

Abstract
Prevailing evidence has established the fundamental role of microenvironmental conditions in tumorigenesis. However, the ability to identify, interrupt, and translate the underlying cellular and molecular mechanisms into meaningful therapies remains limited, due in part to a lack of organotypic culture systems that accurately recapitulate tumor physiology. Integration of tissue engineering with microfabrication technologies has the potential to address this challenge and mimic tumor heterogeneity with pathological fidelity. Specifically, this approach allows recapitulating global changes of tissue-level phenomena, while also controlling microscale variability of various conditions including spatiotemporal presentation of soluble signals, biochemical and physical characteristics of the extracellular matrix, and cellular composition. Such platforms have continued to elucidate the role of the microenvironment in cancer pathogenesis and significantly improve drug discovery and screening, particularly for therapies that target tumor-enabling stromal components. This review discusses some of the landmark efforts in the field of micro-tumor engineering with a particular emphasis on deregulated tissue organization and mass transport phenomena in the tumor microenvironment.
Keywords: Cancer, tumor microenvironment, stroma, transport, tissue engineering, biomaterials
1. Introduction
Despite sustained progress in our knowledge of biological signaling events regulating tumor malignancy, the clinical prognosis for many cancer types has scarcely changed since 1950 (Howlader 2012). Increasing experimental evidence suggests that this discrepancy may be due in part to an under-appreciation of physical phenomena contributing to disease progression. As a result, cancer biologists are increasingly collaborating with physical scientists and engineers to study physicochemical characteristics of solid tumors and their role in modulating intracellular signaling (Michor 2011). Particular emphasis is placed on analyzing microenvironmental changes that fundamentally influence tumor progression and therapy, including aberrant mechanical properties (e.g. cell forces, matrix stiffness), transport phenomena (e.g. mass and energy transfer, fluid dynamics), and growth/reaction kinetics (e.g. metabolism, signaling, proliferation). For example, quantifying therapy-associated mass transport, reaction rates, and the resulting cellular growth kinetics may help to better understand the evolution of drug resistance and inform more efficacious dosing regiments (Foo 2012; Podlaha 2012). To determine such variables, new experimental platforms are needed that accurately recapitulate salient characteristics of the tumor microenvironment.
By integrating strategies from tissue engineering, microfabrication, and cancer biology, biologically-inspired culture models (a.k.a. tumor surrogates) enable studies of physicochemical disease dynamics across multiple scales (Ghajar 2010; Wlodkowic 2010). Tissue engineering technologies, including biomaterials, scaffold fabrication techniques, and bioreactor design, allow facile manipulation of tissue-level phenomena such as culture dimensionality, cell-cell and cell-extracellular matrix (ECM) interactions, and soluble factor transport and signaling. Complementing these tools, microfabrication principles can exert exquisite control over the chemical and physical environment on the cellular scale, for example via incorporation of microfluidic channels or precise variation of mechanical and topographical properties, respectively (Huh 2011; Walker 2004). Hence, the combination of tissue engineering and microfabrication affords the development of novel in vitro approaches to quantitatively assess constitutive microenvironmental features that are frequently neglected by conventional tissue culture methods or obscured by the complexity of in vivo models (Burdett 2010; Griffith & Swartz 2006; Khademhosseini 2006). Moreover, these platforms may interface in real-time with sensitive analytical instrumentation, such as confocal microscopy or mass spectrometry, for unprecedented access to cellular and biomolecular dynamics (Enders 2010; Paguirigan 2010).
Here, we briefly introduce prominent characteristics of the tumor microenvironment, describe current state-of-the-art in vitro technologies to quantify these phenomena, and provide a perspective on future opportunities for micro-engineered tumor platforms in cancer research and clinical application with the ultimate goal of illuminating the multiscale regulation of cancer pathogenesis and therapy response.
2. Biological and physical hallmarks of the tumor microenvironment
Carcinomas comprise the majority of all solid tumors, and their malignancy is driven not only by the genetic transformation of epithelial cells, but also by the remodeling of contiguous stromal tissue to foster growth, metastasis, and therapy resistance (Bissell & Radisky 2001; Mueller 2004; Joyce & Pollard 2009). This tumor stroma derives from reciprocal tumor-host cell crosstalk, whereby tumor cell-secreted growth factors and cytokines stimulate the otherwise quiescent host cells to initiate a variety of processes, including desmoplasia and angiogenesis (Kim 2005; Rønnov-Jessen 2009; Hanahan & Coussens 2012; Otranto 2012). The ensuing physiological changes resemble wound healing processes; however, tumor-associated stromal cells undermine tissue homeostasis rather than normalizing it, eliciting the analogy that tumors function as ‘wounds that do not heal’ (Dvorak 1986; Coussens & Werb 2002). As a result, an asymmetric distribution of physical and chemical cues emerges, comprising spatiotemporally altered matrix mechanical properties and mass transport (Fukumura & Jain 2007; Hanahan & Coussens 2012; Otranto 2012). Below, we describe these pathological consequences of tumor-stroma interactions from both biological and physical science perspectives and summarize microscale tissue engineering strategies to explore the underlying mechanisms.
2.1 Deranged tissue organization
2.1.1 Dimensionality and ECM characteristics
Structural ramifications of malignant transformation begin with the transition from 2D to 3D tissue architecture caused by the hyper-proliferation of epithelial cells. Healthy epithelia are generally organized in polarized sheet structures, with a basolateral surface supported by a basement membrane, an apical lumen with secretory function, and tight junctions between neighboring cells in a confluent pseudo-monolayer (Fig. 1). Disruption of this arrangement by aberrant proliferation, loss of E-cadherin-mediated cell-cell contact, or secretion of matrix-degrading enzymes such as matrix metalloproteinases (MMPs) subverts epithelial homeostasis and promotes carcinogenesis (Beliveau 2010; Nelson 2005; Bissell 2011; Chen 2009). Independent of other environmental conditions, this dimensional shift affects tumor cell growth (Weaver 1997; Wang 1998), migration (Fraley 2010; Zaman 2006), signaling (Fischbach 2009), and drug response (Dhiman 2005; Fischbach 2007). Conversely, restoration of appropriate geometrical context, or inhibition of integrin receptors that mediate 3D cellular engagement to the ECM, can normalize the malignant phenotype of transformed epithelial populations, in part by interrupting bidirectional cross-talk between growth factor and cell adhesion receptors (Wang 1998; Kenny 2003; Weigelt 2010). For these reasons, appropriate choice of culture dimensionality is critical for the development of and interpretation of results collected with biomimetic tissue constructs in cancer research (Pampaloni 2007; Griffith & Swartz 2006; Nelson & Bissell 2006).
Figure 1. Biological and physical changes inherent to the tumor microenvironment.
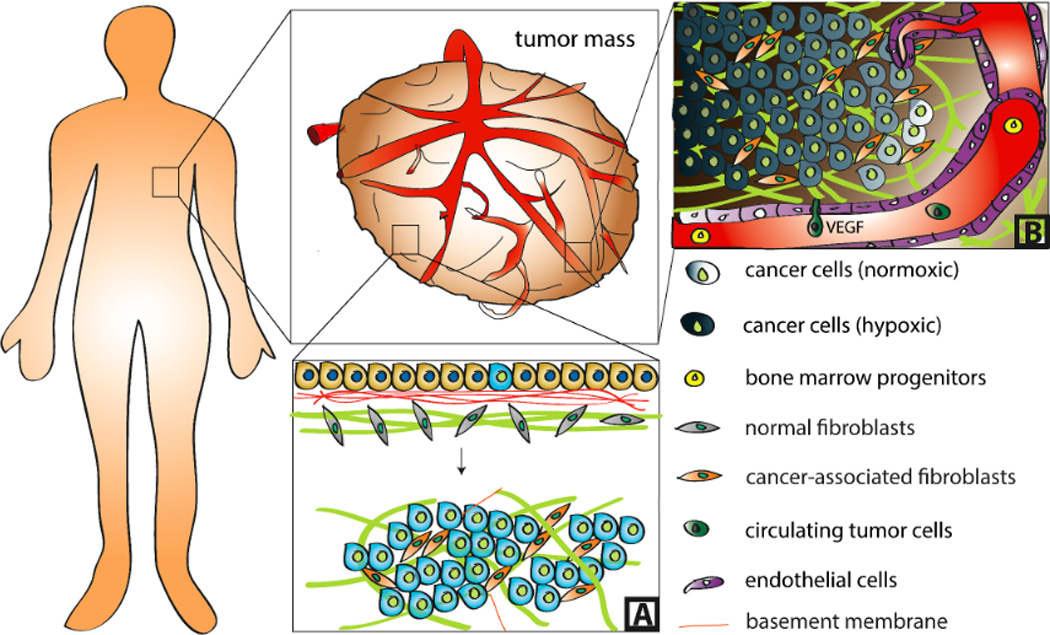
Tumors are characterized by aberrant tissue organization (A) and mass transport (B). While normal epithelia form sheets that adhere to a layer of basement membrane, which is anchored to the underlying stroma, malignant transformation perturbs this architecture and leads to the development of a 3D tumor mass. During this process, basement membrane integrity is compromised and ECM composition, conformation, and mechanical properties are altered due in part to transformation of normal fibroblasts into cancer-associated fibroblasts (A). Excessive tumor cell proliferation results in diffusion-limited oxygen and waste transport and consequential development of hypoxia and acidosis. This induces secretion of pro-angiogenic factors including VEGF that activate neovascularization via angiogenesis and recruitment of bone marrow progenitors. These newly formed vessels, in turn, enable tumor growth and metastasis by providing vascular conduits for nutrients and extravasated circulating tumor cells, respectively (B).
Moreover, ECM properties, such as elasticity, nano- and microstructure, as well as composition affect tumorigenesis and need to be considered when designing matrices for in vitro culture models (Paszek 2005; Discher 2005; Carey 2012). Namely, desmoplastic matrix remodeling by tumor-associated myofibroblasts entails the assembly of a highly dense ECM (Fig. 1), whose physicochemical attributes enhance malignancy through morphogenic deregulation, tumor cell proliferation, vascular recruitment, and myofibroblastic differentiation (Kalluri 2006; Otranto 2012; Chandler and Seo 2012). Mediated by increased deposition, unfolding, and crosslinking of fibrillar adhesion proteins (e.g. fibronectin, collagen type I), desmoplastic stiffening contributes to epithelial transformation in part through integrin-mediated increases in cell contractility (Chandler 2011, Levental 2009). This change in cell contractility, in turn, can directly and indirectly alter gene expression via altering transcription factor activity and the release of matrix-bound, pro-tumorigenic growth factors (e.g., transforming growth factor beta; TGFbeta), respectively (Hinz 2009; Mammoto 2009). Similarly, changes in the distribution of pore sizes, chemical composition, and fiber arrangement due to myofibroblastic remodeling can control aspects of tumor cell phenotype such as adhesion, mechanics, and motility (Zaman 2006; Pathak 2011, 2012). Recapitulating these diverse physical aspects of biological matrices comprises a critical design criterion for tissue-engineered tumor models.
2.1.2 Engineered models to evaluate the effect of tissue dimensionality and ECM interactions
Microfabrication techniques in conjunction with natural or artificial ECM molecules afford investigations into the role of 3D cell-cell and cell-ECM interactions (Stenzel 1974; Roy 2010; Pederson 2003; Carey 2012). Originally pioneered on planar surfaces, micropatterning of natural ECM molecules (e.g. fibronectin) onto cell culture substrates by polydimethylsiloxane (PDMS)-based microcontact printing or soft lithography has been extensively utilized to interrogate topographical and biochemical effects on individual cells and small cell populations (Khademhosseini 2006; Whitesides 2001). For example, such studies were used to decouple the effects of cell shape, spreading, and receptor ligation on cell fate (Tien 2002). However, the sagging and swelling of PDMS in solution limits homogeneous patterning of large surface areas as necessary for proteomic or transcriptional analysis of protein expression. Parylene patterning can address this challenge and has enabled seeding of individual cells or small cell clusters on fibronectin-functionalized surfaces of varying area to examine the role of direct cell-cell contact on tumor cell behavior (Tan 2009). Although both techniques have provided numerous insights regarding cell-matrix and cell-cell interactions, by maintaining monolayer cell culture, they neglect authentic tissue architecture. In contrast, fully-enclosed 3D tissue culture has proved its utility for mimicking the structural environment of tumor-associated stroma and subsequent effects on disease progression. For example, organotypic culture arrays comprising lithographically-defined epithelial tissue structures sandwiched between layers of collagen were used to study geometrical influences on neoplastic progression (Nelson 2008). Likewise, hydrogel-based 3D microwell arrays have emerged as a robust platform to examine cell culture space and intercellular interactions, including their effects on drug response (Charnley 2009, Håkanson 2012). One particular advantage of microscale 3D matrices is the reduction of diffusion barriers to allow homogeneous, temporally-controlled stimulation of cell populations (Raghavan 2010). Furthermore, such systems can be incorporated with lithographically defined microchannels to mimic cellular confinement within 3D tissue, an approach which has revealed that lower ECM porosity due to increased stiffness may promote cell migration (Pathak 2012).
In order to explore the role of matrix composition, structure, and mechanical properties in cancer pathogenesis, biomaterials from regenerative medicine and drug delivery can be adapted to microfabricated culture systems discussed above (Tibbitt 2009), including complex mixtures of biological ECM (e.g., Matrigel®, a basement membrane preparation isolated from Engelbreth-Holm-Swarm murine sarcoma) and isolated ECM components from animal tissue (e.g., collagen I, fibronectin) (Patterson 2010). These natural biomaterials offer unique advantages due to their inherent provision of cell binding sites and the fact that porosity, fiber structure, and stiffness can be readily tailored via adjusting gelling conditions such as temperature, concentration, gel thickness, and media composition (Carey 2012; Wheeldon 2010; Roy 2010). Nevertheless, natural polymers suffer from batch to batch variations and provide limited parameter space for the above-described physical variables. In fact, their modulation often simultaneously alters other materials properties that independently affect cell behavior (e.g. concentration changes affect stiffness but also density of adhesion ligands and porosity) (Miron-Mendoza 2012; Savina 2009). Artificially combining functional protein building blocks via protein engineering strategies provides an attractive route to overcome these shortcomings and generate biological materials systems with unprecedented control over cell adhesion, proteolytic degradation, or stiffness (Romano 2011). However, the fabrication of such molecules is highly specialized and therefore frequently not amenable for routine applications.
Biologically-inspired partially or fully synthetic hydrogels may help to overcome some of the inherent limitations of natural biomaterials. They can be produced in large quantities, a highly reproducible manner, and are increasingly used by both the cancer biology and engineering community to selectively tune the physicochemical composition and structure of cell culture matrices. For example, synthetically modified fibrin or hyaluronic acid have been developed previously to guide endothelial cell behavior for regenerative approaches (Ehrbar 2005; Hanjaya-Putra 2011), but are also relevant in the context of cancer as the deposition of these ECM components is increased during tumorigenesis (Nagy 2012; Slevin 2002). Modifying natural ECM components takes advantage of the inherent cell affinity to the polymer backbone, while introducing selective variations of matrix mechanical properties and degradation via synthetic side chains (Hanjaya-Putra 2012; Anathanarayanan 2011). Polyacrylamide gels covalently coated with ECM components provide another system to independently control elastic moduli and ECM ligand presentation and revealed mechanoregulatory mechanisms of perturbed epithelial tissue homeostasis (Paszek 2005). These substrates are widely utilized by many labs for 2D studies, but as they cannot be remodeled by cells, translation to 3D culture formats or animal studies (i.e., for verification of in vitro data) is limited. Furthermore, recent findings indicate that stiffness-related changes in porosity of polyacrylamide gels may affect the conformation of covalently attached cell adhesion proteins, which, in turn, could also regulate cell behavior (Trappmann 2012). Poly(ethylene glycol) (PEG)-based scaffolds provide an attractive alternative as not only the stiffness of these materials can be readily adjusted, but also the density of conformation-independent cell adhesion sites and/or proteolytically degradable crosslinkers ultimately enabling studies of cellular invasion in 3D culture formats (Gill 2012). Modifications of PEG-based materials in which molecular moieties have been introduced to enable in situ photo-degradation or crosslinking are particularly exciting because this approach affords an opportunity to determine cellular response to dynamic changes of matrix stiffness that may result from tumor development or drug treatments (Kloxin 2009; Li 2006).
2.2 Cancer, A mass transport problem
2.2.1 Current knowledge
Within the tumor stroma, convective and diffusive forces are intrinsic mediators of the biochemical landscape that sustains neoplastic development (Kenny 2006; Netti 2000; Ramanujan 2002). The predominant conduits of mass transport, microvascular networks define the spatial distribution of oxygen, nutrients, endocrine signals, and therapeutic drugs. Moreover, as a consequence of diffusion-limited nutrient and waste transport, the absence of a functional vasculature regulates the development of hypoxia and acidosis during tumor initiation and at interior regions of advanced cancers (Fig. 1). These microenvironmental conditions promote malignancy by driving the selection of cells that mitigate oxidative stress via autonomous energy production, typically through glycolysis (Damert 1997; Cochran 2006; Scott 2011). Metabolic rewiring, known as the ‘Warburg effect’, can further enhance the proliferative, metastatic, and therapy-resistant behavior of the adapted cell population. Furthermore, hypoxia is known to dramatically alter the secretion profile of tumor cells, particularly by inducing pro-angiogenic factors (e.g. vascular endothelial growth factor [VEGF], interleukin-8 [IL-8], basic fibroblast growth factor [bFGF]) that promote angiogenesis and vasculogenesis via the recruitment of adjacent blood vessels and bone marrow-derived progenitor cells, respectively (Hanahan & Folkman 1996). This process of sustained neovascularization constitutes a hallmark capability of cancer and, by surmounting nutrient limitations and providing vascular conduits, enables tumor growth and metastasis (Hanahan & Weinberg 2000). However, compared to healthy vasculature, tumor-associated blood vessels exhibit increased leakiness, tortuosity, instability via absence of mural cells, and subsequent aberrant fluid mechanical forces (Hashizume 2000; Nagy 2008, 2012). As a result, they fail to normalize the hypoxia/acidosis of the tumor core, but instead cause spatial and temporal variations of these conditions (Dvorak 2010). Notably, by contributing to elevated fluid pressure in the tumor, dysfunctional vasculature compromises drug delivery and modulates interstitial flow at the tumor margins (Stohrer 2000). Pressure gradients and subsequent drainage from the tumor perimeter directly and indirectly guide the behavior of tumor and stromal cells by redistributing morphogen and chemokine gradients and by mechanically-inducing stromal differentiation (Haessler 2011; Shieh 2011; Shields 2007). From a physical science perspective, these integrated fluid mechanical forces establish the prevailing biochemical profile of the tumor microenvironment and direct disease progression (Swartz 2012). Likewise, tools to deconvolute convection-diffusion-reaction kinetics, paracrine signaling, vascular supply, and interstitial fluid mechanics are required for the comprehensive study of pathophysiological transport processes, including metabolic maintenance, drug delivery, and evolution of therapy resistance (Basanta 2012).
2.2.2 Biomimetic approaches to study tumor-associated transport phenomena
Whereas conventional in vitro technologies lack spatiotemporal variations of soluble chemicals, and animal studies prohibit quantitative control over mass transport, microfabricated tissues can effectively access these critical pathological parameters (Stroock 2010). Therefore, researchers have employed microfabrication technologies to mimic physiological transport processes, including perfusion for metabolic maintenance, diffusion-controlled soluble factor gradients, convective transport through microfluidic conduits, and more recently, tissue-engineered biomimetic vasculature (Whitesides 2006; Tourovskaia 2005; Walker 2004). For example, microfabrication technologies have surmounted diffusion-limited metabolic maintenance in 3D culture by the development of microscale perfusion bioreactors and by providing access to scaffold geometries within the Krogh length (i.e., to control hypoxia-associated effects in culture studies) (Choi 2007; Du 2011). From the profusion of tissue-engineered, microfabricated assays with well-controlled tissue structure and transport conditions, surprising data has emerged. For example, studies comparing 2D-cultured tumor cells with tumor cells incorporated within microfabricated hydrogel disks have revealed that changes in tissue dimensionality may be similarly if not even more important to the proangiogenic capability of tumor cells as oxidative stress (Verbridge 2010). Moreover, by the tuning of physical and chemical stimuli as well as cellular complexity, microfabricated platforms afford a unique opportunity to examine both the isolated and integrated effects of multiplexed parameters.
Another aspect to consider is that cell responses to gradients of soluble factors, for example, may be different in 2D vs. 3D culture systems, and not only depend on the specific morphogen applied, but also its combination with other molecules as well as the specific spatial and temporal dynamics of exposure. Two-dimensional microfluidic models have proved their utility in the generation of stable, linear concentration gradients to independently evaluate directional responses to biomolecular assymetries, such as chemotaxis and morphogenesis (Jeon 2002; Selimović 2011; Saadi 2007; Dertinger 2001; Georgescu 2008; Mak 2011). Three-dimensional microfluidic gradient generators with single- or multi-layered factor gradients will additionally be able to distinguish biochemical synergies in a physiologically relevant tissue context, which is essential for the study of tumor cell migration and perturbed morphogenesis (Choi 2007; Chung 2009, 2010; Shamloo 2008, 2010; Shin 2011).
While most of the current approaches are still relatively low-throughput, automated 3D microfluidic co-culture arrays may revolutionize this limitation. Such systems will be critical for economically more effective low-volume, high-throughput drug screening, and have already proved efficacious for interrogating tumor-stromal signaling within 3D culture formats without typical drug-diffusion limitations (Bauer 2010; Domenech 2012; Jongpaiboonkit 2008; Montanez-Sauri 2011). By coupling biomaterials with microfabrication, further 3D platforms have emerged to study cellular responses to spatial distribution of physical and chemical stimuli. For example, collagen-based microfluidic channels enable studies of endothelial sprouting in response to VEGF gradients (Chung 2009) and the presence of pericytes (Zheng 2012), and aqueous two-phase systems allow patterning of cells as well as delivery of biochemical factors (Frampton 2011). Together, these systems can recapitulate the heterogeneity of tumor and stromal cell-secreted factors, drug distribution and reaction kinetics, or the gradients of hypoxia and acidosis described above.
In the particular case of tumor angiogenesis, tissue-engineered microvascular constructs will undoubtedly advance the development of artificial tumor surrogates and can be fabricated by a variety of techniques (Fig. 2). Whereas some models employ surface-cultured endothelial monolayers to study tumor-endothelial signaling and angiogenic invasion independent of transport effects, other assays generate fully-enclosed endothelial networks in remodelable matrices (Zheng 2012; Miller 2012; Chrobak 2006; Raghavan 2010; Shamloo 2012; Verbridge 2010; Wong 2010; Liu 2011). For the fabrication of the latter, microfluidic conduits may be incorporated into biomimetic hydrogels through a variety of approaches including pull-through technique, sacrificial fiber molds, modular assembly and soft lithography (Golden & Tien 2007; Du 2011; Nichol 2009; Price 2009, 2011). Incorporating such microfabricated, endothelial-coated vessels into engineered tumor models will undeniably improve in vitro studies of sprouting angiogenesis, endothelial barrier function, and drug delivery (Zheng 2012, Chung 2009, Zervantonakis 2011). In fact, pulsed injections of fluorophores or fluorescently-labeled proteins as mimics of small and large therapeutic molecules allows the measurement of their diffusivity and permeability from microengineered vasculature and permits the analysis of changes in leakiness due to exposure to excess concentrations of proangiogenic molecules (Zheng 2012). Likewise, these devices can be utilized in the quantification of diffusion-reaction metabolic processes in 3D and determination of cell growth rate constants, which are significantly different from suspension and monolayer culture (Choi 2007).
Figure 2. Microfabrication techniques to recapitulate vascular networks in vitro.

Cylindrical 3D networks can be incorporated into hydrogels using carbohydrate glass as sacrificial templates. Labeled human umbilical vein endothelial cells (HUVECs) seeded into these channels form perfusable vascular networks that can be induced to sprout (arrowheads), while incorporation of a second labeled cell type (e.g.10T1/2 cells) into the hydrogel bulk permits recapitulation of stromal tissue. Scale bars represent 1 mm and 200 µm (Miller 2012) (A). Similarly, lithographically patterned microfluidic devices may be used to generate functional vascular networks within remodelable hydrogels. In particular, incorporation of pericytes (indicated by α-SMA staining) into the bulk leads to the recruitment of these cells to HUVEC-lined vessels (indicated by CD31 staining) and their corresponding stabilization. Scale bar, 100 µm (Zheng 2012) (B). Microdevices composed of two parallel endothelial cell-coated channels (GFP-labeled HUVECs in top and dsRed-labeled HUVECs in bottom channel) separated by an invadable collagen matrix allow studies of vascular anastomosis in response to VEGF gradients as well as interstitial flow (dashed arrow) and axial shear flow (solid arrow). Scale bar, 100 µm (Song 2012) (C). Microvascular structures generated in microfluidic type I collagen gels using a pull-through technique can be stabilized by cyclic AMP (cAMP)-elevating agents as shown via perfusion of endothelial cell-coated vessels with fluorescently labeled BSA; *indicates focal leaks. Scale bar, 100 µm (Wong 2010) (D). Culture of stromal cells (10T1/2) and endothelial cells (HMVEC) in adjacent channels separated by a 3D collagen scaffold allows studying the effect of bi-directional paracrine signaling on invasion and sprouting behavior of these cells, respectively. 10T1/2 invaded at an enhanced rate in the presence of HMVECs. Additionally, HMVECs formed sprouts towards the blank collagen scaffold located in the left channel (red arrowheads), whereas vessels were stabilized in regions directed towards 10T1/2s (Chung 2009) (E). Images are modified and reproduced with permissions from the publishers.
In addition to solute delivery, microvascular models can be used to introduce a permissive mechanical regulator of the microenvironment, namely convective transport via interstitial flow (Fukumura 2008; Griffith & Swartz 2006). In fact, interstitial flow can affect tumor progression via multiple mechanisms; for example, through biasing the distribution of cell-secreted factors, forcing mechanical aberrations of the tumor microenvironment by promoting stroma remodeling, and regulating endothelial sprouting (Haessler 2011; Polacheck 2011; Shieh 2011; Shields 2007; Song 2011). Integrated in tumor-engineered platforms selective perfusion of microvascular network recreates inherent pathological properties such as hypoxia/acidosis, compromised drug delivery, edema, and in some cases the direct visualization of migration and intravasation. By quantifying hitherto unexplored responses to transport processes, this unprecedented control of multiple-parameter in vitro models is indispensable for the improved understanding of tumor growth and clinical treatment.
3. Future opportunities
Over the past decade or so, reductionist approaches have helped to decipher the biological and physicochemical complexity of tumors, but as the discipline of tumor engineering matures and technologies improve, we encounter the question of what can be gained by adding complexity. We propose applications for microengineered tumor-mimetic assays in drug development and delivery strategies, simulation of whole-body physiologically-based pharmacokinetic models, and screening of rationally-designed therapeutic regimens. Furthermore, experimental integration of convective-diffusion-reaction processes will provide unprecedented access to quantitative parameters that are instrumental to the validation of comprehensive computational models describing the intra- and extracellular signaling networks underlying tumorigenesis.
3.1 Engineering of functional, fully-human and/or patient-specific micro-tissues
The immediate advantage of complex microengineered tumor models derives from the opportunity to observe interactions between various constituents of the tumor microenvironment in real time with implications for both basic research and drug development. Whereas certain physicochemical parameters may be independently important, it is likely that combinatorial effects accentuate or attenuate their functional significance. Thus, robust platforms that can controllably add or subtract layers of physiological complexity will be indispensable for interrogating variates and co-variates in cancer therapy. For example, the emergence of drug resistant subpopulations may rely on microenvironmental signatures dictated by the cellular and acellular heterogeneity of the tumor mass; identification of the underlying molecular mechanisms has the potential to identify novel therapeutic targets that may improve current therapeutic interventions (Gerlinger 2012; Ene 2011; Nakasone 2012). Likewise, tumor-engineered assays will provide unprecedented ability to disambiguate the indirect effects of targeted cancer drugs. As one poignant example, the underwhelming clinical benefit of VEGF-inhibiting, anti-angiogenic therapies has received considerable attention (Bergers & Hanahan 2008; Fukumura 2007). Low therapeutic efficacy is due in part to a poor understanding of the mechanisms underlying vessel growth and drug response as well as the forced upregulation of alternative proangiogenic factors (e.g., bFGF and IL-8) that render vessels VEGF-independent (Hanahan & Folkman 1996; Folkman 2007; Bergers & Benjamin 2003; Pàez-Ribes 2009; Goel 2011). To explore these questions, our lab is developing microengineered tumor models to determine cooperative parameters that may control endothelial cell sprouting, including cell density, vessel geometry, and endothelial cell autocrine signaling mechanisms (Verbridge, accepted). As biomimetic vascular assays continue improving, the pros and cons of anti-angiogenic therapy and drug delivery (e.g. mechanisms of actions, delivery mode, dosing intervals) will become more apparent and therapies can be adjusted (Carmeliet & Jain 2000, 2011; Jain 2009; Goel 2011).
3.2 Validation of whole-body PBPK-PD models
Knowledge and treatment of the primary tumor site only locally palliates a more pernicious systemic disorder. Through metastasis and colonization, circulating tumor cells can infiltrate distant tissues, often with morbid consequences; similarly, organ distribution and side effects are critical considerations during drug development. Therefore, pre-clinical tools that contribute to holistic understanding of both disease progression and treatment will benefit the development of better pharmaceuticals and dosing strategies. To this end, microengineered tumor cultures may integrate with current “body-on-a-chip” technologies for comprehensive culture models of disease progression across multiple tissues (Esch 2011; Sin 2004; Sung 2010; Vivaraidya 2004). Such systems have already been used to validate whole-body physiologically-based pharmacokinetic (PB-PK) models and improve estimation of critical parameter such as Theile modulus or Peclet number, dimensionless coefficients that describe relative rates of transport and reaction kinetics, and measurement of variability and co-variation with disease or treatment among different organs (Sung 2010). These systems will potentially contribute to the prediction of metastatic homing, dosing regimens, and side-effects, or selection of patients for specific clinical trials (Scott 2012). For example, priming an in vitro tumor with downstream osteogenic or pulmonary compartments to resemble the pre-metastatic niche may help determine mechanisms of organ homing, including characteristics or organ-specific tropisms such as stages of lung inflammation due to smoking (Fig. 3A–B). In addition, microfluidic PB-PK models can inform pharmacodynamic computational models and assist in the extrapolation of such models to animal and human predictions (Fig. 3C). While such devices have been used in toxicity and dosing studies (Esch 2011; Sung 2010), testing of new cancer therapies may also benefit from this technology. Particularly, it could help to better understand the current disconnect between promising preclinical data and lack of clinical efficacy of new compounds, as well as inform dosing strategies and combination therapies to optimize biological response.
Figure 3. Integration of microengineered tumor cultures with “body-on-a-chip” models.
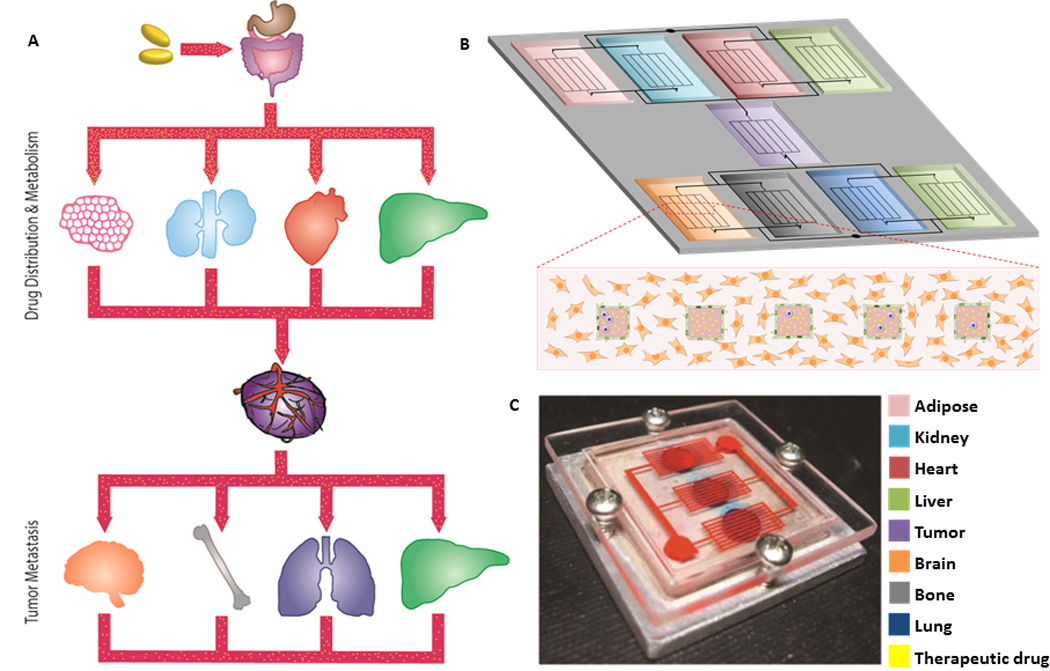
Cancer progression and therapy are controlled by systemic interactions. For example, endocrine signaling between tumors and distant tissues such as brain, bone, lungs, and liver regulate the tropism of certain cancers to particular sites (e.g. breast cancer to bone). Additionally, the efficacy of anti-cancer therapies is regulated by systemic distribution and metabolism in various tissues and organs such as adipose tissue, kidney, heart and liver. Therefore, connecting multiple organ components via vascular conduits in ‘cancer on a chip’ models has the potential to improve our understanding of metastatic homing mechanisms of tumor cells as well as drug therapy (A–B). Already, microscale cell culture analogs have been used to improve the predictability of pharmacokinetic-pharmacodynamic models in vitro (Sung 2010) (C). This platform, comprising bone, liver, and tumor compartments, was used to assess the toxicity of 5-fluorouracil in a 3D tissue context with multi-organ interactions. The combination of in vitro experimental data and mathematical PB-PK modeling may improve screening for drug development, as well as inform our understanding of cancer pathogenesis during therapy. Images are reproduced with permission from the publisher.
3.3 In vitro platforms for integrative mathematical oncology
A final application for tissue-engineered tumor surrogates lies in the realization of comprehensive mathematical cancer models (Anderson 2008). By selective incorporation of individual or multiplexed physiological components of the tumor microenvironment, in vitro tumor models can be used to determine critical in silico parameters, including correlation or interaction coefficients between various elements (Chakrabarti 2012; Borau 2011; Kam 2009; 2012; Stylianopoulos 2010). For example, it is known that integrin engagement, matrix stiffness, and growth factor receptor signaling are intimately linked, but quantitative parameters and modeling of co-variation are yet undetermined. Tissue-engineered models could test predictive behaviors and confirm computational hypotheses regarding disease dynamics. One example where such systems may prove beneficial is the modeling of tumor evolution, by which malignant or resistant populations propagate by somatic selection in resource-constrained tissues (Anderson 2005, 2006, 2009; Quaranta 2008). Computational models of this process show that microenvironmental conditions are key determinants of population phenotype. This observation is consistent with emerging appreciation for the local heterogeneity within primary tumor site and in distant metastases, and it has spawned new hypotheses for therapeutic dosing regiments based on ecological principles (Basanta 2012, 2012; Maruskyk 2012; Navin 2011; Snuderl 2011). However, such simulations are difficult to examine biologically. In vitro systems that include diverse cell populations and heterogeneous physicochemical environment would help validate the mathematical models and perhaps elucidate novel parameters to improve such systems.
4. Conclusions
Microfabrication techniques have the potential to yield important new insights into the pathogenesis of cancer. While great progress has been made in developing model systems for deciphering mechanisms of tumorigenesis, improved throughput and applicability by non-engineers (e.g. cancer biologists and clinicians) will further enhance the significance of these platforms. Moreover, exploring currently unrealized opportunities of recapitulating cancer complexity will help in elucidating molecular, cellular, tissue, and systems level phenomena that could be explored in the future for more efficacious clinical therapies.
Acknowledgements
Funding was provided by a NSF graduate research fellowship for P. DelNero as well as the National Cancer Institute (RC1CA146065, R21CA161532, R01CA173083 and by the Cornell Center on the Microenvironment & Metastasis through Award Number U54CA143876). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Ananthanarayanan B, Kim Y, Kumar S. Biomaterials. 2011;32(31):7913–7923. doi: 10.1016/j.biomaterials.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson AR. Math. Med. Biol. 2005;22:163–186. doi: 10.1093/imammb/dqi005. [DOI] [PubMed] [Google Scholar]
- 3.Anderson AR, Weaver AM, Cummings PT, Quaranta V. Cell. 2006;127:905–915. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 4.Anderson AR, Quaranta V. Nat. Rev. Cancer. 2008;8:227–234. doi: 10.1038/nrc2329. [DOI] [PubMed] [Google Scholar]
- 5.Anderson AR, Rejniak K, Gerlee P, Quaranta V. J. Math. Biol. 2009;58:579–624. doi: 10.1007/s00285-008-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basanta D, Scott JG, Fishman MN, Ayala G, Hayward SW, Anderson AR. Br. J. Cancer. 2012;106:174–181. doi: 10.1038/bjc.2011.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basanta D, Gatenby R, Anderson AR. Mol. Pharm. 2012;9:914–921. doi: 10.1021/mp200458e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer M, Su G, Beebe DJ, Friedl A. Integr. Biol. 2010;2:371–378. doi: 10.1039/c0ib00001a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beliveau A, Mott JD, Lo A, Chen EI, Koller AA, Yaswen P, Muschler J, Bissell MJ. Genes Dev. 2010;24(24):2800–2811. doi: 10.1101/gad.1990410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergers G, Benjamin LE. Nat. Rev. Cancer. 2003;3(6):401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 11.Bergers G, Hanahan D. Nat. Rev. Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bissell MJ, Radisky D. Nat. Rev. Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bissell MJ, Hines WC. Nat. Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borau C, Kamm RD, García-Aznar JM. Phys. Biol. 2011;8:066008. doi: 10.1088/1478-3975/8/6/066008. [DOI] [PubMed] [Google Scholar]
- 15.Burdett E, Kasper K, Mikos A, Ludwig JA. Tissue Eng. Part B Rev. 2010;16(3):351–359. doi: 10.1089/ten.TEB.2009.0676. [DOI] [PubMed] [Google Scholar]
- 16.Carey SP, Kraning-Rush CM, Williams RM, Reinhart-King C. Biomaterials. 2012;33(16):4157–4165. doi: 10.1016/j.biomaterials.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmeliet P, Jain RK. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Jain RK. Nat. Rev. Drug Discov. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 19.Chakrabarti A, Verbridge S, Stroock AD, Fischbach C, Varner JD. Ann. Biomed. Eng. 2012;40(11):2488–2500. doi: 10.1007/s10439-012-0655-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandler EM, Saunders MP, Yoon CJ, Gourdon D, Fischbach C. Phys. Biol. 2011;8 doi: 10.1088/1478-3975/8/1/015008. 015008. [DOI] [PubMed] [Google Scholar]
- 21.Chandler EM, Seo BR, et al. Proc. Natl. Acad. Sci. U S A. 2012;109(25):9786–9791. doi: 10.1073/pnas.1121160109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charnley M, Textor M, Khademhosseini A, Lutolf MP. Integr. Biol. 2009;1:625–634. doi: 10.1039/b918172p. [DOI] [PubMed] [Google Scholar]
- 23.Chen A, Cuevas I, Kenny PA, Miyake H, Mace K, Ghajar C, Boudreau A, Bissell MJ, Boudreau N. Cancer Res. 2009;69:6721–6729. doi: 10.1158/0008-5472.CAN-08-4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi NW, Cabodi M, Held B, Gleghorn JP, Bonassar LJ, Stroock AD. Nat. Mater. 2007;6:908–915. doi: 10.1038/nmat2022. [DOI] [PubMed] [Google Scholar]
- 25.Chrobak KM, Potter DR, Tien J. Microvas. Res. 2006;71:185–196. doi: 10.1016/j.mvr.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Chung S, Sudo R, Mack PJ, Wan CR, Vickerman V, Kamm RD. Lab Chip. 2009;9:269–275. doi: 10.1039/b807585a. [DOI] [PubMed] [Google Scholar]
- 27.Chung S, Sudo R, Vickerman V, Zervantonakis IK, Kamm RD. Ann. Biomed. Eng. 2010;38:1164–1177. doi: 10.1007/s10439-010-9899-3. [DOI] [PubMed] [Google Scholar]
- 28.Cochran DM, Fukumura D, Ancukiewicz M, Carmeliet P, Jain RK. Ann. Biomed. Eng. 2006;34:1247–1258. doi: 10.1007/s10439-006-9134-4. [DOI] [PubMed] [Google Scholar]
- 29.Coussens LM, Werb Z. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Damert A, Machein M, Breier G, Fujita Q, Plate KH. Cancer Res. 1997;57(17):3860–3864. [PubMed] [Google Scholar]
- 31.Dertinger SKW, Chiu DT, Jeon NL, Whitesides GM. Anal. Chem. 2001;73:1240–1246. [Google Scholar]
- 32.Dhiman HK, Ray AR, Panda AK. Biomaterials. 2005;26(9):979–986. doi: 10.1016/j.biomaterials.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 33.Discher DE, Janmey P, Wang YL. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 34.Domenech M, Bjerregaard R, Bushman W, Beebe DJ. Integr. Biol. 2012;4:142–152. doi: 10.1039/c1ib00104c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du Y, Ghodousi Mm, Qi H, Haas N, Xiao W, Khademhosseini A. Biotechnol. Bioeng. 2011;108:1693–1703. doi: 10.1002/bit.23102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dvorak HF. N. Engl. J. Med. 1986;315(26):1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 37.Dvorak HF. Curr. Opin. Hematol. 2010;17(3):225–229. doi: 10.1097/MOH.0b013e3283386638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ehrbar M, Metters A, Zammaretti P, Hubbell JA, Zisch AH. J. Control Release. 2005;101(1–3):93–109. doi: 10.1016/j.jconrel.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 39.Enders JR, Marasco CC, Kole A, Nguyen B, Sevugarajan S, Seale KT, Wikswo JP, McLean JA. IET Syst. Biol. 2010;4:416–427. doi: 10.1049/iet-syb.2010.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ene CI, Fine H. Cancer cell. 2011;20:695–697. doi: 10.1016/j.ccr.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 41.Esch MB, King TL, Shuler ML. Annu Rev Biomed Eng. 2011;13:55–72. doi: 10.1146/annurev-bioeng-071910-124629. [DOI] [PubMed] [Google Scholar]
- 42.Fischbach C, Chen R, Matsumoto T, Schmelzle T, Brugge JS, Polverini PJ, Mooney DJ. Nat. Methods. 2007;4:6–11. doi: 10.1038/nmeth1085. [DOI] [PubMed] [Google Scholar]
- 43.Fischbach C, Kong HJ, Hsiong SX, Evangelista MB, Yuen W, Mooney DJ. Proc. Natl. Acad. Sci. U S A. 2009;106:399–404. doi: 10.1073/pnas.0808932106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Folkman J. Nat. Rev. Drug Discov. 2007;6(4):273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 45.Foo J, Chmielecki J, Pao W, Michor F. J. Thorac. Oncol. 2012;7(10):1583–1593. doi: 10.1097/JTO.0b013e31826146ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fraley SI, Feng Y, Krishnamurthy R, Kim DH, Celedon A, Longmore GD, Wirtz D. Nat. Cell Biol. 2010;12:598–604. doi: 10.1038/ncb2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frampton JP, Lai D, Sriram H, Takayama S. Biomed. Microdevices. 2011;13:1043–1051. doi: 10.1007/s10544-011-9574-y. [DOI] [PubMed] [Google Scholar]
- 48.Fukumura D, Jain RK. J. Cell. Biochem. 2007;101:937–949. doi: 10.1002/jcb.21187. [DOI] [PubMed] [Google Scholar]
- 49.Fukumura D, Jain RK. Microvasc. Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukumura D, Jain RK. APMIS. 2008;116:695–715. doi: 10.1111/j.1600-0463.2008.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Georgescu W, Jourquin J, Estrada L, Anderson AR, Quaranta V, WIkswo JP. Lab Chip. 2008;8:238–244. doi: 10.1039/b716203k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghajar CM, Bissell MJ. Tissue Eng. Part A. 2010;16:2153–2156. doi: 10.1089/ten.tea.2010.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gill BJ, Gibbons DL, Roudsari LC, Saik JE, Rizvi ZH, Roybal JD, Kurie JM, West JL. Cancer Res. 2012;72(22):6013–6023. doi: 10.1158/0008-5472.CAN-12-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Physiol Rev. 2011;91(3):1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Golden AP, Tien J. Lab Chip. 2007;7:720–725. doi: 10.1039/b618409j. [DOI] [PubMed] [Google Scholar]
- 57.Griffith LG, Swartz M. Nat. Rev. Mol. Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 58.Haessler U, Teo JCM, Foretay D, Renaud P, Swartz M. Integr. Biol. 2011;4(4):401–409. doi: 10.1039/c1ib00128k. [DOI] [PubMed] [Google Scholar]
- 59.Håkanson M, Kobel S, Lutolf MP, Textor M, Cukierman E, Charnley M. PLoS One. 2012;7(2):e40141. doi: 10.1371/journal.pone.0040141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanahan D, Folkman J. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 61.Hanahan D, Coussens LM. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 62.Hanahan D, Weinberg RA. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 63.Hanjaya-Putra D, Bose V, Shen YI, Yee J, Khetan S, Fox-Talbot K, Steenbergen C, Burdick JA, Gerecht S. Blood. 2011;118(3):804–815. doi: 10.1182/blood-2010-12-327338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanjaya-Putra D, Wong KT, Hirotsu K, Khetan S, Burdick JA, Gerecht S. Biomaterials. 2012;33:6123–6131. doi: 10.1016/j.biomaterials.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Am. J. Pathol. 2000;156:1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hinz B. Curr. Rheumatol. Rep. 2009;11:120–126. doi: 10.1007/s11926-009-0017-1. [DOI] [PubMed] [Google Scholar]
- 67.Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975–2009. Vintage 2009 Populations; 2012. [Accessed 13 November 2012]. http://seer.cancer.gov/csr/1975_2009_pops09. [Google Scholar]
- 68.Huh D, Hamilton G, Ingber DE. Trends Cell Biol. 2011;21:745–754. doi: 10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jain RK. N. Engl. J. Med. 2009;360:2669–2671. doi: 10.1056/NEJMcibr0902054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jongpaiboonkit L, King WJ, Lyons GE, Paguirigan AL, Warrick JW, Beebe DJ, Murphy WL. Biomaterials. 2008;29:3346–3356. doi: 10.1016/j.biomaterials.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Joyce J, Pollard JW. Nat. Rev. Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kalluri R, Zeisberg M. Nat. Rev. Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 73.Kam Y, Karperien A, Weidow B, Estrada L, Anderson AR, Quaranta V. BMC Res. Notes. 2009;2:130. doi: 10.1186/1756-0500-2-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kenny PA, Bissell MJ. Int J Cancer. 2003;107(5):688–695. doi: 10.1002/ijc.11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kenny PA, Nelson CM, Bissell MJ. Scientist. 2006;20(4):30. [PMC free article] [PubMed] [Google Scholar]
- 76.Khademhosseini A, Langer R. Biomaterials. 2007;28:5087–5092. doi: 10.1016/j.biomaterials.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 77.Khademhosseini A, Langer R, Borenstein J, Vacanti JP. Proc. Natl. Acad. Sci. U S A. 2006;103:2480–2487. doi: 10.1073/pnas.0507681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim JB, Stein R, O’Hare MJ. Tumor Biol. 2005;26:173–185. doi: 10.1159/000086950. [DOI] [PubMed] [Google Scholar]
- 79.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324(5923):59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Q, Wang J, Shahani S, Sun DD, Sharma B, Elisseeff JH, Leong KW. Biomaterials. 2006;27(7):1027–1034. doi: 10.1016/j.biomaterials.2005.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu Y, Markov D, Wikswo JP, McCawley LJ. Biomed. Microdevices. 2011;13:837–846. doi: 10.1007/s10544-011-9554-2. [DOI] [PubMed] [Google Scholar]
- 83.Mak M, Reinhart-King CA, Erickson D. PLoS One. 2011;6:e20825. doi: 10.1371/journal.pone.0020825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LE, Ingber DE. Nature. 2009;457(7233):1103–1108. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marusyk A, Almendro V, Polyak K. Nat. Rev. Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 86.Michor F, Liphardt J, Ferrari M, Widom J. Nat. Rev. Cancer. 2011;11:657–670. doi: 10.1038/nrc3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen DH, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, Bhatia SN, Chen CS. Nat. Mater. 2012;11:768–774. doi: 10.1038/nmat3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miron-Mendoza M, Lin X, Ma L, Ririe P, Petroll WM. Exp. Eye Res. 2012;99:36–44. doi: 10.1016/j.exer.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Montanez-Sauri SI, Sung KE, Puccinelli JP, Pehlke C, Beebe DJ. J. Lab. Autom. 2011;16:171–185. doi: 10.1016/j.jala.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mueller MM, Fusenig NE. Nat. reviews. Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 91.Nagy J, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Angiogenesis. 2008;11:109–119. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nagy JA, Dvorak AM, Dvorak HF. Cold Spring Harb. Perspect. Med. 2012;2(2):a006544. doi: 10.1101/cshperspect.a006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nagy J, Dvorak HF. Clin. Exp. Metastasis. 2012;29(7):657–662. doi: 10.1007/s10585-012-9500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, Park J, Sinha P, Bissell MJ, Frengen E, Werb Z, Egeblad M. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nelson CM, Bissell MJ. Semin. Cancer Biol. 2005;15:342–352. doi: 10.1016/j.semcancer.2005.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nelson CM, Bissell MJ. Annu. Rev. Cell. Dev. Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nelson CM, Inman JL, Bissell MJ. Nat. Protocols. 2008;3:674–678. doi: 10.1038/nprot.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Netti P, Berk D, Swartz M, Grodzinsky J, Jain RK. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 100.Nichol JW, Khademhosseini A. Soft Matter. 2009;5:1312–1319. doi: 10.1039/b814285h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Otranto M, Sarrazy V, Hinz B, Gabbiani G. Cell. 2012;6(3):203–219. doi: 10.4161/cam.20377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paguirigan AL, Puccinelli JP, Su X, Beebe DJ. Assay Drug Dev. Technol. 2010;8:591–601. doi: 10.1089/adt.2010.0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pampaloni F, Reynaud EG, Stelzer EH. Nat. Rev. Mol. Cell Biol. 2007;8(10):839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- 105.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 106.Pathak A, Kumar S. PLoS One. 2011;6(3):e18423. doi: 10.1371/journal.pone.0018423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pathak A, Kumar S. Proc. Natl. Acad. Sci. U S A. 2012;109:10334–10339. doi: 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Patterson J, Martino MM, Hubbell J. Mat. Today. 2010;13:14–22. [Google Scholar]
- 109.Pederson AW, Ruberti JW, Messersmith PB. Biomaterials. 2003;24(26):4881–4890. doi: 10.1016/s0142-9612(03)00369-7. [DOI] [PubMed] [Google Scholar]
- 110.Podlaha O, Riester M, De S, Michor F. Trends Genet. 2012;28:155–163. doi: 10.1016/j.tig.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Polacheck WJ, Charest JL, Kamm RD. Proc. Natl. Acad. Sci. U S A. 2011;108:11115–11120. doi: 10.1073/pnas.1103581108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Price GM, Tien J. Microdevices in Biology and Medicine: Microdevices in Biology and Medicine. Artech House; 2009. pp. 235–248. [Google Scholar]
- 113.Price GM, Tien J. Methods. 2011;671:281–293. doi: 10.1007/978-1-59745-551-0_17. [DOI] [PubMed] [Google Scholar]
- 114.Quaranta V, Rejniak K, Gerlee P, Anderson AR. Semin. Cancer Biol. 2008;18:338–348. doi: 10.1016/j.semcancer.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Raghavan S, Shen CJ, Desai RA, Sniadecki NJ, Nelson CM, Chen CS. J. Cell Sci. 2010;123:2877–2883. doi: 10.1242/jcs.055079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ramanujan S, Biener G, Herman BA. Biophys. J. 2002;83:1650–1660. doi: 10.1016/S0006-3495(02)73933-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rønnov-Jessen L, Bissell MJ. Trends Mol. Med. 2009;15:5–13. doi: 10.1016/j.molmed.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Roy R, Boskey A, Bonassar LJ. J. Biomed. Mater. Res. A. 2010;93(3):843–851. doi: 10.1002/jbm.a.32231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Saadi W, Rhee SW, Lin F, Vahidi B, Chung BG, Jeon NL. Biomed. Microdevices. 2007;9:627–635. doi: 10.1007/s10544-007-9051-9. [DOI] [PubMed] [Google Scholar]
- 120.Savina IN, Dainiak M, Jungvid H, Mikhalovsky SV, Galaev IY. J Biomater Sci Polym Ed. 2009;20(12):1781–1795. doi: 10.1163/156856208X386390. [DOI] [PubMed] [Google Scholar]
- 121.Scott JG, Basanta D, Chinnaiyan P, Canoll P, Swanson KR, Anderson AR. Neuro Oncol. 2011;13:1262–1264. doi: 10.1093/neuonc/nor083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Scott J, Kuhn P, Anderson AR. Nat. Rev. Cancer. 2012;12:445–446. doi: 10.1038/nrc3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Selimović S, Sim WY, Kim SB, Jang YH, Lee WG, Khabiry M, Bae H, Jambovane S, Hong JW, Khademhosseini A. Anal. Chem. 2011;83:2020–2028. doi: 10.1021/ac2001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shamloo A, Ma N, Poo MM, Sohn LL, Heilshorn SC. Lab Chip. 2008;8:1292–1299. doi: 10.1039/b719788h. [DOI] [PubMed] [Google Scholar]
- 125.Shamloo A, Heilshorn SC. Lab Chip. 2010;10:3061–3068. doi: 10.1039/c005069e. [DOI] [PubMed] [Google Scholar]
- 126.Shamloo A, Ph D, Xu H, Heilshorn S. Tissue Eng. Part A. 2012;18:320–330. doi: 10.1089/ten.tea.2011.0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shieh AC, Rozansky Ha, Hinz B, Swartz M. Cancer Res. 2011;71:790–800. doi: 10.1158/0008-5472.CAN-10-1513. [DOI] [PubMed] [Google Scholar]
- 128.Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Cancer Cell. 2007;11:526–538. doi: 10.1016/j.ccr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 129.Shin Y, Jeon JS, Han S, Jung GS, Shin S, Lee SH, Sudo R, Kamm RD, Chung S. Lab Chip. 2011;11:2175–2181. doi: 10.1039/c1lc20039a. [DOI] [PubMed] [Google Scholar]
- 130.Sin A, Chin KC, Jamil MF, Kostov Y, Rao G, Shuler ML. Biotechnol. Prog. 2004;20:338–345. doi: 10.1021/bp034077d. [DOI] [PubMed] [Google Scholar]
- 131.Slevin M, Kumar S, Gaffney J. J. Biol. Chem. 2002;277:41046–41059. doi: 10.1074/jbc.M109443200. [DOI] [PubMed] [Google Scholar]
- 132.Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, Louis DN, Iafrate AJ. Cancer Cell. 2011;20:810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 133.Song JW, Munn LL. Proc. Natl. Acad. Sci. U S A. 2011;2011:1–6. [Google Scholar]
- 134.Song JW, Bazou D, Munn LL. Integr. Biol. 2012;4(8):857–862. doi: 10.1039/c2ib20061a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stenzel KH, Miyata T, Rubin AL. Annu. Rev. Biopys. Bioeng. 1974;3(0):231–253. doi: 10.1146/annurev.bb.03.060174.001311. [DOI] [PubMed] [Google Scholar]
- 136.Stohrer M, Boucher Y, Stangassinger M, Jain RK. Cancer Res. 2000:4251–4255. [PubMed] [Google Scholar]
- 137.Stroock A, Fischbach C. Tissue Eng. Part A. 2010;16:2143–2146. doi: 10.1089/ten.tea.2009.0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stylianopoulos T, Diop-Frimpong B, Munn LL, Jain RK. Biophys. J. 2010;99:3119–3128. doi: 10.1016/j.bpj.2010.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sung JH, Esch MB, Shuler ML. Expert Opin. Drug Metab. Toxicol. 2010;6(9):1063–1081. doi: 10.1517/17425255.2010.496251. [DOI] [PubMed] [Google Scholar]
- 140.Sung JH, Kam C, Shuler ML. Lab Chip. 2010;10:446–455. doi: 10.1039/b917763a. [DOI] [PubMed] [Google Scholar]
- 141.Swartz M, Lund AW. Nat. Rev. Cancer. 2012;12:210–219. doi: 10.1038/nrc3186. [DOI] [PubMed] [Google Scholar]
- 142.Tan CP, Seo BR, Brooks DJ, Chandler EM, Craighead HG, Fischbach C. Integr. Biol. 2009;1:587–594. doi: 10.1039/b908036h. [DOI] [PubMed] [Google Scholar]
- 143.Tibbitt MW, Anseth KS. Biotechnol. Bioeng. 2009;103:655–663. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tien J, Nelson CM, Chen CS. Proc. Natl. Acad. Sci. U S A. 2002;99:1758–1762. doi: 10.1073/pnas.042493399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tourovskaia A, Figueroa-Masot X, Folch A. Lab Chip. 2005;5:14–19. doi: 10.1039/b405719h. [DOI] [PubMed] [Google Scholar]
- 146.Trapmann B, Gautrot JE, Connelly JT, Strange DG, Li Y, Oyen ML, Cohen Stuart MA, Boehm H, Li B, Vogel V, Spatz JP, Watt FM, Huck WT. Nat. Mater. 2012;11:642–629. doi: 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- 147.Verbridge SS, Choi NW, Zheng Y, Brooks DJ, Stroock AD, Fischbach C. Tissue Eng. Part A. 2010;16:2133–2141. doi: 10.1089/ten.tea.2009.0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Verbridge SS, Chakrabarti A, Del Nero P, Kwee B, Varner JD, Stroock AD, Fischbach C. J. Biomed. Mat. Res. Part A. 2012 doi: 10.1002/jbm.a.34587. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Viravaidya K, Sin A, Shuler ML. Biotechnol. Prog. 2004;20(1):316–323. doi: 10.1021/bp0341996. [DOI] [PubMed] [Google Scholar]
- 150.Walker GM, Zeringue HC, Beebe DJ. Lab Chip. 2004;4:91–97. doi: 10.1039/b311214d. [DOI] [PubMed] [Google Scholar]
- 151.Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ. Proc. Natl. Acad. Sci. U S A. 1998;95:14821–14826. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. J. Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wheeldon I, Ahari AF, Khademhosseini A. J. Lab. Autom. 2010;15(6):440–448. doi: 10.1016/j.jala.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Whitesides GM. Nature. 2006;442(7101):368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 155.Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Ann. Biomed. Eng. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- 156.Wlodkowic D, Cooper JM. Curr. Opin. Chem. Biol. 2010;14:556–567. doi: 10.1016/j.cbpa.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 157.Wong KHK, Truslow JG, Tien J. Biomaterials. 2010;31:4706–4714. doi: 10.1016/j.biomaterials.2010.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zaman MH, Trapani LM, Sieminski AL, Mackellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. Proc. Natl. Acad. Sci. U S A. 2006;103:10889–10894. doi: 10.1073/pnas.0604460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zervantonakis IK, Kothapalli CR, Chung S, Sudo R, Kamm RD. Biomicrofluidics. 2011;5:13406. doi: 10.1063/1.3553237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, López JA, Stroock AD. Proc. Natl. Acad. Sci. U S A. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]