

Abstract
Vascular endothelial growth factor (VEGF) is critical for angiogenesis, but also has pleiotropic effects on several nonvascular cells. Our aim was to investigate the role of VEGF in brown adipose tissue (BAT). We show that VEGF expression increases 2.5-fold during differentiation of cultured murine brown adipocytes and that VEGF receptor-2 is phosphorylated, indicating VEGF signaling. VEGF increased proliferation in brown preadipocytes in vitro by 70%, and blockade of VEGF signaling using anti-VEGFR2 antibody DC101 increased brown adipocyte apoptosis, as determined by cell number and activation of caspase 3. Systemic VEGF neutralization in mice, accomplished by adenoviral expression of soluble Flt1, resulted in 7-fold increase in brown adipocyte apoptosis, mitochondrial degeneration, and increased mitophagy compared to control mice expressing a null adenovirus. Absence of the heparan sulfate-binding VEGF isoforms, VEGF164 and VEGF188, resulted in abnormal BAT development in mice at E15.5, with fewer brown adipocytes and lower mitochondrial protein compared to wild-type littermates. These results suggest a role for VEGF in brown adipocytes and preadipocytes to promote survival, proliferation, and normal mitochondria and development.—Bagchi, M., Kim, L A., Boucher, J., Walshe, T. E., Kahn, C. R., D'Amore, P. A. Vascular endothelial growth factor is important for brown adipose tissue development and maintenance.
Keywords: sFlt1, apoptosis, mitochondria, proliferation
Mammals have two types of fat, white adipose tissue (WAT) and brown adipose tissue (BAT). WAT serves as a storage depot for excess calories in the form of triglycerides. In addition, it functions as an endocrine organ, releasing free fatty acids and adipokines, such as leptin and adiponectin (1), that act on various organs, including the brain, liver, and muscle. BAT is specialized to produce heat as a physiological protection against cold. It also acts as a defense against obesity by metabolizing lipids to generate heat instead of storing them as triglycerides (2). BAT is, in fact, the major organ in mammals for nonshivering thermoregulation. Although small rodents, hibernating animals, and human infants were all known to have BAT, it was thought to be absent in adults until recently, when several studies provided convincing evidence for the presence of metabolically active BAT depots in adult humans (for a review, see ref. 3). BAT activity has been shown to be inversely correlated with body mass index, age, and blood glucose level (4). In fact, 50 g of maximally activated BAT could account for up to 20% of the body's daily caloric expenditure (5).
The ability of brown adipocytes to generate heat is due to the high number of mitochondria and expression of mitochondria uncoupling protein 1 (UCP1). UCP1, a proton transporter localized in the inner mitochondrial membrane, pumps protons from the intermembrane space into the mitochondrial matrix, uncoupling the electron transport chain and allowing the electrochemical energy generated from respiration to dissipate as heat (6). UCP1 is activated by free fatty acids and inhibited by purine nucleotides (7). Thus, BAT produces heat following food intake, a response known as diet-induced thermogenesis. Obesity results from excess caloric intake over energy expenditure over a prolonged period of time (8). For optimal thermogenic activity, BAT requires an extensive vascularization to ensure sufficient supplies of oxygen and substrates, as well as rapid heat dissipation. During acclimation to chronic cold or high-fat diet, both vascular density and vascular flow are increased to accommodate the rise in thermogenesis by brown adipocytes (9).
Vascular endothelial growth factor A (VEGF-A; hereafter referred to as VEGF) is a potent angiogenic factor that induces endothelial cell proliferation, migration, survival, and vessel permeability (for a review, see ref. 10). Vascular development is dependent on tight regulation of VEGF, as mice lacking a single VEGF allele die by embryonic day 9.5 (E9.5) with severe vascular defects (11, 12). Similarly, overexpression of VEGF results in embryonic lethality (13). In the adult, VEGF serves to maintain normal vasculature, and inhibition of VEGF signaling has been shown to induce capillary regression and malfunction in several organs, including choroid plexus, thyroid, pancreatic islets, and epididymal WAT (14–16). VEGF binds to the tyrosine kinase receptors VEGFR2 and VEGFR1, as well as to the coreceptor neuropilin-1. Whereas VEGFR1 and VEGFR2 both display high affinity for VEGF-A, VEGFR2 has 10-fold stronger tyrosine kinase activity and is the primary signaling receptor for VEGF, initiating several signaling cascades, including activation of phosphatidylinositol 3-kinase (PI3K)/Akt, mitogen-activated protein kinase (MAPK), and PKC to induce endothelial cell migration and proliferation and vascular permeability (for a review, see refs. 17, 18).
Initially thought to target only endothelial cells, recent findings have indicated pleiotropic roles for VEGF on a wide range of nonendothelial cell types. VEGF is essential for the maintenance of choroid plexus structure and integrity, including ependymal cell function (16). VEGF also promotes Müller cell survival through autocrine signaling, has a paracrine neuroprotective effect on photoreceptors (15), and functions in an autocrine loop to promote the survival of kidney podocytes (19), as well as the maintenance of the retinal pigmented epithelium (20), skeletal muscle (21), and bone differentiation (22).
Alternative splicing of the VEGF gene produces 8 isoforms, with VEGF 121, 165, and 189 (120, 164, and 188 in mice and rats) being the most abundant and well studied. The isoforms differ in their ability to bind heparan sulfate proteoglycan and neuropilin-1 (18). Charged domains in VEGF164 and VEGF188 mediate their binding to heparan sulfate on the cell surface and in the extracellular matrix, where they may be sequestered and function locally. VEGF120 lacks these charged domains, so is freely diffusible and, thus, better able to form gradients. The distribution of VEGF isoforms varies among tissues, suggesting that different isoforms may play distinct roles in vascular development and in the adult (23). This notion was substantiated by the distinct phenotypes of mice that were engineered to express single isoforms through targeted deletion of specific regions of the VEGF gene (24, 25). Both white and brown adipocytes have been shown to express VEGF in vivo and in vitro (26–28). However, neither the distribution of VEGF isoforms nor their function in adipose tissue has been studied. In addition, although VEGF expression in BAT and its role in promoting angiogenesis during cold acclimation have been described (29), the expression of VEGF receptors by brown adipocytes has not been investigated. This study evaluated the role of VEGF and its isoforms in brown adipose and tested the hypothesis that, in addition to supporting adipose angiogenesis, VEGF may play a functional role in adipocytes.
MATERIALS AND METHODS
Animals
C57BL/6J wild-type and VEGF120/120 mice were used in these studies. C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). VEGF120 mice were generated by targeted deletion of exon 6 and 7 of the VEGF gene; these mice express only the VEGF120 splice isoform (24). VEGF120/120 embryos were generated by breeding C57BL/6J VEGF120/+ mice. Timed-pregnant females were euthanized at E15.5. All mice were maintained on a regular chow diet and kept on a 12-h light-dark cycle. Adult mice were euthanized by carbon dioxide inhalation; embryos were euthanized by decapitation. All protocols for animal use were reviewed and approved by the Schepens Eye Research Institutional Animal Care and Use Committee in accordance with the U.S. National Institutes of Health guidelines.
Adenovirus-mediated systemic VEGF neutralization
Mice were anesthetized using ketamine/xylazine and injected with ∼7.5 × 1010 pfu of soluble fms-like tyrosine kinase 1 (sFlt) virus (ad-sFlt1), in 100 μl via the tail vein. At 5 d postinjection, serum was collected by submandibular bleeding, and circulating sFlt1 levels were determined by ELISA (R&D Biosystems, Minneapolis, MN, USA). Animals with circulating levels of sFlt1 of 200 ng/ml or higher were included in the study. Ad-null-infected mice showed no detectable sFlt1. Each group included 8 mice. Mice were euthanized 7 d postinjection, and tissues were dissected. Three mice from each group were perfused for electron microscopy, as described. For histology, the tissues were placed directly in 4% paraformaldehyde (PFA) and fixed for 48 h.
Brown preadipocyte culture and differentiation
Brown preadipocytes were maintained as subconfluent cultures in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin (Lonza, Basel, Switzerland). To induce differentiation, cells were cultured for 48 h beyond confluence in the above growth medium, which was then replaced with brown adipose differentiation medium, consisting of growth medium supplemented with 20 nM insulin, 1 μM dexamethasone, 0.5 mM IBMX, 1 nM triiodothyronine, and 0.125 mM indomethacin (all from Sigma-Aldrich, St. Louis, MO, USA). After 48 h, the differentiation medium was replaced with growth medium supplemented with 1 nM triiodothyronine and 20 nM insulin. Cells were maintained in the above media until the cells were fully differentiated (8 d). Media were replaced every other day.
Proliferation assay
Cells were plated at a density of 1.0 × 104 cells/well in a 24-well plate in growth medium and allowed to attach overnight. The following day, the cells were rinsed once with serum-free DMEM and refed DMEM with 1% FBS with or without 10 ng/ml VEGF. Cells were counted at 24-h intervals for 5 d, using a Coulter Counter (Beckman Coulter, Brea, CA, USA). Each experimental time point was assayed in triplicate.
Apoptosis and terminal transferase dUTP nick end labeling (TUNEL) assay
Fully differentiated brown adipocytes were treated with 10 nM tumor necrosis factor α (TNF-α; Genway Biotech, San Diego, CA, USA) and 10 μg/ml cycloheximide (CHX; Sigma-Aldrich) for 6 h in the presence or absence of VEGF-blocking antibody DC101 (15 μg/ml). Cells were rinsed twice in phosphate-buffered saline (PBS), trypsinized, centrifuged, and processed for fluorescence-activated cell sorter (FACS) analysis using Cytofix/Cytoperm Fixation Permeabilization Kit (BD Biosciences, San Jose, CA, USA), following the manufacturer's instructions, for detection of cleaved caspase-positive cells with Alexa Fluor 488-conjugated cleaved caspase 3 antibody (1:100; Cell Signaling Technologies, Danvers, MA, USA) and analyzed using BD LSR II FACS (BD Biosciences).
Apoptotic BAT cells were detected in paraffin sections using the In Situ Cell Death Detection TMR Red Kit (Roche Diagnostics, Indianapolis, IN, USA), following the manufacturer's instructions. DNase treatment was performed as a positive control, incubation without TdT enzyme was conducted as a negative control.
RNA analysis
Total RNA was isolated from adipocytes using RNA aqueous 4PCR kit (Life Technologies, Grand Island, NY, USA), following the manufacturer's instructions. Reverse transcription was performed on 1 μg RNA using SuperScript III (Life Technologies), as per the manufacturer's instructions. For qPCR and RT-PCR, 10 ng of cDNA was amplified in each reaction using 500 nM forward and reverse primers (Tables 1 and 2) and SYBR Green Master mix (Roche Diagnostics) on the Roche LightCycler 480 II. PCR cycles were as follows: an initial denaturation step (95°C, 10 min), followed by 40 cycles of 95°C for 15 s; 60°C for 30 s, and 72°C for 1 min. Samples were run in triplicates and subjected to melting curve analysis to confirm amplification specificity. Samples were normalized to TATA box-binding protein (TBP) and expressed as relative fold change using the ΔΔCt method of relative quantification.
Table 1.
Mouse qPCR primers
| Target gene | Forward primer | Reverse primer |
|---|---|---|
| PGC1α | AGCCGTGACCACTGACAACGAG | GCTGCATGGTTCTGAGTGCTAAG |
| VEGF | GCACATAGAGAGAATGAGCTTCC | CTCCGCTCTGAACAAGGCT |
| VEGF18831 | GCCAGCACATAGAGAGAATGAGC | AACAAGGCTCACAGTGAACGCT |
| VEGF164 | GCCAGCACATAGAGAGAATGAGC | CAAGGCTCACAGTGATTTTCTGG |
| VEGF120 | GCCAGCACATAGAGAGAATGAGC | CGGCTTGTCACATTTTTCTGG |
| TBP | ACCCTTCACCAATGACTCCTATG | TGACTGCAGCAAATCGCTTGG |
| UCP1 | GGCATTCAGAGGCAAATCAGCT | CAATGAACACTGCCACACCTC |
Table 2.
Mouse RT-PCR primers
| Target gene | Forward primer | Reverse primer | Amplicon size |
|---|---|---|---|
| GAPDH | GTGGCAAAGTGGAGATTGTTGCC | GATGATGACCCGTTTGGCTCC | 291 |
| Nrp1 | TCAGGACCATACAGGAGATGG | TGACATCCCATTGTGCCAAC | 619 |
| UCP1 | TATCATCACCTTCCCGCTG | GTCATATGTTACCAGCTCTG | 505 |
| VEGF | CCTCCGAAACCATGAACTTTCTGCTC | CAGCCTGGCTCACCGCCTTGGCTT | 665, 593, 461 |
| VEGFR1 | GAGAGCATCTATAAGGCAGCGGATT | CACGTTTACAATGAGAGTGGCAGTG | 456 |
| VEGFR2 | TACACAATTCAGAGCGATGTGTGGT | CTGGTTCCTCCAATGGGATATCTTC | 499 |
For absolute quantification of VEGF isoforms, a standard curve was constructed for each of the 3 isoforms by amplifying serial dilutions (103 to 109 ng DNA/reaction) of a plasmid coding for a single isoform, using primers specific for each isoform (30, 31). The level of each isoform was calculated relative to the standard curve. Each sample and dilution was run in triplicate, as described above. Results are expressed as means ± se.
Western blot analysis
For analysis of proteins, cells were homogenized in RIPA lysis buffer (Sigma-Aldrich). Protein concentration was measured using a BCA assay (Bio-Rad Laboratories, Hercules, CA, USA). Equivalent amounts of protein were separated by SDS-PAGE under reducing condition, transferred to Immobilon-P membrane (Millipore, Billerica, MA, USA), and immunoblotted with the appropriate antibodies. Proteins were visualized with SuperSignal West Extended Duration Substrate (Pierce Biotechnologies, Rockford, IL, USA). Membranes were stripped for 30 min at 50°C and reprobed when necessary.
Antibodies
Antibodies used for Western blot analysis included anti-phospho-Y951-VEGFR2 (1:300), VEGFR2 (1:1000), cleaved caspase 3 (1:1000), whole caspase 3 (1:1000), phospho-S473-Akt (1:250), and pan-Akt (1:1000) from Cell Signaling Technologies, anti-endomucin (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-β-tubulin (1:500; Abcam, Cambridge, MA, USA). Secondary antibodies used included horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) from donkey (GE Healthcare, Little Chalfont, UK). Antibodies used for immunohistochemistry (IHC) to UCP1 (1:500), Cox IV (1:1000), and F4/80 (1:100) were purchased from Abcam; light-chain 3B (LC3B; 1:200) was from Cell Signaling Technology; cleaved caspase 3 (1:100) and perilipin (1:200) were from Sigma-Aldrich. Secondary antibodies for IHC were biotinylated anti-rat IgG and anti-rabbit IgG, both raised in goat (Vector Laboratories, Burlingame, CA, USA), and for immunofluorescence were Alexa Fluor-488 conjugated anti-rat and Dylight 549-conjugated anti-rabbit IgG (Invitrogen).
Immunohistochemistry
Embryos and BAT were fixed overnight in 4% PFA in PBS and embedded in paraffin, sectioned, and stained with hematoxylin & eosin (H&E) and/or Masson's trichrome. For IHC, sections were rehydrated, deparaffinized, pretreated by boiling in citrate buffer (pH 6.0), and incubated with 1% H2O2 to block endogenous peroxidase activity, then blocked in blocking buffer (3% rabbit serum) for 30 min. Samples were incubated with appropriate antibodies in blocking buffer overnight at 4°C in a humidified chamber, then washed and incubated with biotinylated secondary antibody for 1 h, followed by additional washes. Antibody localization signal was visualized using avidin-biotin-horseradish peroxidase and 3,3′-diaminobenzidine (DAB) substrate (Vectastain Elite ABC kit; Vector Laboratories) and counterstained with hematoxylin for labeling cell nuclei. Isotope-matched IgGs served as negative controls. For immunofluorescence of differentiated adipocytes, cells differentiated on sterilized coverslips were fixed in 4% PFA (10 min), washed, blocked (30 min), incubated overnight with appropriate antibodies, counterstained with 4′-6-diamidino-2-phenylindole (DAPI) for labeling cell nuclei, and mounted using 1:1 PBS and glycerol.
Electron microscopy
At 7 d after adenovirus injections, Ad-sFlt1 and ad-null mice were anesthetized with ketamine/xylazine and fixed via aortic perfusion with 10 ml sodium cacodylate buffer (0.1 M, pH 7.4), followed by 10 ml fixative (paraformaldehyde and glutaraldehyde, 2.5% each in 0.1 M sodium cacodylate buffer, pH 7.4; Electron Microscopy Sciences, Hatfield, PA, USA). The BAT was dissected and postfixed in the same fixative for 72 h, treated with osmium tetroxide and uranyl acetate, embedded in propyleneoxide, and visualized using a Tecnai G2 Spirit BioTWIN transmission electron microscope (TEM; FEI, Hillsboro, OR, USA).
Statistical analysis
Values are expressed as mean ± se (unless specified), and statistical analysis was performed using an unpaired Student's t test (P<0.001; P<0.01; P<0.05; ns, P>0.05).
RESULTS
Abnormal BAT morphology and mitochondrial protein expression in VEGF120/120 embryos
The profile of VEGF isoform expression in BAT and WAT was determined by qPCR. Absolute quantification of VEGF isoform expression using isoform-specific VEGF primers revealed that heparan sulfate-binding isoforms, VEGF164 and VEGF188, comprised >99% of total VEGF, whereas VEGF120 accounted for <0.5% in both white and brown adipose depots. In BAT, VEGF164 comprised 45% and VEGF188 contributed to 55% of the total VEGF, whereas subcutaneous and perigonadal had 84 and 36% VEGF164, respectively, and 16 and 64% VEGF188, respectively (Fig. 1A).
Figure 1.
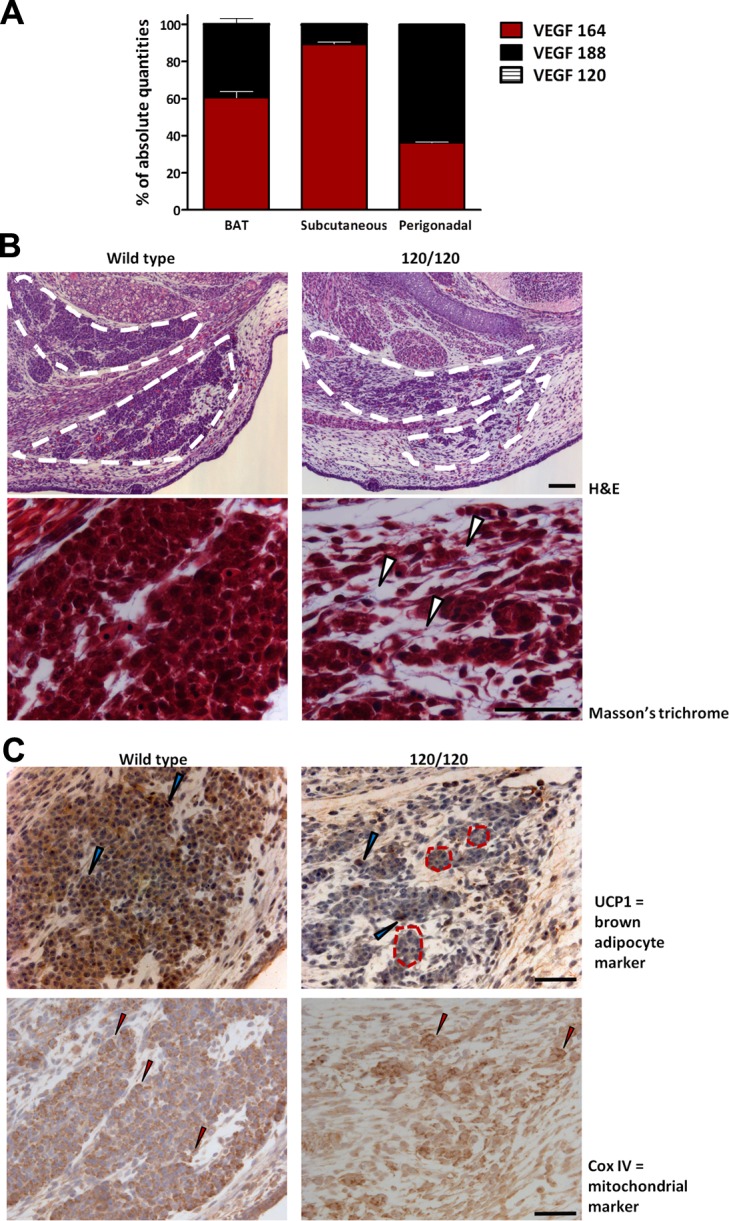
Heparan sulfate-binding VEGF isoforms play a role in BAT development. A) Absolute amount of each VEGF isoform was determined by quantitative PCR using isoform-specific primers and serial dilutions of a known amount of standard. Total VEGF mRNA was the sum of the 3 isoforms. Percentage of each isoform was calculated on the basis of the total VEGF, thus obtained. B) Top panel: morphology of brown fat in wild-type (left panel) and VEGF120/120 (right panel) embryos at E15.5 revealed by H&E staining shows reduced embryonic BAT (dotted white outline) in the interscapular region of VEGF120/120 embryo compared to wild type. Bottom panel: Masson's trichrome staining (higher magnification) revealed disorganized tissue structure. Small clusters of embryonic brown adipocytes are surrounded by abundant extracellular matrix with some collagen deposition (arrowheads). C) Top panel: IHC localization of UCP1, a brown adipocyte marker, shows robust expression in wild type (left panel) but reduced and mosaic expression in 120/120 brown adipocytes (right panel). Dashed red lines indicate clusters of brown adipocytes in 120/120 in which most cells have a lower expression compared to wild type, with a few cells exhibiting strong expression (blue arrowheads). Bottom panel: IHC localization of the mitochondrial marker Cox IV with strong expression in almost all cells of the wild-type BAT and lower expression in most adipocytes of VEGF 120/120; very few cells exhibited strong expression (red arrowheads) in VEGF 120/120. Scale bars = 50 μm.
Given the dominance of locally acting VEGF isoforms in adipose depots, we hypothesized that mice lacking VEGF164 and VEGF188 would have abnormal adipose tissue. To investigate this possibility, we examined adipose tissue in mice that were genetically engineered to express only VEGF120 (VEGF120/120), but at a level comparable to total VEGF in wild-type mice (24). Because VEGF120/120 mice die during late gestation and WAT forms only after birth, the analyses were confined to BAT, which develops in the interscapular region at around E15 (32). As E15.5 was the latest time point at which VEGF120/120 mouse could be obtained, the structure of BAT from VEGF120/120 and wild-type E15.5 littermates was examined, using H&E and Masson's trichrome staining.
There was a significant reduction in the volume of BAT (Fig. 1B; outlined area) in VEGF120/120 embryos compared to their wild-type littermates. Whereas wild-type brown adipocytes were arranged in compact clusters, VEGF120/120 brown adipocytes were disorganized and scattered in the interscapular region, with much smaller clusters of adipocytes and some collagen deposition (white arrowheads). Typical of this stage in development, wild-type brown adipocytes were cuboidal in shape and contained tiny lipid droplets. In contrast, there was no apparent lipid accumulation in the VEGF120/120 adipocytes.
To gauge the functional integrity of the embryonic BAT, we examined the distribution of UCP1, which is essential for brown adipocyte thermogenesis, and Cox IV protein, a member of the electron transport chain and a well-established mitochondrial marker. Wild-type embryonic brown adipocytes stained strongly for UCP1, whereas there was a significant decrease in the number of VEGF120/120 cells expressing UCP1 (Fig. 1C). In the VEGF120/120 embryo, clusters of presumptive brown adipocytes (Fig. 1C; outlined area) contained, primarily, cells with low expression of UCP1 compared to wild type and a few cells with strong expression (Fig. 1C; blue arrowheads). Cox IV expression was also reduced in the presumptive brown adipocytes of the VEGF120/120 embryo relative to that of wild type. While wild-type brown adipocytes strongly expressed Cox IV protein, most VEGF120/120 presumptive brown adipocytes expressed low to undetectable levels, and only a few cells displayed high level of Cox IV expression (red arrowheads). The relative absence of mitochondrial markers indicates that brown adipocyte differentiation was impaired in the absence of VEGF164 and VEGF188.
There was no difference in adipocyte apoptosis between the VEGF120/120 and wild-type mice at E15.5 (data not shown) nor was there a measurable difference in cell proliferation, which was visualized by IHC with Ki67 (data not shown).
VEGF expression increases during brown adipocyte differentiation
In light of the impaired BAT development in the absence of heparan sulfate-binding VEGF isoforms, we hypothesized that VEGF may play a significant role in BAT development and function, by acting on the vasculature, on the adipocytes, or on both. Because of perinatal lethality of the VEGF120/120 embryos, this model could not be used to study VEGF expression and function in mature brown adipocytes. Therefore, we utilized immortalized murine brown preadipocyte cells, derived from neonatal brown adipocyte precursors, which have been shown to differentiate into brown adipocytes in vitro (33). Brown preadipocytes were grown differentiated as described previously. More than 95% of cells had multiple lipid droplets by d 6, morphologically resembling their in vivo counterparts. The differentiation was confirmed by cell staining and immunohistochemical and gene expression analyses. In vivo, the differentiation of brown adipocytes is accompanied by increased mitochondrial biogenesis. Accordingly, mitochondria in the in vitro differentiated brown cells were visualized using CMTM Rosamine MitoTracker, a mitochondria-specific vital dye, and lipid droplets were localized using immunohistochemical staining for the distribution of perilipin (Fig. 2A). Markers of adipocyte differentiation, such as peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α), UCP1, and β3-adrenoreceptor (34, 35), increased significantly over the course of differentiation (Fig. 2B). Specific to our interest, VEGF levels were increased by 2.5- to 3-fold over the course of differentiation (Fig. 2C). Of the 3 VEGF isoforms, VEGF164 was the most abundant, whereas low levels of VEGF188 and VEGF120 were also detected. VEGF164 constituted 90% of total VEGF at d 0 and rose to 98% of total VEGF by d 8. While levels of VEGF120 also increased during the course of differentiation, they decreased from ∼10% of the total at d 0 to ∼1% at d 8. These data suggest that VEGF expression may be coordinately regulated with the process of brown adipocyte differentiation (Fig. 2D). VEGFR2 was expressed by both brown preadipocytes and differentiated brown adipocytes. VEGFR1 and Nrp1 were also present in both undifferentiated cells and differentiated brown adipocytes (Fig. 2E). The expression of VEGF receptors in both preadipocytes and differentiated brown adipocytes raises the possibility of an autocrine role for VEGF in these cells.
Figure 2.

VEGF expression increases during brown adipocyte differentiation. A) Lipid droplets were identified using immunofluorescent localization of perilipin (left panel). Mitochondria in live cells were visualized using CMTM Rosamine MitoTracker and revealed the large number of mitochondria (middle panel), merge (right panel). Nuclei were visualized by DAPI. Scale bar represents 50 μm. B) Expression of adipocyte markers PGC1α (left panel), UCP1 (center panel), and β3 adrenergic receptors (right panel), was quantified using qPCR. *P < 0.05, **P < 0.01, ***P < 0.001. C) The increase in VEGF expression during brown adipocyte differentiation was correlated with that of brown adipocyte markers. *P < 0.05. D) Absolute quantities of VEGF isoforms using qPCR during brown adipocyte differentiation show VEGF164 to be the most abundant isoform E) VEGF receptor expression during brown adipocyte differentiation was determined by RT-PCR and indicated strong VEGFR2 expression in both preadipocytes and differentiated adipocytes.
VEGF signals through VEGFR2 and is a mitogen for brown preadipocytes
VEGFR2 activation was assessed by examining the phosphorylation of known tyrosine residues in unstimulated brown preadipocytes in regular culture conditions. Western blot analysis of preadipocyte lysates revealed phosphorylation of VEGFR2 at tyrosine 951. This phosphorylation was lost in the absence of serum but returned when VEGF was added to serum-free culture medium, and again abrogated when the VEGFR2 neutralizing antibody, DC101, was added along with VEGF in the serum-free medium, indicating that VEGF signals through VEGFR2 in brown preadipocytes (Fig. 3A).
Figure 3.
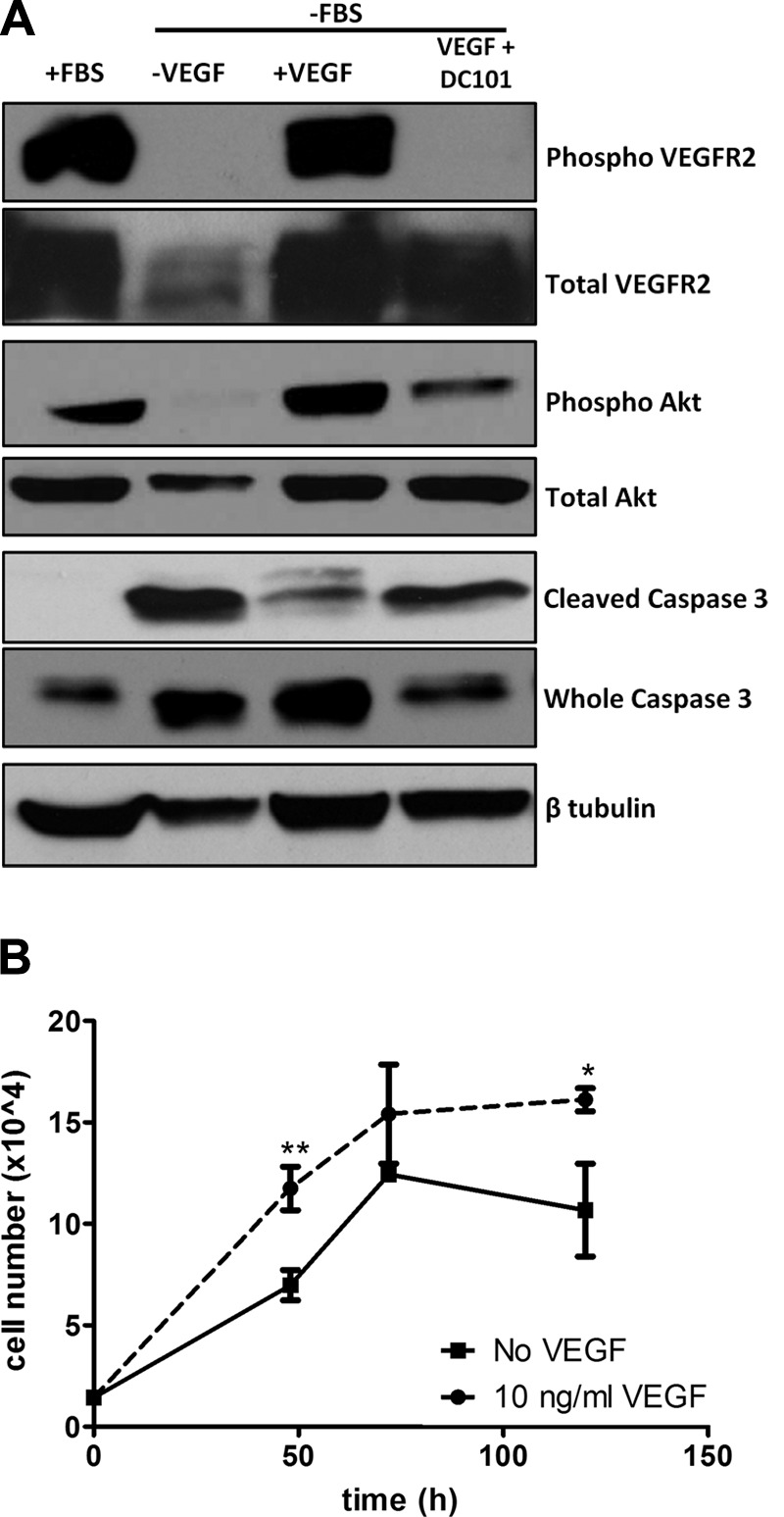
VEGF mediates brown preadipocyte survival and proliferation. A) Phosphorylation of VEGFR2 at Y951 revealed active VEGF signaling and phosphorylation of Akt at S453 demonstrated PI3K/Akt activation. Brown preadipocytes cultured serum free for 12 h were protected from apoptosis by addition of 25 ng/ml VEGF, as determined by the reduced expression of cleaved (activated) caspase 3. This protection was reversed on the addition of DC101, a VEGFR2-blocking antibody (15 μg/ml). B) Addition of VEGF (10 ng/ml) led to an increase in the number of brown preadipocytes compared to 1% FBS alone. *P < 0.05, **P < 0.01.
The PI3K-Akt pathway is reported to be downstream of the VEGF–VEGFR2 signaling cascade and participates in important cellular processes, such as proliferation and survival (36). Akt activation was thus examined in the above samples. Akt was phosphorylated in cells that were grown in normal growth medium, and serum withdrawal resulted in a nearly complete loss of Akt phosphorylation (Fig. 3A). The addition of VEGF to serum-free medium resulted in robust Akt phosphorylation, and the addition of DC101 along with VEGF reduced this phosphorylation by 80% (Fig. 3A), indicating that VEGF signaling activated Akt in brown preadipocytes.
To investigate the function of VEGF in brown preadipocytes, cell proliferation was assessed. Preadipocytes were cultured in low serum in the presence or absence of 10 ng/ml VEGF and counted every alternate day. VEGF led to a stimulation of cell proliferation when compared to untreated cells, with an increase in cell number of 70% at 48 h (Fig. 3B), indicating that VEGF functions as a mitogen for brown preadipocytes.
VEGF acts as a survival factor for brown preadipocytes and adipocytes
Brown preadipocytes are sensitive to serum starvation (growth factor withdrawal), a condition known to induce apoptosis. To determine whether VEGF signaling was playing a role in brown preadipocyte survival, cells were maintained in regular medium without serum, in the presence or absence of VEGF or DC101. After 12 h of serum starvation, ∼1/3 of the cells were observed to be detached. Whole-cell lysates were collected following 12 h of treatment and analyzed by Western blot analysis. Cleavage of the inactive proenzyme caspase 3 to its smaller (17–19 and 12 kDa) proteolytically active forms is considered a marker of apoptosis, as the cleaved fragments are critical executioners of apoptosis (37, 38). As expected, nutrient deprivation resulted in a robust activation of caspase 3, demonstrated by the presence of the cleaved 17- to 19-kDa fragments (Fig. 3A). Culture of the cells in serum-free medium, but in the presence of VEGF, resulted in 70% reduction in caspase 3 cleavage and activation, indicative of reduced apoptosis. The addition of DC101 along with VEGF, thereby blocking VEGF signaling, resulted in an 80% increase in levels of cleaved caspase 3 compared to cells in VEGF alone, indicating that VEGF-VEGFR2 signaling was necessary for preadipocyte survival.
In light of the above observation, we hypothesized that VEGF might serve a similar function for brown adipocytes, which also express VEGFR2. While nutrient deprivation does not lead to death of mature brown adipocytes, TNF-α, in combination with low (10 μg/ml) doses of CHX, has been shown to induce apoptosis in brown adipocytes, with a maximal effect between 4 and 6 h (39–41). Brown adipocytes, differentiated in vitro, were treated with 10 nM TNF-α and 10 μg/ml CHX for 6 h in the presence or absence of DC101. Quantification of cell number revealed that whereas TNF-α/CHX alone resulted in a 7% reduction in the number of cells compared to untreated brown adipocytes, treatment with DC101 plus TNF-α/CHX resulted in a 14% loss of brown adipocytes (Fig. 4A). This level of cell death is significant as adipocytes are relatively resistant to apoptosis. For an additional measure of survival, the percentage of cells with caspase 3 activation was measured by FACS analysis. There was a 5-fold increase in the number of adipocytes with cleaved caspase 3 after treatment with TNF-α/CHX and DC101 compared to treatment with TNF/CHX alone (Fig. 4B), indicating that blockade of VEGF signaling leads to increased brown adipocyte apoptosis.
Figure 4.
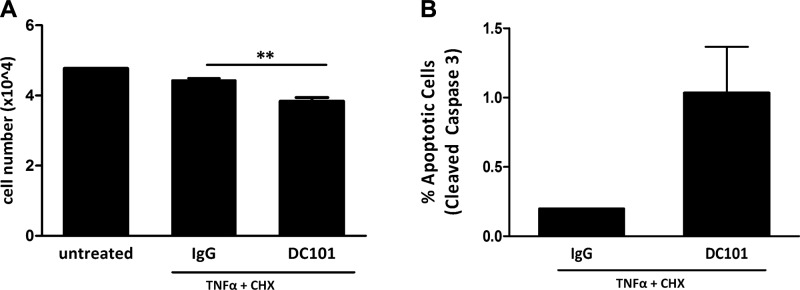
Blockade of VEGF signaling sensitizes brown adipocytes to apoptosis. Differentiated brown adipocytes at d 7 were treated with 10 nM TNF-α and 10 μg/ml CHX for 6 h to induce apoptosis in the presence or absence of DC101, a VEGFR2-blocking antibody (15 μg/ml). A) Treatment with DC101 in the presence of TNF-α/CHX led to reduced survival and fewer brown adipocytes compared to TNF-α/CHX alone. **P < 0.01. B) Detection of apoptotic adipocytes by FACS analysis for Alexa Fluor 488-conjugated cleaved (activated) caspase 3 revealed increased caspase 3 activation in the presence of DC101. P = 0.067.
VEGF neutralization reduces BAT vascular density and is associated with fibrosis and inflammation of BAT in vivo
Given our data indicating a role for VEGF in BAT development in vivo and in brown adipocyte survival in vitro, we investigated a possible function for VEGF in brown adipose in the adult. To block VEGF signaling in vivo mice were injected intravenously with an adenovirus expressing sFlt1, which acts as a soluble decoy receptor for VEGF and results in systemic VEGF neutralization, or an empty adenovirus for control. To examine capillary density in BAT of mice following systemic VEGF neutralization, BAT sections were probed with antisera against the endothelial-specific glycoprotein endomucin, which is expressed by the endothelium of capillaries and veins but not arteries (42, 43). At 7 d following injection of the adenovirus, there was a marked reduction in microvessel density in the Ad-sFlt1-injected mice compared to Ad-null-injected mice, detected by endomucin staining on BAT sections (Fig. 5A). Furthermore, endomucin levels analyzed by Western blotting were decreased by 50% in the BAT of Ad-sFlt 1-injected mice compared to control mice (Fig. 5B), confirming that VEGF neutralization led to a reduction in vascular density of BAT.
Figure 5.

Systemic VEGF neutralization leads to reduced BAT vascular density and is associated with increased fibrosis and inflammation. A) Capillary density in BAT of sFlt1-expressing and control mice was revealed by immunofluorescence staining for endomucin. Nuclei were labeled with DAPI. There were fewer capillaries (arrowheads) in the BAT of sFlt1-expressing mice compared to controls. B) Total endomucin protein levels in whole-tissue lysates from BAT, detected by Western blot analysis (left panel) and quantification of endomucin levels (right panel), normalized to β tubulin, confirmed the reduced vasculature; *P < 0.05. C) Collagen deposition in BAT of sFlt1-expressing and control mice, detected by Masson's trichrome staining, revealed increased collagen (arrowheads), indicative of fibrosis. D) Immunohistochemical localization of F4/80 revealed infiltrating macrophages (black arrowheads) in the BAT of ad-sFlt1-expressing mice but none in ad-Null mice. F4/80-positive cells were detected adherent to the vessel walls of sFlt1-expressing mice (red arrowheads), suggesting local inflammation. Dotted black line denotes the endothelial lining. L, lumen. Scale bars = 50 μm.
Reduced vascular density results in local hypoxia, which can, in turn, lead to inflammation and fibrosis in adipose tissue (44). Collagen I, the predominant collagen in adipose tissue (45), was detected by Masson's trichrome staining, which stains muscle red, nuclei dark purple, and collagen bright blue. Collagen appeared as thick blue depositions around adipocytes and blood vessels (yellow arrowheads) in the sFlt1-expressing mice compared to thin collagen sheets in the wild-type mice (Fig. 5C).
IHC localization of F4/80 revealed the presence of a large number of macrophages in the BAT of sFlt1-expressing mice (black arrowheads), whereas no macrophages were observed in the BAT of control mice (Fig. 5D). The features of immune cell infiltration, including adhesion (Fig. 5D, red arrowheads) and extravasation of the F4/80-positive cells, suggest that VEGF neutralization resulted in local inflammation in BAT.
VEGF neutralization leads to brown adipocyte apoptosis in vivo
Because blockade of VEGF signaling resulted in increased apoptosis in vitro, we wanted to investigate the effect of VEGF neutralization on brown adipocyte survival in vivo. Apoptosis was first assessed by IHC localization of activated (cleaved) caspase 3 in BAT. Many adipocytes were stained positively for cleaved caspase 3 in the BAT of Ad-sFlt1-injected mice compared to only a few positive cells in Ad-null-injected mice (Fig. 6A).
Figure 6.

VEGF neutralization leads to brown adipocyte apoptosis in vivo. A) Apoptosis detected by immunohistochemical localization of activated (cleaved) caspase 3. Top panel: adipocytes with activated caspase 3 (arrowhead) were rare in the BAT of control mice; low magnification (left panel) and high-magnification (right panel) views. Middle and lower panels: apoptotic cells, with cleaved caspase 3 in the cytosol surrounding lipid droplets, occurred frequently in clumps or singly near vessels in the BAT of sFlt1-expressing mice. L, lumen; LD, lipid droplet. Scale bars = 50 μm. B) TUNEL staining demonstrating that BAT of mice expressing sFlt1 have more TUNEL-positive cells (arrowheads) compared to control mice. Scale bar = 100 μm. C) TUNEL-positive adipocytes (white arrowheads) and endothelial cells (red arrowheads) were identified by costaining with endomucin; only TUNEL-positive adipocytes were counted for quantification. Scale bar = 50 μm. D) Quantification of TUNEL-positive cells in each section reveals more than a 6-fold increase in brown adipocyte apoptosis in sFlt1-expressing mice vs. controls. *P < 0.05.
To examine apoptosis by another approach and to quantify apoptotic cells, TUNEL assay was performed (Fig. 6B). TUNEL staining revealed similarly large number of apoptotic (TUNEL+) cells in the BAT of sFlt1-expressing mice compared to null. To distinguish adipocytes from vascular cells, BAT sections were costained with endomucin to identify the endothelium (Fig. 6C). There was a 6- to 7-fold increase in the number of apoptotic adipocytes in the BAT of sFlt1-expressing mice compared to control BAT (Fig. 6D). These data indicate that VEGF acts as a survival factor for brown adipocytes in vivo.
Effect of VEGF neutralization on mitochondria and brown adipocyte morphology in vivo
As mitochondria are central to the thermogenic function of brown adipose, we assessed the effect of VEGF neutralization on mitochondrial ultrastructure. Ultrastructural analysis of BAT taken from control mice at 7 d after Ad-null injection revealed that each adipocyte contained numerous mitochondria that were uniform in shape and size, displayed dense, tightly packed cristae that spanned the width of each mitochondrion (Fig. 7A). In contrast, mitochondria in brown adipocytes from sFlt1-expressing mice often lacked cristae or had cristae that were incomplete, irregular in arrangement, and/or did not span the mitochondrion (Fig. 7A, red arrowheads). Incomplete or absent cristae are associated with mitochondrial degeneration and inactivity. Observation of the widespread mitochondrial degeneration in Ad-sFlt1 injected mice raised the possibility that the overall number of mitochondria may also be different following VEGF neutralization. The number of mitochondria (including the ones with incomplete cristae) in TEM sections of Ad-sFlt1 and control BAT were counted. Brown adipocytes of Ad-sFlt1 injected mice had 30% fewer mitochondria compared to control mice (Fig. 7B). The mitochondrial anomalies in brown adipocytes of sFlt1-expressing mice are consistent with the concept that VEGF neutralization leads to reduced brown adipocyte function.
Figure 7.

VEGF neutralization results in reduced mitochondria and disrupted ultrastructure. A) Electron micrograph of brown adipocytes showing abundant mitochondria, lipid droplets (LD), and a nearby vessel (L). BAT of sFlt1-expressing mice revealed reduced number of mitochondria and a majority of the mitochondria showed obvious signs of degeneration, such as incomplete or involuting cristae (red arrowhead) or complete lack of cristae (yellow asterisks). Mitochondria in the BAT of control mice were typical of functional brown adipocytes: they were numerous and densely packed, with complete double membranous cristae. High magnification depicted in bottom panel. B) Average number of mitochondria per field indicates a significant decrease in the number of mitochondria in sFlt1-expressing mice. Scale bars = 500 nm; P = 0.00007.
Electron microscopic examination of the brown adipocytes of sFlt1-expressing mice also revealed autophagosomes engulfing double-membraned organelles reminiscent of mitochondria; these structures were not observed in the control mice (Fig. 8A). The individual autophagic vesicles appeared either electron-dense or light, likely reflecting the stage of autophagic degradation. During autophagy, LC3B protein undergoes post-translational modification and is converted from form I to a faster migrating form II, which become associated with autophagic vesicles (46), thus serving as a marker of autophagy (47). To further characterize the observed mitochondrial autophagy (mitophagy), LC3B expression was assessed in BAT using IHC. A pattern of light brown puncta distributed throughout the cytosol of individual cells was detected in both control and sFlt1-expressing mice, reflecting a basal level of autophagy. However, the BAT of Ad-sFlt1-injected mice contained many adipocytes with denser dark brown cytoplasmic puncta, indicating a greater abundance of autophagosomes (Fig. 8B). This staining pattern was rarely observed in Ad-null mice, an observation that is consistent with the ultrastructural analysis that did not reveal mitophagy in BAT from control mice. Increased mitophagy is associated with reduced lipolysis in WAT (48) and reduced thermogenesis in BAT (49). The observations of altered mitochondrial structure in BAT in association with VEGF neutralization support a role for VEGF in the maintenance of brown adipocyte integrity.
Figure 8.

VEGF neutralization results in brown adipocyte mitophagy. A) Electron micrograph of brown adipocytes from Ad-sFlt1-injected mice showing autophagy of mitochondria (red arrowhead). Left panel: early stage of mitophagy in which the intact mitochondria is being engulfed by an autophagosome. Note that the engulfed mitochondria appears healthy, with complete cristae, suggesting that mitophagy in sFlt1-expressing mice is not a clearance pathway for abnormal mitochondria but a separate mechanism for reduction in mitochondria. Right panel: advanced stage of mitophagy in which the autophagosome has fused with a lysosome and material has degraded, resulting in a lower density compared to the surrounding tissue. The double membranous structure present in the autophagolysosome is indicative of a mitochondria. Scale bars = 500 nm. B) Immunohistochemical localization of the autophagic marker LC3B. The punctate staining indicates autophagosomes with which the LC3B is associated. There is increased puncta in BAT of the sFlt1-expressing mice compared to control mice. N, nucleus; LD, lipid droplet. Scale bars = 50 μm. C) Brown adipocyte histology, analyzed by H&E, demonstrates the typical adipocyte morphology of multiple small lipid droplets in each cell of the BAT from control mice (left), whereas Ad-sFlt1-expressing mice had significantly larger lipid droplets (yellow arrowhead) not normally observed in lean mice. Scale bars = 50 μm.
Whereas brown adipocytes from the control mice had numerous small lipid droplets that are typical of BAT histology, brown adipocytes of sFlt1-expressing mice exhibited much larger lipid droplets (Fig. 8C), a phenotype that is indicative of increased lipid content and unusual in normal brown adipocytes of lean mice. This morphological difference was readily observed during histological analysis by H&E, as well as during ultrastructural analysis.
DISCUSSION
VEGF expression in BAT and WAT has been reported earlier, and it has been demonstrated that VEGF plays a role in adipose tissue angiogenesis (26, 50, 51); however, the function of VEGF in brown adipocytes has not been explored.
In this study, VEGF expression increased concomitantly with brown adipocyte differentiation. While it has been reported that VEGF is expressed in whole BAT tissue (53) as well as in brown preadipocytes and adipocytes (26, 54), there had been no quantitative measure of the change in total VEGF or its isoforms during the course of differentiation before this. The observations that VEGF164 comprised the majority of VEGF in differentiated brown adipocytes and that VEGF120 was largely absent were in agreement with our in vivo observations. However, there was virtually no detectable VEGF188 in cultured adipocytes, an observation that has been previously reported in rat brown adipocytes differentiated in vitro (54) and appears to be a limitation of in vitro culture. The expression of VEGFR2 in undifferentiated preadipocytes and differentiated brown adipocytes coupled with the observation of VEGFR2 phosphorylation is novel and indicates active autocrine VEGF signaling in cultured brown preadipocytes and adipocytes. In support of this notion, the addition of VEGF induced preadipocyte proliferation in vitro, and VEGF was a survival factor for both preadipocytes and mature adipocytes in vitro.
Although there are reports of the effect of VEGF neutralization on BAT angiogenesis during cold acclimation, this study assessed the role of VEGF in unstimulated BAT and brown adipocytes by systemic VEGF neutralization in mice housed at normal temperature and fed a regular chow diet. Systemic VEGF neutralization resulted in brown adipocyte apoptosis as early as 7 d postinjection, suggesting that VEGF could have a direct effect on mature brown adipocyte survival in vivo. Our laboratory has found that VEGF isoform expression in various tissues reflects the proximity between the VEGF source and its targets (16, 55). The predominance of heparan sulfate-binding VEGF isoforms in BAT is consistent with the hypothesis that VEGF functions in brown adipocytes in an autocrine and/or juxtacrine manner. Our observations in vitro further validate this notion: cultured brown adipocytes in which apoptosis was induced by treatment with TNF-α and CHX showed a higher level of cell death when VEGF signaling was blocked with DC101. This is physiologically relevant because levels of TNF-α is significantly higher in adipose tissue of obese animals, is known to induce apoptosis in brown adipocytes, and this study suggests that the cytotoxic effects of TNF-α may be aggravated in the absence of VEGF. While a role for VEGF in brown adipocyte and preadipocyte apoptosis has not been reported before this, VEGF has been demonstrated to be a survival factor for other highly specialized cell types, such as photoreceptors, the retinal pigmented epithelium, Müller cells, and podocytes (15, 19, 20). Apoptosis in brown preadipocytes is regulated by insulin through PI3K/Akt signaling (39, 40). We have demonstrated that VEGF also signals in these cells through VEGFR2, and the PI3K/Akt pathway is activated.
Examination of the ultrastructure of BAT from sFlt1-expressing mice revealed abnormal mitochondria with partial or complete loss of cristae, a phenotype that is commonly associated with reduced mitochondrial activity and degeneration (56, 57). Adipocytes in these mice also displayed mitophagy along with an increase in autophagosomes, as judged by the presence of LC3B form II, phenomena absent in control BAT. A possible explanation for this is that systemic VEGF neutralization results in dysfunctional or inactive brown adipocytes in which the mitochondria are not employed in fatty acid oxidation, and are thus targeted for autophagy to conserve cellular resources and maintain cellular homeostasis (49). In the absence of efficient lipid hydrolysis, the excess triglycerides are stored within the brown adipocytes of the sFlt1-expressing mice, resulting in large fused droplets atypical of normal brown adipocytes. Consistent with this observation, it has recently been demonstrated that adipose tissue-specific VEGF expression results in “browning” of WAT, increased BAT mass and function, and protection against high-fat diet and insulin resistance (52, 58). In addition, it has been proposed that mitophagy regulates brown-white transdifferentiation of adipocytes and negatively correlates with brown adipocyte fate (48). Our findings complement this report, and there is the possibility that the fused lipid droplets, fewer mitochondria, and increased mitophagy observed on VEGF neutralization are early events in brown to white transdifferentiation. Alternatively and/or additionally, the observed mitochondrial degeneration may be an indirect effect of a nutrient-poor environment due to capillary dropout after systemic VEGF neutralization.
Patterns of VEGF isoform expression varies among tissues, both during development and in the adult (23). Examination of VEGF isoform expression in BAT revealed the VEGF to consist of ∼60 and 40% of VEGF164 and VEGF188, respectively, with VEGF120 accounting for <1%. VEGF120 is freely diffusible, while VEGF188 remains largely matrix and cell associated and is thought to act locally, and VEGF164 has intermediate properties. The absence of VEGF164 and VEGF188 led to significant morphological abnormalities in the embryonic brown adipocytes, including a marked reduction in number and disorganization relative to embryonic wild-type brown adipose, with no detectable difference in adipocyte proliferation or apoptosis at the time point examined. In light of the in vitro observation that VEGF is a mitogen and survival factor for preadipocytes, it is possible that the BAT anomalies in the absence of VEGF120/120 are the result of decreased survival or proliferation of embryonic brown preadipocytes earlier in development. UCP1 expression was markedly reduced in the brown adipocytes of VEGF120/120 mice, with most cells displaying little or no expression, a phenotype indicative of incomplete differentiation. Mitochondrial content, marked by the expression of Cox IV protein, was also significantly reduced, indicating impaired mitochondrial biogenesis. This finding is consistent with the observation in adult BAT, that systemic VEGF neutralization resulted in massive mitochondrial degeneration and reduced mitochondrial number.
The formation of a vasculature is essential for proper organ development and function and is tightly regulated by various proangiogenic and antiangiogenic factors. As for virtually all other tissues, adipogenesis and angiogenesis are spatially and temporally coupled (59), and VEGF has been shown to mediate most of the angiogenesis in adipose tissue (60, 61). In the adult, adipose tissue is one of the most plastic organs; an earlier study reported that sFlt1-mediated systemic VEGF neutralization resulted in significant capillary regression in adipose tissue after 14 d of injection, along with few other organs, such as the pancreas and thyroid (14). In this study, systemic VEGF neutralization resulted in ∼50% reduction in vascular density within 7 d of Ad-sFlt1 injection, underscoring the plasticity of adipose vasculature and was also associated with inflammation and fibrosis, presumably because of hypoxia, resulting from the capillary dropout. Interest in VEGF as a potential target for weight control has led to significant work on the effect of VEGF neutralization in WAT. Recent studies demonstrated a dichotomous role of VEGF in adipose tissue: in the early response to a high-fat diet, WAT-specific transgenic VEGF expression confers protection from metabolic insults, leads to a browning of white adipocytes, and increased BAT mass (52, 58), whereas blockade of VEGF signaling during obesity and metabolic dysfunction leads to improved insulin sensitivity and metabolic health.
Our work has identified an additional function of VEGF in BAT: its direct beneficial effects on brown adipocytes to promote their survival, proliferation, and maintenance of mitochondria. Our findings add to the growing list of pleiotropic effects of VEGF, some of which were described earlier. As anti-VEGF therapies are currently in use, it is important to know of the effects of VEGF neutralization in nontarget organs in the adult.
Acknowledgments
This work was supported by U.S. National Institutes of Health grants EY15435 (to P.A.D.) and K12-EY16335 (to L.K.).
The authors acknowledge Dhanesh Amarnani for his technical assistance in the sFlt1 experiments.
Footnotes
- BAT
- brown adipose tissue
- CHX
- cycloheximide
- DAB
- 3,3′-diaminobenzidine
- DAPI
- 4′,6′-diamino-2-phenylindole
- DMEM
- Dulbecco's modified Eagle's medium
- FACS
- fluorescence-activated cell sorter
- FBS
- fetal bovine serum
- H&E
- hematoxylin and eosin
- IHC
- immunohistochemistry
- IgG
- immunoglobulin G
- LC3B
- light-chain 3B
- MAPK
- mitogen-activated protein kinase
- PBS
- phosphate-buffered saline
- PFA
- paraformaldehyde
- PGC1α
- peroxisome proliferator-activated receptor γ coactivator 1α
- PI3K
- phosphatidylinositol 3-kinase
- sFlt1
- soluble fms-like tyrosine kinase 1
- TEM
- transmission electron microscope
- TNF-α
- tumor necrosis factor α
- TUNEL
- terminal transferase dUTP nick end labeling
- UCP1
- uncoupling protein 1
- VEGF
- vascular endothelial growth factor
- VEGFR1/2
- vascular endothelial growth factor receptor 1/2
- WAT
- white adipose tissue;
REFERENCES
- 1. Rosen E. D., Spiegelman B. M. (2006) Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444, 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nedergaard J., Cannon B. (2010) The changed metabolic world with human brown adipose tissue: therapeutic visions. Cell Metab. 11, 268–272 [DOI] [PubMed] [Google Scholar]
- 3. Cypess A. M., Kahn C. R. (2010) The role and importance of brown adipose tissue in energy homeostasis. Curr. Opin. Ped. 22, 478–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cypess A. M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A. B., Kuo F. C., Palmer E. L., Tseng Y. H., Doria A., Kolodny G. M., Kahn C. R. (2009) Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rothwell N. J., Stock M. J. (1979) A role for brown adipose tissue in diet-induced thermogenesis. Nature 281, 31–35 [DOI] [PubMed] [Google Scholar]
- 6. Farmer S. R. (2008) Molecular determinants of brown adipocyte formation and function. Genes Dev. 22, 1269–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouillaud F., Arechaga I, Petit P. X., Raimbault S., Levi-Meyrueis C., Casteilla L., Laurent M., Rial E., Ricquier D. (1994) A sequence related to a DNA recognition element is essential for the inhibition by nucleotides of proton transport through the mitochondrial uncoupling protein. Eur. Mol. Biol. Org. J. 13, 1990–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seale P., Lazar M. (2009) A brown fat in humans: turning up the heat on obesity. Diabetes 58, 1482–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Korac A, Buzadzic B, Petrovic V., Vasilijevic A., Jankovic A., Micunovic K., Korac B. (2008) The role of nitric oxide in remodeling of capillary network in rat interscapular brown adipose tissue after long-term cold acclimation. Histol. Histopathol. 23, 441–450 [DOI] [PubMed] [Google Scholar]
- 10. Nieves B. J., D'Amore P. A., Bryan B. A. (2009) The function of vascular endothelial growth factor. BioFactors 35, 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carmeliet P., Ferreira V., Breier G, Pollefeyt S., Kieckens L., Gertsenstein M., Fahrig M., Vandenhoeck A., Harpal K., Eberhardt C., Declercq C., Pawling J., Moons L., Collen D., Risau W., Nagy A. (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380, 435–439 [DOI] [PubMed] [Google Scholar]
- 12. Ferrara N., Carver-Moore K., Chen H., Dowd M., Lu L., O'Shea K. S., Powell-Braxton L., Hillan K. J., Moore M. W. (1996) Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380, 439–442 [DOI] [PubMed] [Google Scholar]
- 13. Miquerol L., Langille B. L., Nagy A. (2000) Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development 127, 3941–3946 [DOI] [PubMed] [Google Scholar]
- 14. Kamba T., Tam B. Y., Hashizume H., Haskell A., Sennino B., Mancuso M. R., Norberg S. M., O'Brien S. M., Davis R. B., Gowen L. C., Anderson K. D., Thurston G., Joho S., Springer M. L., Kuo C. J., McDonald D. M. (2006) VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am. J. Physiol. Heart Circ. Physiol 290, H560–H576 [DOI] [PubMed] [Google Scholar]
- 15. Saint-Geniez M., Maharaj A. S., Walshe T. E., Tucker B. A., Sekiyama E., Kurihara T., Darland D. C., Young M. J., D'Amore P. A. (2008) Endogenous VEGF is required for visual function: evidence for a survival role on Müller cells and photoreceptors. PloS One 3, e3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maharaj A. S. R., Walshe T. E., Saint-Geniez M., Venkatesha S., Maldonado A. E., Himes N. C., Matharu K. S., Karumanchi S. A., D'Amore P. A. (2008) VEGF and TGF-β are required for the maintenance of the choroid plexus and ependyma. J. Exp. Med. 205, 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferrara N., Gerber H.-P., LeCouter J. (2003) The biology of VEGF and its receptors. Nat. Med. 9, 669–676 [DOI] [PubMed] [Google Scholar]
- 18. Patel-Hett S., D'Amore P. A. (2011) Signal transduction in vasculogenesis and developmental angiogenesis. Intl. J. Dev. Biol. 55, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guan F., Villegas G., Teichman J., Mundel P., Tufro A. (2006) Autocrine VEGF-A system in podocytes regulates podocin and its interaction with CD2AP. Am. J. Physiol. Renal Physiol. 291, F422–F428 [DOI] [PubMed] [Google Scholar]
- 20. Ford K. M., Saint-Geniez M., Walshe T., Zahr A., D'Amore P. A. (2011) Expression and role of VEGF in the adult retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 52, 9478–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bryan B. A., Walshe T. E., Mitchell D. C., Havumaki J. S., Saint-Geniez M., Maharaj A. S., Maldonado A. E., D'Amore P. A. (2008) Coordinated vascular endothelial growth factor expression and signaling during skeletal myogenic differentiation. Mol. Biol. Cell 19, 994–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mayer H., Bertram H., Lindenmaier W., Korff T., Weber H., Weich H. (2005) Vascular endothelial growth factor (VEGF-A) expression in human mesenchymal stem cells: autocrine and paracrine role on osteoblastic and endothelial differentiation. J. Cell. Biochem. 95, 827–839 [DOI] [PubMed] [Google Scholar]
- 23. Ng Y. S., Rohan R., Sunday M. E., Demello D. E., D'Amore P. A. (2001) Differential expression of VEGF isoforms in mouse during development and in the adult. Dev. Dyn. 220, 112–121 [DOI] [PubMed] [Google Scholar]
- 24. Carmeliet, Ng, P., Y-S., Nuyens D., D'Amore P., Shima D. (1999) Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF 164 and VEGF 188. Nat. Med. 5, 495–502 [DOI] [PubMed] [Google Scholar]
- 25. Stalmans I., Ng Y. S., Rohan R., Fruttiger M.., Bouché A., Yuce A., Fujisawa H., Hermans B., Shani M.., Jansen S., Hicklin D., Anderson D. J., Gardiner T., Hammes H. P., Moons L., Dewerchin M., Collen D., Carmeliet P., D'Amore P. A. (2002) Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J. Clin. Invest. 109, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Asano A., Irie Y., Saito M. (2001) Isoform-specific regulation of vascular endothelial growth factor (VEGF) family mRNA expression in cultured mouse brown adipocytes. Mol. Cell. Endocrinol. 174, 71–76 [DOI] [PubMed] [Google Scholar]
- 27. Claffey K. P., Wilkison W. O., Spiegelman B. M. (1992) Vascular endothelial growth factor. Regulation by cell differentiation and activated second messenger pathways. J. Biol. Chem. 267, 16317–16322 [PubMed] [Google Scholar]
- 28. Miyazawa-Hoshimoto S., Takahashi K., Bujo H., Hashimoto N., Yagui K., Saito Y. (2005) Roles of degree of fat deposition and its localization on VEGF expression in adipocytes. Am. J. Physiol. Endocrinol. Metab. 288, E1128–E1136 [DOI] [PubMed] [Google Scholar]
- 29. Xue Y., Petrovic N., Cao R., Larsson O., Lim S., Chen S., Feldmann H. M., Liang Z., Zhu Z., Nedergaard J., Cannon B., Cao Y. (2009) Hypoxia-independent angiogenesis in adipose tissues during cold acclimation. Cell Metab. 9, 99–109 [DOI] [PubMed] [Google Scholar]
- 30. Saint-Geniez M., Maldonado A. E., D'Amore P. A. (2006) VEGF expression and receptor activation in the choroid during development and in the adult. Invest. Ophthalmol. Vis. Sci. 47, 3135–3142 [DOI] [PubMed] [Google Scholar]
- 31. Zhang L., Yang N., Mohamed-Hadley A., Rubin S. C., Coukos G. (2003) Vector-based RNAi, a novel tool for isoform-specific knock-down of VEGF and anti-angiogenesis gene therapy of cancer. Biochem. Biophys. Res. Commun. 303, 1169–1178 [DOI] [PubMed] [Google Scholar]
- 32. Barnard T. (1969) The ultrastructural differentiation of brown adipose tissue in the rat. J. Ultrastruct. Res. 29, 311–322 [DOI] [PubMed] [Google Scholar]
- 33. Klein J., Fasshauer M., Klein H. H., Benito M., Kahn C. R. (2002) Novel adipocyte lines from brown fat: a model system for the study of differentiation, energy metabolism, and insulin action. Bioassays 24, 382–388 [DOI] [PubMed] [Google Scholar]
- 34. Collins S., Cao W., Robidoux J. (2004) Learning new tricks from old dogs: beta-adrenergic receptors teach new lessons on firing up adipose tissue metabolism. Mol. Endocrinol. 18, 2123–2131 [DOI] [PubMed] [Google Scholar]
- 35. Jimenez M., Léger B., Canola K., Lehr L., Arboit P., Seydoux J., Russell A. P., Giacobino J. P., Muzzin P., Preitner F. (2002) Beta(1)/beta(2)/beta(3)-adrenoceptor knockout mice are obese and cold-sensitive but have normal lipolytic responses to fasting. FEBS Lett. 530, 37–40 [DOI] [PubMed] [Google Scholar]
- 36. Zachary I. (2003) VEGF signalling: integration and multi-tasking in endothelial cell biology. Biochem. Soc. Trans. 31, 1171–1177 [DOI] [PubMed] [Google Scholar]
- 37. Fernandes-Alnemri T., Litwack G., Alnemri E. S. (1994) CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J. Biol. Chem. 269, 30761–30764 [PubMed] [Google Scholar]
- 38. Nicholson D. W., Ali A., Thornberry N. A., Vaillancourt J. P., Ding C. K., Gallant M., Gareau Y., Griffin P. R., Labelle M., Lazebnik Y. A., Munday N. A., Raju S. M., Smulson M. E., Yamin T.-T., Yu V. L., Miller D. K. (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376, 37–43 [DOI] [PubMed] [Google Scholar]
- 39. Boucher J., Macotela Y., Bezy O., Mori M. A., Kriauciunas K., Kahn C. R. (2010) A kinase-independent role for unoccupied insulin and IGF-1 receptors in the control of apoptosis. Sci. Signal. 3, ra87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miranda S., González-Rodríguez A., Revuelta-Cervantes J., Rondinone C. M., Valverde A. M. (2010) Beneficial effects of PTP1B deficiency on brown adipocyte differentiation and protection against apoptosis induced by pro- and anti-inflammatory stimuli. Cell. Signal. 22, 645–659 [DOI] [PubMed] [Google Scholar]
- 41. Nisoli E., Briscini L., Tonello C., De Giuli-Morghen C., Carruba M. O. (1997) Tumor necrosis factor-a induces apoptosis in rat brown adipocytes. Cell 771–778 [DOI] [PubMed] [Google Scholar]
- 42. Kuhn A., Brachtendorf G., Kurth F., Sonntag M., Samulowitz U., Metze D., Vestweber D. (2002) Expression of endomucin, a novel endothelial sialomucin, in normal and diseased human skin. J. Invest. Dermatol. 119, 1388–1393 [DOI] [PubMed] [Google Scholar]
- 43. Dela Paz N. G., D'Amore P. A. (2009) Arterial versus venous endothelial cells. Cell Tissue Res. 335, 5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Halberg N., Khan T., Trujillo M. E., Wernstedt-Asterholm I., Attie A. D., Sherwani S., Wang Z. V., Landskroner-Eiger S., Dineen S., Magalang U. J., Brekken R. A., Scherer P. E. (2009) Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 29, 4467–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Napolitano L. (1963) The differentiation of white adipose cells. An electron microscope study. J. Cell Biol. 18, 663–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tanida I., Ueno T., Kominami E. (2004) Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 279, 47704–47710 [DOI] [PubMed] [Google Scholar]
- 47. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singh R., Xiang Y., Wang Y., Baikati K., Cuervo A. M., Luu Y. K., Tang Y., Pessin J. E., Schwartz G. J., Czaja M. J. (2009) Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest. 119, 3329–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Milosevic M., Ukropina M., Čakić-Milošević M., Korać A. (2008) Autophagy of mitochondria in brown adipocytes of chemically thyroidectomised rats. EMC 2008 14th European Microscopy Congress 1–5 September 2008, 3, 113–114 [Google Scholar]
- 50. Xue Y., Cao R., Nilsson D., Chen S., Westergren R., Hedlund E. M., Martijn C., Rondahl L., Krauli P., Walum E., Enerbäck S., Cao Y. (2008) FOXC2 controls Ang-2 expression and modulates angiogenesis, vascular patterning, remodeling, and functions in adipose tissue. Proc. Natl. Acad. Sci. U. S. A. 105, 10167–10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fredriksson J. M., Nikami H., Nedergaard J. (2005) Cold-induced expression of the VEGF gene in brown adipose tissue is independent of thermogenic oxygen consumption. FEBS Lett. 579, 5680–5684 [DOI] [PubMed] [Google Scholar]
- 52. Elias I., Franckhauser S., Ferré T., Vilà L., Tafuro S., Muñoz S., Roca C., Ramos D., Pujol A., Riu E., Ruberte J., Bosch F. (2012) Adipose tissue overexpression of vascular endothelial growth factor protects against diet-induced obesity and insulin resistance. Diabetes 61, 1801–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Asano A., Morimatsu M., Nikami H., Yoshida T., Saito M. (1997) Adrenergic activation of vascular endothelial growth factor mRNA expression in rat brown adipose tissue: implication in cold-induced angiogenesis. Biochem. J. 328, 179–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tonello C., Giordano A., Cozzi V., Cinti S., Stock M. J., Carruba M. O., Nisoli E. (1999) Role of sympathetic activity in controlling the expression of vascular endothelial growth factor in brown fat cells of lean and genetically obese rats. FEBS Lett. 442, 167–172 [DOI] [PubMed] [Google Scholar]
- 55. Saint-Geniez M., Kurihara T., Sekiyama E., Maldonado A. E., D'Amore P. A. (2009) An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc. Natl. Acad. Sci. U. S. A. 106, 18751–18756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cinti S. (2007) The adipose organ. In Nutrition and Health: Adipose Tissue and Adipokines in Health and Disease (Fantuzzi G., Mazzone T., eds) pp. 3–19, Humana Press, New York [Google Scholar]
- 57. Cigolini M., Cinti S., Bosello O., Brunetti L., Björntorp P. (1986) Isolation and ultrastructural features of brown adipocytes in culture. J. Anat. 145, 207–216 [PMC free article] [PubMed] [Google Scholar]
- 58. Sun K., Wernstedt Asterholm I., Kusminski C. M., Bueno A. C., Wang Z. V., Pollard J. W., Brekken R. A., Scherer P. E. (2012) Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc. Natl. Acad. Sci. U. S. A. 109, 5874–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Crandall D. L., Hausman G. J., Kral J. G. (1997) A review of the microcirculation of adipose tissue: anatomic, metabolic, and angiogenic perspectives. Microcirculation 4, 211–232 [DOI] [PubMed] [Google Scholar]
- 60. Zhang Q. X., Magovern C. J., Mack C. A., Budenbender K. T., Ko W., Rosengart T. K. (1997) Vascular endothelial growth factor is the major angiogenic factor in omentum: mechanism of the omentum-mediated angiogenesis. J. Surg. Res 67, 147–154 [DOI] [PubMed] [Google Scholar]
- 61. Fukumura D., Ushiyama A., Duda D. G., Xu L., Tam J., Krishna V., Chatterjee K., Garkavtsev I., Jain R. K. (2003) Paracrine regulation of angiogenesis and adipocyte differentiation during in vivo adipogenesis. Circ. Res. 93, e88–e97 [DOI] [PMC free article] [PubMed] [Google Scholar]