

Abstract
The signaling molecule adenosine has been implicated in attenuating acute lung injury (ALI). Adenosine signaling is terminated by its uptake through equilibrative nucleoside transporters (ENTs). We hypothesized that ENT-dependent adenosine uptake could be targeted to enhance adenosine-mediated lung protection. To address this hypothesis, we exposed mice to high-pressure mechanical ventilation to induce ALI. Initial studies demonstrated time-dependent repression of ENT1 and ENT2 transcript and protein levels during ALI. To examine the contention that ENT repression represents an endogenous adaptive response, we performed functional studies with the ENT inhibitor dipyridamole. Dipyridamole treatment (1 mg/kg; EC50=10 μM) was associated with significant increases in ALI survival time (277 vs. 395 min; P<0.05). Subsequent studies in gene-targeted mice for Ent1 or Ent2 revealed a selective phenotype in Ent2−/− mice, including attenuated pulmonary edema and improved gas exchange during ALI in conjunction with elevated adenosine levels in the bronchoalveolar fluid. Furthermore, studies in genetic models for adenosine receptors implicated the A2B adenosine receptor (Adora2b) in mediating ENT-dependent lung protection. Notably, dipyridamole-dependent attenuation of lung inflammation was abolished in mice with alveolar epithelial Adora2b gene deletion. Our newly identified crosstalk pathway between ENT2 and alveolar epithelial Adora2b in lung protection during ALI opens possibilities for combined therapies targeted to this protein set.—Eckle, T., Hughes, K., Ehrentraut, H., Brodsky, K. S., Rosenberger, P., Choi, D.-S., Ravid, K., Weng, T., Xia, Y., Blackburn, M. R., Eltzschig, H. K. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury.
Keywords: A2B, dipyridamole, hypoxia-inducible factor, CD73, CD39, apyrase, ecto-nucleotidase
Acute lung injury (ALI) is an inflammatory lung disease that causes acute respiratory distress syndrome in patients (1). It is defined by its short onset and the presence of noncardiogenic pulmonary edema, leading to attenuated gas exchange and massive lung inflammation (2–4). Although there is currently no specific therapy available, management consists of aggressive treatment of the cause, vigilant supportive care, and the prevention of nosocomial infections. Thus, ALI represents a clinically important consequence of sepsis, trauma, surgery, and critical illness, significantly contributing to morbidity and mortality of patients requiring critical care support. Despite optimal management, mortality remains very high, ranging between 35 and 60% (2). Reportedly, ∼200,000 patients develop ALI annually in the United States, leading to 75,000 deaths and accounting for up to 3.6 million hospital days (5). Although there have been only a few detailed, in-person interviews and examinations to obtain follow-up data on survivors of ALI, a recent study evaluated 109 ALI survivors up to 5 yr after discharge from the intensive care unit. This study demonstrated that exercise limitation, physical and psychological sequelae, decreased physical quality of life, and increased costs and use of health care services are important legacies of severe lung injury (6). Together, these findings highlight the urgent need and biomedical significance of finding novel treatment approaches for ALI. Among the hallmarks of ALI is massive accumulation of inflammatory cells in different compartments of the lungs (7), in conjunction with cytokine release and inflammatory activation of recruited or resident cells (8, 9). Other characteristics include a disruption of the alveolar-capillary barrier function, resulting in extensive pulmonary edema and attenuated gas exchange (2, 10–13).
Extracellular adenosine is a signaling molecule that has been implicated in lung protection during ALI. It functions through activation of G-protein-coupled cell surface receptors (Adora1, Adora2a, Adora2b, and Adora3). For example, previous studies implicated the Adora2b adenosine receptor in attenuating ALI by improving alveolar fluid transport (14), promoting alveolar-capillary barrier function (15–18) or attenuating lung inflammation during ALI (19). During inflammatory conditions, extracellular adenosine stems predominantly from the breakdown of precursor nucleotides such as ATP, ADP, and AMP (4, 20–26), which are being released by multiple cell types, such as endothelial cells, inflammatory cells, or pulmonary epithelial cells (3, 12, 27–31). Indeed, mice with genetic deficiencies causing attenuated extracellular nucleotide phosphohydrolysis (cd39−/− or cd73−/− mice) experience a more severe disease course during ALI (11, 32, 32) and other inflammatory diseases (33). In the setting of ALI, extracellular adenosine signaling is terminated by uptake of adenosine from the extracellular toward the intracellular compartment (4). This adenosine transport activity is predominantly mediated by equilibrative nucleoside transporters (34–37). They represent transmembranous channels that allow adenosine to freely cross the extracellular cell membrane after its gradient. Previous studies have implicated repression of equilibrative nucleoside transporter (ENT) 1 in enhancing vascular adenosine responses during hypoxia and ischemia (34, 37) or ENT2 during hypoxia-induced inflammation of the intestine (35). At present, the functional roles of ENTs during ALI are essentially unknown. In the present studies we hypothesized that ENT repression could lead to increased bronchoalveolar (BAL) adenosine levels and increased activation of adenosine receptors, thereby promoting lung protection. To address this hypothesis, we combined genetic approaches in gene-targeted mice for Ent1 or Ent2 with pharmacological studies. These studies suggest that termination of pulmonary adenosine signaling is predominantly mediated by Ent2 and reveal a previously unknown crosstalk pathway between ENT2 and alveolar epithelial Adora2b adenosine receptors in promoting lung protection during ALI.
MATERIALS AND METHODS
Experimental animals
Wild-type (WT; BL6C57), VeCadherinCre [B6.Cg-Tg(Cdh5-cre)7Mlia/J; ref. 38], and GermlineCre [B6.C-Tg(CMV-cre)1Cgn/J; ref. 39] mice were purchased from The Jackson Laboratory, Bar Harbor, ME, USA). Sftpc-Cre mice were obtained from Brigid Hogan (Duke University, Durham, NC, USA; ref.40). To obtain tissue-specific Adora2b−/− mice, we crossed Adora2bf/f (Ozgene) mice with the appropriate Cre mouse. To obtain whole-body Adora2b−/−, we bred Adora2bf/f mice with the GermlineCre mouse. To obtain Adora2bf/f Spc-Cre (alveolar epithelial specific) or Adora2bf/f VeCadherinCre (endothelial specific) mice, we bred Adora2bf/f mice with the Sftpc-Cre or VeCadherinCre mouse, respectively. Ent1−/− mice were obtained from the D.-S.C. laboratory (Mayo Clinic College of Medicine, Rochester, MN, USA). Ent2−/− mice were purchased from Lexicon (The Woodlands, TX, USA). Mice were bred and maintained in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (U.S. National Institutes of Health, Bethesda, MD, USA). Experimental protocols were approved by the institutional review board at the University of Colorado (Denver, CO, USA) and were in accordance with the U.S. National Institutes of Health Protection of Animals guidelines for the use of live animals.
ALI model
At 8–12 wk old, age- and weight-matched male mice were anesthetized with pentobarbital (70 mg/kg) before the procedure and placed on a temperature-controlled heated table (RT; Effenberg, Munich, Germany) with a rectal thermometer probe attached to a thermal feedback controller to maintain body temperature at 37°C. In addition, all animals were monitored with an electrocardiogram (Hewlett Packard, Böblingen, Germany). Fluid replacement was performed with normal saline, 0.05 ml/h i.p. After tracheotomy, the tracheal tube was connected to a mechanical ventilator with pediatric tubing (Servo 900C; Siemens AG, Munich, Germany). Mice were ventilated in a pressure-controlled ventilation mode at different inspiratory pressure levels (15, 35, and 45 mbar) for different time periods (1–4 h). All animals were ventilated with an inspired oxygen concentration of 100% [fraction of inspired oxygen (Fio2) of 1]. In a subset of experiments, animals were ventilated under deep anesthesia at 35 mbar until a cardiac standstill was observed in the surface electrocardiogram, and mice were subsequently euthanized. To investigate the effect of ENT inhibition on survival, mice were pretreated with dipyridamole (1 mg/kg i.p.) 1 h before the procedure.
Dipyridamole
Dipyridamole at concentrations ≤ 10 μM impairs uptake of adenosine from the extracellular space (41). This drug may also inhibit adenosine deaminase, attenuate cyclic phosphodiesterase activity (thereby increasing cGMP), relax directly smooth muscle, and interact with the prostaglandin system (41). However, all of these latter activities occur at concentrations > 10 μM (41). Because ≤1 mg/kg i.p. dipyridamole results in serum concentrations ≤ 10 μM (42), this dose was used in our studies to effectively inhibit both ENT1 and ENT2 (IC50 for ENTs: ENT1=5.0±0.9 nM and ENT2=356±13 nM). Therefore, the above target serum concentration of ≤10 μM will effectively inhibit both transporters (43).
Tissue harvest
At the indicated time points, mice were anesthetized and killed by exsanguination. BAL fluid was obtained after the lung was lavaged with 0.5 ml of saline 3 times. BAL fluid supernatant was snap-frozen after centrifugation at 1000 g for 5 min at 4°C. Pulmonary tissue was flushed with 10 ml of saline via the right ventricle, snap frozen in liquid nitrogen, and stored at −80°C (10, 14).
RNA isolation and real-time polymerase chain reaction (PCR)
Total RNA was extracted from tissue by TRIzol, followed by cDNA synthesis using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. Quantitative reverse transcriptase (RT)-PCR (ABI 7900HT; Applied Biosystems, Carlsbad, CA, USA) was performed to measure relative mRNA levels for various transcripts, with Power SYBR Green PCR Master Mix (Applied Biosystems) containing 1 μM sense and 1 μM antisense primers.
Primers for real-time RT-PCR
Real-time RT-PCR was performed with murine QuantiTect Primer Assay for β-actin (QT01136772; Qiagen, Valencia, CA, USA) or the following primers (Invitrogen, Carlsbad, CA, USA): mouse ENT1 sense CTTGGGATTCAGGGTCAGAA or antisense ATCAGGTCACACGACACCAA; mouse ENT2 sense CATGGAAACTGAGGGGAAGA or antisense GTTCCAAAGGCCTCACAGAG; human ENT1 sense CTTGGGCTTGGAGAACAC or antisense AAGGCACCTGGTTTCTGTC; and human ENT2 sense CTTCCATACCCACTCTCTCACC or antisense GAGAGAGAGGGGATTGGGTC.
Immunoblotting and immunohistochemistry experiments
Rabbit polyclonal anti-ENT1 antibody (ab48607; Abcam Inc., Cambridge, MA, USA) and rabbit polyclonal anti-ENT2 antibody (ab48595; Abcam) were used to determine Ent1 or Ent2 protein content from whole lungs. For this purpose, we ventilated C57BL/6J mice (Charles River Laboratories, Wilmington, MA, USA) with the ventilator settings indicated in the figure legends. Animals were euthanized, and the remaining blood was removed from the pulmonary circulation by injection of 1 ml of phosphate-buffered saline (PBS) into the right ventricle. The lungs were excised and immediately frozen at −80°C until immunoblotting. For this purpose, tissues were homogenized and lysed for 10 min in ice-cold lysis buffer (107 polymorphonuclear neutrophils (PMNs)/500 μl; 150 mM NaCl; 25 mM Tris, pH 8.0; 5 mM EDTA; 2% Triton X-100; and 10% mammalian tissue protease inhibitor cocktail; Sigma-Aldrich, St. Louis, MO, USA) and collected in microfuge tubes. After spinning at 14,000 g to remove cell debris, the pellet was discarded. Proteins were solubilized in reducing Laemmli sample buffer and heated to 90°C for 5 min. Samples were resolved on a 12% polyacrylamide gel and transferred to nitrocellulose membranes. The membranes were blocked for 1 h at room temperature in PBS supplemented with 0.2% Tween 20 and 4% bovine serum albumin (BSA). The membranes were incubated in 10 μg/ml Ent1 or Ent2 antibody for 1 h at room temperature, followed by 10-min washes in PBS. The membranes were then incubated in 1:3000 donkey anti-rabbit horseradish peroxidase. The wash was repeated, and proteins were detected by enhanced chemiluminescence.
Measurement of BAL fluid albumin content
The albumin content of BAL fluid supernatants was measured with a mouse albumin enzyme-linked immunosorbent assay (ELISA) quantitation set according to the manufacturer's instructions (Bethyl Laboratories, Montgomery, TX, USA). Samples were diluted 1:10,000 (10, 14).
Blood gas analysis
To assess pulmonary gas exchange, blood gas analyses were performed in subsets of experiments by obtaining arterial blood via cardiac puncture. In short, a lateral thoracotomy was performed to access the left ventricle, and blood was obtained via cardiac puncture. Analysis was performed immediately after collection with an i-Stat Analyzer (Abbott Laboratories, Abbott Park, IL, USA), and the arterial partial pressure of oxygen was measured, in addition to arterial partial carbon dioxide pressure and pH values (10, 14).
Measurement of adenosine concentrations in BAL fluid
Nucleosides were measured as described previously (44). In brief, to measure the nucleoside levels in BAL fluid, mice were anesthetized with 2.5% Avertin, and the lungs were lavaged 4 times with 0.3 ml of PBS containing 10 μM dipyridamole (Sigma-Aldrich), 10 μM deoxycoformycin (adenosine deaminase inhibitor; R&D Systems Inc., Minneapolis, MN, USA), and 10 μM αβ-methylene ADP (Sigma-Aldrich). BAL fluid was then centrifuged to remove cells and debris. To measure nucleoside levels, 200 μl BAL supernatant was analyzed by reverse-phase high-performance liquid chromatography (HPLC) as described previously (45). Representative peaks were identified and quantified using external standard curves.
Cell culture and treatments
Calu-3 human airway epithelial cells were cultured as described previously (11, 14). Human primary alveolar epithelial cells (HPAEpiCs; ScienCell Research Laboratories, Carlsbad, CA, USA) were cultured according to the supplier's instructions.
Analysis of β-galactosidase (β-Gal) expression in lungs of Adora2b reporter mice
To localize Adora2b in lung tissues, we analyzed β-Gal expression in Adora2b-knockout/β-Gal-knock-in mice (46). Lungs were harvested after perfusion fixation in 4% paraformaldehyde-0.1 M phosphate buffer and postfixed in the same fixative for 3 h, followed by cryoprotection in 20% sucrose in 0.1 M phosphate buffer (pH 7.2) overnight at 4°C. With use of a cryostat, 16-μm sections were collected onto Superfrost Plus slides (Thermo Fisher Scientific, Waltham, MA, USA). The slides were washed 3 times for 10 min each in 0.1 M PBS before incubation in blocking solution (2% normal goat serum, 1% BSA, and 0.3% Triton in PBS) for 1 h at room temperature. The samples were then incubated overnight at 4°C in chicken anti-β-Gal (1:500; Aves Labs Inc., Tigard, OR, USA) diluted in blocking solution. After 3 washes in PBS, samples were incubated for 2 h at room temperature with Alexa Fluor 568 goat anti-chicken IgG (1:400; Invitrogen) diluted in blocking buffer. Slides were then washed twice in PBS, followed by a 10-minute wash in 0.1 M phosphate buffer, and coverslipped using Vectashield (Vector Laboratories, Burlingame, CA, USA).
Quantification of immunohistochemical staining
The amount of antibody staining was quantified by using Photoshop-based image analysis (Adobe Systems, Mountain View, CA, USA; refs. 47, 48). All samples were analyzed in triplicate. The final immunostaining intensity (arbitrary units) was determined by subtracting the intensity of the negative control.
Myeloperoxidase (MPO) assay
Tissue samples were measured with a mouse MPO ELISA kit (Hycult Biotech, Plymouth Meeting, PA, USA). MPO levels were normalized to protein concentrations afterward.
Cytokine protein levels
Interleukin-6 (IL-6) lung tissue levels were determined using commercially available ELISA kits for mouse (R&D Systems) according to the manufacturer's instructions (10, 14).
Software and statistical analysis
Data analysis and plotting were performed using Prism 4.0c (GraphPad Software, San Diego, CA, USA). Statistical analyses were performed with an unpaired t test, 1-way analysis of variance (ANOVA) or 2-way ANOVA and the Bonferroni multiple comparison posttest correction as indicated. All data are expressed as means ± sd.
RESULTS
Pulmonary adenosine transporters ENT1 and ENT2 are repressed during ALI
Previous studies had demonstrated a functional role of endogenously generated adenosine in attenuating acute inflammatory events of the lungs (8, 49–51). During ALI, adenosine signaling is terminated by uptake of adenosine from the intracellular toward the extracellular compartment through adenosine transporters, particularly through ENT1 and ENT2. We hypothesized that ENT-type adenosine transporters could function to fine-tune extracellular adenosine levels during ALI. To address their contribution, we examined ENT1/ENT2 transcript and protein levels during ALI. In these studies, we used a previously described model of mechanical ventilation to induce ALI (10, 11, 14, 52, 53). We exposed mice over 0–4 h of injurious mechanical ventilation and assessed pulmonary ENT1 and ENT2 transcript and protein levels (Fig. 1). Indeed, we observed time-dependent repression of ENT1 and ENT2 transcript and protein levels. Moreover, ENT protein repression was more pronounced in mice exposed to higher inspiratory pressure levels, indicating that ENT repression is more dramatic with more severe degrees of ALI. Taken together, these findings demonstrate that endogenous levels of adenosine transporters ENT1 and ENT2 are repressed during ALI induced by mechanical ventilation.
Figure 1.
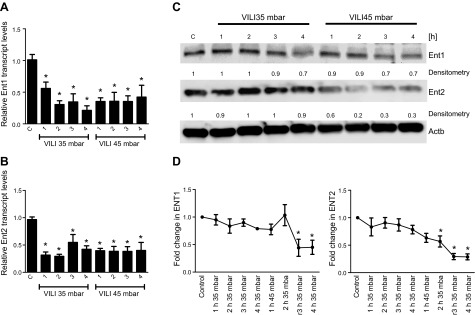
ENT1 and ENT2 repression during ALI. A−C) To examine the influence of mechanical ventilation on pulmonary ENT expression patterns, WT mice (BL6C57) were exposed to ventilator-induced lung injury (VILI; pressure-controlled mechanical ventilation at an inspiratory pressure of 35 and 45 mbar with an inspired oxygen concentration of 100% and exposure time of 1−4 h) and analyzed for ENT1 or ENT2 transcript or protein levels. A, B) Lungs were harvested at indicated time points, total RNA was isolated, and ENT1 (A) or ENT2 (B) mRNA levels were determined by real-time RT-PCR. Data were calculated relative to the internal housekeeping gene [β-actin (Actb)] and are expressed as mean ± sd fold change compared with that for the control (0 min of ventilation) at each indicated time. *P < 0.05; n = 4. C) Frozen lung tissue was lysed, and proteins were resolved by SDS-PAGE. Resultant Western blots were probed with anti-ENT1 or anti-ENT2 antibody. A representative blot of 3 is displayed. C, control. D) Quantification of Western blots by densitometry.
Pharmacologic inhibition of ENTs with dipyridamole promotes survival time during ALI
Based on the above studies demonstrating repression of ENT1 and ENT2 during ALI, we considered that ALI-induced ENT repression could be part of an endogenous protective pathway to increase pulmonary adenosine levels and adenosine-mediated lung protection. To address this hypothesis, we next pursued pharmacological studies using the nonspecific ENT inhibitor dipyridamole. Indeed, we had shown in previous studies that dipyridamole treatment is associated with effective inhibition of ENT1 and ENT2 and elevations of extracellular adenosine levels (34, 35, 37). Therefore, we pretreated mice with dipyridamole (1 mg/kg i.p.; EC50=10 μM) or vehicle before ALI induction. Indeed, we found that this treatment was associated with elevated levels of BAL adenosine, as measured via HPLC (Fig. 2A). We subsequently exposed the deeply anesthetized animals to ALI induced by pressure-controlled ventilation and assessed survival time, defined at the time from the onset of the experimental procedure until the occurrence of a flat line in the surface electrocardiogram. Consistent with the notion that ENT repression or inhibition could enhance lung-protective adenosine signaling effects, we observed that mice treated with dipyridamole experienced significantly longer survival times in this model. Indeed, median survival was prolonged from 277 to 395 min after dipyridamole treatment (P<0.05; Fig. 2A). Similarly, pulmonary edema, as assessed by albumin leakage into the BAL fluid, was significantly attenuated in dipyridamole-treated mice (Fig. 2C). Taken together, these studies indicate that dipyridamole-elicited inhibition of ENTs provides robust lung protection during ALI induced by mechanical ventilation.
Figure 2.
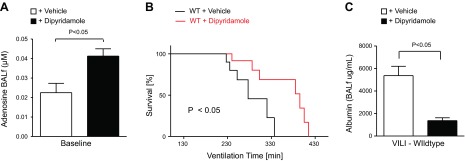
Ventilator-induced lung injury (VILI) in mice pretreated with dipyridamole. A) Measurement of BAL fluid (BALf) adenosine levels by HPLC in WT mice treated with 1 mg/kg body weight of dipyridamole. Note elevated BAL adenosine levels in mice treated with dipyridamole. B) WT mice were exposed to VILI after dipyridamole pretreatment (1 mg/kg body weight), and survival times were determined during VILI. Mechanical ventilation was applied using pressure-controlled settings (inspiratory pressure of 35 mbar and inspired oxygen concentration 100%; respiratory rate and inspiratory/expiratory ratio were adjusted to maintain normal pH) until a cardiac standstill was observed in the surface electrocardiogram. Note significantly prolonged survival after dipyridamole treatment (P<0.05; n=8). C) Albumin concentration in the BAL fluid was determined by ELISA. Note significantly decreased albumin concentration in the BAL fluid of dipyridamole-treated mice (P<0.05 over vehicle; n=4).
Selective deletion of Ent2 is associated with attenuated pulmonary edema and improved gas exchange during ALI
Based on the above findings showing that intraperitoneal dipyridamole treatment is associated with prolonged survival during ALI, we next pursued genetic studies to address the relative contributions of individual ENTs. Mice with global genetic deletion of Ent1 or Ent2 have been described previously (34, 54). Indeed, both mouse lines reproduce normally and show no obvious immunological disease manifestations when kept in a pathogen-free environment (34). To address the relative contributions of ENT1 or ENT2 during ALI, we performed a head-to-head comparison of Ent1−/− mice and Ent2−/− mice during ALI induced by mechanical ventilation. Whereas gene-targeted mice for Ent1 experienced elevations of albumin leakage similar to those of littermate controls matched in age, gender, and weight, Ent2−/− mice were profoundly protected (Fig. 3A). As an additional outcome parameter, we performed studies to address lung function during ALI. Here, we used the ratio of the arterial partial pressure of oxygen (Pao2) to the Fio2 as a readout for gas exchange during ALI. Indeed, we observed that after 3 h of mechanical ventilation at an inspiratory pressure level of 45 mbar the Pao2/Fio2 gradient was dramatically attenuated in WT mice or mice with genetic deletion of Ent1, reaching levels <200 mmHg, consistent with the definition of moderate acute respiratory distress syndrome in patients (1). In contrast, Ent2−/− mice were profoundly protected from development of ALI-associated alterations in gas exchange (Fig. 3B). We subsequently exposed the deeply anesthetized Ent2−/− mice or littermate controls matched in age, gender, and weight to ALI induced by pressure-controlled ventilation and assessed survival time, defined as the time from the onset of the experimental procedure until the occurrence of a flat line in the surface electrocardiogram. Consistent with a functional role of Ent2 deletion in lung protection during ALI, we observed significantly longer survival times in Ent2−/− mice (P<0.05; Fig. 3C). Taken together, these findings provide the first genetic evidence for a functional role of ENT2 in lung protection during ALI.
Figure 3.

Ventilator-induced lung injury (VILI) in mice gene-targeted for ENT1 or ENT2. A, B) Ent1−/− mice, Ent2−/− mice, or littermate controls were mechanically ventilated using pressure-controlled ventilation with an inspired oxygen concentration of 100% over 180 min at 45 mbar. A) Albumin concentration in the BAL fluid was determined by ELISA. Note the significantly decreased albumin concentration in the BAL fluid of Ent2−/− mice (P<0.05 over WT or Ent1−/− mice; n=6). B) To assess pulmonary gas exchange, blood gas analyses were performed by obtaining arterial blood via cardiac puncture. Analysis was performed immediately, and the Pao2/Fio2 ratio was determined. Results are means ± sd (n=6). C) Ent2−/− mice or littermate controls matched in age, weight, and gender were exposed to VILI, and survival times were determined. Mechanical ventilation was applied using pressure-controlled settings (inspiratory pressure of 35 mbar and inspired oxygen concentration 100%; respiratory rate and inspiratory/expiratory ratio were adjusted to maintain normal pH) until a cardiac standstill was observed in the surface electrocardiogram. Note significantly prolonged survival in Ent2−/− mice (P<0.05, n=8).
Ent2 as important regulator of adenosine levels in the alveolar space
After having shown in gene-targeted mice for individual Ents that selective deletion of Ent2 is associated with lung protection from pulmonary edema and improved gas exchange during ALI, we subsequently pursued studies to examine the functional role of adenosine signaling in the above observations. For this purpose, we first examined adenosine levels in the BAL fluid in Ent2−/− mice. Indeed, we observed that BAL adenosine levels were dramatically increased in Ent2−/− mice (Fig. 4A). To understand the contribution of ENTs to adenosine levels within the alveolar space, we also examined the relative transcript levels of ENT1 and ENT2 in pulmonary epithelial cells. Here we found that ENT2 transcript levels were 16-fold higher than ENT1 levels in Calu-3 pulmonary epithelial cells (P<0.05; Fig. 4B). Similarly, ENT2 transcript or protein levels in HPAEpiCs were significantly higher than their ENT1 expression (Fig. 4C). Moreover, ENT2 protein levels were higher than ENT1 levels in Calu-3 pulmonary epithelial cells (Fig. 4D). Taken together, these findings implicate a functional role for ENT2 in regulating adenosine levels in the BAL fluid.
Figure 4.
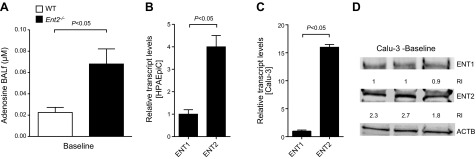
Function of pulmonary ENT2. A) BAL fluid (BALf) from WT or Ent2−/− mice was obtained by lavaging the lungs 3 times with 0.5 ml of PBS containing 200 μM dipyridamole and 1 μg/ml deoxycoformycin. Adenosine concentrations were measured by HPLC (n=6). B, C) Comparative gene expression of pulmonary ENT1 or ENT2. Confluent HPAEpiCs (B) or Calu-3 pulmonary epithelial cells (C) were analyzed for ENT1 or ENT2 transcript levels by real-time RT-PCR relative to housekeeping gene β-actin (ActB). Results are means ± sd (n=3). D) ENT1 and ENT2 levels in Calu-3 cells at baseline assessed by Western blotting.
Alveolar epithelial Adora2b adenosine receptors contribute to ENT-dependent lung protection during ALI
After having shown that Ent2 deletion is associated with elevated BAL adenosine levels, we pursued studies to explore the functional contribution of adenosine receptors in ENT-mediated lung protection. In previous studies, we had performed a head-to-head comparison of Adora1−/−, Adora2a−/−, Adora2b−/−, and Adora3−/− mice during ALI induced by pressure-controlled mechanical ventilation. Indeed, these investigations revealed a selective phenotype in Adora2b−/− mice, characterized by dramatic increases in lung inflammation (14). Based on these findings, we hypothesized that the lung protection observed after pharmacological inhibition of ENTs or the genetic deletion of Ent2 is mediated by adenosine signaling through Adora2b. Previous studies showed that Adora2b expression in the lung is predominantly localized to alveolar epithelial cells (55). To address the expression of Adora2b during ALI, we exposed previously described Adora2b gene reporter mice to ALI induced by mechanical ventilation (46, 56). Indeed, immunohistochemical studies in these mice revealed predominant alveolar epithelial expression of the Adora2b gene with robust increases in Adora2b expression after injurious mechanical ventilation (Fig. 5). To understand the functional role of Adora2b signaling in ENT-dependent lung protection, we subsequently performed dipyridamole treatment in genetic models for Adora2b. As a first step, we exposed mice with global Adora2b deletion (22, 57, 58) to ALI after pretreatment with dipyridamole (Fig. 6). PMN trafficking was significantly enhanced in Adora2b−/− mice compared with that in control WT mice (Fig. 6). Next, we exposed mice with a selective deletion of Adora2b on endothelial cells (Adora2bf/f VeCadCre) or alveolar epithelial cells (Adora2bf/f Spc-Cre) to ALI after dipyridamole pretreatment (Fig. 6). Here we found enhanced PMN trafficking in Adora2bf/f Spc-Cre compared with that in WT controls, similar to that in Adora2b−/− mice. However, Adora2bf/f VeCadCre mice revealed a phenotype identical to that of the WT controls. Thus, dipyridamole pretreatment seemed to have no affect on PMN trafficking into the lungs of Adora2b−/− or Adora2bf/f Spc-Cre mice, as assessed by pulmonary MPO levels. However, dipyridamole pretreatment in mice with deletion of the Adora2b on endothelial cells (Adora2bf/f VeCadCre) was as protective from neutrophil trafficking into the lungs during ALI as seen in WT animals. These findings indicated that protection by ENT inhibition was indeed Adora2b dependent but did not include Adora2b on the vasculature. Moreover, an observed protection from inflammatory cytokine elevations that we observed in Ent2−/− mice (IL-6; Fig. 7A) or after dipyridamole pretreatment (Fig. 7B) was completely abolished in Adora2bf/f Spc-Cre mice (Fig. 7C). Taken together, these findings implicate adenosine Adora2b receptors expressed on alveolar epithelial cells in mediating the lung protection observed with pharmacological ENT inhibition or genetic Ent2 deletion.
Figure 5.

Adora2b expressed on lung epithelia. A) Adora2b reporter mice (Adora2b-knockout/β-Gal-knock-in mice) were exposed to 3 h of high-pressure ventilation (45 mbar) or underwent normoventilation (3 h at 15 mbar). Lungs were stained for β-Gal as an indicator of the activity of the Adora2b gene promoter, which drives expression of the reporter gene (original view, ×400; a representative image of 3 is shown). Arrows indicate Adora2b staining. Data are representative of 3 experiments showing similar results. B) Quantification of immunohistochemical staining. Amount of antibody staining was quantified by using Photoshop-based image analysis (47, 48). All samples were analyzed in triplicate. Final immunostaining intensity [arbitrary units (AU)] was determined by subtracting the intensity of the negative control (C). VILI, ventilator-induced lung injury.
Figure 6.

Tissue-specific function of Adora2b during ALI. Adora2b−/−, Adora2bf/f VeCadCre, or Adora2bf/f Spc-Cre mice or corresponding age-, weight-, and gender-matched littermate controls (Cre-expressing mice) were exposed to ventilator-induced lung injury (VILI; pressure-controlled mechanical ventilation at an inspiratory pressure of 45 mbar for 3 h with an inspired oxygen concentration of 100%). Mice were pretreated with dipyridamole (1 mg/kg of body weight i.p.), and pulmonary neutrophil accumulation was quantified using MPO ELISA. Note significant increase in MPO activity in mice with global Adora2b deletion or in mice with a lack of Adora2b on alveolar epithelia only. Results are means ± sd (P<0.05; n=6).
Figure 7.
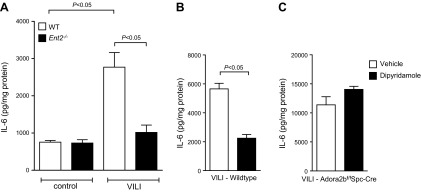
Dipyridamole relies on alveolar epithelial Adora2b. WT (BL6C57) or Ent2−/− mice were exposed to ventilator induced lung injury (VILI; pressure-controlled mechanical ventilation at an inspiratory pressure of 45 mbar with an inspired oxygen concentration of 100% and exposure time of 180 min). A) IL-6 levels in WT or Ent2−/− mice were evaluated in lung tissue homogenates using a mouse ELISA. Results are means ± sd (n=6). B, C) WT (B) or Adora2bf/f Spc-Cre (C) mice were pretreated with dipyridamole before the onset of VILI, and IL-6 levels were evaluated in lung tissue homogenates using a mouse ELISA. Results are means ± sd (n=6). Note abolished anti-inflammatory effect of dipyridamole during VILI in mice lacking Adora2b on lung epithelial cells only.
DISCUSSION
ALI is among the leading causes of the morbidity and mortality of critical illness (5). However, there are presently no pharmacological approaches available that provide specific means of lung protection. Thus, the search for novel therapeutic approaches to dampen lung inflammation and pulmonary edema during ALI is an area of intense investigation. Based on previous studies implicating adenosine signaling in lung protection during ALI (4, 8), we performed studies to address the functional role of adenosine transporters, which were implicated in terminating pulmonary adenosine signaling via adenosine uptake, in murine models of ALI. Initial studies revealed that mRNA expression of the 2 dominant adenosine transporters Ent1 and Ent2 are repressed during ALI, whereas pharmacological ENT inhibition was associated with lung protection during ALI induced by mechanical ventilation. Subsequent studies of ALI in gene-targeted mice for Ent1 or Ent2 demonstrated selective protection in Ent2−/− mice. Although Ent2−/− mice had higher BAL adenosine levels, studies in global or tissue-specific deletion models of the Adora2b adenosine receptor indicated that ENT2-mediated lung protection involves alveolar epithelial Adora2b signaling. Taken together, these studies suggest that ENT repression and concomitant Adora2b-signaling provides lung protection during ALI. Moreover, these findings provide a rational for testing ENT inhibitors such as dipyridamole in the treatment of patients experiencing ALI.
Our studies are consistent with previous findings suggesting that adenosine signaling can be targeted for lung protection. For example, previous studies had shown that oxygenation may inhibit protective adenosine signaling effects during ALI caused by the inhalation of combined toxins from gram-positive and gram-negative bacteria (8). These studies demonstrate that when mice are maintained at lower oxygen levels during ALI, hypoxia-elicited increases in adenosine signaling through the Adora2a provides lung protection. Notably, these findings challenge the common practice of use of high levels of oxygenation in the treatment of patients with ALI. Moreover, they also demonstrate that if high levels of oxygen may be necessary to maintain the function of vital organs, adenosine receptor agonists could be efficiently supplemented to overcome the detrimental effects of hyperoxia. These findings are based on a landmark study from the same research group showing that endogenous adenosine through activation of Adora2a receptors dampens inflammation in a wide range of inflammatory models (51). In addition, hypercapnia has recently been shown to also cause stabilization of proinflammatory gene programs, such as nuclear factor-κB, which could contribute to lung injury in this model (59, 60).
Consistent with the above findings showing that hypoxia-mediated adenosine responses are protective during ALI (8, 49, 50), previous studies had shown that hypoxia mediates transcriptional repression of ENT1 and ENT2 and concomitant protection from hypoxia-induced inflammation via protective adenosine signaling effects (35–37, 61, 62). For example, adenosine uptake by endothelial cells is attenuated after hypoxia exposure or small interfering RNA-mediated repression of ENT1 (37). Hypoxia-mediated decreases of adenosine transport of intestinal epithelial cells are predominantly mediated by hypoxia-elicited ENT2 repression (35). Indeed, the ENT1 and ENT2 promoter contain binding sites for the transcription factor hypoxia-inducible factor (HIF). HIF is a heterodimeric transcription factor with its α-subunit HIF1A or HIF2A being stabilized during conditions of limited oxygen availability (63, 64). Many parallels between the HIF pathway and inflammatory transcriptional pathways have been described (65, 66), including studies showing that HIF signaling can dampen hypoxia-elicited inflammation (67–70) or provide tissue adaptation to hypoxia (20, 71–73). Thus, previous studies had shown that binding of HIF to the ENT1 or ENT2 promoter causes transcriptional repression of ENT1 and ENT2, thereby leading to attenuated expression of ENT1 or ENT2 and decreased cellular adenosine uptake (35, 37). Moreover, studies in mice with tissue-specific deletion of Hif1a revealed higher expression of Ent1 after Hif1a deletion (37). Other studies have shown in models of ischemia and reperfusion injury that ENT repression is associated with elevated levels of extracellular adenosine and concomitant organ protection during ischemia and reperfusion injury (25, 34). It is conceivable that the observed ENT repression during ALI could be mediated by hypoxia-elicited transcriptional pathways (such as HIF stabilization; ref. 7), leading to increased extracellular adenosine levels and signaling events through Adora2b.
The present findings implicate the adenosine transport inhibitor dipyridamole in tissue protection from ALI. These findings are important from a translational perspective, because dipyridamole is approved for usage in patients (74, 75). For example, dipyridamole can be clinically used as a coronary vasodilator to inhibit adenosine uptake and cause adenosine-mediated coronary vasodilatation, which can be used to unmask coronary artery stenosis during pharmacological stress echocardiography (4). Similarly to the adenosine-uptake inhibitor dipyridamole, previous studies had demonstrated that several other pharmacological compounds function through indirect enhancement of extracellular adenosine signaling. For example, the anti-inflammatory properties of methotrexate and sulfasalazine result from increased production of extracellular adenosine (76–80). Notably, both medications function by molecular mechanisms, resulting in increased adenosine production from nucleotide phosphohydrolysis (76). In contrast, the present studies implicate mechanisms downstream of extracellular adenosine signaling, namely adenosine transport, in mediating tissue protection. Moreover, there are also studies implicating adenosine metabolism [to inosine via the adenosine deaminase (81) or to adenosine monophosphate via the adenosine kinase (82, 83)] in mediation of tissue protection from hypoxia-induced inflammation. Moreover, several elegant studies show that in addition to extracellular adenosine generation from ATP/AMP, there are alternative pathways for the extracellular generation of adenosine and concomitant tissue protection, e.g., adenosine production via the extracellular 2′,3′-cAMP-adenosine pathway (84–86). Thus, dipyridamole-mediated inhibition of adenosine transporters represents one of several pharmacological approaches that could be pursued to increase extracellular adenosine levels and concomitant tissue protection.
Taken together, the present studies suggest that termination of extracellular adenosine signaling via pulmonary adenosine transporters, in particular by ENT2, can be targeted to enhance extracellular adenosine signaling via alveolar Adora2b receptors. This crosstalk pathway is important from a translational perspective, as the adenosine transport inhibitor dipyridamole has been used safely in patients, and its indications could be expanded toward ALI treatment (74, 75). Future challenges will involve taking these findings from bench to bedside and addressing the safety and efficiency of dipyridamole treatment in patients with ALI. However, it is important to point out that some studies suggest that dipyridamole is an even more potent inhibitor of human ENTs than rodent ENTs (87). Therefore, the beneficial effects of dipyridamole in the present study may actually underestimate the potential beneficial effects in patients. That is to say, the use of dipyridamole (a drug already approved for human use per se; refs. 74, 75) would potentially be even more effective (with fewer off-target effects) in humans than in mice.
Acknowledgments
The authors acknowledge Kristann Magee, Stephanie Bonney, and Megan Bonney for technical assistance.
This research was supported by U.S. National Heart Institute grants R01-DK097075, R01-HL0921, R01-DK083385, R01-HL098294, and P01-HL114457-01 to H.K.E.; K08-HL102267 to T.E.; and R01-HL070952 to M.R.B.; and by a Crohn's and Colitis Foundation of America grant to H.K.E.
Footnotes
- β-Gal
- β-galactosidase
- ALI
- acute lung injury
- ANOVA
- analysis of variance
- BAL
- bronchoalveolar lavage
- BSA
- bovine serum albumin
- ELISA
- enzyme-linked immunosorbent assay
- ENT
- equilibrative nucleoside transporter
- Fio2
- fraction of inspired oxygen
- HPAEpiC
- human primary alveolar epithelial cell
- HIF
- hypoxia inducible factor
- HPLC
- high-performance liquid chromatography
- IL-6
- interleukin-6
- MPO
- myeloperoxidase
- Pao2
- arterial partial pressure of oxygen
- PBS
- phosphate-buffered saline
- PCR
- polymerase chain reaction
- PMN
- polymorphonuclear neutrophil
- RT
- reverse transcriptase
- WT
- wild type
REFERENCES
- 1. Ranieri V. M., Rubenfeld G. D., Thompson B. T., Ferguson N. D., Caldwell E., Fan E., Camporota L., Slutsky A. S. (2012) Acute respiratory distress syndrome: the Berlin definition. JAMA 307, 2526–2533 [DOI] [PubMed] [Google Scholar]
- 2. Ware L. B., Matthay M. A. (2000) The acute respiratory distress syndrome. N. Engl. J. Med. 342, 1334–1349 [DOI] [PubMed] [Google Scholar]
- 3. Eckle T., Koeppen M., Eltzschig H. K. (2009) Role of extracellular adenosine in acute lung injury. Physiology (Bethesda) 24, 298–306 [DOI] [PubMed] [Google Scholar]
- 4. Eltzschig H. K., Sitkovsky M. V., Robson S. C. (2012) Purinergic signaling during inflammation. N. Engl. J. Med. 367, 2322–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rubenfeld G. D., Caldwell E., Peabody E., Weaver J., Martin D. P., Neff M., Stern E. J., Hudson L. D. (2005) Incidence and outcomes of acute lung injury. N. Engl. J. Med. 353, 1685–1693 [DOI] [PubMed] [Google Scholar]
- 6. Herridge M. S., Tansey C. M., Matte A., Tomlinson G., Diaz-Granados N., Cooper A., Guest C. B., Mazer C. D., Mehta S., Stewart T. E., Kudlow P., Cook D., Slutsky A. S., Cheung A. M. (2011) Functional disability 5 years after acute respiratory distress syndrome. N. Engl. J. Med. 364, 1293–1304 [DOI] [PubMed] [Google Scholar]
- 7. Eltzschig H. K., Carmeliet P. (2011) Hypoxia and inflammation. N. Engl. J. Med. 364, 656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thiel M., Chouker A., Ohta A., Jackson E., Caldwell C., Smith P., Lukashev D., Bittmann I., Sitkovsky M. V. (2005) Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 3, e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin T. R. (2002) Neutrophils and lung injury: getting it right. J. Clin. Invest. 110, 1603–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eckle T., Fullbier L., Grenz A., Eltzschig H. K. (2008) Usefulness of pressure-controlled ventilation at high inspiratory pressures to induce acute lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L718–L724 [DOI] [PubMed] [Google Scholar]
- 11. Eckle T., Fullbier L., Wehrmann M., Khoury J., Mittelbronn M., Ibla J., Rosenberger P., Eltzschig H. K. (2007) Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J. Immunol. 178, 8127–8137 [DOI] [PubMed] [Google Scholar]
- 12. Reutershan J., Vollmer I., Stark S., Wagner R., Ngamsri K. C., Eltzschig H. K. (2008) Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 23, 473−482 [DOI] [PubMed] [Google Scholar]
- 13. Van Linden A., Eltzschig H. K. (2007) Role of pulmonary adenosine during hypoxia: extracellular generation, signaling and metabolism by surface adenosine deaminase/CD26. Expert Opin. Biol. Ther. 7, 1437–1447 [DOI] [PubMed] [Google Scholar]
- 14. Eckle T., Grenz A., Laucher S., Eltzschig H. K. (2008) A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J. Clin. Invest. 118, 3301–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou Y., Schneider D. J., Morschl E., Song L., Pedroza M., Karmouty-Quintana H., Le T., Sun C. X., Blackburn M. R. (2011) Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J. Immunol. 186, 1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu Q., Harrington E. O., Newton J., Casserly B., Radin G., Warburton R., Zhou Y., Blackburn M. R., Rounds S. (2010) Adenosine protected against pulmonary edema through transporter- and receptor A2-mediated endothelial barrier enhancement. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L755–L767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou Y., Mohsenin A., Morschl E., Young H. W., Molina J. G., Ma W., Sun C. X., Martinez-Valdez H., Blackburn M. R. (2009) Enhanced airway inflammation and remodeling in adenosine deaminase-deficient mice lacking the A2B adenosine receptor. J. Immunol. 182, 8037–8046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eltzschig H. K., Collard C. D. (2004) Vascular ischaemia and reperfusion injury. Br. Med. Bull. 70, 71–86 [DOI] [PubMed] [Google Scholar]
- 19. Schingnitz U., Hartmann K., Macmanus C. F., Eckle T., Zug S., Colgan S. P., Eltzschig H. K. (2010) Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J. Immunol. 184, 5271–5279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eltzschig H. K., Eckle T. (2011) Ischemia and reperfusion—from mechanism to translation. Nat. Med. 17, 1391–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hart M. L., Grenz A., Gorzolla I. C., Schittenhelm J., Dalton J. H., Eltzschig H. K. (2011) Hypoxia-inducible factor-1α-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J. Immunol. 186, 4367–4374 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Hart M. L., Gorzolla I. C., Schittenhelm J., Robson S. C., Eltzschig H. K. (2010) SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J. Immunol. 184, 4017–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hart M. L., Jacobi B., Schittenhelm J., Henn M., Eltzschig H. K. (2009) Cutting Edge: A2B Adenosine receptor signaling provides potent protection during intestinal ischemia/reperfusion injury. J. Immunol. 182, 3965–3968 [DOI] [PubMed] [Google Scholar]
- 24. Hart M. L., Henn M., Kohler D., Kloor D., Mittelbronn M., Gorzolla I. C., Stahl G. L., Eltzschig H. K. (2008) Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 22, 2784–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hart M. L., Much C., Gorzolla I. C., Schittenhelm J., Kloor D., Stahl G. L., Eltzschig H. K. (2008) Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology 135, 1739.e3−1750.e3 [DOI] [PubMed] [Google Scholar]
- 26. Hart M. L., Kohler D., Eckle T., Kloor D., Stahl G. L., Eltzschig H. K. (2008) Direct treatment of mouse or human blood with soluble 5′-nucleotidase inhibits platelet aggregation. Arterioscler. Thromb. Vasc. Biol. 28, 1477–1483 [DOI] [PubMed] [Google Scholar]
- 27. Eltzschig H. K., Macmanus C. F., Colgan S. P. (2008) Neutrophils as sources of extracellular nucleotides: functional consequences at the vascular interface. Trends Cardiovasc. Med. 18, 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Faigle M., Seessle J., Zug S., El Kasmi K. C., Eltzschig H. K. (2008) ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS One 3, e2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eltzschig H. K., Weissmuller T., Mager A., Eckle T. (2006) Nucleotide metabolism and cell-cell interactions. Methods Mol. Biol. 341, 73–87 [DOI] [PubMed] [Google Scholar]
- 30. Eltzschig H. K., Eckle T., Mager A., Kuper N., Karcher C., Weissmuller T., Boengler K., Schulz R., Robson S. C., Colgan S. P. (2006) ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 99, 1100–1108 [DOI] [PubMed] [Google Scholar]
- 31. Eltzschig H. K., Ibla J. C., Furuta G. T., Leonard M. O., Jacobson K. A., Enjyoji K., Robson S. C., Colgan S. P. (2003) Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 198, 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eltzschig H. K. (2009) Adenosine: an old drug newly discovered. Anesthesiology 111, 904–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsukamoto H., Chernogorova P., Ayata K., Gerlach U. V., Rughani A., Ritchey J. W., Ganesan J., Follo M., Zeiser R., Thompson L. F., Idzko M. Deficiency of CD73/ecto-5′-nucleotidase in mice enhances acute graft-versus-host disease. Blood 119, 4554−4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grenz A., Bauerle J. D., Dalton J. H., Ridyard D., Badulak A., Tak E., McNamee E. N., Clambey E., Moldovan R., Reyes G., Klawitter J., Ambler K., Magee K., Christians U., Brodsky K. S., Ravid K., Choi D. S., Wen J., Lukashev D., Blackburn M. R., Osswald H., Coe I. R., Nurnberg B., Haase V. H., Xia Y., Sitkovsky M., Eltzschig H. K. (2012) Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J. Clin. Invest. 122, 693–710 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Morote-Garcia J. C., Rosenberger P., Nivillac N. M., Coe I. R., Eltzschig H. K. (2009) Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology 136, 607–618 [DOI] [PubMed] [Google Scholar]
- 36. Loffler M., Morote-Garcia J. C., Eltzschig S. A., Coe I. R., Eltzschig H. K. (2007) Physiological roles of vascular nucleoside transporters. Arterioscler. Thromb. Vasc. Biol. 27, 1004–1013 [DOI] [PubMed] [Google Scholar]
- 37. Eltzschig H. K., Abdulla P., Hoffman E., Hamilton K. E., Daniels D., Schonfeld C., Loffler M., Reyes G., Duszenko M., Karhausen J., Robinson A., Westerman K. A., Coe I. R., Colgan S. P. (2005) HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med. 202, 1493–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alva J. A., Zovein A. C., Monvoisin A., Murphy T., Salazar A., Harvey N. L., Carmeliet P., Iruela-Arispe M. L. (2006) VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 235, 759–767 [DOI] [PubMed] [Google Scholar]
- 39. Schwenk F., Baron U., Rajewsky K. (1995) A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Okubo T., Hogan B. L. (2004) Hyperactive Wnt signaling changes the developmental potential of embryonic lung endoderm. J. Biol. 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ko K. R., Ngai A. C., Winn H. R. (1990) Role of adenosine in regulation of regional cerebral blood flow in sensory cortex. Am. J. Physiol. 259, H1703–H1708 [DOI] [PubMed] [Google Scholar]
- 42. Kim H. H., Sawada N., Soydan G., Lee H. S., Zhou Z., Hwang S. K., Waeber C., Moskowitz M. A., Liao J. K. (2008) Additive effects of statin and dipyridamole on cerebral blood flow and stroke protection. J. Cereb. Blood Flow Metab. 28, 1285–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ward J. L., Sherali A., Mo Z. P., Tse C. M. (2000) Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J. Biol. Chem. 275, 8375–8381 [DOI] [PubMed] [Google Scholar]
- 44. Wakamiya M., Blackburn M. R., Jurecic R., McArthur M. J., Geske R. S., Cartwright J., Jr., Mitani K., Vaishnav S., Belmont J. W., Kellems R. E., Finegoldt M. J., Montgomery C. A., Jr., Bradley A., Caskey C. T. (1995) Disruption of the adenosine deaminase gene causes hepatocellular impairment and perinatal lethality in mice. Proc. Natl. Acad. Sci. U. S. A. 92, 3673–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blackburn M. R., Volmer J. B., Thrasher J. L., Zhong H., Crosby J. R., Lee J. J., Kellems R. E. (2000) Metabolic consequences of adenosine deaminase deficiency in mice are associated with defects in alveogenesis, pulmonary inflammation, and airway obstruction. J. Exp. Med. 192, 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang D., Zhang Y., Nguyen H. G., Koupenova M., Chauhan A. K., Makitalo M., Jones M. R., St Hilaire C., Seldin D. C., Toselli P., Lamperti E., Schreiber B. M., Gavras H., Wagner D. D., Ravid K. (2006) The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J. Clin. Invest. 116, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lahm A., Uhl M., Lehr H. A., Ihling C., Kreuz P. C., Haberstroh J. (2004) Photoshop-based image analysis of canine articular cartilage after subchondral damage. Arch. Orthop. Trauma Surg. 124, 431–436 [DOI] [PubMed] [Google Scholar]
- 48. Lehr H. A., Mankoff D. A., Corwin D., Santeusanio G., Gown A. M. (1997) Application of Photoshop-based image analysis to quantification of hormone receptor expression in breast cancer. J. Histochem. Cytochem. 45, 1559–1565 [DOI] [PubMed] [Google Scholar]
- 49. Sitkovsky M., Lukashev D. (2005) Regulation of immune cells by local-tissue oxygen tension: HIF1α and adenosine receptors. Nat. Rev. Immunol. 5, 712–721 [DOI] [PubMed] [Google Scholar]
- 50. Sitkovsky M. V., Lukashev D., Apasov S., Kojima H., Koshiba M., Caldwell C., Ohta A., Thiel M. (2004) Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol. 22, 657–682 [DOI] [PubMed] [Google Scholar]
- 51. Ohta A., Sitkovsky M. (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414, 916–920 [DOI] [PubMed] [Google Scholar]
- 52. Koeppen M., Eckle T., Eltzschig H. K. (2011) Pressure controlled ventilation to induce acute lung injury in mice. J. Vis. Exp. 51, 2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koeppen M., McNamee E. N., Brodsky K. S., Aherne C. M., Faigle M., Downey G. P., Colgan S. P., Evans C. M., Schwartz D. A., Eltzschig H. K. (2012) Detrimental role of the airway mucin Muc5ac during ventilator-induced lung injury. [E-pub ahead of print] Mucosal Immunol. doi:10.1038/mi.2012.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Choi D. S., Cascini M. G., Mailliard W., Young H., Paredes P., McMahon T., Diamond I., Bonci A., Messing R. O. (2004) The type 1 equilibrative nucleoside transporter regulates ethanol intoxication and preference. Nat. Neurosci. 7, 855−861 [DOI] [PubMed] [Google Scholar]
- 55. Cagnina R. E., Ramos S. I., Marshall M. A., Wang G., Frazier C. R., Linden J. (2009) Adenosine A2B receptors are highly expressed on murine type II alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L467−L474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grenz A., Osswald H., Eckle T., Yang D., Zhang H., Tran Z. V., Klingel K., Ravid K., Eltzschig H. K. (2008) The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 5, e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eckle T., Faigle M., Grenz A., Laucher S., Thompson L. F., Eltzschig H. K. (2008) A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood 111, 2024–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eckle T., Krahn T., Grenz A., Kohler D., Mittelbronn M., Ledent C., Jacobson M. A., Osswald H., Thompson L. F., Unertl K., Eltzschig H. K. (2007) Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 115, 1581–1590 [DOI] [PubMed] [Google Scholar]
- 59. Oliver K. M., Lenihan C. R., Bruning U., Cheong A., Laffey J. G., McLoughlin P., Taylor C. T., Cummins E. P. (2012) Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J. Biol. Chem. 287, 14004–14011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Taylor C. T., Cummins E. P. (2011) Regulation of gene expression by carbon dioxide. J. Physiol. 589, 797–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Casanello P., Torres A., Sanhueza F., Gonzalez M., Farias M., Gallardo V., Pastor-Anglada M., San Martin R., Sobrevia L. (2005) Equilibrative nucleoside transporter 1 expression is downregulated by hypoxia in human umbilical vein endothelium. Circ. Res. 97, 16–24 [DOI] [PubMed] [Google Scholar]
- 62. Chaudary N., Naydenova Z., Shuralyova I., Coe I. R. (2004) Hypoxia regulates the adenosine transporter, mENT1, in the murine cardiomyocyte cell line, HL-1. Cardiovasc. Res. 61, 780–788 [DOI] [PubMed] [Google Scholar]
- 63. Taylor C. T., McElwain J. C. (2010) Ancient atmospheres and the evolution of oxygen sensing via the hypoxia-inducible factor in metazoans. Physiology (Bethesda) 25, 272–279 [DOI] [PubMed] [Google Scholar]
- 64. Cummins E. P., Taylor C. T. (2005) Hypoxia-responsive transcription factors. Pflügers Arch. 450, 363−371 [DOI] [PubMed] [Google Scholar]
- 65. Cummins E. P., Berra E., Comerford K. M., Ginouves A., Fitzgerald K. T., Seeballuck F., Godson C., Nielsen J. E., Moynagh P., Pouyssegur J., Taylor C. T. (2006) Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc. Natl. Acad. Sci. U. S. A. 103, 18154–18159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Taylor C. T. (2008) Interdependent roles for hypoxia inducible factor and nuclear factor-κB in hypoxic inflammation. J. Physiol. 586, 4055–4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cummins E. P., Seeballuck F., Keely S. J., Mangan N. E., Callanan J. J., Fallon P. G., Taylor C. T. (2008) The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 134, 156–165 [DOI] [PubMed] [Google Scholar]
- 68. Aherne C. M., Collins C. B., Masterson J. C., Tizzano M., Boyle T. A., Westrich J. A., Parnes J. A., Furuta G. T., Rivera-Nieves J., Eltzschig H. K. (2012) Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut 61, 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grenz A., Dalton J. H., Bauerle J. D., Badulak A., Ridyard D., Gandjeva A., Aherne C. M., Brodsky K. S., Kim J. H., Tuder R. M., Eltzschig H. K. (2011) Partial netrin-1 deficiency aggravates acute kidney injury. PLoS One 6, e14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rosenberger P., Schwab J. M., Mirakaj V., Masekowsky E., Mager A., Morote-Garcia J. C., Unertl K., Eltzschig H. K. (2009) Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 10, 195–202 [DOI] [PubMed] [Google Scholar]
- 71. Eckle T., Hartmann K., Bonney S., Reithel S., Mittelbronn M., Walker L. A., Lowes B. D., Han J., Borchers C. H., Buttrick P. M., Kominsky D. J., Colgan S. P., Eltzschig H. K. (2012) Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 18, 774–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Koeppen M., Harter P. N., Bonney S., Bonney M., Reithel S., Zachskorn C., Mittelbronn M., Eckle T. (2012) Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology 116, 1245–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Eckle T., Kohler D., Lehmann R., El Kasmi K. C., Eltzschig H. K. (2008) Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation 118, 166–175 [DOI] [PubMed] [Google Scholar]
- 74. Dixon B. S., Beck G. J., Vazquez M. A., Greenberg A., Delmez J. A., Allon M., Dember L. M., Himmelfarb J., Gassman J. J., Greene T., Radeva M. K., Davidson I. J., Ikizler T. A., Braden G. L., Fenves A. Z., Kaufman J. S., Cotton J. R., Jr., Martin K. J., McNeil J. W., Rahman A., Lawson J. H., Whiting J. F., Hu B., Meyers C. M., Kusek J. W., Feldman H. I. (2009) Effect of dipyridamole plus aspirin on hemodialysis graft patency. N. Engl. J. Med. 360, 2191–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sacco R. L., Diener H. C., Yusuf S., Cotton D., Ounpuu S., Lawton W. A., Palesch Y., Martin R. H., Albers G. W., Bath P., Bornstein N., Chan B. P., Chen S. T., Cunha L., Dahlof B., De Keyser J., Donnan G. A., Estol C., Gorelick P., Gu V., Hermansson K., Hilbrich L., Kaste M., Lu C., Machnig T., Pais P., Roberts R., Skvortsova V., Teal P., Toni D., Vandermaelen C., Voigt T., Weber M., Yoon B. W. (2008) Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N. Engl. J. Med. 359, 1238–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Morabito L., Montesinos M. C., Schreibman D. M., Balter L., Thompson L. F., Resta R., Carlin G., Huie M. A., Cronstein B. N. (1998) Methotrexate and sulfasalazine promote adenosine release by a mechanism that requires ecto-5′-nucleotidase-mediated conversion of adenine nucleotides. J. Clin. Invest. 101, 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cronstein B. N. (1997) The mechanism of action of methotrexate. Rheum. Dis. Clin. North Am. 23, 739–755 [DOI] [PubMed] [Google Scholar]
- 78. Cronstein B. N., Naime D., Ostad E. (1994) The antiinflammatory effects of methotrexate are mediated by adenosine. Adv. Exp. Med. Biol. 370, 411–416 [DOI] [PubMed] [Google Scholar]
- 79. Cronstein B. N., Naime D., Ostad E. (1993) The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Invest. 92, 2675–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cronstein B. N., Eberle M. A., Gruber H. E., Levin R. I. (1991) Methotrexate inhibits neutrophil function by stimulating adenosine release from connective tissue cells. Proc. Natl. Acad. Sci. U. S. A. 88, 2441–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Eltzschig H. K., Faigle M., Knapp S., Karhausen J., Ibla J., Rosenberger P., Odegard K. C., Laussen P. C., Thompson L. F., Colgan S. P. (2006) Endothelial catabolism of extracellular adenosine during hypoxia: the role of surface adenosine deaminase and CD26. Blood 108, 1602–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Morote-Garcia J. C., Rosenberger P., Kuhlicke J., Eltzschig H. K. (2008) HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 111, 5571–5580 [DOI] [PubMed] [Google Scholar]
- 83. Sitkovsky M. V. (2008) Damage control by hypoxia-inhibited AK. Blood 111, 5424–5425 [DOI] [PubMed] [Google Scholar]
- 84. Jackson E. K., Gillespie D. G. (2013) Extracellular 2′,3′-cAMP-adenosine pathway in proximal tubular, thick ascending limb, and collecting duct epithelial cells. Am. J. Physiol. Renal Physiol. 304, F49–F55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jackson E. K., Ren J., Gillespie D. G., Dubey R. K. (2010) Extracellular 2′,3′-cyclic adenosine 5′-monophosphate is a potent inhibitor of preglomerular vascular smooth muscle cell and mesangial cell growth. Hypertension 56, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jackson E. K., Ren J., Mi Z. (2009) Extracellular 2′,3′-cAMP is a source of adenosine. J. Biol. Chem. 284, 33097–33106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yao S. Y., Ng A. M., Muzyka W. R., Griffiths M., Cass C. E., Baldwin S. A., Young J. D. (1997) Molecular cloning and functional characterization of nitrobenzylthioinosine (NBMPR)-sensitive (es) and NBMPR-insensitive (ei) equilibrative nucleoside transporter proteins (rENT1 and rENT2) from rat tissues. J. Biol. Chem. 272, 28423–28430 [DOI] [PubMed] [Google Scholar]