation of a high molecular weight polyphosphate from Neisseria gonorrhoeae. J Exp Med. 1983;157:2049–60. doi: 10.1084/jem.157.6.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tinsley CR, Manjula BN, Gotschlich EC. Purification and characterization of polyphosphate kinase from Neisseria meningitidis. Infect Immun. 1993;61:3703–10. doi: 10.1128/iai.61.9.3703-3710.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumble KD, Kornberg A. Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem. 1995;270:5818–22. doi: 10.1074/jbc.270.11.5818. [DOI] [PubMed] [Google Scholar]
- 45.Doggen CJM, Rosendaal FR, Meijers JCM. Levels of intrinsic coagulation factors and the risk of myocardial infarction among men: Opposite and synergistic effects of factors XI and XII. Blood. 2006;108:4045–51. doi: 10.1182/blood-2005-12-023697. [DOI] [PubMed] [Google Scholar]
- 46.Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105:269–73. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 47.Halbmayer WM, Mannhalter C, Feichtinger C, Rubi K, Fischer M. Factor XII (Hageman factor) deficiency: a risk factor for development of thromboembolism. Incidence of factor XII deficiency in patients after recurrent venous or arterial thromboembolism and myocardial infarction. Wien Med Wochenschr. 1993;143:43–50. [PubMed] [Google Scholar]
- 48.Kuhli C, Scharrer I, Koch F, Ohrloff C, Hattenbach L-O. Factor XII deficiency: a thrombophilic risk factor for retinal vein occlusion. Am J Ophthalmol. 2004;137:459–64. doi: 10.1016/j.ajo.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 49.Girolami A, Randi ML, Gavasso S, Lombardi AM, Spiezia F. The occasional venous thromboses seen in patients with severe (homozygous) FXII deficiency are probably due to associated risk factors: a study of prevalence in 21 patients and review of the literature. J Thromb Thrombolysis. 2004;17:139–43. doi: 10.1023/B:THRO.0000037670.42776.cd. [DOI] [PubMed] [Google Scholar]
- 50.Endler G, Marsik C, Jilma B, Schickbauer T, Quehenberger P, Mannhalter C. Evidence of a U-shaped association between factor XII activity and overall survival. J Thromb Haemost. 2007;5:1143–8. doi: 10.1111/j.1538-7836.2007.02530.x. [DOI] [PubMed] [Google Scholar]
- 51.Zito F, Drummond F, Bujac SR, Esnouf MP, Morrissey JH, Humphries SE, Miller GJ. Epidemiological and genetic associations of activated factor XII concentration with factor VII activity, fibrinopeptide A concentration, and risk of coronary heart disease in men. Circulation. 2000;102:2058–62. doi: 10.1161/01.cir.102.17.2058. [DOI] [PubMed] [Google Scholar]
- 52.Colhoun HM, Zito F, Norman Chan N, Rubens MB, Fuller JH, Humphries SE. Activated factor XII levels and factor XII 46C>T genotype in relation to coronary artery calcification in patients with type 1 diabetes and healthy subjects. Atherosclerosis. 2002;163:363–9. doi: 10.1016/s0021-9150(02)00022-9. [DOI] [PubMed] [Google Scholar]
- 53.Grundt H, Nilsen DWT, Hetland Ø, Valente E, Fagertun HE. Activated factor 12 (FXIIa) predicts recurrent coronary events after an acute myocardial infarction. Am Heart J. 2004;147:260–6. doi: 10.1016/j.ahj.2003.07.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.