

Abstract
The Na+/I− symporter (NIS) is a plasma membrane glycoprotein that mediates active I− transport in the thyroid, the first step in the biosynthesis of the iodine-containing thyroid hormones T3 and T4. Several NIS mutants have been identified as a cause of congenital I− transport defect (ITD), and their investigation has yielded valuable mechanistic information on NIS. Here we report a thorough characterization of the ITD-causing NIS mutation in which the sixth intracellular loop residues 439–443 are missing. This mutant protein was intracellularly retained, incompletely glycosylated, and intrinsically inactive. Engineering 5 Ala at positions 439–443 partially recovered cell surface targeting and activity (∼15%). Strikingly, NIS with the sequence 439-AANAA-443, in which Asn was restored at position 441, was targeted to the plasma membrane and exhibited ∼95% the transport activity of WT NIS. Based on our NIS homology model, we propose that the side chain of N441, a residue conserved throughout most of the SLC5 family, interacts with the main chain amino group of G444, capping the α-helix of transmembrane segment XII and thus stabilizing the structure of the molecule. Our data provide insight into a critical interhelical interaction required for NIS folding and activity.—Li, W., Nicola, J. P., Amzel, L. M., Carrasco, N. Asn441 plays a key role in folding and function of the Na+/I− symporter (NIS).
Keywords: plasma membrane targeting, helix capping, interhelical interaction, congenital hypothyroidism, protein folding
The Na+/I− symporter (NIS) is a plasma membrane glycoprotein that plays a key role in thyroid physiology and pathophysiology. NIS mediates the active transport of I− in the thyroid gland and other tissues, including salivary glands, stomach, intestine, and lactating breast (1–5). I− is an essential constituent of the thyroid hormones T3 and T4 (triiodothyronine and thyroxine, respectively), which are critical for the development and maturation of the central nervous system, lungs, and skeletal muscle in the fetus and newborn, as well as for intermediary metabolism in virtually all tissues starting in childhood and continuing throughout adulthood (6).
Our group cloned NIS and has since characterized it extensively (2, 7–21). Our experimentally tested NIS secondary structure model shows a protein with 13 transmembrane segments (TMSs), an extracellularly facing amino terminus, and a cytosolic carboxyl terminus (see Fig. 1A and refs. 10, 14). Although NIS is highly N-glycosylated, glycosylation is not critical for NIS targeting to the plasma membrane or function (10). NIS activity results from coupling the inward translocation of Na+ down its electrochemical gradient to the inward transport of I− against its electrochemical gradient, with an electrogenic 2 Na+:1 I− stoichiometry (8). The driving force for this process is the inwardly directed Na+ gradient generated by the Na+/K+ ATPase. Significantly, we have demonstrated that NIS translocates different substrates with different stoichiometries, as NIS-mediated transport of the environmental pollutant perchlorate (ClO4−) or perrhenate (ReO4−) is electroneutral, i.e., 1 Na+:1 ClO4−/ReO4− (16, 21).
Figure 1.

Characterization of expression and activity of Δ439–443 NIS. A) NIS secondary structure model. Cylinders represent the 13 TMS, in Roman numerals; branches indicate N-linked glycosylation sites. The amino acid sequence deleted in the ITD-causing Δ439–443 NIS mutant in IL-6 is depicted in blue. B) Steady state I− transport assays in nontransfected (Nt), wild-type (WT), or Δ439–443 NIS cDNA-transiently transfected COS-7 cells. Cells were incubated with 20 μM I− in the absence (dark bars) or presence (light bars) of 80 μM ClO4− for 45 min at 37°C, as described in Materials and Methods. Results are expressed as means ± sd (pmol I−/μg DNA). Values are representative of ≥3 different experiments; each experiment was performed in triplicate. C) Representative FACS analysis of total (top panel) and plasma membrane (bottom panel, surface) NIS expression in COS-7 cells nontransfected (solid gray) or transfected with WT (blue line) or Δ439–443 NIS (red line). Cells were stained with anti-HA Ab, followed by R-phycoerythrin-conjugated anti-rat Ab. D) Immunofluorescence analysis of permeabilized COS-7 cells expressing WT or Δ439–443 NIS. Cells were stained with anti-human NIS Ab, followed by anti-rabbit Alexa 488-conjugated Abs (green). Nuclei were stained with DAPI (blue). White scale bar: 20 μm. E) I− uptake in MVs prepared from WT or Δ439–443 NIS-expressing COS-7 cells. MVs were incubated with 40 μM I− in the absence (dark bars) or presence (light bars) of 80 μM ClO4−. Results are expressed as means ± sd (pmol I−/mg protein). The experiment shown is representative of 3 independent experiments; each experimental point was performed in triplicate.
I− transport defect (ITD) is a rare autosomal recessive disorder caused by naturally occurring mutations in the NIS gene (22, 23). Patients with ITD lack the ability to actively accumulate I− in thyroid follicular cells, thus leading to congenital hypothyroidism. The general clinical picture of ITD consists of a variable degree of hypothyroidism; reduced to absent thyroid uptake of radioiodide or pertechnetate, as determined by scintigraphy; and low I− saliva to plasma ratio (24, 25). To date, 14 ITD-causing NIS mutations have been identified: −54C>T, V59E, G93R, R124H, Δ142–323, Q267E, C272X, Δ287–288, T354P, G395R, Δ439–443, frameshift 515X, Y531X, and G543E (22, 26).
Studies of different ITD-causing NIS mutations in our laboratory have provided key mechanistic information on NIS maturation, trafficking, and structure/function relations (16–20, 27, 28). A detailed analysis of the T354P mutation revealed that the hydroxyl group at the β-carbon of the residue at position 354 is essential for NIS function and that other β-hydroxyl-containing residues in TMS IX are involved in Na+ binding/translocation (18). Similarly, the investigation of the G93R mutation led to the discovery that position 93 in TMS III is critical for substrate selectivity and stoichiometry and that changes from an outwardly to an inwardly open conformation during the transport cycle use G93 as a pivot (16). Recently the R124H NIS mutant was shown to be intracellularly retained, yet intrinsically active, and R124 was demonstrated to play a structural role by bridging together IL-2 and 6, thus allowing a local folding required for NIS maturation and trafficking to the plasma membrane to occur (15).
The ITD-mutant Δ439–443 NIS was first described in an Italian patient with congenital hypothyroidism carrying a homozygous mutation c.1314_1328del (29). Amino acids 439–443 are putatively located in IL-6 connecting TMSs XI and XII (see Fig. 1A). When expressed in COS-7 cells, Δ439–443 NIS did not transport I−, suggesting that the deletion is the direct cause of ITD (29). Here we have carried out a thorough molecular analysis of the Δ439–443 NIS deletion. We found that Δ439–443 NIS is retained intracellularly and intrinsically inactive, as determined by I− transport assays in membrane vesicles. Engineering 5 Ala at the positions of the missing amino acids significantly improved cell surface targeting and partially recovered I− transport. Furthermore, we identified N441 as a critical amino acid involved in NIS folding. Our results suggest that the side chain of N441, a conserved residue in most members of the SLC5 family of transporters, interacts with the main chain amino group of G444, as part of an N-terminal helix-capping motif.
MATERIALS AND METHODS
Cell culture and transient transfection
COS-7 and Madin-Darby canine kidney (MDCK-II) cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gemini BioProducts, West Sacramento, CA, USA), 2 mM l-glutamine, and antibiotics.
Cells were transfected with 4 μg plasmid using Lipofectamine Reagent enhanced with Plus Reagent (Invitrogen) according to the manufacturer's instructions. Flow cytometry analysis was performed 1 d after transfection. Cells were assayed for I− uptake, cell surface biotinylation, and immunoblot and immunofluorescence analysis 48 h posttransfection.
Stable MDCK-II polyclonal populations were selected and propagated in growth medium containing 1 g/L G418 (Mediatech, Herndon, VA, USA) as described previously (30).
Site-directed mutagenesis
Mutations in human NIS containing an amino terminus hemagglutinin (HA) epitope tag cloned into pcDNA3.1 vector were introduced using QuikChange Site-Directed Mutagenesis kit (Strategene, La Jolla, CA, USA) as reported previously (31). Individual mutagenic oligonucleotides were generated using Strategene's QuickChange primer design program. All constructs were sequenced to verify the specific nucleotide substitutions.
Preparation of membrane vesicles
Membrane vesicles were prepared as described previously (9, 20). Briefly, cells were resuspended in ice-cold 250 mM sucrose, 1 mM EGTA, and 10 mM Hepes (pH 7.5) containing protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA) and disrupted with a motor-driven teflon-pestle homogenizer. The homogenate was centrifuged at 500 g for 15 min at 4°C, and the supernatant was ultracentrifuged at 100,000 g for 1 h at 4°C. The resulting pellet was resuspended in ice-cold 250 mM sucrose, 1 mM MgCl2, and 10 mM Hepes (pH 7.5).
I− transport in cells and membrane vesicles
I− transport assays in COS-7 or MDCK-II cells was performed as described previously (16, 32). Briefly, cells were incubated in Hanks' balanced salt solution (HBSS) containing 20 μM I− supplemented with 100 μCi/μmol carrier-free Na125I (PerkinElmer, Boston, MA, USA) for 45 min at 37°C. NIS-specific I− uptake was assessed in the presence of the NIS inhibitor ClO4− (80 μM). Reactions were terminated by aspirating the radioactive solution and washing with ice-cold HBSS. Accumulated 125I− was extracted with ice-cold ethanol and then quantified in a Wizard γ counter (PerkinElmer). DNA was determined by the diphenylamine method after trichloroacetic acid precipitation (1). Iodide uptake was expressed as picomoles of I− per μg DNA (pmol/μg DNA) and standardized by NIS expression, as analyzed by fluorescence activated cell sorting (FACS) under permeabilized conditions.
For I−-dependent kinetic analysis, cells were incubated for 2 min in HBSS buffer containing 2.5–320 μM I−. Initial-rate data were analyzed by nonlinear regression using the following equation for I−-dependent I− uptake: v = (Vmax * [I−])/(Km + [I−]). Background obtained from nontransfected cells was subtracted. Data were analyzed with Gnuplot software (http://www.gnuplot.info).
I− uptake in membrane vesicles was performed as described previously (9, 20). Aliquots containing 100 μg of protein were assayed for 125I− uptake by incubating for 3 min at room temperature with an equal volume of a solution containing 80 μM I− (supplemented with 100 μCi/μmol carrier-free Na125I), 1 mM MgCl2, 10 mM Hepes (pH 7.5), 2 mM methimazole, and 200 mM NaCl. Reactions were terminated by the addition of 4 ml of ice-cold quenching solution containing 1 mM Tris-HCl (pH 7.5), 250 mM KCl, and 1 mM methimazole followed by rapid filtration through wet 0.22-μm nitrocellulose filters (EMD Millipore, Billerica, MA, USA). Radioactivity retained by membrane vesicles (MVs) was determined as described above.
Deglycosylation assays
Membrane vesicles (10 μg) were deglycosylated with endo-β-acetylglucosaminidase H (Endo H; New England Biolabs, Ipswich, MI, USA) in 50 mM sodium citrate (pH 5.5) at 37°C for 2 h. As negative control, membrane vesicles were incubated in the same buffer without enzyme. After treatment, samples were subjected to immunoblot analysis.
Immunoblotting
SDS-PAGE, electrotransference to nitrocellulose membranes, and immunoblotting were performed as described previously (9, 33). Membranes were incubated with 4 nM of an affinity-purified polyclonal anti-human NIS Ab directed against the last 13 residues of the cytosolic NIS carboxyl terminus (2). Equal loading or purification controls were assessed by stripping and reprobing the same blot with 0.25 μg/ml monoclonal anti-α-tubulin Ab (Sigma-Aldrich, St. Louis, MO, USA) or 0.5 μg/ml monoclonal anti-Na+/K+ ATPase α1-subunit Ab (Affinity BioReagents, Golden, CO, USA). Horseradish peroxidase (HRP)-linked secondary anti-mouse and anti-rabbit Abs were obtained from Jackson ImmunoResearch (West Grove, PA, USA) and Amersham Biosciences (Piscataway, NJ, USA), respectively. Proteins were visualized using the enhanced chemiluminescence Western blot detection system (Amersham Biosciences). Band intensities were measured densitometrically using the ImageJ image software (U.S. National Institutes of Health, Bethesda, MD, USA).
Flow cytometry and cell sorting
Cells were stained using indirect immunofluorescence procedures (19, 20). Briefly, paraformaldehyde-fixed cells were incubated in PBS containing 0.2% BSA (Sigma-Aldrich) for nonpermeabilized conditions or an additional 0.2% saponin for permeabilized conditions with 3 nM anti-HA rat monoclonal antibody (Roche Applied Science) or 1:50 anti-human NIS VJ1 mouse monoclonal antibody directed against an undefined extracellular conformational epitope of NIS (34). After washing, cells were incubated with 50 nM of Alexa-488-conjugated goat anti-mouse antibody or R-phycoerythrin (PE)-conjugated goat anti-rat antibody (Life Technologies, Grand Island, NY, USA). The fluorescence of 104 cells/tube was assayed in FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). All data were analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
Cell surface biotinylation
Biotinylation of cell surface proteins was performed as described previously (18, 19). Briefly, transfected cells were incubated with 1 mg/ml of the membrane-impermeable biotin reagent sulfo-NHS-SS-biotin (Pierce Chemical, Rockford, IL, USA), which covalently interacts with extracellular primary amines. Cells were lysed, and biotinylated proteins were precipitated overnight with streptavidin-coated beads (Pierce Chemical). Beads were washed and adsorbed proteins were eluted with sample buffer containing 10 mM dithiothreitol at 75°C for 5 min and analyzed by immunoblot.
Immunofluorescence
Cells seeded onto glass coverslips were fixed in 2% paraformaldehyde, quenched with 50 mM NH4Cl in PBS containing 1 mM CaCl2 and 0.1 mM MgCl2 (PBS/CM), and permeabilized or not in PBS/CM supplemented with 0.2% (v/v) Triton X-100, and 5% (v/v) goat serum (Life Technologies). Cells were immunostained with 4 nM polyclonal anti-human NIS antibody, 3 nM anti-HA antibody or 1:50 anti-NIS VJ1 antibody in PBS/CM containing 0.2% BSA. For analysis under permeabilized conditions an additional 0.1% Triton was added. Secondary staining proceeded with 50 nM anti-rabbit Alexa-488 and anti-mouse Alexa-594 antibodies (Life Technologies). Coverslips were mounted with a DAPI-containing mounting medium (Vector Laboratories, Berlingame, CA, USA), and images were acquired on a Bio-Rad Radiance 2000 scanning laser confocal microscope. Image editor software Adobe Photoshop (Adobe, San Jose, CA, USA) was used to prepare digitized images.
Modeling
The homology model of residues 50–476 of NIS has been described previously (16). Modeling of the deletion and the different amino acid substitutions was carried out using PyMol (Schrödinger, Portland, OR, USA).
Statistical analysis
Statistical analysis was performed by GraphPad Prism (GraphPad Software, San Diego, CA, USA) from ≥3 independent experiments. Multiple group analysis was conducted by 1-way ANOVA. As posttest, the Newman-Keuls multiple comparison test was used. Comparisons between 2 groups were made using unpaired 2-tailed Student's t test. Differences were considered statistically significant at values of P < 0.05.
RESULTS
Δ439–443 NIS is intracellularly retained
According to our experimentally tested NIS secondary structure model, the missing residues of Δ439–443 NIS are located in intracellular loop 6 (IL-6), which connects TMSs XI and XII (Fig. 1A). COS-7 cells were transfected with wild-type (WT) or Δ439–443 NIS cDNA and assayed for I− transport. As reported previously (29), at steady state, no perchlorate (ClO4−)-inhibited I− accumulation was observed in Δ439–443 NIS-transfected cells, in contrast to control WT NIS-expressing cells (Fig. 1B and refs. 7, 9, 19, 27, 28). We assessed whether the absence of I− transport in Δ439–443 NIS-expressing cells was due to faulty targeting of the transporter to the plasma membrane. Flow cytometry (FACS) showed that Δ439–443 NIS was not targeted to the cell surface (Fig. 1C). When FACS was performed under permeabilized conditions to determine total NIS protein expression, we observed staining in both WT and Δ439–443 NIS-expressing cells. In contrast, when the experiments were performed in nonpermeabilized cells to ascertain NIS expression at the plasma membrane only, using an anti-HA Ab directed against an extracellular HA tag engineered onto the amino terminus of NIS, staining was evident exclusively in WT NIS-transfected cells (Fig. 1C). In addition, immunofluorescence analysis under permeabilized conditions showed that, consistent with our FACS results, Δ439–443 NIS was retained intracellularly (Fig. 1D).
Δ439–443 NIS is intrinsically inactive
I− transport in intact cells is only observed when NIS is targeted to the plasma membrane (35). Δ439–443 NIS is not active in transiently transfected COS-7 cells ultimately because it is retained intracellularly. However, the lack of I− transport in these cells does not necessarily mean that Δ439–443 NIS is intrinsically inactive. Therefore, we prepared MVs from COS-7 cells expressing either WT or Δ439–443 NIS and assayed them for active I− accumulation. As expected, MVs from WT NIS-expressing cells exhibited ClO4−-inhibitable I− transport (Fig. 1E and ref. 20). In contrast, no significant I− accumulation was detected in MVs from Δ439–443 NIS-expressing cells (Fig. 1E). These results show that, in addition to impaired trafficking to the cell surface, Δ439–443 NIS may either be intrinsically inactive or have undetectable activity.
Insertion of 5 Ala partially rescues NIS plasma membrane targeting
To investigate the role of the deleted amino acids (aa 439–443) in NIS function, we engineered a mutant containing 5 Ala (5A NIS) at the positions of the missing amino acids. We generated permanently transfected MDCK-II cells expressing either WT, Δ439–443, or 5A NIS and assayed them for I− transport. In agreement with our data in COS-7 cells, Δ439–443 NIS-expressing MDCK-II cells did not transport I−, in contrast to WT NIS-expressing cells (Fig. 2A). Remarkably, 5A NIS-expressing cells displayed ClO4−-inhibitable I− transport, albeit at a modest level (Fig. 2A). Δ439–443 and 5A NIS were expressed at levels comparable to those of WT NIS, as shown by FACS under permeabilized conditions (Fig. 2B, total). However, under nonpermeabilized conditions, 5A NIS was found to be partially targeted to the cell surface, whereas Δ439–443 NIS was retained intracellularly (Fig. 2B, surface), as in COS-7 cells. Consistent with these findings, cell surface biotinylation experiments showed that, as expected, the 75- to 100-kDa mature NIS polypeptide was at the plasma membrane, whereas no biotinylated NIS polypeptides were detected in Δ439–443 NIS-transfected cells (Fig. 2C, third lane). Moreover, a modest amount of mature 5A NIS was demonstrated at the plasma membrane, in agreement with the partial recovery of I− transport mediated by this mutant.
Figure 2.
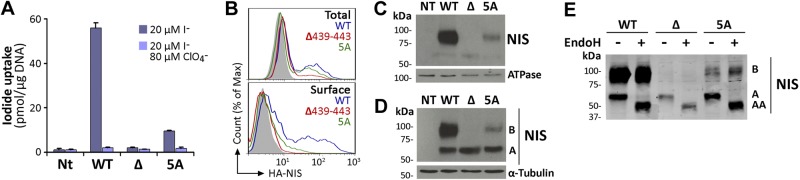
Insertion of 5 Ala partially rescues NIS plasma membrane targeting. A) Steady-state I− uptake in nontransfected (NT), and permanently expressing WT, Δ439–443 (Δ), or 5A NIS MDCK-II cells. Cells were incubated with 20 μM I− in the absence (dark bars) or presence (light bars) of 80 μM ClO4−. Results are expressed as means ± sd (pmol I−/μg DNA). Values are representative of ≥3 different experiments; in each experiment activity was analyzed in triplicate. B) Representative FACS analysis of NIS expression under permeabilized (top panel, total) and nonpermeabilized (bottom panel, surface) conditions. Cells were stained with anti-HA Ab, followed by R-phycoerythrin-conjugated anti-rat Ab. Nontransfected cells are represented in solid gray; WT NIS in blue line; Δ439–443 NIS in red line, and 5A NIS in green line. C) Immunoblot analysis of cell surface-biotinylated proteins from nontransfected and WT, Δ439–443, or 5A NIS-expressing MDCK-II cells, performed with anti-human NIS Ab (top panel). The plasma membrane marker Na+/K+ ATPase was used as a positive control (bottom panel). D) Immunoblot analysis of membrane fractions from WT, Δ439–443, or 5A NIS-transfected MDCK-II cells performed with anti-human NIS Ab (upper panel). The NIS electrophoretic pattern includes immaturely glycosylated (∼60 kDa, band A), and mature or fully glycosylated (∼100 kDa, band B) polypeptides. The housekeeping protein α-tubulin was used as loading control (bottom panel). E) Immunoblot analysis of membrane proteins treated (+) or not (−) with Endo H, and immunoblotted with anti-human NIS Ab. Right side of the blot indicates the relative electrophoretic mobilities of the corresponding NIS polypeptides (AA: ∼50, A: ∼60, B: ∼100 kDa), depending on glycosylation status.
To investigate the impaired plasma membrane targeting of Δ439–443 NIS, we first assessed whether Δ439–443 NIS was properly glycosylated. Membrane fractions from permanently transfected MDCK-II cells were assayed by Western blot using an anti-NIS Ab. As reported (15, 20), the electrophoretic pattern of WT NIS comprised immaturely glycosylated (∼60 kDa, band A) and mature or fully glycosylated (∼100 kDa, band B) polypeptides (Fig. 2D). The mature NIS polypeptide was absent in Δ439–443 NIS-expressing cells, suggesting that this mutant NIS species was not fully processed (Fig. 2D). Significantly, we barely observed the fully glycosylated NIS polypeptide in 5A NIS-expressing cells.
To investigate Δ439–443 NIS glycosylation further, we used endo-β-acetylglucosaminidase H (Endo H) treatment. As proteins that mature beyond the medial-Golgi are Endo H resistant, sensitivity to Endo H indicates that the protein in question has not been processed past the medial-Golgi. Endo H caused the disappearance of partially glycosylated polypeptides from WT, Δ439–443, and 5A NIS (Fig. 2E, band A) and a simultaneous appearance of the ∼50 kDa nonglycosylated NIS polypeptide (Fig. 2E, band AA). However, as expected, the mature WT and 5A NIS polypeptides were Endo H resistant (Fig. 2E, band B). These findings show that, whereas Δ439–443 NIS is initially glycosylated, it is ultimately retained in intracellular compartments prior to the medial-Golgi, in contrast to some 5A NIS molecules, which mature fully and reach the plasma membrane.
Deletion A439–P443 affects the extracellular conformational epitope VJ1
As expected, WT NIS-expressing MDCK-II cells, under nonpermeabilized conditions, showed a plasma membrane-associated immunofluorescence staining pattern with anti-HA Ab. (Fig. 1 and Fig. 3, staining in green). In contrast, there was no staining in Δ439–443 NIS-expressing cells when probed under nonpermeabilized conditions, in agreement with the notion that Δ439–443 NIS is retained intracellularly. Indeed, staining was observed in Δ439–443 NIS-expressing cells only under permeabilized conditions, as demonstrated previously in COS-7 cells (Fig. 1D). In addition, 5A NIS was mainly expressed intracellularly and barely detected at the plasma membrane (Fig. 3).
Figure 3.

Deletion Δ439–443 affects the conformational epitope VJ1. Immunofluorescence analysis of permeabilized or nonpermeabilized permanently transfected MDCK-II cells stained with anti-HA or anti-human NIS VJ1 Abs, followed by R-phycoerythrin-conjugated anti-mouse (red) or Alexa 488-conjugated anti-rat (green) Abs, respectively. Under permeabilized conditions, pictures were taken at 2 different resolutions (×600 and ×1200). Scale bars = 20 μm.
Interestingly, staining with the anti-human NIS VJ1 Ab, which recognizes an extracellularly facing conformational epitope, revealed fewer positive Δ439–443 and 5A NIS- than WT NIS-expressing cells (Fig. 3, staining in red), even though the total number of cells expressing each mutant and WT NIS was similar, as shown by HA Ab staining. These findings raised the possibility that the anti-NIS VJ1 Ab has a lower affinity for Δ439–443 and 5A NIS than for WT NIS as a result of conformational changes affecting the VJ1 epitope, a notion quantitatively investigated below.
Engineering of N441 into 4-Ala NIS fully restores targeting of NIS to the plasma membrane
To determine the importance of the amino acids deleted in Δ439–443 NIS for targeting to the plasma membrane and function, we engineered cDNA constructs individually restoring each native amino acid at positions 441 and 442 into a 4-Ala (4A) NIS background to generate MDCK-II cells expressing N441/4A or T442/4A NIS. N441/4A NIS-mediated I− transport was comparable to that by WT NIS, whereas T442/4A NIS displayed ∼60% of WT NIS uptake (Fig. 4A). Total expression levels of all 3 NIS proteins were similar; however, T442/4A NIS showed reduced plasma membrane targeting, as assessed by FACS analysis (Fig. 4B). Initial rates of I− transport revealed that the Km values for I− of N441/4A and T442/4A (23±3 and 30±4 μM, respectively) were similar to that of WT NIS (27±3 μM), whereas that of 5A NIS was 4-fold higher (101±14 μM), indicating that 5A NIS has a lower apparent affinity for I− than the other 3 NIS proteins (Fig. 4C). Moreover, the presence of Asn at position 441 suffices to restore not only NIS targeting to the cell surface but also its Km for I−.
Figure 4.

N441 in 4A NIS fully rescues protein targeting to the plasma membrane. A) Steady state I− uptake in nontransfected (Nt) and permanently expressing WT, N441/4A, or T442/4A NIS MDCK-II cells. Cells were incubated with 20 μM I− in the absence (dark bars) or presence (light bars) of 80 μM ClO4−. Results are expressed as means ± sd (pmol I−/μg DNA). Values are representative of ≥3 different experiments; in each experiment activity was analyzed in triplicate. B) Representative FACS histograms showing NIS expression under permeabilized (top panel, total) and nonpermeabilized (bottom panel, surface) conditions. Cells were stained with anti-HA Ab, followed by R-phycoerythrin-conjugated anti-rat Ab. Nontransfected cells are represented in solid gray and cells stably expressing WT, N441/4A, or T442/4A are indicated in blue, green, and red, respectively. C) Initial rates of I− uptake were determined at the indicated concentrations of I−. Calculated curves (smooth lines) were generated using the equation v = (Vmax*[I−])/(Km+[I−]) adjusted to consider background data obtained in nontransfected cells. Shown is a representative experiment of 3 independent experiments. D) Relative affinity of anti-human NIS-VJ1 Ab for WT NIS and NIS mutants. Permanently expressing MDCK-II cells were immunostained under permeabilized conditions with anti-human NIS-VJ1 or anti-HA Abs, followed by Alexa 488-conjugated anti-mouse or R-phycoerythrin-conjugated anti-rat Abs, respectively. The ratio of mean fluorescence intensity in the positive population of cell stained either with anti-human NIS VJ1 or anti-HA Abs was calculated for each mutant. The ratio obtained for WT NIS expressing cells was arbitrarily defined as 1. *P < 0.05 (ANOVA, Newman-Keuls test). E) Immunoblot analysis of membrane fractions from WT and nonglycosylated (TQ) NIS-transfected COS-7 cells performed with anti-human NIS Ab. Relative electrophoretic mobilities of the corresponding NIS polypeptides (AA: ∼50, A: ∼60, B: ∼100 kDa) are indicated. F) FACS histograms showing NIS expression under permeabilized conditions. Cells were stained with anti-HA or anti-human NIS-VJ1 Abs. Nontransfected cells are represented in solid gray and COS-7 cells expressing either WT or TQ NIS are indicated by blue and red lines.
To explore the possible reduced affinity of anti-VJ1 Ab for Δ439–443 and 5A NIS (Fig. 3), MDCK-II cells were stained with anti-VJ1 and anti-HA Abs and analyzed by FACS under permeabilized conditions. Evaluation of the ratio of mean fluorescence intensity of positive cells stained with VJ1 over HA quantitatively confirmed a reduced affinity of anti-VJ1 Ab for Δ439–443 NIS (Fig. 4D). Insertion of 5A residues modestly restored the conformational epitope VJ1. Interestingly, engineering Asn-441, but not Thr-442, completely recovered the affinity of the conformational Ab, suggesting that Asn-441 plays a significant role in NIS folding. No differences in staining efficiency were observed between an anti-human NIS Ab directed against the NIS carboxyl terminus and an anti-HA Ab (not shown).
The anti-VJ1 Ab recognizes an extracellular conformational epitope proposed to contain amino acids from the last extracellular loop (34). As this loop is glycosylated at 2 positions (N489 and N502), and Δ439–443 NIS is partially glycosylated, we investigated whether the carbohydrate moieties constitute part of the epitope recognized by the Ab. We engineered a NIS mutant where Asn residues present in the 3 core glycosylation sites N225, N489, and N502 were replaced by Gln to yield nonglycosylated NIS (NIS TQ; Fig. 4E, band AA). COS-7 cells were transiently transfected with WT NIS and NIS TQ cDNAs and anti-VJ1 staining was assessed by FACS under permeabilized condition. There was no difference in mean fluorescence intensity of positive cells (Fig. 4F), indicating that the carbohydrate moieties are not part of the VJ1 epitope.
N441 participates in helix N-capping of TMS XII
In α-helices, the backbone carbonyl groups make H-bonds with the main-chain amino groups of the fourth residue farther along in the helix. This structural characteristic leaves the initial 4 amide hydrogens of the helix with unsatisfied H-bonding potential. Therefore, these amino groups are satisfied by side-chain H-bond acceptor groups of polar residues preceding the beginning of the helix, thus terminating and stabilizing the α-helix at the amino-terminal end (36, 37). Similarly, a complementary arrangement is also found at the carboxyl terminus of α-helices. This motif, termed helix capping, has been shown to play an important role in the folding, stability, and function of proteins (38, 39). The functional relevance of helix capping is highlighted by mutations associated with inherited disorders such as neurodegenerative diseases or diabetes mellitus (40, 41).
We have recently generated a NIS homology model (16) using as a template the crystal structure of the Vibrio parahaemolyticus Na+/galactose symporter (vSGLT), the only structure determined of any protein belonging to the SLC5 family (42). On the basis of our NIS homology model, we propose that the main chain amino group of G444 at the beginning of TMS XII is capped by the carboxamide side-chain of N441 (Fig. 5A). To test this hypothesis, we substituted N441 with Gln, an amino acid reported to participate in N-capping (37), in the background of 4A NIS (Q441/4A NIS). This mutant was transiently transfected into COS-7 cells and assayed for I− transport. At steady state, Q441/4A NIS exhibited the same levels of I− transport and cell surface expression as those of WT NIS (Fig. 5B, C). In stark contrast, when Gln was substituted at position 440 rather than 441 (Q440/4A NIS), the resulting protein did not transport I− (Fig. 5B) and barely reached the cell surface (Fig. 5C). This may be so because the distance between Q440 and the amide group of G444 in TMS XII may be too long to allow for the formation of an H bond, leaving the helix backbone amino group unsatisfied. Consistent with these observations, when we introduced an Asn at position 440 (N440/4A NIS), the same phenomenon occurred (Fig. 5B). The only mutants that reached the plasma membrane, as shown by immunfluorescence, were those in which the N-capping amino acids Asn or Gln were located at position 441 (Fig. 5D), in agreement with our FACS data. These results are consistent with the notion that N441 participates in N-capping of TMS XII.
Figure 5.
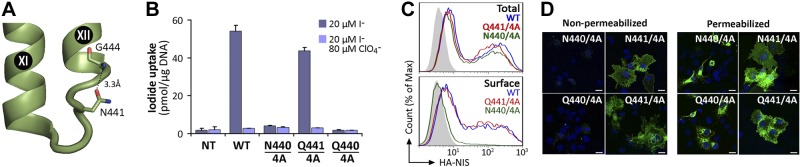
N441 participates in N-capping of TMS XII. A) Close-up of NIS homology model based on the structure of vSGLT showing IL-6 connecting TMS XI and XII. Structural and biochemical data indicate the participation of N441 in N-terminal helix-capping of TMS XII. The carboxamide side chain of N441 may form an H bond (dotted line) with the main chain NH group of G444. B) Steady-state I− uptake in nontransfected (Nt) and transiently transfected COS-7 cells expressing WT, N440/4A, Q441/4A, and Q440/4A NIS. Cells were incubated with 20 μM I− in the absence (dark bars) or presence (light bars) of 80 μM ClO4−. Results are expressed as means ± sd (pmol I−/μg DNA). Values are representative of ≥3 different experiments; in each experiment, activity was analyzed in triplicate. C) Representative FACS histograms showing NIS expression under permeabilized (top panel, total) and nonpermeabilized (bottom panel, surface) conditions. Cells were stained with anti-HA Ab, followed by R-phycoerythrin-conjugated anti-rat Ab. Nontransfected cells are represented in solid gray; cells expressing WT, Q441/4A and N440/4A NIS are indicated by blue, red, and green lines. D) Immunofluorescence analysis of nonpermeabilized (left panels) and permeabilized (right panels) transiently transfected COS-7 cells stained with anti-HA Ab, followed by R-phycoerythrin-conjugated anti-rat Ab (green). Cell nuclei were stained with DAPI (blue). Scale bars = 20 μm.
Conserved residue N441 is required for NIS targeting to the plasma membrane
NIS belongs to the SLC5A family of transporters, which couple the inward “uphill” translocation of their substrates to the inward “downhill” transport of Na+. Interestingly, N441 is highly conserved throughout the family (Fig. 6A). To assess the role of this residue, we generated the N441A and N441Q NIS mutants in the WT NIS background and assayed I− uptake in transiently transfected COS-7 cells. N441A NIS only retained less than half of the transport activity displayed by WT NIS, whereas N441Q NIS exhibited an activity comparable to that of WT NIS, consistent with the structural similarity and capping properties of Asn and Gln (Fig. 6B). Although Asn is preferred to Gln at the N-cap position (37), Gln is structurally and chemically perfectly suited to participate in helix N-capping; it is just less frequently observed than Asn. Plasma membrane targeting of N441A NIS was partially impaired, even though its expression levels were comparable to those of N441Q and WT NIS (Fig. 6C), which explains the lower I− accumulation by N441A NIS. In conclusion, our findings strongly suggest that N441 participates in TMS XII stabilization by capping the helix. This proposal is supported by the observation that, in the experimental structure of vSGLT (42), the template used for the NIS homology model, Thr452, the equivalent residue to Asn441, is directly involved in an N-capping interaction.
Figure 6.
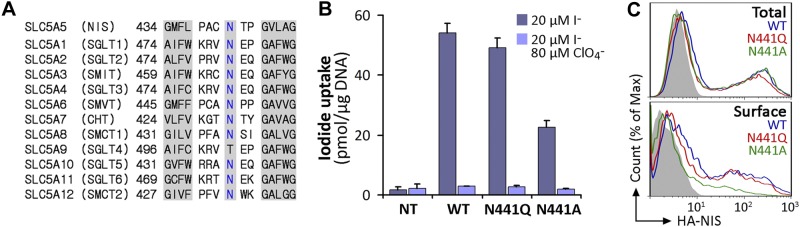
Conserved residue N441 is required for NIS targeting to the plasma membrane. A) Sequence alignment of IL-6 of SLC5A family members. The N441 position in NIS and corresponding residues in other members are indicated in blue. Left and right shaded sequences represent fragments of TMS XI and XII, respectively, according to the crystal structure of vSGLT. B) Steady-state I− uptake in nontransfected (Nt) or transiently transfected COS-7 cells expressing WT, N441Q, or N441A NIS. Cells were incubated with 20 μM I− in the absence (dark bars) or presence (light bars) of 80 μM ClO4−. Results are expressed as means ± sd (pmol I−/μg DNA). Values are representative of ≥3 different experiments; in each experiment activity was analyzed in triplicate. C) Representative FACS histograms showing NIS expression under permeabilized (top panel, total) and nonpermeabilized (bottom panel, surface) conditions. Cells were stained with anti-HA Ab, followed by R-phycoerythrin-conjugated anti-rat Ab. Nontransfected cells are represented in solid gray; WT NIS, blue line; N441Q NIS, red line; and N441A NIS, green line.
DISCUSSION
ITD is an uncommon autosomal-recessive condition caused by naturally occurring mutations in the SLC5A5 gene, which encodes NIS (23). The identification of disease-causing NIS mutations has been made possible by the availability of the NIS cDNA sequence (7). To date, 14 such mutations have been reported (−54C>T, V59E, G93R, R124H, Δ142–323, Q267E, Δ287–288, C272X, T354P, Δ439–443, G395R, G543E, frameshift 515X, and Y531X). The investigation of NIS mutations has been an extremely valuable approach to elucidate structure and function relations in NIS. Some of these ITD mutants, including V59E, G93R, R124H, Q267E, T354P, G395R, and G543E, have been studied in detail and have provided key mechanistic information on NIS (15–20, 27, 28). For example, an analysis of the T354P substitution revealed that this position requires an OH group at the β-carbon and led us to study other β-OH-containing residues in TMS IX. We have shown that these amino acids are involved in Na+ binding and translocation, and we have proposed a structural homology between the leucine transporter from Aquifex aeolicus (LeuT; ref. 43) and NIS, even though they belong to different families and there is no primary sequence homology between them (18). A remarkable surprise was that the V. parahaemolyticus Na+/galactose symporter, which belongs to the same family as NIS, actually displays the same fold as LeuT (42, 44). Furthermore, we have recently reported that position 93 in NIS is critical for substrate selectivity and stoichiometry (16).
Five of the previously studied NIS mutants (V59E, G93R, T354P, G395R, and Q267E) are fully glycosylated and properly targeted to the plasma membrane (16–19, 27, 28). In sharp contrast, the NIS mutants R124H and G543E were found to mature only partially and be retained in intracellular organelles instead of being expressed at the cell surface (15, 20). Here we report on the extensive characterization of the ITD-causing Δ439–443 NIS mutant. As previously shown (29), we observed that cells expressing the mutant Δ439–443 NIS did not display any I− accumulation (Figs. 1B, 2A), a finding consistent with the idea that the mutation was the direct cause of ITD. In addition, we determined that the Δ439–443 NIS mutant matures only partially and is retained intracellularly (Figs. 1C, 2B). We also showed that Δ439–443 NIS failed to accumulate I− in membrane vesicles (Fig. 1E), indicating that the deletion causes the protein to be intrinsically inactive.
To understand the role played by the deleted amino acids in NIS structure, we engineered a mutant containing 5 Ala residues (5A NIS) replacing the amino acids missing in Δ439–443 NIS. Surprisingly, we found that 5A NIS was partially targeted to the cell surface and displayed modest I− uptake levels, indicating that restoring the missing amino acid segment, even if it consists of 5 Ala, brings partial NIS activity back (Fig. 2A). Although we demonstrated that Δ439–443 NIS was incompletely glycosylated, some 5A NIS molecules were completely glycosylated (Fig. 2C–E). However, the incomplete glycosylation of Δ439–443 NIS is unlikely to be the direct cause of the protein's intracellular retention, since studies carried out in our laboratory have demonstrated that glycosylation is not essential for NIS targeting to the plasma membrane or transport activity (10). Therefore, our findings suggest that additional structural changes other than those causing impaired glycosylation are likely to be present in Δ439–443 NIS.
Significantly, the anti-NIS VJ1 Ab, which recognizes an extracellular conformational epitope proposed to contain amino acids from the last extracellular loop (34), seems to display a lower affinity for Δ439–443 than for WT NIS (Figs. 3 and 4D). Given that the last extracellular loop of NIS is glycosylated at 2 positions, we investigated whether the carbohydrate moieties constitute part of the epitope recognized by the Ab and demonstrated that this was not the case (Fig. 4F). An altered conformation of the VJ1 epitope in Δ439–443 NIS may be caused by the shorter distance between TMS XI and XII in the mutant, thus explaining the lower affinity of the anti-VJ1 Ab for Δ439–443 NIS relative to WT NIS. Consistent with this notion, we detected a higher affinity of the anti-VJ1 Ab for 5A NIS than for Δ439–443 NIS, and virtually the same affinity for N441/4A NIS and WT NIS (Fig. 4D). Thus, introducing N441 alone was enough to restore the immunorecognition of the protein by the anti-VJ1 Ab. Strikingly, N441/4A NIS exhibited targeting to the plasma membrane and I− transport properties comparable to WT NIS (Fig. 4A, B), suggesting a significant structural role for N441, a highly conserved residue in all members of the SLC5A family of proteins except for SGLT4 (Fig. 6A). It is noteworthy that the C440A substitution had no effect on NIS function and targeting to the plasma membrane (15). Moreover, the N441A substitution significantly decreased I− uptake and targeting to the plasma membrane, whereas replacing Asn441 with Gln, a residue that may participate in helix capping, yielded a protein that behaved similarly to WT NIS (Figs. 6B, C). Although Gln is not as preferred as Asn at the N-cap position (37), structurally and chemically it is perfectly suited to participate in helix N-capping; it is just less frequently observed than Asn. Disruption of stability by mutations that affect helix capping has been reported in proteins such as HNF1-α, resulting in monogenic diabetes mellitus, and in a prion protein fragment (40, 41). Helix capping motifs have also been invoked in the stabilization of the TMS VI of several GPCRs (39, 45).
Our NIS homology model based on the structure of the bacterial Na+/galactose symporter (vSGLT; ref. 16) shows that IL-6 between TMS XI and XII in WT NIS is ∼12 Å in length. We propose that the amide hydrogen in the main chain at the beginning of TMS XII (G444) is capped by the side chain of N441 (Fig. 5A). In contrast, the corresponding distance is markedly shorter in Δ439–443 NIS. Bringing the amino terminus of TMS XII and the carboxyl terminus of TMS XI together requires large changes in the orientation of the 2 helices with respect to each other, to the rest of the molecule, and to the membrane. With these changes the protein is neither targeted to the cell surface nor functional (Fig. 1 and Fig. 7), whereas in the 5A mutant, the consecutive Ala residues may form a helix that increases the distance between TMS XI and XII to 5–7 Å, shorter than the original 12 Å but long enough to allow the protein to be partially folded and minimally targeted to the plasma membrane, albeit with a lower apparent affinity for I− than WT NIS (Figs. 2, 4C, and 7). For their part, both N441/4A and Q441/4A NIS mimic WT NIS, likely because in these mutants TMS XII is capped. In contrast, neither N440/4A nor Q440/4A NIS is targeted to the plasma membrane (Figs. 5, 7), possibly because in these mutants the distance between N440 or Q440 and TMS XII may be too long for N-capping to occur.
Figure 7.
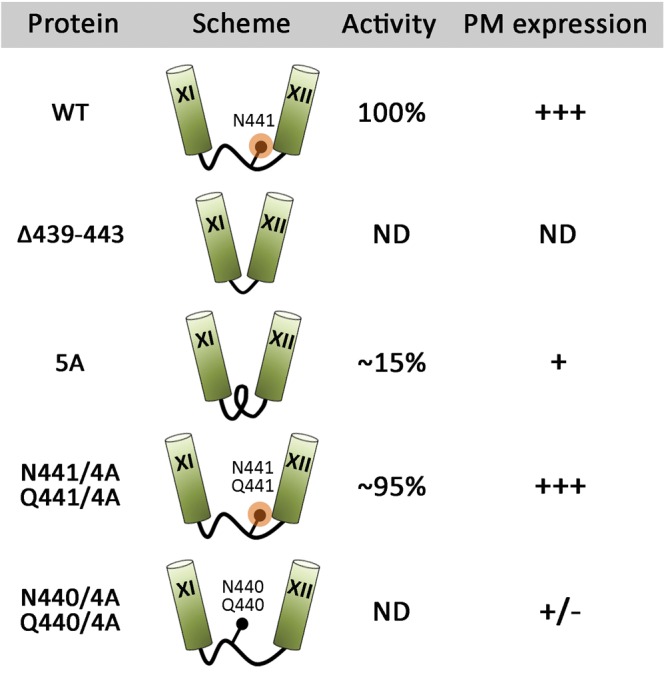
Summarized schematic representation of the results. Cylinders represent TMS, connecting black lines intracellular loop 6, black circles the indicated amino acid residues, and orange circles N-capping. Activity denotes I− transport; PM, plasma membrane; ND, nondetectable.
Acknowledgments
The authors thank Dr. Sabine Costagliola (Free University of Brussels, Brussels, Belgium) for providing the anti-human NIS VJ1 monoclonal antibody. The authors are grateful to Dr. George Rose (Johns Hopkins University, Baltimore, MD, USA) for his insightful comments and to the members of the N.C. laboratory for critical reading of the manuscript.
J.P.N. was supported by the Brown-Coxe postdoctoral fellowship from Yale University School of Medicine. This work was supported by U.S. National Institutes of Health grant DK-41544 (N.C.).
The authors declare no conflicts of interest.
Footnotes
- Endo H
- endo-β-acetylglucosaminidase H
- FACS
- fluorescence activated cell sorting
- HA
- hemagglutinin
- HBSS
- Hanks' balanced salt solution
- ITD
- I− transport defect
- MDCK-II
- Madin-Darby canine kidney
- MV
- membrane vesicle
- NIS
- Na+/I− symporter
- TMS
- transmembrane segment
- WT
- wild-type
REFERENCES
- 1. Nicola J. P., Basquin C., Portulano C., Reyna-Neyra A., Paroder M., Carrasco N. (2009) The Na+/I− symporter mediates active iodide uptake in the intestine. Am. J. Physiol. Cell Physiol. 296, C654–C662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tazebay U. H., Wapnir I. L., Levy O., Dohan O., Zuckier L. S., Zhao Q. H., Deng H. F., Amenta P. S., Fineberg S., Pestell R. G., Carrasco N. (2000) The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat. Med. 6, 871–878 [DOI] [PubMed] [Google Scholar]
- 3. Wapnir I. L., van de Rijn M., Nowels K., Amenta P. S., Walton K., Montgomery K., Greco R. S., Dohan O., Carrasco N. (2003) Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J. Clin. Endocrinol. Metab. 88, 1880–1888 [DOI] [PubMed] [Google Scholar]
- 4. Altorjay A., Dohan O., Szilagyi A., Paroder M., Wapnir I. L., Carrasco N. (2007) Expression of the Na+/I- symporter (NIS) is markedly decreased or absent in gastric cancer and intestinal metaplastic mucosa of Barrett esophagus. BMC Cancer 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicola J. P., Reyna-Neyra A., Carrasco N., Masini-Repiso A. M. (2012) Dietary iodide controls its own absorption through post-transcriptional regulation of the intestinal Na+/I− symporter. J. Physiol. 590, 6013–6026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dohan O., De la Vieja A., Paroder V., Riedel C., Artani M., Reed M., Ginter C. S., Carrasco N. (2003) The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr. Rev. 24, 48–77 [DOI] [PubMed] [Google Scholar]
- 7. Dai G., Levy O., Carrasco N. (1996) Cloning and characterization of the thyroid iodide transporter. Nature 379, 458–460 [DOI] [PubMed] [Google Scholar]
- 8. Eskandari S., Loo D. D., Dai G., Levy O., Wright E. M., Carrasco N. (1997) Thyroid Na+/I− symporter: mechanism, stoichiometry, and specificity. J. Biol. Chem. 272, 27230–27238 [DOI] [PubMed] [Google Scholar]
- 9. Levy O., Dai G., Riedel C., Ginter C. S., Paul E. M., Lebowitz A. N., Carrasco N. (1997) Characterization of the thyroid Na+/I− symporter with an anti-COOH terminus antibody. Proc. Natl. Acad. Sci. U. S. A. 94, 5568–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levy O., De la Vieja A., Ginter C. S., Riedel C., Dai G., Carrasco N. (1998) N-linked glycosylation of the thyroid Na+/I− symporter (NIS). Implications for its secondary structure model. J. Biol. Chem. 273, 22657–22663 [DOI] [PubMed] [Google Scholar]
- 11. De La Vieja A., Dohan O., Levy O., Carrasco N. (2000) Molecular analysis of the sodium/iodide symporter: impact on thyroid and extrathyroid pathophysiology. Physiol. Rev. 80, 1083–1105 [DOI] [PubMed] [Google Scholar]
- 12. Dohan O., De la Vieja A., Carrasco N. (2000) Molecular study of the sodium-iodide symporter (NIS): a new field in thyroidology. Trends Endocrinol. Metab. 11, 99–105 [DOI] [PubMed] [Google Scholar]
- 13. Dohan O., Baloch Z., Banrevi Z., Livolsi V., Carrasco N. (2001) Rapid communication: predominant intracellular overexpression of the Na(+)/I(−) symporter (NIS) in a large sampling of thyroid cancer cases. J. Clin. Endocrinol. Metab. 86, 2697–2700 [DOI] [PubMed] [Google Scholar]
- 14. Riedel C., Levy O., Carrasco N. (2001) Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J. Biol. Chem. 276, 21458–21463 [DOI] [PubMed] [Google Scholar]
- 15. Paroder V., Nicola J. P., Ginter C. S., Carrasco N. (2013) The iodide transport defect-causing mutation R124H: a δ-amino group at position 124 is critical for maturation and trafficking of the Na+/I− symporter (NIS). J. Cell Sci. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paroder-Belenitsky M., Maestas M. J., Dohan O., Nicola J. P., Reyna-Neyra A., Follenzi A., Dadachova E., Eskandari S., Amzel L. M., Carrasco N. (2011) Mechanism of anion selectivity and stoichiometry of the Na+/I− symporter (NIS). Proc. Natl. Acad. Sci. U. S. A. 108, 17933–17938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reed-Tsur M. D., De la Vieja A., Ginter C. S., Carrasco N. (2008) Molecular characterization of V59E NIS, a Na+/I− symporter mutant that causes congenital I- transport defect. Endocrinology 149, 3077–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De la Vieja A., Reed M. D., Ginter C. S., Carrasco N. (2007) Amino acid residues in transmembrane segment IX of the Na+/I− symporter play a role in its Na+ dependence and are critical for transport activity. J. Biol. Chem. 282, 25290–25298 [DOI] [PubMed] [Google Scholar]
- 19. De La Vieja A., Ginter C. S., Carrasco N. (2004) The Q267E mutation in the sodium/iodide symporter (NIS) causes congenital iodide transport defect (ITD) by decreasing the NIS turnover number. J. Cell Sci. 117, 677–687 [DOI] [PubMed] [Google Scholar]
- 20. De la Vieja A., Ginter C. S., Carrasco N. (2005) Molecular analysis of a congenital iodide transport defect: G543E impairs maturation and trafficking of the Na+/I− symporter. Mol. Endocrinol. 19, 2847–2858 [DOI] [PubMed] [Google Scholar]
- 21. Dohan O., Portulano C., Basquin C., Reyna-Neyra A., Amzel L. M., Carrasco N. (2007) The Na+/I symporter (NIS) mediates electroneutral active transport of the environmental pollutant perchlorate. Proc. Natl. Acad. Sci. U. S. A. 104, 20250–20255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spitzweg C., Morris J. C. (2010) Genetics and phenomics of hypothyroidism and goiter due to NIS mutations. Mol. Cell. Endocrinol. 322, 56–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fujiwara H. (1997) Congenital hypothyroidism caused by a mutation in the Na+/I− symporter. Nat. Genet. 17, 122. [DOI] [PubMed] [Google Scholar]
- 24. Wolff J. (1983) Congenital goiter with defective iodide transport. Endocr. Rev. 4, 240–254 [DOI] [PubMed] [Google Scholar]
- 25. Stanbury J. B., Chapman E. M. (1960) Congenital hypothyroidism with goiter: absence of an iodide-concentrating mechanism. Lancet 1, 1162–1165 [DOI] [PubMed] [Google Scholar]
- 26. Nicola J. P., Nazar M., Serrano-Nascimento C., Goulart-Silva F., Sobrero G., Testa G., Nunes M. T., Munoz L., Miras M., Masini-Repiso A. M. (2011) Iodide transport defect: functional characterization of a novel mutation in the Na+/I− symporter 5′-untranslated region in a patient with congenital hypothyroidism. J. Clin. Endocrinol. Metab. 96, E1100–1107 [DOI] [PubMed] [Google Scholar]
- 27. Dohan O., Gavrielides M. V., Ginter C., Amzel L. M., Carrasco N. (2002) Na(+)/I(−) symporter activity requires a small and uncharged amino acid residue at position 395. Mol. Endocrinol. 16, 1893–1902 [DOI] [PubMed] [Google Scholar]
- 28. Levy O., Ginter C. S., De la Vieja A., Levy D., Carrasco N. (1998) Identification of a structural requirement for thyroid Na+/I− symporter (NIS) function from analysis of a mutation that causes human congenital hypothyroidism. FEBS Lett. 429, 36–40 [DOI] [PubMed] [Google Scholar]
- 29. Tonacchera M., Agretti P., de Marco G., Elisei R., Perri A., Ambrogini E., De Servi M., Ceccarelli C., Viacava P., Refetoff S., Panunzi C., Bitti M. L., Vitti P., Chiovato L., Pinchera A. (2003) Congenital hypothyroidism due to a new deletion in the sodium/iodide symporter protein. Clin. Endocrinol. (Oxf.) 59, 500–506 [DOI] [PubMed] [Google Scholar]
- 30. Paroder V., Spencer S. R., Paroder M., Arango D., Schwartz S., Jr., Mariadason J. M., Augenlicht L. H., Eskandari S., Carrasco N. (2006) Na(+)/monocarboxylate transport (SMCT) protein expression correlates with survival in colon cancer: molecular characterization of SMCT. Proc. Natl. Acad. Sci. U. S. A. 103, 7270–7275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nicola J. P., Nazar M., Mascanfroni I. D., Pellizas C. G., Masini-Repiso A. M. (2010) NF-kappaB p65 subunit mediates lipopolysaccharide-induced Na(+)/I(−) symporter gene expression by involving functional interaction with the paired domain transcription factor Pax8. Mol. Endocrinol. 24, 1846–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Purtell K., Paroder-Belenitsky M., Reyna-Neyra A., Nicola J. P., Koba W., Fine E., Carrasco N., Abbott G. W. (2012) The KCNQ1-KCNE2 K+ channel is required for adequate thyroid I- uptake. FASEB J. 26, 3252–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nicola J. P., Velez M. L., Lucero A. M., Fozzatti L., Pellizas C. G., Masini-Repiso A. M. (2009) Functional toll-like receptor 4 conferring lipopolysaccharide responsiveness is expressed in thyroid cells. Endocrinology 150, 500–508 [DOI] [PubMed] [Google Scholar]
- 34. Pohlenz J., Duprez L., Weiss R. E., Vassart G., Refetoff S., Costagliola S. (2000) Failure of membrane targeting causes the functional defect of two mutant sodium iodide symporters. J. Clin. Endocrinol. Metab. 85, 2366–2369 [DOI] [PubMed] [Google Scholar]
- 35. Kaminsky S. M., Levy O., Salvador C., Dai G., Carrasco N. (1994) Na(+)-I- symport activity is present in membrane vesicles from thyrotropin-deprived non-I(−)-transporting cultured thyroid cells. Proc. Natl. Acad. Sci. U. S. A. 91, 3789–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Richardson J. S., Richardson D. C. (1988) Amino acid preferences for specific locations at the ends of alpha helices. Science 240, 1648–1652 [DOI] [PubMed] [Google Scholar]
- 37. Aurora R., Rose G. D. (1998) Helix capping. Protein Sci. 7, 21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhukovsky E. A., Mulkerrin M. G., Presta L. G. (1994) Contribution to global protein stabilization of the N-capping box in human growth hormone. Biochemistry 33, 9856–9864 [DOI] [PubMed] [Google Scholar]
- 39. Schulz A., Bruns K., Henklein P., Krause G., Schubert M., Gudermann T., Wray V., Schultz G., Schoneberg T. (2000) Requirement of specific intrahelical interactions for stabilizing the inactive conformation of glycoprotein hormone receptors. J. Biol. Chem. 275, 37860–37869 [DOI] [PubMed] [Google Scholar]
- 40. Narayana N., Phillips N. B., Hua Q. X., Jia W., Weiss M. A. (2006) Diabetes mellitus due to misfolding of a beta-cell transcription factor: stereospecific frustration of a Schellman motif in HNF-1alpha. J. Mol. Biol. 362, 414–429 [DOI] [PubMed] [Google Scholar]
- 41. Gallo M., Paludi D., Cicero D. O., Chiovitti K., Millo E., Salis A., Damonte G., Corsaro A., Thellung S., Schettini G., Melino S., Florio T., Paci M., Aceto A. (2005) Identification of a conserved N-capping box important for the structural autonomy of the prion alpha 3-helix: the disease associated D202N mutation destabilizes the helical conformation. Int. J. Immunopathol. Pharmacol. 18, 95–112 [DOI] [PubMed] [Google Scholar]
- 42. Faham S., Watanabe A., Besserer G. M., Cascio D., Specht A., Hirayama B. A., Wright E. M., Abramson J. (2008) The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321, 810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl–dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 44. Krishnamurthy H., Piscitelli C. L., Gouaux E. (2009) Unlocking the molecular secrets of sodium-coupled transporters. Nature 459, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neumann J. M., Couvineau A., Murail S., Lacapere J. J., Jamin N., Laburthe M. (2008) Class-B GPCR activation: is ligand helix-capping the key? Trends Biochem. Sci. 33, 314–319 [DOI] [PubMed] [Google Scholar]