

Abstract
The costimulatory receptor Slamf6 partially controls lupus-related autoimmunity in congenic Sle1b mice; for instance, the presence of the protein isoform Slamf6-H1 in Sle1b.Slamf6-H1 mice mitigates disease. Here, we report that young Sle1b mice, but not Sle1b.Slamf6-H1 or B6 mice, contain a memory T-helper cell subset identified by ]mt]2-fold increase in expression of 17 genes, chief among which is Spp1, encoding the cytokine osteopontin (OPN). These T follicular helper (TFH) cells, including OPN+ TFH cells, expand concomitantly with severity of the disease. By contrast, Sle1b.Slamf6-H1 or Sle1b.SAP−/− mice do not develop autoantibodies and the number of TFH cells is 5 times lower than in age-matched Sle1b mice. By comparing Sle1b and Sle1b.OPN−/− mice, we find that the lack of OPN expression impedes early autoantibody production. Furthermore, on the adoptive transfer of Sle1b.OPN−/− CD4+ T cells into bm12 recipients autoantibody production and germinal center formation is reduced compared to recipients of Sle1b.OPN+/+ CD4+ T cells. We propose a model in which OPN provides a survival signal for a precursor TFH cell subset, which is a key factor in autoimmunity. Keszei, M., Detre, C., Castro, W., Magelky, E., O'Keeffe, M., Kis-Toth, K., Tsokos, G. C., Wang, N., Terhorst, C. Expansion of an osteopontin-expressing T follicular helper cell subset correlates with autoimmunity in B6.Sle1b mice and is suppressed by the H1-isoform of the Slamf6 receptor.
Keywords: Ly108, SAP, systemic lupus erythematosus, OPN, TFH
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by a wide spectrum of disease manifestations, chief among which are autoantibody production and kidney failure. In the lupus-prone congenic mouse strains, B6.Sle1b (Sle1b) and B6.129chr1b, short DNA segments derived from the NZW or 129 strains, respectively, are embedded in the B6 genome (1, 2). Genetic studies of Sle1b and B6.129chr1b mice indicate that polymorphisms of several of the 9 genes encoding the mouse signaling lymphocytic activation molecule family (Slamf) cell surface receptors on chromosome 1 are implicated in the phenotype of these two lupus-prone mouse strains (1, 2). At least two haplotypes are detected in inbred strains: Slamf-haplotype 1 (e.g., in B6) and Slamf-haplotype 2 (e.g., in 129, NZW, BALB/c) (1). Autoimmunity is thought to develop in Sle1b and B6.129chr1b mice because of an epistatic interplay between one or more Slamf-haplotype 2 genes with several B6 genes, e.g., Yaa (1, 2). Consistent with this model, autoantibody induction in congenic B6.129chr1b mice is likely to be dependent on the presence of the Slamf6 gene (3), because congenic Slamf6−/− (129.B6) mice, which contain an almost identical 129 DNA segment, do not develop lupus-related autoimmunity (3). The mouse Slamf6 gene was first reported to encode two distinct proteins, namely Slamf6-1 and Slamf6-2, generated by alternative exon usage (1–3). In Sle1b (NZW.B6) mice (1, 4), the ratio of these two protein isoforms is altered compared with B6, and this ratio affects early B-cell development, leading to accelerated autoantibody responses. Although the outcomes of this study suggest that the Slamf6.1 and Slamf6.2 alleles are positive and negative regulators of autoimmunity in Sle1b mice, the contribution of the Slamf6 gene to immune tolerance is more complex. Surprisingly, an additional protein isoform termed Slamf6-H1 exists, which is only expressed in Slamf-haplotype 1 mice, e.g., B6, but not in mice that contain the Slamf-haplotype 2 DNA segment on chromosome 1, e.g., 129, NZW or Sle1b. Furthermore, Sle1b mice that are hemizygous for a Slamf6-H1-expressing BAC-based transgene, i.e., Sle1b.Slamf6-H1 mice, developed a markedly reduced CD4+ T-cell-dependent autoimmunity (3).
To elucidate the role of Slamf6-H1 in autoantibody production of Sle1b mice in this work, we use global gene expression analyses to compare cells isolated from Sle1b, Sle1b.Slamf6-H1, and wt B6 mice. Surprisingly, 17 genes are up-regulated in Sle1b CD4+ T cells compared to Sle1b.Slamf6 H1 or wt B6 CD4+ T cells. Cell surface marker analyses determined that a subset of memory PD1+ CD4+ T cells, which contain T follicular helper (TFH) cells, is expanded in Sle1b but not in Sle1b.Slamf6-H1 or B6 mice, and that this expansion correlates with an increase in disease activity. Not only do PD1+ CXCR5+ SLAMF-associated protein (SAP)+ TFH cells express the cytokine osteopontin (OPN), the number of OPN+ TFH cells increases with the severity of disease. Conversely, spontaneous autoantibody production observed in OPN−/− Sle1b mice is lower than in OPN+/+ Sle1b littermates. When CD4+ T cells isolated from Sle1b mice were transferred into coisogenic bm12 recipients, autoantibodies developed concomitantly with an expansion of TFH cells. By contrast, on the transfer of Sle1b.OPN−/− CD4+ T cells, neither expansion of TFH cells nor autoantibody production was observed. Taken together, our data suggest a key role for OPN+ TFH cells in lupus-related autoimmunity in mice.
MATERIALS AND METHODS
Mice
B6 and B6.C-H-2bm12/KhEg (bm12) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Sle1b (5) mice were provided by L. Morel (University of Florida, Gainesville, FL, USA). Sle1b.Slamf6-H1 mice (formerly Sle1b.BACLy108-H1) were generated as described previously (3). Sle1b.SAP−/− mice were generated by crossing B6.SAP−/− (6) and Sle1b strains employing PCR-based microsatellite analysis and genotyping, as described previously (3, 6). OPN−/− [B6.129S6 (Cg)-Spp1tm1Blh/J (N10+N2F5)] mice obtained from the Jackson Laboratory were crossed with Sle1b mice. All procedures were conducted according the guideline of the Beth Israel Deaconess Medical Center (BIDMC) Institutional Animal Care and Use Committee.
Microarray analysis
CD4+ T cells (Miltenyi Biotech, Auburn, CA, USA) were isolated from 12-wk-old mice, 6 animals/group. From each group, RNA was isolated using a total RNA isolation kit (Qiagen, Valencia, CA, USA) and was hybridized onto 3 HT MG-430 PM Affymetrix microarrays by pooling 2 samples/array. The Affymetrix GeneChip Array Station HT system (Affymetrix, Santa Clara, CA, USA) was used for labeling, washing, and staining of the probes. Samples were analyzed using the HT scanner.
Bioinformatics
The Department of Biostatistics and Computational Biology at Dana-Farber Cancer Institute (DFCI; Boston, MA, USA) performed bioinformatics analyses. Array quality was assessed using the R/Bioconductor package (7). Raw data files were processed using the robust multiarray average (RMA) algorithm (8). We used Linear Models for Microarray Data (limma; ref. 9) to test for differential gene expression in the contrasts of interest before results were adjusted for multiple testing using the Benjamini and Hochberg method (10). Gene set enrichment analysis (GSEA) was performed with the preranked implementation of the GSEA software package (11) using the moderated t-statistics from limma (9) to determine rank order. All raw and processed microarray data were deposited into the National Center for Biotechnology Information (NCBI; Bethesda, MD, USA) Gene Expression Omnibus database (accession no. GSE31733; http://www.ncbi.nlm.nih.gov/geo).
ELISA
Serum titers of anti-chromatin and anti-ssDNA IgG were determined as described previously (3); for total mouse IgG titers, a kit from Fitzgerald Industries International (Acton, MA, USA) was used. To determine mIL-2 and mOPN, ELISA kits were purchased from BD Biosciences (San Jose, CA, USA) and R&D Systems (Minneapolis, MN, USA), respectively. Cytokine concentrations in culture supernatants were determined according to the manufacturer's protocol.
Cell proliferation assay
To assess proliferation of CD4+ T cells, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; thiazolyl blue) was purchased from Sigma (St. Louis, MO, USA; M5655) and used according to the manufacturer's protocol.
Flow cytometry
Single-cell suspensions of splenocytes were stained with FITC, biotin, or PE-labeled monoclonal antibodies directed against CD4 (L3T4), CD44 (IM7), CD62L (MEL-14), CD69 (H1.2F3), PD-1 (RMP1-30), GL7 (GL-7), Fas (Jo2), B220 (RA3-6B2), CXCR5 (2G8), CD200 (OX90), BTLA (8F4), and Ly9 (30C7) purchased from Biolegend (San Diego, CA, USA), eBioscience (San Diego, CA, USA), or BD Pharmingen (San Diego, CA, USA). Dead cells were excluded using DAPI (Invitrogen, Carlsbad, CA, USA).
The number of OPN+ and Bcl6+ cells was determined by intracellular staining with affinity-purified polyclonal Goat IgG (R&D Systems) or anti-Bcl6 (K112-91; BD Bioscences) after permeabilizing the cells (Biolegend kit). Analyses were performed with a 5-laser BD LSRII Flow cytometer (BD Biosciences). For cell sorting, splenocytes were stained as above and processed with a FACS Aria cell sorter (BD Biosciences).
In some immunofluorescence experiments, splenocyte suspensions were enriched with CD4+ T cells by negative selection (Miltenyi Biotech selection kit) or were depleted of B cells (Mouse Pan B kit, Dynabeads; Invitrogen).
Quantitative PCR
RNA was isolated with TRIzol (Invitrogen). mRNA levels of OPN (probes: Mm00436767_m1) and Bcl-6 (probes: Mm00477633_m1) were measured by a TaqMan Gene Expression Assay with the ABI Prism 7000 SDS platform (Applied Biosystems, Foster City, CA, USA).
RESULTS
Gene expression signature defines the presence of an Spp+ CD4+ T-cell subset in young Sle1b mice
To elucidate the manner by which the Slamf6-H1 protein isoform suppresses T-cell-dependent autoimmunity in Sle1b mice (3), CD4+ T cells were purified from 12-wk-old Sle1b, Sle1b.Slamf6-H1, or B6 mice, and a global gene expression profile was analyzed. Only 17 genes were highly up-regulated in Sle1b CD4+ T cells as compared with the same cells derived from Sle1b.Slamf6-H1 or B6 mice (Fig. 1A and Supplemental Table S1). These genes included Spp1 (encoding OPN), Sostdc1, Stfa3, and IL1R2 (CD121b), and to a lesser extent, Pdcd1 (PD-1). Moreover, expression of several interferon-signature genes was specifically increased in the Sle1b CD4+ T cells (Supplemental Fig. S1 and Supplemental Table S1). Although a similar set of genes was found in a memory CD4+ T-cell subset isolated from senescent (>16-mo-old) B6 mice (13), there are a number of differences between the two subsets. For instance, expression of the transcription factor c/EBPα (Supplemental Fig. S1), which was found in senescent mice (13), is not increased in Sle1b CD4+ T cells.
Figure 1.

Expansion of a memory CD4+ T-cell subset in Sle1b mice, but not in B6 and Sle1b.Slamf6-H1 mice, as judged by gene-expression microarray analyses. A–C) Analyses of splenic CD4+ T cells isolated by negative selection from B6, Sle1b, and Sle1b.Slamf6-H1 mice. B6, Sle1b, and Sle1b.Slamf6-H1 CD4+ T cells (12 wk old) were activated with plate-bound αCD3 (0.1 μg/ml) for 0 h (A), 4 h (B), or 24 h (C). Each dot represents a normalized gene expression ratio derived from the average of 3 microarrays (6 mice) per group. Red dots indicate genes previously found in a set of genes that were up-regulated in PD-1+ memory cells of >15-mo-old B6 mice (13). D–F) Validation of the Sle1b-specific CD4+ T-cell expression signature. Data sets were validated using GSEA (11) in the B6 vs. Sle1b (top panels) and the Sle1b.Slamf6-H1 vs. Sle1b (bottom panels) contrasts at 0, 4, and 24 h after αCD3 stimulation. Enrichment score (y axis) reflects the degree to which a gene set is overrepresented at the top or bottom of a ranked list of genes. Vertical lines below the enrichment plot indicate the position of individual PD-1+ CD4+-specific genes (13) in the rank-ordered data set. Statistical significance of the enrichment of the memory phenotype PD-1+ CD4+-specific dataset in Sle1b CD4+ T cells is estimated by false discovery rate (FDR) q value.
The increase in expression of these 17 genes in Sle1b CD4+ T cells was not sustained by an in vitro stimulation with a monoclonal antibody directed against CD3; the gene-expression signature was markedly reduced after 4 h (Fig. 1B, E) and after 24 h, only a trace amount of these genes was detectable (Fig. 1C, F). The enrichment score and the position of individual CD4+ T-cell-specific genes in the rank-ordered data set (Fig. 1D–F) both support these conclusions. Taken together, the observations indicate that a specific gene expression signature defines a subset of memory CD4+ T cells in young lupus-prone Sle1b mice, which is absent in Sle1b.Slamf6-H1 or B6 mice.
Expansion of PD-1+ CXCR5+ TFH cells in Sle1b mice correlates with disease progression
Because TFH cells are thought to be major players in T-cell-dependent humoral immunity, we next evaluated whether both memory CD4+ T cells and TFH cells were expanded in Sle1b mice (14). First, as judged by flow cytometry, in Sle1b mice, the PD-1+ CD4+ memory T-cell subset expansion correlated with the progression from a prelupus to a lupus state (Fig. 2A). Furthermore, expansion of the PD-1+ CD4+ T cells was far less in nonlupus mice, i.e., Sle1b.Slamf6-H1 (P=0.03) or B6 (P=0.03), of the same age. (Fig. 2B). Second, the percentage of CXCR5+ PD-1+ CD4+ cells in 6-mo-old Sle1b mice was higher than in B6 or Sle1b.Slamf6-H1 mice (Fig. 2C), suggesting that a majority of the PD1+ CD44+ CD4+ memory cells are TFH cells. Third, CXCR5+ PD-1hi cells were already detectable in 12-wk-old Sle1b mice, but not in young B6 or Sle1b.Slamf6-H1 animals (Fig. 2D). Collectively, the data demonstrate that a majority of the memory CD4+ T cells in Sle1b mice are CXCR5+PD-1+ TFH cells and that the expansion of these TFH cells is suppressed by the presence of the Slamf6-H1 transgene.
Figure 2.
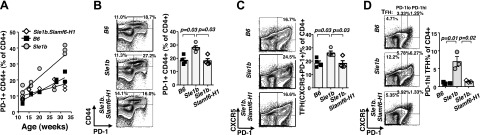
Number of PD-1+ CD44+ CXCR5+ CD4+ T cells in Sle1b mice increases with age as compared to Sle1b.Slamf6-H1 and B6 mice. A) Percentages of CD4+ PD-1+ CD44+ splenic T cells from various age groups of B6, Sle1b, and Sle1b.Slamf6-H1 females determined by flow cytometry. Parameters of linear regression: B6 S (slope) = 0.71, r2=0.91, P < 0.0001; Sle1b S = 1.15, r2 = 0.71, P = 0.002; Sle1b.Slamf6-H1 S = 0.36, r2=0.6, P = 0.003. B, C) Splenocytes of 6-mo-old female mice were labeled with the indicated surface markers and analyzed by flow cytometry. Gray bars show mean values of data points of the 4 individual mice per group; error bars indicate se. Contour plots represent each indicated group. Statistical analysis was performed by the Mann-Whitney nonparametric, 2-tailed test. D) Percentages of CXCR5+ PD-1hi and CXCR5+ PD-1lo TFH cells in the spleens of 12-wk-old female B6, Sle1b, and Sle1b.BACSlamf6-H1 mice were determined by flow cytometry, as described in Materials and Methods. Gray bars show mean values of data points of the 3 individual mice per group; error bars indicate se. Contour plots represent each indicated group.
Autoantibody production and TFH cell expansion in Sle1b mice are dependent on the presence of the adapter SAP
One hallmark of mature committed TFH cells is the expression of the SLAMF-specific single SH2-domain adapter SAP (Sh2d1a; 15, 16). Several studies indicate that SAP-dependent signaling promotes the optimal interaction between TFH and B cells in the germinal center reaction (17–21). To test the hypothesis that in Sle1b mice, both autoantibody titers and TFH-cell expansion would be affected by the absence of the adapter Sle1b.SAP−/− mice were generated. Indeed, the anti-chromatin and anti-ssDNA IgG titers in the serum of a cohort of 6- to 8-mo-old Sle1b.SAP−/− mice were as low as in B6 mice and were dramatically reduced as compared to age-matched Sle1b mice (Fig. 3A, B). In addition, the number of germinal center B (GCB) cells and CD69+B220+ B cells in the spleen of Sle1b.SAP−/− mice was as low as in B6 mice and was reduced compared to Sle1b mice (Fig. 3C, D). However, the total number of B cells, as well as the number of follicular and marginal zone B cells, was identical in Sle1b.SAP−/−, Sle1b, and B6 mice (data not shown). The spontaneous expansion of memory CD4+ cells and of TFH cells observed in Sle1b mice was absent in Sle1b.SAP−/− mice and B6 mice (Fig. 3E, F).
Figure 3.

Expansion of CXCR5+ PD-1+ TFH cells in lupus-prone Sle1b mice is dependent on SAP. A, B) Anti-chromatin (A) and anti-ssDNA (B) IgG levels in the serum of 6- to 8-mo-old female mice were determined as described in Materials and Methods. EU, ELISA unit. B6, n = 16; Sle1b, n = 15; Sle1b.SAP−/−, n = 14. C–F) Numbers of germinal center B cells (B220+ GL7hi Fashi; C) and percentages of CD69+-activated B220+ B cells (D), PD-1+ CD4+ (E), and CXCR5+PD-1+ TFH cells (F) in the spleens of 6- to 8-mo-old female mice were determined by flow cytometry. Gray bars show mean values; error bars indicate se. Contour plots represent each indicated group. B6, n = 5; Sle1b, n = 6; Sle1b.SAP−/−, n = 7 (n=3 in D). Data represent 2 independent experiments. Statistical analysis was performed by the Mann-Whitney nonparametric, 2-tailed test.
Taken together, the outcomes of these experiments demonstrate that the expansion of memory PD-1+ CD4+ T cells and PD-1+ CXCR5+ TFH cells in Sle1b mice is SAP dependent and correlates with the disease manifestations, i.e., autoantibody production and the number of GCB cells. Thus, both the introduction of Slamf6-H1 and the absence of SAP reduce the spontaneous expansion of TFH cells in Sle1b mice, the latter being consistent with a role of SAP in cognate interactions between TFH and B cells.
TFH cells express the cytokine OPN
To determine whether memory and TFH cells express OPN, CD4+ T cells isolated from 12-mo-old Sle1b mice were separated by FACS into PD1+ CXCR5+ CD4+ TFH cells, PD-1− memory cells, and naive CD4+ T cells. On the basis of quantitative TaqMan PCR, TFH cells in the spleen of from 12-mo-old Sle1b mice coexpressed Spp1 and Bcl6 mRNA at markedly higher levels than PD1+ CD44+ CD4+ T or naive CD4+ T cells (Fig. 4A). This was confirmed by Western blot analysis of the OPN protein in lysates of the same cells (Fig. 4B).
Figure 4.
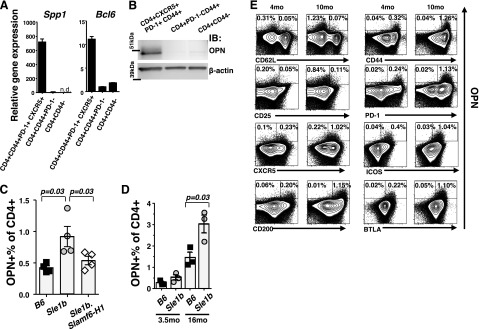
Increased expression of OPN in PD-1+ CD44+ CXCR5+ TFH cells isolated from Sle1b mice, as compared to Sle1b.Slamf6-H1 and B6 mice. A) Spp1 and Bcl6 gene expression was determined by quantitative TaqMan PCR. Splenocytes from two 12-mo-old female Sle1b mice were pooled, and the 3 CD4+ T-cell subsets were isolated by FACS using antibodies as indicated. RNA was isolated and used for TaqMan PCR, as described in Materials and Methods. B) Western blot analysis with an anti-OPN antibody. Pooled splenocytes obtained from three 16-mo-old female Sle1b mice were separated by negative selection with MACS and by FACS with the indicated surface markers. C) Percentage of OPN+ cells in splenic CD4+ T cells isolated from 6-mo-old B6, Sle1b, and Sle1b.Slamf6-H1 female mice (means±se). D) Percentage of OPN+ T cells in the spleen of B6 and Sle1b female mice of the indicated ages (means±se). E) Multicolor intracellular staining of OPN in combination with the indicated surface markers using the same samples as in C. Dot plot of CD4+ OPN+ cells is overlaid on contour plots of the total CD4+ staining. Flow cytometric analyses of CD4+ T cells isolated by MACS-negative selection from 2 pooled spleens of 4- or 16-mo-old Sle1b female mice. All stainings with anti-OPN and the indicated surface markers in each group were generated from the same sample. Data are representative of 3 independent experiments.
As judged by cytoplasmic staining with a monoclonal antibody directed against OPN, the percentage of CD4+ cells that express OPN was ∼1% in Sle1b mice, twice the percentages found in Sle1b.BACSlamf6-H1 or B6 mice of the same age (Fig. 4C). Between the ages of 3.5 and 16 mo, the percentage of OPN+ CD4+ T cells expanded to the same extent in Sle1b and B6 mice (Fig. 4D). Furthermore, the OPN+ CD4+ T cells in aged Sle1b mice are memory T-helper cells that express CD44 and PD1 (Fig. 4E). A significant percentage of the OPN+ CD4+ memory T cells are TFH cells, as they express CXCR5, ICOS, CD200, and BTLA (Fig. 4E and ref. 18) in an age-dependent manner (Fig. 4E and Supplemental Fig. S2).
Taken together, the results support the concept that the number of OPN-expressing TFH cells increases in conjunction with autoantibody production in Sle1b mice.
OPN plays a major role in the TFH-cell-induced autoimmunity of Sle1b mice, as judged by the analyses of OPN−/− Sle1b mice
The observations reported here suggested that OPN+ TFH cells might partake in the pathogenesis of lupus-related autoimmunity in the Sle1b mouse, a hypothesis that is indirectly supported by the finding that polymorphisms of the SPP1 gene in patients with SLE are associated with the pathogenesis of the disease (22, 23). To test this hypothesis, we generated OPN−/− Sle1b mice in which the major CD4+ T-cell subsets were identical to those found in Sle1b mice (Supplemental Fig. S3A) and αCD3-induced in vitro proliferation of OPN−/− Sle1b and Sle1b CD4+ T cells was identical (Supplemental Fig. S3B). However, in the serum of 9- to 12-wk-old OPN−/−Sle1b mice, both anti-chromatin autoantibodies (Fig. 5A) and total IgG titers (Fig. 5B) were significantly reduced compared to Sle1b mice. However, anti-chromatin IgG autoantibodies in OPN−/− Sle1b mice reached similar levels as in Sle1b mice at the age of 14–19 wk (Fig. 5A), and OPN−/− Sle1b mouse serum IgG levels were similar to those in Sle1b mice by the age of 14–19 wk (Fig. 5B). Thus, OPN−/− Sle1b mice exhibit a delayed onset of spontaneous autoantibody and IgG production in comparison to Sle1b mice.
Figure 5.

Delayed onset of humoral autoimmunity in Sle1b.OPN−/− mice. Anti-chromatin IgG (A) and total IgG (B) levels in the serum of B6, Sle1b, and Sle1b.OPN−/− mice were determined with ELISA. EU, ELISA unit. Age 6–7 wk: B6, n = 4; Sle1b, n = 9; Sle1b.OPN−/−, n = 4. Age 9–12 wk: B6, n = 6; Sle1b, n = 6; Sle1b.OPN−/−, n = 4. Age 14–19 wk: B6, n = 3; Sle1b, n = 4; Sle1b.OPN−/−, n = 4.
To test our hypothesis that OPN plays a T-cell-intrinsic role in the pathogenesis of lupus-related autoimmunity, we directly compared TFH expansion with autoantibody induction. To this end, pools of CD4+ T cells purified from B6, Sle1b or Sle1b.OPN−/− mice were transferred into B6 coisogenic bm12 recipients, which carry a 3-aa mutation in MHC class II. In accordance with our previous findings (3), peripheral CD4+ T cells derived from Sle1b mice induced significantly higher anti-ssDNA and anti-chromatin IgG autoantibody levels in bm12 recipients than B6 CD4+ T cells (Fig. 6A, B). By contrast, Sle1b.OPN−/− CD4+ T cells induced substantially lower autoantibody levels than Sle1b cells (Fig. 6A, B), which was also reflected by the significantly reduced numbers of GCB cells and plasma cells in the recipients of Sle1b.OPN−/− cells (Fig. 6C, D).
Figure 6.
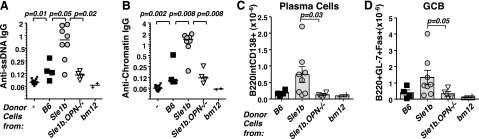
OPN regulates T-helper-cell-induced humoral autoimmunity in the Sle1b → bm12 transfer model. Pooled donor CD4+ T cells (7×106) isolated from lymph nodes and spleens of ≥3 B6, Sle1b, or Sle1b.OPN−/− mice were transferred into bm12 recipients. In control experiments, either no cells or bm12 CD4+ T cells were transferred. Recipient mice were analyzed 4 wk after the transfers, as described previously (3). A, B) Anti-ssDNA (A) and anti-chromatin IgG (B) in the serum was determined by ELISA. C, D) Splenocytes were stained with the indicated surface markers and analyzed by flow cytometry. Gray bars indicate mean values; error bars show se. B6 → bm12, n = 4; Sle1b → bm12, n = 8, Sle1b.OPN−/− → bm12, n = 4. Statistical analyses used the Mann-Whitney nonparametric, 2-tailed test.
Consistent with our hypothesis that OPN plays a role in the development or maintenance and expansion of TFH cells, we found that expansion of CD4+ CXCR5+ PD-1+ cells derived from Sle1b.OPN−/− mice in the bm12 recipients was dramatically reduced compared to the expansion of Sle1b TFH cells (Fig. 7A). Taken together, we conclude that CD4+ T-cell intrinsic expression of osteopontin is requisite for TFH-cell-dependent humoral autoimmunity in the bm12 lupus model.
Figure 7.

OPN supports the sustained survival of donor CD4+ and TFH cells in bm12 recipients. CD4+ T cells were analyzed 4 wk post-transfer into bm12 recipients, as described in Fig. 5. A) Flow cytometric comparison of TFH cells isolated from bm12 recipients after the transfer of B6, Sle1b, or Sle1b.OPN−/−. Gray bars indicate mean values of data points; error bars indicate se. B) Tracking of donor Sle1b and Sle1b.OPN−/− CD4+ T cells by flow cytometry employing a Slamf-haplotype 2 specific antibody. Bottom panel represents staining of the spleen of the recipient with the highest percentage of engraftment. C) Statistical analysis of the quantitation of Slamf-haplotype 2 donor cells by the Mann-Whitney nonparametric, 2-tailed test. D) Donor Sle1b CD4+ T cells are CXCR5+ PD-1+, as judged by flow cytometry. E) Intracellular flow cytometry staining of OPN in Sle1b donor (Slamf-haplotype 2) and recipient CD4+ T cells. Bottom dot plot represents a spleen with the highest percentage of engraftment. F) Statistical analysis of E.
To track the fate of the donor Sle1b CD4+ T cells in bm12 mice, we took advantage of the ability to distinguish between host (bm12, Slamf-haplotype 1) and graft (Sle1b, Slamf-haplotype 2) CD4+ T cells using a monoclonal antibody (30C7) that recognizes the extracellular domains of the Slamf-haplotype 2 isoform of Slamf3 (CD229) (24). Flow cytometric analyses with 30C7 revealed that an average 7% of the CD4+ splenic T cells in the Sle1b → bm12 mice were Sle1b donor cells and that almost all of the donor cells were positive for the TFH markers CXCR5 and PD-1. (Fig. 7A–D). Surprisingly, the donor Sle1b.OPN−/− CD4+ T cells were scarcely detectable or completely missing in the Sle1b.OPN−/−→ bm12 cohort 4 wk after transfer (Fig. 7B, C). As expected the donor-derived Sle1b CD4+ TFH cells produced OPN, whereas Sle1b.OPN−/− CD4+ T cells did not (Fig. 7E, F).
At 3 or 6 d after the transfer into bm12 recipients, the proliferation rate of donor Sle1b and Sle1b.OPN−/− CD4+ T cells was the same (Fig. 8A–E). Furthermore, at 6 d after transfer, all activated donor CD4+ T cells following multiple rounds of division (CFSElo) up-regulated PD-1 and expressed CXCR5 at intermediate or high levels in both the Sle1b → bm12 and the Sle1b.OPN−/− → bm12 cohorts (Fig. 8A–E). During the period between the second and the fourth week post-transfer, the numbers of Sle1b.OPN−/− CD4+ T donor cells diminished, in contrast to the Sle1b CD4+ T donor cells (Fig. 7).
Figure 8.
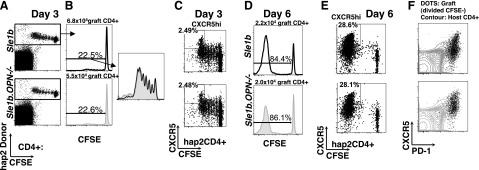
Analyses of Sle1b or Sle1b.OPN−/− donor CD4+ T cells in bm12 recipients 3 and 6 d post-transfer. CFSE-labeled CD4+ T cells (5×106) isolated from Sle1b or Sle1b.OPN−/− mice were transferred i.p. into bm12 mice. A–C) CD4+ T cells were isolated from the spleen 3 d after transfer and analyzed by flow cytometry. A) Donor cells were identified by haplotype 2 (Sle1b)-specific anti-Slamf3 antibody staining. B) In vivo donor cell proliferation was judged by CFSE dilution in CD4+ hap2Slamf3+ gated donor cells (left panels). Region of CFSElo (proliferated) cells were magnified for convenience (right panel; black line, Sle1b donor; gray filled curve, Sle1b.OPN−/− donor). C) CXCR5 up-regulation on the surface of donor cells. D–F) CD4+ T cells were isolated from the spleen 6 d after transfer and analyzed by flow cytometry. We determined the number of CD4+ hap2Slamf3+ donor cells (D; gating strategy as in Fig. 7B), donor CD4+ T-cell proliferation as judged by CFSE (D, E), and CXCR5 and PD-1 expression on the surface of donor cells (F).
In summary, CD4+ T cells from both Sle1b.OPN−/− and Sle1b donors convert to TFH cells within 6 d when transferred to bm12 recipients. Long-term (4-wk) survival per expansion of committed Sle1b TFH cells, however, does not occur in the absence of OPN. The data support the concept that OPN plays a role in maintenance of a subset of TFH cells.
DISCUSSION
SLE is a complex disease with a diverse range of symptoms caused by a number of genetic and environmental factors (25). However, it is well established that a breach of tolerance against nuclear autoantigens is one of the major factors in the pathophysiology of the disease, which involves a role for T-helper cells (25, 26). Previously, we found that CD4+ T cells from Sle1b mice support the breach of B-cell peripheral tolerance (3) and that the Slamf6 gene regulates this process, especially through a novel isoform, Slamf6-H1, which is absent in Sle1b mice. Autoantibody production is reduced in Sle1b mice, which are hemizygous for a transgene expressing the Slamf6-H1 isoform (Sle1b.Slamf6.H1; ref. 3).
Here, we report that in young autoimmune-prone Sle1b mice, but not in Sle1b.Slamf6.H1 mice, a memory T-helper-cell subset exists that heretofore only had been detected in aged B6 mice. This subset, which is distinguished by overexpression of 17 genes, contains TFH cells. As in the lupus-prone Roquinsan/san mouse (32), in Sle1b mice, the increased number of TFH cells also correlates with the severity of the lupus-like disease, and in both cases, the Slamf-specific adapter SAP is indispensable for autoantibody production. Because the presence of B cells susceptible to autoreactivity is prerequisite for the development of the lupus-like phenotype in Sle1b mice (31, 4), we hypothesize that TFH cells are necessary, but not sufficient, to trigger a complete humoral autoimmunity in the Sle1b model. Other factors, such as mutual activating interactions with autoreactive B cells and/or a type I interferon-producing autoinflammatory milieu, are necessary for their full development.
Unexpectedly, while expression of the Spp1 gene, which encodes OPN, was highly up-regulated in CD4+ T cells derived from Sle1b mice, it was not in Sle1b.Slamf6-H1 or Sle1b.SAP−/− CD4+ T cells. The observation that Slamf6-H1 suppresses the spontaneous expansion of a unique OPN+ TFH-cell subset is consistent with this Slamf6 isoform suppressing autoantibody production (3). We hypothesize that this suppression is caused by a TFH-cell intrinsic mechanism, which relies on an interplay between this isoform and the 3 other known isoforms: Slamf6-1 (33), Slamf6-2 (33), and Slamf6-3 (34), possibly by interfering with binding of the SAP docking sites Slamf6-1, Slamf6-2, and Slamf6-3 (35, 36, 37). Alternatively, the presence of Slamf6-H1 might affect the intricate balance between SAP binding and recruitment of the tyrosine phosphatase SHP-1 into the immune synapse (36, 38).
Anti-chromatin autoantibody levels indicate that there is a delayed onset of humoral autoimmunity in Sle1b.OPN−/− mice compared to the OPN-expressing Sle1b strain. This finding is consistent with the observation of a delayed polyclonal B-cell activation and autoantibody production in the lupus-prone Faslpr/lpr mice that had been crossed with a OPN−/− mouse (41). To directly confirm that OPN plays an important role in the aberrant TFH expansion in Sle1b mice, we employed the bm12 transfer model (3, 12). In contrast to the gradual emergence of autoantibodies and lymphocyte activation 3 to 4 wk post-transfer of Sle1b CD4+ T cells into bm12 recipients, no disease manifestations were observed on the transfer of Sle1b.OPN−/− or B6 CD4+ T cells. This model turned out to be particularly relevant, since a large percentage of the Sle1b CD4+ T cells rapidly acquired the CXCR5+PD-1+ TFH-cell phenotype. Interestingly, donor Sle1b and Sle1b.OPN−/− CD4+ T cells proliferated at the same rate and equally up-regulated CXCR5 and PD-1 expression in the acute response (d 3 and 6), but OPN-deficient donor cells disappeared during the 4-wk-long chronic stimulation. In line with this observation, short-term hapten immunization induces similar responses in OPN-deficient and OPN-sufficient Sle1b mice (data not shown). Thus, it would appear that the OPN+ TFH population is more linked to the chronic autoimmune phenotype than an acute hapten-specific immune response.
Our findings are consistent with the literature and extend previous observations, namely that in the absence of OPN, the survival and/or maintenance of CD4+ T cells in EAE is impaired (39, 40). Furthermore, in vitro assays suggested that OPN stimulates proliferation of B cells (41), which, in turn, could stimulate expansion of autoimmune TFH cells (31). The notion that OPN could perhaps play a role in the pathogenesis of SLE is consistent with the finding of SNP variants in SLE patients (22, 23). Taken together, the data suggest that OPN could be a potential target for the treatment of lupus.
Supplementary Material
Acknowledgments
The authors thank Drs. Xuefang Xie and Shiva Gautam (Beth Israel Deaconess Medical Center) and Mick Correll (Dana-Farber Cancer Institute, Boston, MA, USA) for invaluable help with the gene-expression array experiments and the statistical analyses. The authors are grateful to Gongxian Liao and other members of the C.T. laboratory for discussions and for a critical review of the manuscript.
This work was supported by grants from the U.S. National Institutes of Health to C.T. (PO1 AI-065687 and RO1 DK-073339) and to the Harvard Catalyst Statistics consulting service (8UL1TR000170-05).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- GCB
- germinal center B
- GSEA
- gene set enrichment analysis
- OPN
- osteopontin
- SAP
- SLAMF-associated protein
- SLAMF
- signaling lymphocyte activation molecule family
- SLE
- systemic lupus erythematosus
- Sle1b
- B6.Sle1b
- TFH
- T follicular helper
REFERENCES
- 1. Wandstrat A. E., Nguyen C., Limaye N., Chan A. Y., Subramanian S., Tian X. H., Yim Y. S., Pertsemlidis A., Garner H. R., Jr., Morel L., Wakeland E. K. (2004) Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity 21, 769–780 [DOI] [PubMed] [Google Scholar]
- 2. Carlucci F., Cortes-Hernandez J., Fossati-Jimack L., Bygrave A. E., Walport M. J., Vyse T. J., Cook H. T., Botto M. (2007) Genetic dissection of spontaneous autoimmunity driven by 129-derived chromosome 1 loci when expressed on C57BL/6 mice. J. Immunol. 178, 2352–2360 [DOI] [PubMed] [Google Scholar]
- 3. Keszei M., Detre C., Rietdijk S. T., Munoz P., Romero X., Berger S. B., Calpe S., Liao G., Castro W., Julien A., Wu Y. Y., Shin D. M., Sancho J., Zubiaur M., Morse H. C., III, Morel L., Engel P., Wang N., Terhorst C. (2011) A novel isoform of the Ly108 gene ameliorates murine lupus. J. Exp. Med. 208, 811–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kumar K. R., Li L., Yan M., Bhaskarabhatla M., Mobley A. B., Nguyen C., Mooney J. M., Schatzle J. D., Wakeland E. K., Mohan C. (2006) Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science 312, 1665–1669 [DOI] [PubMed] [Google Scholar]
- 5. Morel L., Blenman K. R., Croker B. P., Wakeland E. K. (2001) The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proc. Natl. Acad. Sci. U. S. A. 98, 1787–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu C., Nguyen K. B., Pien G. C., Wang N., Gullo C., Howie D., Sosa M. R., Edwards M. J., Borrow P., Satoskar A. R., Sharpe A. H., Biron C. A., Terhorst C. (2001) SAP controls T-cell responses to virus and terminal differentiation of TH2 cells. Nat. Immunol. 2, 410–414 [DOI] [PubMed] [Google Scholar]
- 7. Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M. A., Rossini J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J. Y., Zhang J. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Irizarry R. A., Bolstad B. M., Collin F., Cope L. M., Hobbs B., Speed T. P. (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smyth G. K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- 10. Benjamini Y., Hochberg Y. (1995) Controlling the false-discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 [Google Scholar]
- 11. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morris S. C., Cohen P. L., Eisenberg R. A. (1990) Experimental induction of systemic lupus erythematosus by recognition of foreign Ia. Clin. Immunol. Immunopathol. 57, 263–273 [DOI] [PubMed] [Google Scholar]
- 13. Shimatani K., Nakashima Y., Hattori M., Hamazaki Y., Minato N. (2009) PD-1+ memory phenotype CD4+ T cells expressing C/EBPα underlie T-cell immunodepression in senescence and leukemia. Proc. Natl. Acad. Sci. U. S. A. 106, 15807–15812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Breitfeld D. (2000) Follicular B helper T cells express CXC chemokine receptor 5, localize to B-cell follicles, and support immunoglobulin production. J. Exp. Med. 192, 1545–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sayos J., Wu C., Morra M., Wang N., Zhang X., Allen D., van., Schaik S., Notarangelo L., Geha R., Roncarolo M. G., Oettgen H., De Vries J. E., Aversa G., Terhorst C. (1998) The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 395, 462–469 [DOI] [PubMed] [Google Scholar]
- 16. Coffey A. J., Brooksbank R. A., Brandau O., Oohashi T., Howell G. R., Bye J. M., Cahn A. P., Durham J., Heath P., Wray P., Pavitt R., Wilkinson J., Leversha M., Huckle E., Shaw-Smith C. J., Dunham A., Rhodes S., Schuster V., Porta G., Yin L., Serafini P., Sylla B., Zollo M., Franco B., Bolino A., Seri M., Lanyi A., Davis J. R., Webster D., Harris A., Lenoir G., de St Basile G., Jones A., Behloradsky B. H., Achatz H., Murken J., Fassler R., Sumegi J., Romeo G., Vaudin M., Ross M. T., Meindl A., Bentley D. R. (1998) Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat. Genet. 20, 129–135 [DOI] [PubMed] [Google Scholar]
- 17. Qi H., Cannons J. L., Klauschen F., Schwartzberg P. L., Germain R. N. (2008) SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 455, 764–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crotty S. (2011) Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 [DOI] [PubMed] [Google Scholar]
- 19. Cannons J. L., Qi H., Lu K. T., Dutta M., Gomez-Rodriguez J., Cheng J., Wakeland E. K., Germain R. N., Schwartzberg P. L. (2010) Optimal germinal center responses require a multistage T cell: B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity 32, 253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Linterman M. A., Rigby R. J., Wong R. K., Yu D., Brink R., Cannons J. L., Schwartzberg P. L., Cook M. C., Walters G. D., Vinuesa C. G. (2009) Follicular helper T cells are required for systemic autoimmunity. J. Exp. Med. 206, 561–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deenick E. K., Chan A., Ma C. S., Gatto D., Schwartzberg P. L., Brink R., Tangye S. G. (2010) Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity 33, 241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Forton A. C., Petri M. A., Goldman D., Sullivan K. E. (2002) An osteopontin (SPP1) polymorphism is associated with systemic lupus erythematosus. Hum. Mutat. 19, 459. [DOI] [PubMed] [Google Scholar]
- 23. D'Alfonso S., Barizzone N., Giordano M., Chiocchetti A., Magnani C., Castelli L., Indelicato M., Giacopelli F., Marchini M., Scorza R., Danieli M. G., Cappelli M., Migliaresi S., Bigliardo B., Sabbadini M. G., Baldissera E., Galeazzi M., Sebastiani G. D., Minisola G., Ravazzolo R., Dianzani U., Momigliano-Richiardi P. (2005) Two single-nucleotide polymorphisms in the 5′ and 3′ ends of the osteopontin gene contribute to susceptibility to systemic lupus erythematosus. Arthritis Rheum. 52, 539–547 [DOI] [PubMed] [Google Scholar]
- 24. Ledbetter J. A., Herzenberg L. A. (1979) Xenogeneic monoclonal antibodies to mouse lymphoid differentiation antigens. Immunol. Rev. 47, 63–90 [DOI] [PubMed] [Google Scholar]
- 25. Tsokos G. C. (2011) Systemic lupus erythematosus. N. Engl. J. Med. 365, 2110–2121 [DOI] [PubMed] [Google Scholar]
- 26. Sobel E. S., Satoh M., Chen Y., Wakeland E. K., Morel L. (2002) The major murine systemic lupus erythematosus susceptibility locus Sle1 results in abnormal functions of both B and T cells. J. Immunol. 169, 2694–2700 [DOI] [PubMed] [Google Scholar]
- 27. Cunninghame Graham D. S., Vyse T. J., Fortin P. R., Montpetit A., Cai Y. C., Lim S., McKenzie T., Farwell L., Rhodes B., Chad L., Hudson T. J., Sharpe A., Terhorst C., Greenwood C. M., Wither J., Rioux J. D. (2008) Association of LY9 in UK and Canadian SLE families. Genes Immun. 9, 93–102 [DOI] [PubMed] [Google Scholar]
- 28. You Y., Wang Z., Deng G. H., Liu Y., Hao F. (2010) Detection and functional evaluation of −262A/T and −188A/G polymorphisms of SLAM gene in patients with systemic lupus erythematosus. J. Rheumatol. 37, 2268–2272 [DOI] [PubMed] [Google Scholar]
- 29. Kim J. R., Mathew S. O., Patel R. K., Pertusi R. M., Mathew P. A. (2010) Altered expression of signaling lymphocyte activation molecule (SLAM) family receptors CS1 (CD319) and 2B4 (CD244) in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 160, 348–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keszei M., Latchman Y. E., Vanguri V. K., Brown D. R., Detre C., Morra M., Arancibia-Carcamo C. V., Paul E., Calpe S., Castro W., Wang N., Terhorst C., Sharpe A. H. (2011) Auto-antibody production and glomerulonephritis in congenic Slamf1-/- and Slamf2-/- [B6.129] but not in Slamf1-/- and Slamf2-/- [BALB/c. 129] mice. Int. Immunol. 23, 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wong E. B., Khan T. N., Mohan C., Rahman Z. S. (2012) The Lupus-prone NZM2410/NZW strain-derived Sle1b sublocus alters the germinal center checkpoint in female mice in a B cell-intrinsic manner. J. Immunol. 189, 5667–5681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vinuesa C. G., Cook M. C., Angelucci C., Athanasopoulos V., Rui L., Hill K. M., Yu D., Domaschenz H., Whittle B., Lambe T., Roberts I. S., Copley R. R., Bell J. I., Cornall R. J., Goodnow C. C. (2005) A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435, 452–458 [DOI] [PubMed] [Google Scholar]
- 33. Peck S. R., Ruley H. E. (2000) Ly108: a new member of the mouse CD2 family of cell surface proteins. Immunogenetics 52, 63–72 [DOI] [PubMed] [Google Scholar]
- 34. Zhong M. C., Veillette A. (2008) Control of T lymphocyte signaling by Ly108, a signaling lymphocytic activation molecule family receptor implicated in autoimmunity. J. Biol. Chem. 283, 19255–19264 [DOI] [PubMed] [Google Scholar]
- 35. Fraser C. C., Howie D., Morra M., Qiu Y., Murphy C., Shen Q., Gutierrez-Ramos J. C., Coyle A., Kingsbury G. A., Terhorst C. (2002) Identification and characterization of SF2000 and SF2001, two new members of the immune receptor SLAM/CD2 family. Immunogenetics 53, 843–850 [DOI] [PubMed] [Google Scholar]
- 36. Calpe S., Wang N., Romero X., Berger S. B., Lanyi A., Engel P., Terhorst C. (2008) The SLAM and SAP gene families control innate and adaptive immune responses. Adv. Immunol. 97, 177–250 [DOI] [PubMed] [Google Scholar]
- 37. Dutta M., Schwartzberg P. L. (2012) Characterization of Ly108 in the thymus: evidence for distinct properties of a novel form of Ly108. J. Immunol. 188, 3031–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kageyama R., Cannons J. L., Zhao F., Yusuf I., Lao C., Locci M., Schwartzberg P. L., Crotty S. (2012) The receptor Ly108 functions as a SAP adaptor-dependent on-off switch for T cell help to B cells and NKT cell development. Immunity 36, 986–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hur E. M., Youssef S., Haws M. E., Zhang S. Y., Sobel R. A., Steinman L. (2007) Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat. Immunol. 8, 74–83 [DOI] [PubMed] [Google Scholar]
- 40. Guan H., Nagarkatti P. S., Nagarkatti M. (2011) CD44 reciprocally regulates the differentiation of encephalitogenic Th1/Th17 and Th2/regulatory T cells through epigenetic modulation involving DNA methylation of cytokine gene promoters, thereby controlling the development of experimental autoimmune encephalomyelitis. J. Immunol. 186, 6955–6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lampe M. A., Patarca R., Iregui M. V., Cantor H. (1991) Polyclonal B cell activation by the Eta-1 cytokine and the development of systemic autoimmune disease. J. Immunol. 147, 2902–2906 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.