

Lipodystrophies are a heterogeneous group of conditions in which individuals never develop or progressively lose adipose tissue in parts or all of their bodies (1,2) (Table 1). In this commentary, we will make the case that 1) defining lipodystrophy is a work in progress; 2) not all forms of lipodystrophy are very rare; 3) lipodystrophy and obesity can occur simultaneously and their metabolic consequences are similar and possibly synergistic; 4) leptin treatment can have impressive therapeutic effects; and 5) these conditions provide useful paradigms to explore the role of the adipose tissue on metabolic homeostasis and to investigate pathways leading from distinct genetic mutations to very different clinical phenotypes.
Table 1.
Phenotypic characterization of lipodystrophy subtypes with inclusion of genotype (if available)
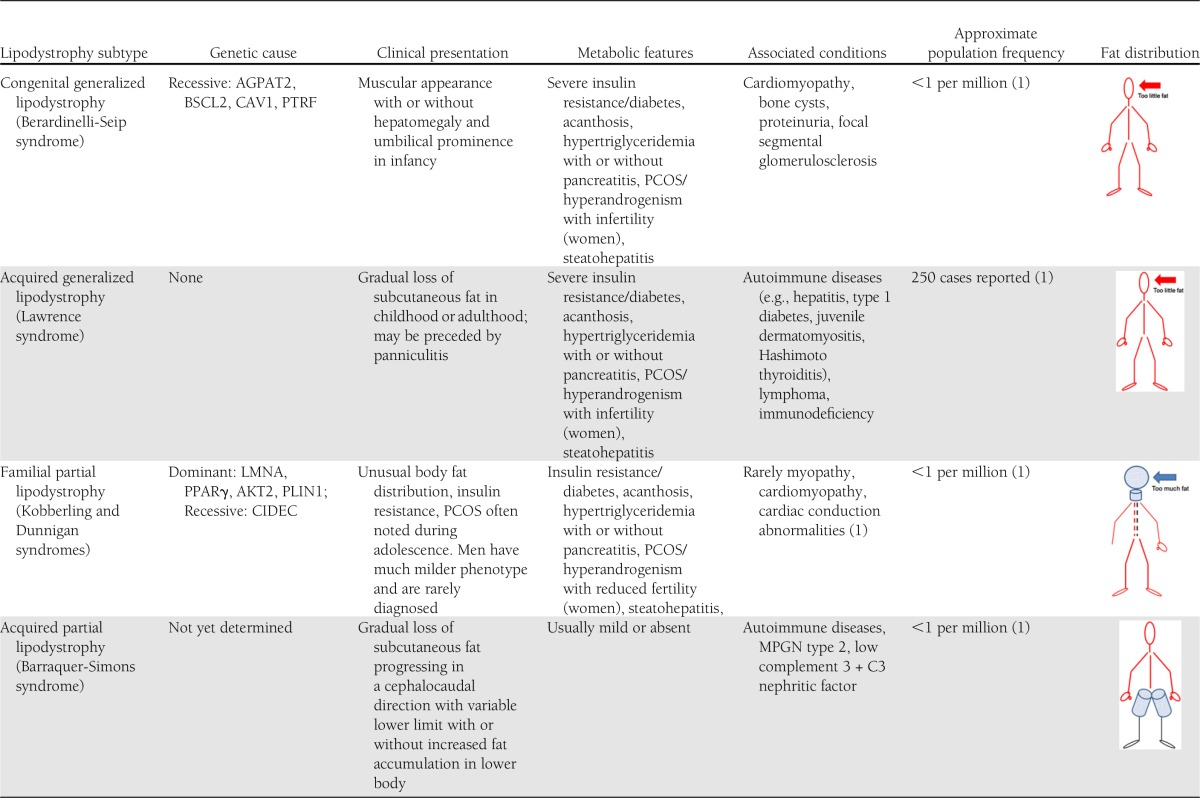

What is lipodystrophy? A recent consensus statement by the American Association of Clinical Endocrinologists acknowledges the difficulty in determining quantitative criteria and concludes that “lipodystrophy is a condition characterized by regional or total loss or absence of subcutaneous fat. This can occur either in the presence or absence of metabolic abnormalities, and with diverse clinical presentations. While generalized forms of lipodystrophy are often diagnosed during childhood or adolescence, some forms of lipodystrophy, particularly familial partial lipodystrophy, may bear some resemblance to common metabolic disorders managed by adult endocrinologists” (3). There are no specific cutoff levels for percent body fat or leptin concentrations, and it often requires time and long-term follow-up to confirm the diagnosis.
Much progress has been made in identifying the genetic etiologies of many lipodystrophy forms (1). How missing or abnormal gene products prevent adipocyte development or cause disappearance of adipose tissue in distinct regions of the human body, however, remains mostly unknown. An excellent example is familial partial lipodystrophy or Dunnigan syndrome, in which numerous mutations of the LMNA gene have been found. LMNA is expressed in a tissue-dependent manner and encodes lamin A/C, a protein, which is important for the integrity and function of the nuclear envelope. It is entirely unclear why some white adipocytes are affected and others are not. Dunnigan syndrome is characterized by accumulation of fat in the neck area and loss of subcutaneous fat only in the limbs or the gluteal region—parts of the body often not revealed on a brief clinical examination (Table 1). Thus, some patients have been misdiagnosed as having Cushing syndrome and others have suffered from delayed recognition and therapy of their marked hypertriglyceridemia and treatment-resistant diabetes associated with severe insulin resistance. Recently, it has been reported that individuals with metabolic syndrome but without classical findings of Dunnigan syndrome had a surprisingly high prevalence of mutations in LMNA and ZMPSTE24 (a gene encoding one of the lamin A processing enzymes) (4). Thus, as often in medicine, it appears that certain genetically determined conditions when associated with milder phenotypes are much more common than initially anticipated.
In this issue of Diabetes Care, Strickland et al. (5) describe a novel form termed “partial lipodystrophy of the limbs” (PLL). In comparison with other forms of lipodystrophy, which are extremely rare (e.g., congenital generalized lipodystrophy has an estimated prevalence of 1 in 10 million), this condition (PLL), similar to lipodystrophy associated with antiretroviral treatment of HIV (6), may affect larger numbers of individuals. The BMI of people with PLL is described to cover a wide range from normal to obese. Affected individuals have disproportionately slender forearms with or without slender calves (and at times thighs) compared with the rest of their bodies. Since huge physiological variations in quantity and distribution of body fat exist among healthy humans, it is necessary to provide evidence that a variant is harmful in order to classify it as pathological. Strickland et al. make the case that patients with PLL are more insulin resistant and have worse glycemia than others with similar degrees of obesity or type 2 diabetes, implying that this lipodystrophy has clinical significance. With greater awareness and more detailed clinical studies including laboratory testing and determination of body composition, it will become evident whether this phenotype is a circumscribed entity. Questions to be addressed are whether adipose tissue in the distal extremities is lost or never gained, whether these patients are leptin deficient or have other adipokine abnormalities, and whether affected individuals benefit from early recognition and intervention to lower their risk of developing diabetes or to more aggressively treat their overt diabetes. Thus far, the etiology, heredity, and prevalence of this condition remain to be determined.
The novel description of a presumably more common form of lipodystrophy is contrasted by the recent identification of an exceedingly rare form, part of an autoinflammatory condition: CANDLE (chronic neutrophilic dermatosis with lipodystrophy and elevated temperature) syndrome (7). Infants as young as 2 weeks of age (and at the latest by 12 months of age) present with skin rashes, accompanied by episodic fevers, anemia, and eventual development of partial lipodystrophy, predominantly affecting the face, wrists, ankles, and distal parts of fingers and toes. Joint contractures may develop early, and affected children fail to thrive. Again, incorrect diagnoses ranging from Lyme disease to cutaneous myelogenous leukemia have led to unnecessary and harmful treatments such as whole-body radiation and prolonged suffering of the affected child. CANDLE syndrome is caused by mutations in the proteasome gene PSMB8, which had been reported earlier by Garg et al. (8) to cause JMP syndrome (joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced childhood onset lipodystrophy) in adults (8,9). A promising treatment trial (clinical trial reg. no. NCT01724580) is underway at the National Institutes of Health to test whether the Janus kinase 1/2 inhibitor baricitinib is beneficial. Janus kinases phosphorylate activated cytokine receptors, which subsequently recruit STAT transcription factors known to modulate gene transcription. Thus, it is proposed that the inflammatory cascade is interrupted.
Making the correct diagnosis may have vital consequences for patients with lipodystrophy. For familial forms of lipodystrophy, genetic counseling is essential, and in all cases, patients should be screened for known comorbidities, including but not limited to metabolic abnormalities such as diabetes, hypertriglyceridemia, and steatohepatitis, as well as cardiomyopathy (10) and kidney disease (11). Making a diagnosis of lipodystrophy may also be critical for medical management. Conventional therapies for hyperlipidemia or diabetes are often ineffective, especially in patients with extreme metabolic disturbances. Because patients with lipodystrophy have deficient adipose tissue, they also have low levels of adipocyte-derived hormones (adipokines). Leptin was the first of these adipokines to be discovered in 1994 (12) and is a major regulator of appetite and metabolism. As a result of leptin deficiency, patients with lipodystrophy have hyperphagia, which exacerbates ectopic lipid deposition and insulin resistance. In 2000, the first patient with lipodystrophy received recombinant leptin to correct leptin deficiency. Since then, between 100 and 200 patients with non–HIV associated lipodystrophy have been treated with leptin replacement worldwide, which has been shown to improve metabolic complications in numerous subtypes, including patients with profound leptin deficiency and others with “relative leptin deficiency” in partial forms of lipodystrophy (13). In these latter forms, leptin levels tend to be unexpectedly low for the degree of adiposity, suggesting that leptin may be differentially secreted by different fat depots. Unfortunately, leptin levels were not available in the patients with the newly described “partial lipodystrophy of the limbs.” Presently, leptin is only available in clinical trials, but if and when it is approved by the U.S. Food and Drug Administration as an orphan drug, it remains to be seen whether it will be an effective, safe, and well-tolerated therapeutic agent for PLL and other forms of lipodystrophy. These include conditions associated with active inflammatory diseases, such as lupus erythematosus, dermatomyositis (14), or the above-mentioned CANDLE syndrome (7). Thus far, leptin treatment has been withheld in inflammatory diseases because of concerns regarding leptin’s potential proinflammatory effects. These have largely been deducted from animal and in vitro model systems and include upregulation of tumor necrosis factor-α (15). To date, leptin replacement has not been shown to promote inflammation in humans, but the number of treated subjects is too small to come to a definite conclusion. Furthermore, leptin has been successfully used in a few patients with quiescent juvenile dermatomyositis without exacerbating the underlying disease. Despite leptin’s beneficial effects on metabolism, some patients express disappointment because leptin treatment does not lead to restoration of adipocytes. Fat cell transplantation and synthetic fillers have become viable options for some individuals, especially for those suffering from severe facial fat loss. Active investigation is also focusing on the potential therapeutic role of adipose stem cells (16).
In summary, greater awareness and correct diagnosis of previously described and novel forms of lipodystrophy may spare patients an arduous voyage through misdiagnoses and unnecessary treatments. Certain lipodystrophy-associated genotypes may in fact be more common in milder clinical conditions, such as metabolic syndrome. A worldwide registry of patients with lipodystrophy and lipodystrophy-related genetic mutations would certainly benefit our concerted efforts in learning about the natural history, developing better diagnostic criteria, and providing safe and effective treatment.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
No potential conflicts of interest relevant to this article were reported.
Footnotes
See Strickland et al., p. 2247
References
- 1.Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011;96:3313–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arioglu E, Rother KI, Reitman ML, Premkumar A, Taylor SI. Lipoatrophy syndromes: when ‘too little fat’ is a clinical problem. Pediatr Diabetes 2000;1:155–168 [DOI] [PubMed] [Google Scholar]
- 3.Handelsman Y, Oral EA, Bloomgarden ZT, et al. The clinical approach to the detection of lipodystrophy - an AACE consensus statement. Endocr Pract 2013;19:107–116 [DOI] [PMC free article] [PubMed]
- 4.Dutour A, Roll P, Gaborit B, et al. High prevalence of laminopathies among patients with metabolic syndrome. Hum Mol Genet 2011;20:3779–3786 [DOI] [PubMed] [Google Scholar]
- 5.Strickland LR, Guo F, Lok K, Garvey WT. Type 2 diabetes with partial lipodystrophy of the limbs: a new lipodystrophy phenotype. Diabetes Care 2013;36:2247–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domingo P, Estrada V, López-Aldeguer J, Villaroya F, Martínez E. Fat redistribution syndromes associated with HIV-1 infection and combination antiretroviral therapy. AIDS Rev 2012;14:112–123 [PubMed] [Google Scholar]
- 7.Liu Y, Ramot Y, Torrelo A, et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum 2012;64:895–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garg A, Hernandez MD, Sousa AB, et al. An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy. J Clin Endocrinol Metab 2010;95:E58–E63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agarwal AK, Xing C, DeMartino GN, et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet 2010;87:866–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lupsa BC, Sachdev V, Lungu AO, Rosing DR, Gorden P. Cardiomyopathy in congenital and acquired generalized lipodystrophy: a clinical assessment. Medicine (Baltimore) 2010;89:245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Musso C, Javor E, Cochran E, Balow JE, Gorden P. Spectrum of renal diseases associated with extreme forms of insulin resistance. Clin J Am Soc Nephrol 2006;1:616–622 [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994;372:425–432 [DOI] [PubMed] [Google Scholar]
- 13.Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 2010;53:27–35 [DOI] [PubMed] [Google Scholar]
- 14.Bingham A, Mamyrova G, Rother KI, et al. Childhood Myositis Heterogeneity Study Group Predictors of acquired lipodystrophy in juvenile-onset dermatomyositis and a gradient of severity. Medicine (Baltimore) 2008;87:70–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Procaccini C, Jirillo E, Matarese G. Leptin as an immunomodulator. Mol Aspects Med 2012;33:35–45 [DOI] [PubMed] [Google Scholar]
- 16.Zeve D, Tang W, Graff J. Fighting fat with fat: the expanding field of adipose stem cells. Cell Stem Cell 2009;5:472–481 [DOI] [PMC free article] [PubMed] [Google Scholar]