

Abstract
Aims
The vascular endothelial growth factor (VEGF) family contains four major isoforms and three receptor subtypes. The expressions of each VEGF isoform in cardiac repair/remodeling after myocardial infarction (MI) remains uncertain and is investigated in the current study.
Methods and Results
Temporal and spatial expressions of VEGF isoforms and VEGFR subtypes were examined in the infarcted rat heart. Sham-operated rats served as controls. We found that the normal myocardium expressed all VEGF isoforms. Following MI, VEGF-A was only increased in the border zone at day 1 and was significantly decreased in the infarcted heart during the 42 day observation period afterwards. VEGF-B was significantly suppressed in the infarcted heart. VEGF-C and VEGF-D were markedly increased in the infarcted heart in both early and late stages of MI. VEGFR-1 and 2 were significantly decreased in the infarcted heart, while VEGFR-3 was significantly increased, which was primarily expressed in blood vessels and myofibroblasts (myoFb).
Conclusions
VEGF isoforms and VEGFR subtypes are differentially expressed in the infarcted heart. Increased VEGF-A in the very early stage of MI suggests the potential role in initiating the cardiac angiogenic response. Suppressed cardiac VEGF-B postMI suggests that it may not be critical to cardiac repair. The presence of enhanced VEGF–C and VEGF-D along with its receptor, VEGFR-3, in various cell types of the infarcted heart suggest that these isoforms may regulate multiple responses during cardiac repair/remodeling.
Keywords: Myocardial infarction, VEGF isoforms, VEGF receptor subtypes, Cardiac repair/remodeling
INTRODUCTION
Ventricular dysfunction appears most commonly in patients with previous myocardial infarction (MI). MI induces loss of contractile myocardium and blood vessels in the infarcted myocardium. Cardiac repair occurring at the site of myocyte loss preserves the structural integrity of the heart and is essential to its recovery. Angiogenic, inflammatory and fibrogenic responses are central to cardiac repair. Studies from our group have shown that newly formed vessels first appear in the border zone (the region between the infarcted and noninfarcted myocardium) following infarction, then extend into the infarcted myocardium [1, 2]. Microvascular density was most evident in the infarcted myocardium within the first two weeks postMI.
Angiogenesis is coincident with inflammatory and fibrogenic responses in the infarcted myocardium. Monocyte-derived macrophages are the major inflammatory cells involved in cardiac repair and phenotypically transformed fibroblasts (myoFb) play a major role in fibrous tissue formation (scar) in the infarcted myocardium [1, 3, 4, 5]. MyoFb are not resident cells in the normal heart, while appear and accumulated in the infarcted myocardium. Cells that account for the appearance of myoFb have been demonstrated to include interstitial fibroblasts and circulating bone marrow-derived progenitor cells. Specific factors that facilitate this differentiation process have been identified. It is presumed that TGF-β1 governs the appearance of the myoFb phenotype.
Factors regulating cardiac repair/remodeling have been extensively investigated and transforming growth factor, fibroblast growth factor and platelet-derived growth factor have been shown to contribute to cardiac repair/remodeling postMI [6, 7]. The VEGF family of growth factors play a key role in angiogenesis and lymphangiogenesis by promoting new vessel formation during embryonic development and after injury and by creating collateral vessels to bypass blocked vessels [8, 9]. The VEGF family is composed of five different isoforms: VEGF-A, B, C, D and placenta growth factor. VEGFs initiate cellular responses by interacting with tyrosine kinase receptors on the cell surface. There are three main VEGF receptor subtypes, VEGFR-1 (Flt-1), VEGFR-2 (KDR/Flk-1) and VEGFR-3(Flt-4). VEGF-A binds to VEGFR-1 and VEGFR-2; VEGF-B to VEGFR-1; and VEGF-C and VEGF-D to VEGFR-2 and VEGFR-3 [10]. Previous studies by us and others have shown that cardiac VEGF-A expression is only increased in the very early stage of MI [1]. This suggests that VEGF-A plays a role in initiating angiogenesis but may not be critical in the later stages of angiogenic response. The potential role of the other VEGF isoforms, VEGF-B, C and D, in cardiac angiogenesis postMI remains unknown, and the first aim of the study is to determine the temporal expression of these isoforms and their receptors in the infarcted heart.
In addition to blood vessels, VEGFR have been observed in other types of cells, such as neurons, cancer cells and epithelial cells [11-13], suggesting that VEGF also regulates other cell processes in addition to angiogenesis and lymphangiogenesis. The second aim of the study, therefore, was to investigate whether VEGF plays a role in other responses of cardiac repair/remodeling postMI, such as inflammation and fibrosis.
METHODS
Animal Model
The study was approved by University of Tennessee Health Science Center Animal Care and Use Committee. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology: Shewan LG and Coats AJ. Ethics in the authorship and publishing of scientific articles. Int J Cardiol 2010;144:1-2.
Eight-week-old male Sprague-Dawley rats (Harlan, Indianapolis, IN) were anesthetized with 1.5% isoflurane, intubated and ventilated with a rodent respirator. The adequacy of anesthesia was determined by tail pinch. The heart was exposed via a left thoracotomy and the left anterior descending artery was ligated with a 7-0 silk suture. The chest was then closed and lungs reinflated using positive end-expiratory pressure [1]. The surgery leads to free wall infarction of the left ventricle. Sham-operated rats served as controls. Rats with MI were sacrificed at postoperative day 1, 3, 7, 14, 28 and 42 (n = 8 rats for each time point) by isoflurane anesthesia followed by heart removal. The heart was kept frozen at -80 °C until use.
RT-PCR
Total RNA was extracted from cardiac tissue using Trizol Reagent (Invitrogen, Carlsbad, CA). The RNA was treated with DNase using TURBO DNA-free kit (Ambion, Austin, TX), and purified with RNeasy Mini Kit (Qiagen Inc. USA, Valencia, CA). The purification, concentration and integrity of the RNA were examined with NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE), and Angilent Bioanalyzer (Angilent Technologies, Foster City, CA), respectively. cDNA was prepared from total RNA using a High Capacity cDNA reverse transcriptase kit (Applied Biosystems Foster City, CA). The gene-specific probes and primer sets for VEGF-A, B, C and D, and VEGFR-1, 2, and 3 were deduced using Universal ProbeLibrary Assay Design software (https://www.roche-applied-science.com). VEGF-A, B, C and D, and VEGFR-1, 2, and 3 mRNA levels were detected and analyzed on an LightCycler 480 System (Roche, Indianapolis, IN) under the following cycling conditions: 1 cycle at 95°C for 5 min, 45 cycles at 95°C for 10 seconds, 60°C for 30 seconds, and 72°C for 10 seconds. The PCR mix contained 0.2μl of 10μM primers, 0.1μl of 10μM Universal library probe, 5μl of LC 480 master mix (2X), 2μl of template cDNA and RNase-free water to 10μl. TATA box-binding protein has been selected as the endogenous quantity control. Fold change was used to compare the difference between groups [7].
Western Blotting
VEGF-A, B, C and D and VEGFR-1, 2 and 3 protein levels were measured by western blot. Briefly, the normal, noninfarcted left myocardium (septum), border zone and infarcted myocardium were dissected and homogenized separately. The supernatants were collected and separated by 10% SDS-PAGE. Protein concentration in each sample was measured in duplicate using Bradford using a BSA standard curve for loading the same amount of protein per sample/lane. After electrophoresis, samples were transferred to PVDF membranes and incubated with antibody against VEGF-A, B and D (Santa Cruz Biotechnology, Santa Cruz, CA), VEGF-C (R&D, Minneapolis, MN), VEGFR-1 (Santa Cruz Biotechnology, Santa Cruz, CA), VEGFR-2 (Cell Signaling Technology, Danvers, MA), and VEGFR-3 (R&D, Minneapolis, MN). Blots were subsequently incubated with peroxidase-conjugated secondary antibody (Sigma, St. Louis, MO). After washing, the blots were developed with an enhanced chemoluminescence method. The amount of protein detected was assessed by means of quantitative densitometry with a computer image analyzing system [7]. However, the most commonly used internal controls including GAPDH, beta actin, and beta tubulin were significantly altered in the infarcted myocardium compared to the control heart and therefore could not be used as internal controls in this study [14]. Thus, eight samples were detected in each group to provide the reliable results.
Fluorescence immunohistochemistry
Vascular endothelial cells, smooth muscle cells, myoFb and macrophages are the primary cells in the infarcted myocardium. Cells expressing VEGFR-3 in the infarcted myocardium were determined by fluorescent immunohistochemistry. For double fluorescence immunohistochemical localization of VEGFR-3 in blood vessels, myoFb or macrophages, cryostat sections (6μm) were prepared and blocked in 1% bovine serum albumin. The mouse anti-α-smooth muscle actin (α-SMC, a marker of myoFb and blood vessels) (Sigma, St. Louis, MO) antibody or mouse anti-ED-1 (a marker of macrophages) (Serotec, Oxford, UK) were applied and revealed using goat anti-mouse IgG-FITC (Sigma, St Louis, MO). Rabbit anti-VEGFR-3 antibody (Santa Cruz, Santa Cruz, CA) was then applied and revealed by Cy3-labeled anti-rabbit IgG (Sigma, St. Louis, MO). Samples were examined with a laser-scanning confocal image system (Zeiss LSM510).
Statistical Analysis
Statistical analysis of data obtained from RT-PCR and western blot was performed using analysis of variance. Values are expressed as mean±SEM with P<0.05 considered significant. Multiple group comparisons among controls and each group were made by Scheffe′s F-test.
RESULTS
Cardiac VEGF-A, B, C and D Gene Expression
VEGF-A, B, C, and D mRNAs were detectable in the control heart (Figure 1). Compared to controls, VEGF-A mRNA in the infarcted myocardium was significantly decreased from day 1 to 42 postMI. VEGF-A mRNA in the border zone was slightly increased at day 1 postMI and then significantly decreased from day 3 to 42. VEGF-A mRNA in the noninfarcted septum remained unchanged during the 42 day observation period (Figure 1).
Figure 1.
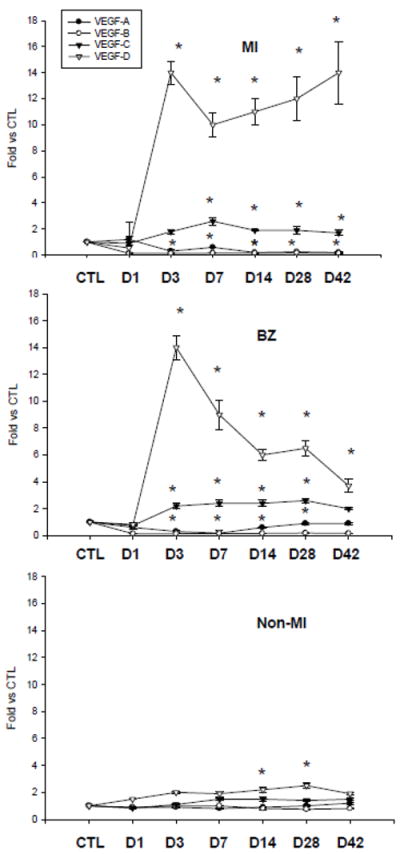
Cardiac VEGF isoform gene expression: Compared to controls, VEGF-A and B mRNA were decreased in the infarcted myocardium (MI) and the border zone (BZ) but remained unchanged in noninfarcted myocardium (Non-MI). VEGF-C mRNA was increased in the infarcted myocardium and border zone from day 3 to 42, but was unchanged in the noninfarcted myocardium. VEGF-D was markedly increased in the infarcted myocardium and border zone in the early and late stages of MI and in the noninfarcted myocardium in the late stage of MI.
Compared to controls, VEGF-B mRNA was significantly reduced in the border zone and infarcted myocardium during the 42 day observation period. VEGF-B mRNA remained unchanged in the noninfarcted myocardium in both early and late stages of MI (Figure 1).
VEGF-C mRNA was significantly increased in the border zone and infarcted myocardium over the course of 42 days postMI. VEGF-C mRNA levels in the noninfarcted myocardium were similar to controls (Figure 1).
Compared to controls, VEGF-D mRNA levels in the border zone and infarcted myocardium were reduced at day 1 postMI, then markedly elevated at day 3 and remained increased over the course of 42 days. Compared to controls, VEGF-D mRNA levels in the noninfarcted myocardium were significantly elevated at day 14 and 28 postMI (Figure 1).
Cardiac VEGF-A, B, C and D Protein Expression
As detected by western blot, VEGF-A, B, C, and D proteins were present in the control heart (Figure 2).
Figure 2.
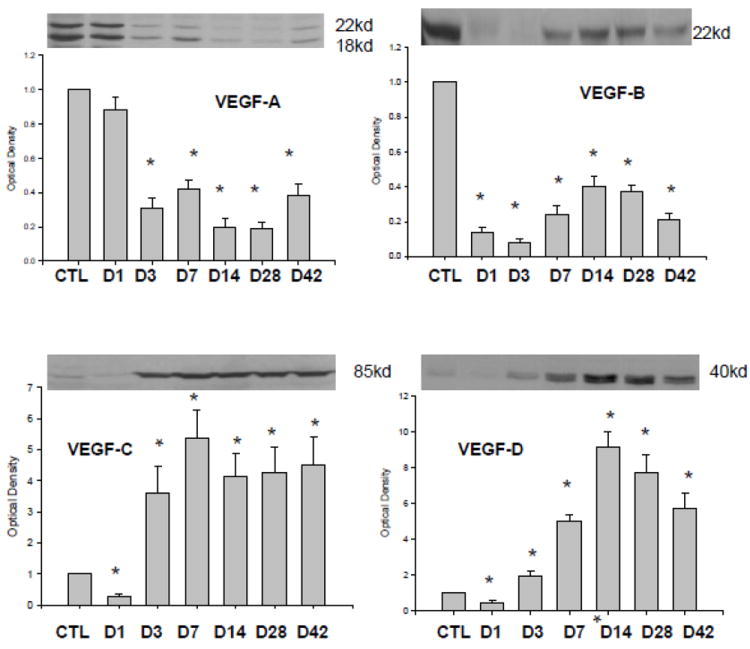
Expression of VEGF isoforms in the infarcted myocardium: Compared to controls, VEGF-A164 (18KD) and VEGF-A188 (22KD) and VEGF-B protein levels were reduced over the course of 42 days of observation period. VEGF-C and VEGF-D expressions were suppressed at day 1 postMI and then significantly increased from day 3 to day 42.
In the infarcted myocardium (Figure 2), VEGF-A and B protein levels were significantly reduced during the 42 day observation period, while VEGF-C and VEGF-D protein levels were reduced at day 1 postMI, then significantly increased at day 3, and remained significantly elevated thereafter.
In the border zone (Figure 3), VEGF-A was significantly increased at day 1 postMI, then reduced from day 3 and remained suppressed thereafter. VEGF-B levels was significantly reduced from day 1 to 14 and returned to normal levels at day 28 and 42. VEGF-C and VEGF-D protein levels were reduced at day 1 and significantly increased thereafter.
Figure 3.
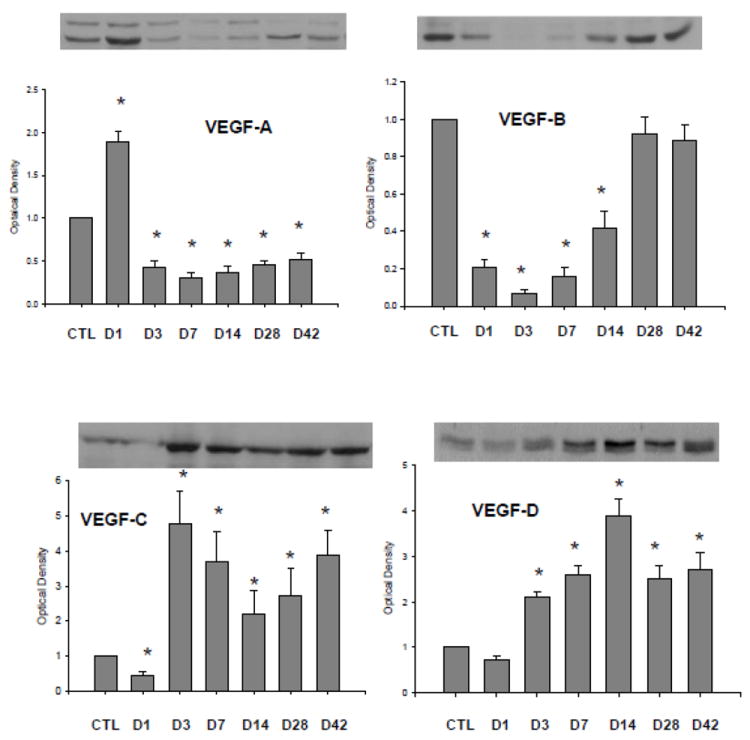
Expression of VEGF isoforms in the border zone: Compared to controls, VEGF-A164, but not VEGF-A188, at day 1 postMI was significantly increased and then declined. VEGF-B protein levels were reduced from day 1 through day 14 and then returned to control level. VEGF-C and VEGF-D expressions were significantly increased from day 3 to day 42.
VEGF-A, B, C, and D protein levels were not different from controls in noninfarcted myocardium (not shown).
Cardiac VEGFR-1, 2, and 3 Gene Expression
VEGFR-1, 2 and 3 mRNAs were normally expressed in the rat heart (Figure 4). After MI, VEGFR-1 mRNA was significantly reduced in the infarcted myocardium during 42 days of observation period. VEGFR-2 mRNA remained unchanged in the infarcted myocardium at day 1 and 3 and then decreased compared to controls. VEGFR-3 was, however, significantly upregulated in the infarcted myocardium at day 1 and remained elevated thereafter (Figure 4).
Figure 4.
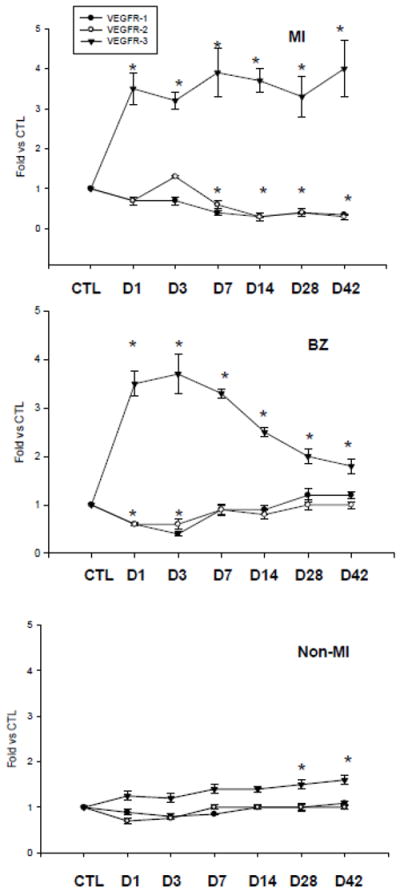
Cardiac VEGFR gene expression: Compared to controls, VEGFR-1 and VEGFR-2 mRNA was decreased or unchanged in the infarcted heart. VEGFR-3 mRNA was significantly increased in the border zone and infarcted myocardium in the early and late stage of MI. VEGFR-3 mRNA was elevated in the noninfarcted myocardium only in the late stage of MI.
VEGFR-1 and 2 mRNAs in the border zone were significantly reduced at day 1 and 3 and then returned to the normal levels. VEGFR-3 mRNA was significantly increased in the border zone at day 1, peaked at day 3 and gradually declined, but remained significantly higher than controls (Figure 4).
Compared to controls, VEGFR-1 and VEGFR-2 mRNA remained unchanged in the noninfarcted myocardium throughout 42 days postMI. VEGFR-3 mRNA level in the noninfarcted myocardium was not different from controls in the early stage of MI, but was significantly increased at day 28 and 42 (Figure 4).
Cardiac VEGFR-1, 2, and 3 Protein Expression
Compared to controls, VEGFR-1 protein level was significantly reduced in the infarcted myocardium from day 1 to 42. VEGFR-1 was also significantly decreased in the border zone from day 1 to 14 and recovered to the normal level at day 28 and 42 (Figure 5).
Figure 5.
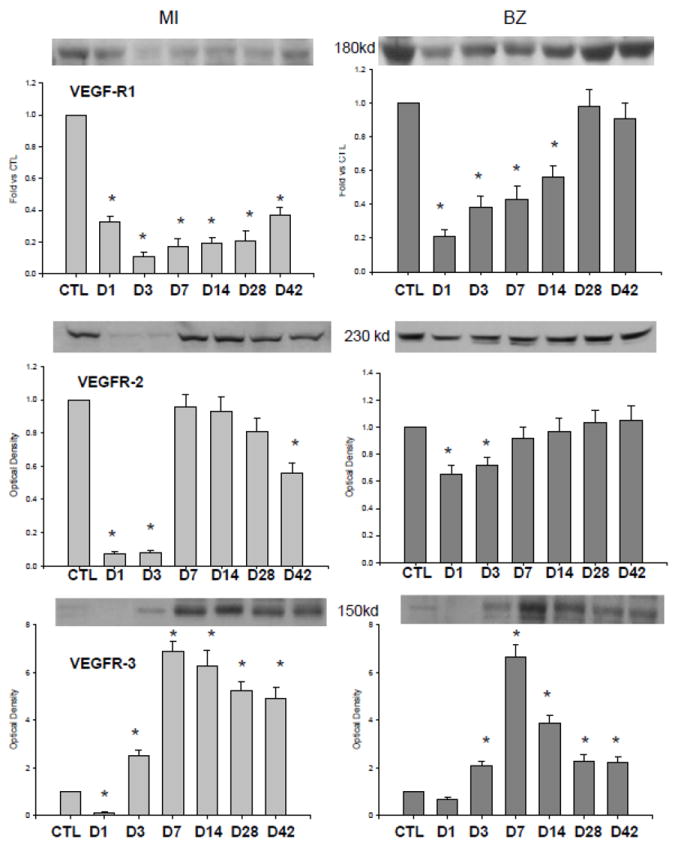
Cardiac expression of VEGFR subtypes. Compared to controls, VEGFR-1 and VEGFR-2 were reduced or unchanged in the infarcted myocardium and border zone. VEGFR-3 was reduced in the infarcted myocardium and border zone at day 1, significantly increased at day 3 and remained elevated thereafter.
VEGFR-2 was significantly suppressed in the infarcted and the border zone at day 1 and 3 and returned to the control levels in the later stages of MI (Figure 5).
Compared to the control heart, VEGFR-3 was reduced in both the infarcted myocardium and border zone at day 1, significantly increased at day 3, peaked at day 7 and gradually declined thereafter, but remained significantly higher than controls.
Cells Expressing VEGFR-3 in the normal and Infarcted Myocardium
As detected by fluorescent immunohistochemistry, α-SMC+ myoFb in the interstitial space and smooth muscle cells in newly formed vessels were evident in the infarcted myocardium (Figure 6). ED-1 staining revealed macrophages accumulated at the infarct site (Figure 7).
Figure 6.
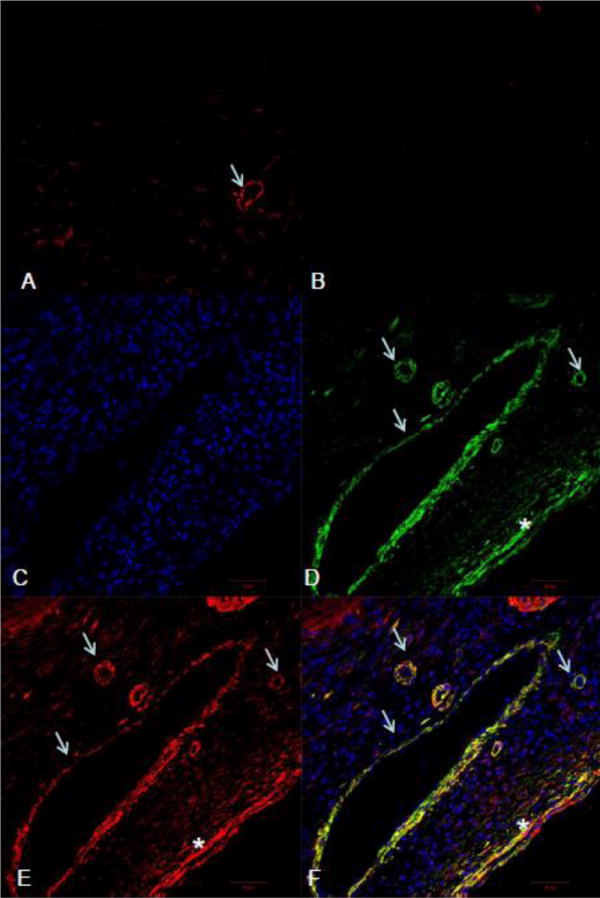
Fluorescent α-SMA and VEGFR-3 double staining in the heart: Immunohistochemistry revealed positive VEGFR-3 staining in blood vessels (arrows) of the normal heart (panel A). α-SMA labeling showed that myoFb (*) and newly formed vessels (arrows) appeared and accumulated in the infarcted myocardium at day 7 postMI (panel D). VEGFR-3 staining revealed positive staining in the same region (panel E), which is colocalized with blood vessels and myoFb (yellow) (panel F). Panel B: negative control; panel C: nuclear dapi staining.
Figure 7.
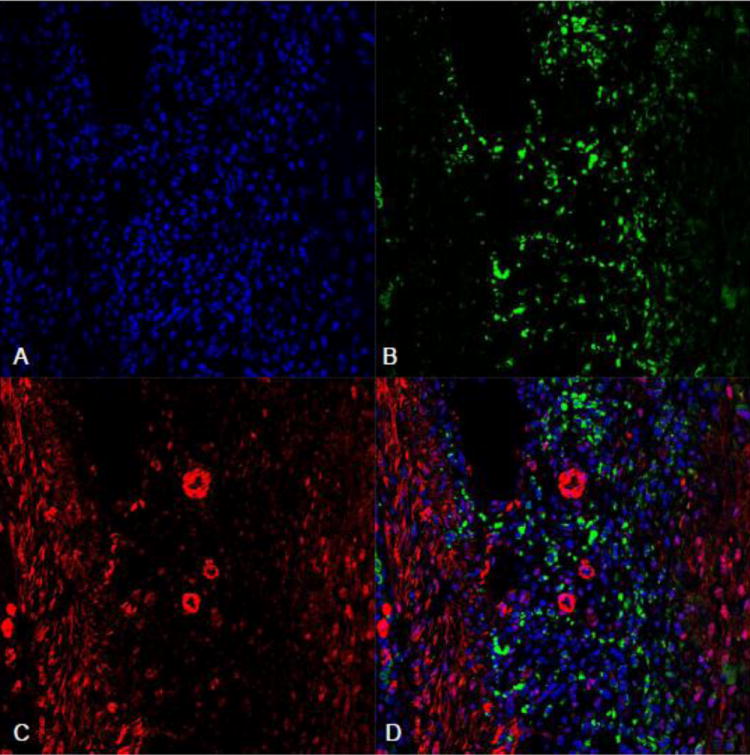
Fluorescent ED-1 and VEGFR-3 double staining in the infarcted myocardium: ED-1+ macrophages (green) were accumulated in the infarcted myocardium at day 7 postMI (panel B). Panel C showed VEGFR-3 staining (red) in the same region. Overlapped images of ED-1 and VEGFR-3 (panel D) showed that VEGFR-3 was negatively expressed in macrophages. Panel A: nuclear dapi staining.
VEGFR-3 labeling was observed in the blood vessels in the normal myocardium (Figure 6). Following MI, positive VEGFR-3 staining was observed in the border zone and infarcted myocardium. Cells expressing VEGFR-3 were primarily myoFb and blood vessels (Figure 6). VEGFR-3 staining was negatively labeled in macrophages (Figure 7).
DISSCUSSION
To our knowledge, this is the first study exploring the expression of the VEGF isoforms and their receptors in the infarcted heart and their potential regulation on cardiac repair/remodeling.
Firstly, our results have shown that all of VEGF isoforms and VEGFR subtypes are expressed in the blood vessels of the normal heart. This observation indicates that VEGF isoforms plays a role in the maintenance of the growth and function of blood vessels in the normal adult heart.
Secondly, our results showed that VEGF isoforms were differentially expressed in the infarcted heart compared to controls. VEGF-A is the most extensively studied member of the VEGF family. It contains three major isoforms in the rat (VEGF120, VEGF164, and VEGF188), arising from alternative splicing of mRNA and the relative abundance of each isoform is similar in the infarcted heart [15]. Our results showed that VEGF-A164 (18KD) and VEGF188 (22KD) expression was elevated only in the border zone at day 1 postMI, but decreased in the later stages. However, VEGF-A164 and VEGF188 expression was significantly decreased in the infarcted and noninfarcted myocardium, confirming our previous findings.
VEGF-A effects are mediated by activation of VEGFR-1 and VEGFR-2. VEGFR-2 signaling appears to mediate nearly all of the known cellular responses [16]. The function of VEGFR-1 is less well defined, but is thought to modulate VEGFR-2 signaling. We found that both VEGFR-1 and VEGFR-2 were remained unchanged or were reduced in the infarcted heart. These findings suggest that VEGF-A may play a role in initiating the angiogenic response in the infarcted heart, but it is not critical for the ongoing angiogenic process. The proangiogenic role of VEGF-A in pathological conditions has been investigated most extensively in tumors [17-19]. VEGF-A serves as the predominant promoter of tumor growth by stimulating vascular endothelial cell growth, survival, and proliferation.
While VEGF-A is important for the formation of blood vessels, VEGF-B seems to play a role only in the maintenance of newly formed blood vessels during pathological conditions [20]. The present study showed that cardiac VEGF-B mRNA and protein levels were persistently reduced following MI. VEGF-B exerts its effects via the VEGFR-1, which was also reduced in the infarcted heart. The importance of the differential expression of VEGF isoforms is not clear, but reduced VEGF-B expression in the infarcted heart suggests that it may not be critical to cardiac angiogenesis postMI.
VEGF-C is known to play a role in angiogenesis and lymphangiogenesis [21]. It also affects the permeability of blood vessels. VEGF-C exerts its effect through VEGFR-2 and VEGFR-3 activation. We observed significantly increased VEGF-C mRNA and protein levels in the infarcted myocardium and border zone in both early and late stages of MI. Enhanced cardiac VEGF-C expression is spatially and temporally coincident with VEGFR-3 expression, suggesting that VEGF-C is involved in ongoing cardiac angiogenesis postMI. The importance of VEGF-C in wound healing has been reported in tissue repair. VEGF-C enhanced angiogenesis and lymphangiogenesis in the wound and significantly accelerated wound healing in comparison to the control wounds in diabetic repair. Blockage of VEGF-C with a specific inhibitor delayed diabetic wound closure [22].
VEGF-D also is known to play a role in stimulating lymphangiogenesis and angiogenesis [23]. It is secreted from the cell as a full-length form with propeptides flanking a central region containing binding sites for VEGFR-2 and VEGFR-3. VEGF-D is normally expressed in various tissues including heart, lung, skeletal muscle, colon and small intestine [24]. In pathological conditions, such as cancer, VEGF-D functions as a mitogen for endothelial cells that promotes tumor growth and metastatic spread [25, 26]. The potential role of VEGF-D in angiogenesis in the repairing tissue is less understood. We showed that VEGF-D mRNA and protein levels were significantly increased in the infarcted myocardium in both early and late stages of MI, which is coincident with enhanced VEGFR-3 expression. Furthermore, VEGFR-3 was expressed in newly formed vessels in the infarcted myocardium, suggesting that VEGF-D is involved in angiogenesis in the infarcted heart.
Thirdly, we investigated whether VEGF-D, the most dramatically increased VEGF isoform in the infarcted heart, may be involved in cardiac repair responses other then angiogenesis. Following MI, macrophages are accumulated in the infarcted myocardium and border zone and are the major inflammatory cells involved in cardiac repair. These inflammatory cells participate in the removal of necrotic cells, and release inflammatory cytokines/growth factors. In addition, fibrous tissue formation is activated in the infarcted myocardium. The fibrogenic component substitutes for lost myocytes in the infarcted myocardium. Cells responsible for fibrous tissue formation at the site of MI consist principally of myoFb [27]. MyoFb are not residential cells in the normal myocardium, but appear at the border zone and the infarct site during the first week postMI, rapidly proliferate and are highly active in fibrous tissue formation via their expression of type I and III fibrillar collagens. Our immunohistochemical study showed that in addition to blood vessels, VEGFR-3 was also highly expressed in myoFb in the infarcted myocardium. These results imply that enhanced VEGF-D is involved in the fibrogenic response and contribute to cardiac repair. Further studies are required to explore specific roles of VEGF-D in cardiac fibrosis.
Multi-factors have been reported to activate the expression of VEGF. Hypoxia stimulates the expression of VEGF-A in human lung cancer cells [28]. Inflammatory cytokines, including interleukin-1beta and tissue necrosis factor-alpha up-regulate VEGF-C expression in fibroblasts [29]. Insulin growth factor increases VEGF-C expression in breast cancer [30]. Further studies are required to determine the stimuli of VEGF-C and VEGF-D expression in the repairing heart.
In summary, this the first study exploring the expression and potential role of VEGF isoforms and VEGFR subtypes in the repair of the infarcted rat heart. VEGF-A was only upregulated at the border zone at day 1, suggesting its potential role in initiating the angiogenic response in the damaged myocardium. VEGF-C, VEGF-D and VEGFR-3 were up-regulated in the infarcted myocardium. VEGFR-3 was primarily expressed in blood vessels and myoFb, suggesting that VEGF-D is not only involved in angiogenesis, but also in fibrogenic response in the infarcted myocardium. Reduced cardiac VEGF-B postMI suggests that this isoform may not be critical in cardiac repair. Further studies are required to better understand the specific roles of VEGF-C and VEGF-D in cardiac repair.
Acknowledgments
This work was supported by NIH Heart, Blood, and Lung Institute (1RO1-HL096503, Yao Sun).
Footnotes
Conflict of Interest: none declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhao T, Zhao W, Chen Y, et al. Vascular endothelial growth factor (VEGF)-A: role on cardiac angiogenesis following myocardial infarction. Microvasc Res. 2010;80:188–94. doi: 10.1016/j.mvr.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao W, Zhao T, Chen Y, et al. Reactive oxygen species promote angiogenesis in the infarcted rat heart. Int J Exp Pathol. 2009;90:621–9. doi: 10.1111/j.1365-2613.2009.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao W, Zhao D, Yan R, Sun Y. Cardiac oxidative stress and remodeling following infarction: role of NADPH oxidase. Cardiovasc Pathol. 2009;18:156–66. doi: 10.1016/j.carpath.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun Y, Cleutjens JP, Diaz-Arias AA, Weber KT. Cardiac angiotensin converting enzyme and myocardial fibrosis in the rat. Cardiovasc Res. 1994;28:1423–32. doi: 10.1093/cvr/28.9.1423. [DOI] [PubMed] [Google Scholar]
- 5.Martinez Rosas M. Cardiac remodeling and inflammation. Arch Cardiol Mex. 2006;76(Suppl 4):S58–66. [PubMed] [Google Scholar]
- 6.Nah DY, Rhee MY. The inflammatory response and cardiac repair after myocardial infarction. Korean Circ J. 2009;39:393–8. doi: 10.4070/kcj.2009.39.10.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao T, Zhao W, Chen Y, et al. Acidic and basic fibroblast growth factors involved in cardiac angiogenesis following infarction. Int J Cardiol. 2011;152:307–13. doi: 10.1016/j.ijcard.2010.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 9.Ferrara N. Vascular endothelial growth factor and the regulation of angiogenesis. Recent Prog Horm Res. 2000;55:15–35. discussion -6. [PubMed] [Google Scholar]
- 10.Yamazaki Y, Morita T. Molecular and functional diversity of vascular endothelial growth factors. Mol Divers. 2006;10:515–27. doi: 10.1007/s11030-006-9027-3. [DOI] [PubMed] [Google Scholar]
- 11.Waldner MJ, Wirtz S, Jefremow A, et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J Exp Med. 2010;207:2855–68. doi: 10.1084/jem.20100438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yi ZY, Feng LJ, Xiang Z, Yao H. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in hepatocellular carcinoma cells. J Invest Surg. 2011;24:67–76. doi: 10.3109/08941939.2010.542272. [DOI] [PubMed] [Google Scholar]
- 13.Piltonen M, Planken A, Leskela O, et al. Vascular endothelial growth factor C acts as a neurotrophic factor for dopamine neurons in vitro and in vivo. Neuroscience. 2011 doi: 10.1016/j.neuroscience.2011.06.084. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson RE, Carroll HP, Harris A, Maher ER, Selby PJ, Banks RE. Housekeeping proteins: a preliminary study illustrating some limitations as useful references in protein expression studies. Proteomics. 2005;5:566–71. doi: 10.1002/pmic.200400941. [DOI] [PubMed] [Google Scholar]
- 15.McColm JR, Geisen P, Hartnett ME. VEGF isoforms and their expression after a single episode of hypoxia or repeated fluctuations between hyperoxia and hypoxia: relevance to clinical ROP. Mol Vis. 2004;10:512–20. [PMC free article] [PubMed] [Google Scholar]
- 16.Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19:2003–12. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 17.Connolly DT, Heuvelman DM, Nelson R, et al. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–8. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albrecht I, Kopfstein L, Strittmatter K, et al. Suppressive effects of vascular endothelial growth factor-B on tumor growth in a mouse model of pancreatic neuroendocrine tumorigenesis. PLoS One. 2010;5:e14109. doi: 10.1371/journal.pone.0014109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ueda Y, Yamagishi T, Samata K, et al. A novel low molecular weight VEGF receptor-binding antagonist, VGA1102, inhibits the function of VEGF and in vivo tumor growth. Cancer Chemother Pharmacol. 2004;54:16–24. doi: 10.1007/s00280-004-0763-8. [DOI] [PubMed] [Google Scholar]
- 20.Joukov V, Kaipainen A, Jeltsch M, et al. Vascular endothelial growth factors VEGF-B and VEGF-C. J Cell Physiol. 1997;173:211–5. doi: 10.1002/(SICI)1097-4652(199711)173:2<211::AID-JCP23>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 21.Orpana A, Salven P. Angiogenic and lymphangiogenic molecules in hematological malignancies. Leuk Lymphoma. 2002;43:219–24. doi: 10.1080/10428190290005964. [DOI] [PubMed] [Google Scholar]
- 22.Saaristo A, Tammela T, Farkkila A, et al. Vascular endothelial growth factor-C accelerates diabetic wound healing. Am J Pathol. 2006;169:1080–7. doi: 10.2353/ajpath.2006.051251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rissanen TT, Markkanen JE, Gruchala M, et al. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res. 2003;92:1098–106. doi: 10.1161/01.RES.0000073584.46059.E3. [DOI] [PubMed] [Google Scholar]
- 24.Achen MG, Jeltsch M, Kukk E, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4) Proc Natl Acad Sci U S A. 1998;95:548–53. doi: 10.1073/pnas.95.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stacker SA, Caesar C, Baldwin ME, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–91. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- 26.Thelen A, Scholz A, Benckert C, et al. VEGF-D promotes tumor growth and lymphatic spread in a mouse model of hepatocellular carcinoma. Int J Cancer. 2008;122:2471–81. doi: 10.1002/ijc.23439. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Weber KT. Angiotensin converting enzyme and myofibroblasts during tissue repair in the rat heart. J Mol Cell Cardiol. 1996;28:851–8. doi: 10.1006/jmcc.1996.0080. [DOI] [PubMed] [Google Scholar]
- 28.Currie MJ, Hanrahan V, Gunningham SP, et al. Expression of vascular endothelial growth factor D is associated with hypoxia inducible factor (HIF-1a) and the HIF-1a target gene DEC1, but not lymph node metastasis in primary human breast carcinomas. J Clin Pathol. 2004;57:829–834. doi: 10.1136/jcp.2003.015644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ristimaki A, Narko K, Enholm B, et al. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 1998;273:8413–8. doi: 10.1074/jbc.273.14.8413. [DOI] [PubMed] [Google Scholar]
- 30.Zhu C, Qi X, Chen Y, et al. PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in IGF-1-induced VEGF-Cupregulation in breast cancer. Cancer Res Clin Oncol. 2011;137:1587–94. doi: 10.1007/s00432-011-1049-2. [DOI] [PubMed] [Google Scholar]