

Abstract
Ezrin, radixin, and moesin (ERM) proteins link cortical actin to the plasma membrane and coordinate cellular events that require cytoskeletal rearrangement, including cell division, migration, and invasion. While ERM proteins are involved in many important cellular events, the mechanisms regulating their function are not completely understood. Our laboratory previously identified reciprocal roles for the sphingolipids ceramide and sphingosine-1-phosphate (S1P) in the regulation of ERM proteins. We recently showed that ceramide-induced activation of PP1α leads to dephosphorylation and inactivation of ERM proteins, while S1P results in phosphorylation and activation of ERM proteins. Following these findings, we aimed to examine known inducers of the SK/S1P pathway and evaluate their ability to regulate ERM proteins. We examined EGF, a known inducer of the SK/S1P pathway, for its ability to regulate the ERM family of proteins. We found that EGF induces ERM c-terminal threonine phosphorylation via activation of the SK/S1P pathway, as this was prevented by siRNA knockdown or pharmacological inhibition of SK. Using pharmacological, as well as genetic, knockdown approaches, we determined that EGF induces ERM phosphorylation via activation of S1PR2. In addition, EGF led to cell polarization in the form of lamellipodia, and this occurred through a mechanism involving S1PR2-mediated phosphorylation of ezrin T567. EGF-induced cellular invasion was also found to be dependent on S1PR2-induced T567 ezrin phosphorylation, such that S1PR2 antagonist, JTE-013, and expression of a dominant-negative ezrin mutant prevented cellular invasion toward EGF. In this work, a novel mechanism of EGF-stimulated invasion is unveiled, whereby S1P-mediated activation of S1PR2 and phosphorylation of ezrin T567 is required.—Orr Gandy, K. A., Adada, M., Canals, D., Carroll, B., Roddy, P., Hannun, Y. A., Obeid, L. M. Epidermal growth factor-induced cellular invasion requires sphingosine-1-phosphate/sphingosine-1-phosphate 2 receptor-mediated ezrin activation.
Keywords: radixin, moesin, actin cytoskeleton, polarization
Ezrin, radixin, and moesin (ERM) make up a portion of the ERM family of proteins, which are involved in linking cortical actin to the plasma membrane, and are therefore involved in the many cellular processes that coordinated changes in cytoskeletal architecture (1). ERM proteins also function as signaling platforms, acting as a scaffold for the necessary arrangement of effector molecules (1). ERM proteins have been implicated in numerous cellular functions, including spindle formation and cytokinesis (2), cell migration, and cell invasion (1). Given the prosurvival and proliferative events in which the ERM family of proteins is involved, it is not surprising that deregulation of ERM proteins have been related to many cancers, even serving as a potential biomarker and prognostic indicator of disease (3). Targeting the ERM family of proteins and the mechanisms that regulate their activity may serve as a novel therapeutic approach for the treatment of various cancers and other proliferative diseases.
The conformation of ERM proteins dictates their activity, such that when the N and C termini are folded onto one another, the protein is inactive and therefore unable to coordinate changes in the cytoskeleton (4). On the other hand, ERM proteins are activated when phosphorylated on a C-terminal threonine residue [ezrin, threonine 567 (T567); radixin, threonine 564 (T564); and moesin, threonine 558 (T558)] following binding to plasma membrane phosphatidylinositol-4,5-bisphosphate (PIP2) (5). While the N terminus of ERM proteins binds the plasma membrane, the phosphorylated C terminus binds cortical actin (6), thereby linking changes in actin to changes in the plasma membrane morphology (6). Many ligands are known to induce the phosphorylation of ERM proteins, including epidermal growth factor (EGF) and platelet derived growth factor (PDGF) (7). Recently, however, novel mechanisms of ERM regulation by sphingolipids were identified in our laboratory (5, 8, 9). ERM proteins were first found to be negatively regulated by cisplatin-mediated activation of acid sphingomyelinase (10) and subsequent production of ceramide. More recently, our laboratory has identified a novel PIP2-independent mechanism of ERM regulation, whereby ERM proteins are dephosphorylated via ceramide-mediated activation of protein phosphatase 1α (PP1α; ref. 5). Moreover, work from our laboratory identified d-erythro-sphingosine-1-phosphate (S1P) as an inducer of ERM phosphorylation and activation (8, 9). Following these findings, we became interested in S1P regulation of ERM proteins and therefore examined known inducers of the sphingosine kinase (SK)/S1P pathway for their ability to regulate phosphorylation and subsequent activation of ERM proteins.
SK, the enzyme responsible for the production of the ERM-activating lipid S1P, undergoes regulation by numerous extracellular ligands, including EGF (11), such that EGF has been shown to activate both SK isoforms, SK1 and SK2, (11, 12) resulting in production of S1P and activation of S1P cell-surface receptors (S1PR1–S1PR5; ref. 13). In this work, we examine the role of the SK/S1P pathway in EGF-mediated ERM phosphorylation. We found that EGF induces phosphorylation of ERM proteins and, pharmacological inhibition or siRNA knockdown of SK inhibited this. Also, S1P signaling through S1PR2 was found to be necessary for EGF-induced ERM phosphorylation. Cellular invasion induced by EGF was also dependent on SK/S1P/S1PR2 activation of ERM proteins. Here, we present a novel mechanism of EGF-induced invasion involving sphingolipid regulation of ERM proteins, as well as clarify a role for S1PR2 in the regulation of cellular invasion. These findings highlight the importance of the SK/S1P pathway as a secondary messaging system and reveal a potential therapeutic target for the treatment of cancers that are or have become resistant to EGF-targeted treatments, such as that which often occurs in non-small cell lung carcinomas (NSCLCs; refs 14, 15).
MATERIALS AND METHODS
Materials
High-glucose Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), rhodamine-phalloidin penicillin-streptomycin, and Superscript III First-Strand Synthesis kit were purchased from Invitrogen (Carlsbad, CA, USA). Essentially fatty acid-free bovine serum albumin (BSA) and monoclonal anti-β-actin antibody were from Sigma-Aldrich (St. Louis, MO, USA). S1P and d-erythro-17-carbon sphingosine (C17-Sph) were from Avanti polar lipids (Alabaster, AL, USA). Anti-phospho-ERM (pERM) antibody was from Cell Signaling Technology (Danvers, MA, USA). Anti VSV-G and HRP-labeled secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Chemiluminescence kit was from Thermo Scientific (Suwanee, GA, USA). Draq5- and 488-conjugated secondary antibodies were purchased from Alexis Biochemicals (San Diego, CA, USA). SYBR Green was purchased from Bio-Rad (Hercules, CA, USA). JTE-013, BML-241, VPC23019, and SKi-II (4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol; ref. 16) were purchased from Cayman Chemical (Ann Arbor, MI, USA). S1P2R-green fluorescent protein (GFP) plasmid construct (RG210163) was purchased from OriGene (Rockville, MD, USA). SKX (amidine-based SK inhibitor) was a kind gift from Dr. Kevin Lynch and Dr. Andrew Bolt (SphynKx Therapeutics, Charlottesville, VA, USA; ref. 17). Tumor Invasion System 24-multiwell plates, calcein AM fluorescent dye, and human recombinant EGF were from BD Biosciences (San Jose, CA, USA).
Cell lines and culture conditions
HeLa cells were originally purchased from American Type Culture Collection (ATCC; Manassas, VA, USA) and maintained as described previously (8). Briefly, DMEM was supplemented with 1% penicillin-streptomycin and 10% FBS, and cells were incubated in standard culture conditions: 37°C, and 5% CO2. When serum-free medium was used, 0.1% BSA, 1% penicillin-streptomycin, and 10mM HEPES were supplemented to DMEM. In all cases, before conducting treatments, cells were serum starved overnight (16–18 h).
Immunoblotting
Immunoblotting was performed as described previously (8, 9). Briefly, after removal of medium, cells were directly lysed in buffer containing 1% (w/v) SDS. Next, cells were sonicated and boiled before proteins were separated via SDS-PAGE (4–15%, Tris-HCl) using the Bio-Rad Criterion system. Proteins were transferred to nitrocellulose membranes and blocked for ≥1 h with 5% nonfat milk in PBS/0.1% Tween 20 (PBS-T). Membranes were incubated with primary antibody pERM diluted 1:1000 or 1:3000 β-actin at 4°C overnight. Secondary antibody incubation occurred for 1 h at room temperature at a 1:5000 dilution. Visualization was carried out per manufacturer protocol.
RNA interference
Gene silencing was carried out as described previously, with minor changes (8). Using siRNA purchased from Qiagen (Valencia, CA, USA), directed against human SK1, SK2, and S1PR2: SK1, 5′-AAGGGCAAGGCCTTGCAGCTC-3′; SK2, Hs_SPHK2_5 FlexiTube siRNA SI00288561 (FlexiTube siRNA, experimentally verified; Qiagen); S1PR2, Hs_EDG5_6 FlexiTube siRNA SI02663227 (FlexiTube siRNA, experimentally verified; Qiagen). Scrambled siRNA was used as a negative control: SCR, negative control siRNA (1027130, Qiagen). All-Star siRNA (Qiagen) was also used a negative control; HeLa cells were seeded at 30% confluence in DMEM with 10% FBS. After 1 d, siRNA transfection was carried with the corresponding siRNA at final concentration of 20 nM using Oligofectamine transfection reagent (Invitrogen) and following the manufacturer's protocol. After 36 h, HeLa cells were serum starved overnight. All treatments were carried out 48 h following siRNA transfection.
Quantitative real-time reverse transcription–polymerase chain reaction (RT-PCR)
Cells were washed with ice-cold PBS, then directly lysed with the lysis buffer provided in the RNA minieasy kit from Qiagen. RNA extraction was carried out as per the manufacturer's protocol. RNA was then quantified using Thermo Scientific NanoDrop. RNA (1 μg) was converted into cDNA using SuperScript III First-Strand Synthesis Systems and following Invitrogen protocol. The cDNA was diluted (1:15), and 5 μl was used per 25-μl reaction. Each 25-μl real-time RT-PCR contained a ratio of 12.5:0.5:0.5:6.5 [SYBR Green/10 μM forward (F) primer/10 μM reverse (R) primer/distilled water]. Using the Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA), PCR reaction conditions were carried out as described previously by our group (8)The following primer sequences were used to detect expression: human β-actin F, 5′-ATTGGCAATGAGCGGTTCC-3′, and human β-actin R, 5′-GGTAGTTTCGTGGCCACA-3′; hSK1 F, 5′-CTGGCAGCTTCCTTGAACCAT-3′, and hSK1 R, 5′-TGTGCAGAGACAGCAGGTTCA-3′; hSK2 F, 5′-TTGCTCAACTGCTCACTG-3′, and hSK2 R, 5′-AGACAGGAAGGAGAAACAG-3′. Using Q-Gene software (http://www.qgene.org), β-actin was used to normalize the obtained threshold cycle values that were shown as mean normalized expression.
C17-Sph labeling
HeLa cells were plated at 150,000 cells/60-mm dish. Once ∼75% confluent, cells were serum starved overnight, 16–18 h. Following starvation, cells were pretreated for 1 h with vehicle or 0.1 μM SKX. Next, 1 μM C17 sphingosine was added and allowed to equilibrate for 20 min. EGF (10 ng/ml) was then added for 2 minn and medium was removed; the reaction was stopped with lipid extraction buffer and sent for analysis at the Medical University of South Carolina shared lipidomics core facility.
Immunofluorescence and confocal microscopy
Laser-scanning confocal and immunofluorescence microscopy analyses were carried out as described previously (8). Grown on poly-d-lysine-coated confocal dishes (MatTek Corp., Ashland, MA, USA), cells were deprived of serum prior to treatments. Cells were fixed, permeabilized, and blocked, then exposed to primary (1:500) and secondary antibodies, according to the manufacturer's protocol. Using a LSM510 META confocal microscope (Carl Zeiss, Inc., Oberkochen, Germany) and a Leica SP8 confocal microcopy system (Leica Microsystems, Wetzlar, Germany), photo images were obtained and analyzed using LSM Image Browser software (http://www.zeiss.com) and Leica software, respectively.
Plasmid constructs and transient transfections
S1P2R-GFP plasmid full-length ezrin cDNA and VSV-G-tag pCB6 plasmids have been previously described (8, 10). Ezrin point mutations were generated as described previously (8). Cells growing on 35- or 100-mm dishes were transfected with 1–3 μg of pCB6-wild-type (WT) ezrin, pCB6-T567 to alanine mutant (T567A) ezrin, or S1P2R-GFP plasmid DNA using Lipofectamine (pCB6 constructs; Invitrogen) or Effectene (pEGFP construct; Qiagen) transfection reagent according to the manufacturer's instructions.
Cellular invasion assays
Cell invasion studies were carried out using the BD Biosciences Tumor Invasion System (345166) according to manufacturer's protocol, with minor changes as described previously (8). Briefly, following serum starvation, cells were removed from culture dish and resuspended at 200,000 cells/ml in 750 μl of appropriate medium, or medium plus chemo attractant was placed in the well of a 24-well plate, followed by 500 μl cell suspension in serum-free medium, with or without 5 μM JTE013, in the apical chamber of the transwell insert. Cells were allowed to invade under normal cell culture conditions for 48 h. Cells were stained with calcein AM and then visualized under fluorescence microscopy; total invading cells were counted using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
RESULTS
EGF induces ERM phosphorylation in an SK-dependent manner
EGF is known to induce ERM phosphorylation, and in order to determine whether this occurs in a SK/S1P-dependent manner, pERM responses to varying doses and times of EGF treatment were characterized (Fig. 1). While using S1P as a positive control for the induction of ERM phosphorylation (8, 18), we established the range of EGF concentrations that activate ERM proteins by treating cells with increasing doses of EGF for 5 min. As little as 1 ng/ml EGF was able to evoke ERM phosphorylation, which increased further with increasing doses of EGF, up to 100 ng/ml (Fig. 1). In addition to examining the doses of EGF that phosphorylate and activate ERM proteins, we examined the temporal regulation of ERM proteins by EGF (Fig. 1). HeLa cells treated with 10 ng/ml EGF displayed acute phosphorylation of ERM proteins beginning as early as 0.5 min and decreasing by ∼15 min (Fig. 1). These data verify previous reports that EGF regulates ERM proteins through T567 phosphorylation (7, 19, 20).
Figure 1.
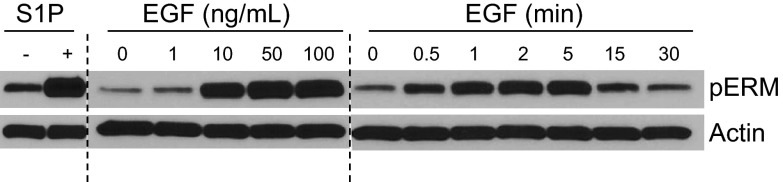
EGF-induced ERM phosphorylation. HeLa cells were serum starved overnight and treated with 100 nM S1P for 5 min, indicated doses of EGF (ng/ml) for 5 min, or 10 ng/ml EGF for the indicated times (min). pERM and actin levels were analyzed via Western blot; blots are representative, n = 3.
Next, in order to assess the role of SK in EGF-mediated ERM phosphorylation, both genetic and pharmacological approaches were used. First, the effect of siRNA-mediated SK down-regulation on EGF-induced pERM was determined. HeLa cells were pretreated with siRNA targeting SK1, SK2, or both SK1 and SK2, and significant knockdown was confirmed by real time PCR (Supplemental Fig. S1 and refs. 8, 21). Following 48 h of SK knockdown, cells were treated with 10 ng/ml EGF for 5 min, and ERM C-terminal phosphorylation was evaluated via Western blot (Fig. 2A). Cells treated with control All Star siRNA displayed robust ERM phosphorylation following 5 min of EGF treatment. In contrast, knockdown of SK2 but not of SK1 diminished EGF-induced pERM. Furthermore, double knockdown of SK1 and SK2 was comparable to the SK2 knockdown alone (Fig. 2A). These data suggest that SK2 and likely not SK1 is required for EGF-induced ERM phosphorylation.
Figure 2.
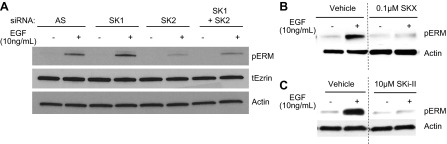
Involvement of sphingosine kinase in EGF-mediated pERM. A) HeLa cells were treated with 20 nM SK1, SK2, or SK1 plus SK2 siRNA for 48 h. After 48 h of siRNA, cells were serum starved, then treated with 10 ng/ml EGF for 5 min. tEzrin, total ezrin. B, C) HeLa cells were serum starved for 16–18 h. Following 1 h pretreatment with DMSO vehicle, 0.1μM SKX (B) or 10 μM SKi-II cells (C) were treated with 10 ng/ml EGF for 5 min. pERM and actin levels were analyzed via Western blot; blots are representative, n = 3.
To further consolidate the role of SK in EGF activation of ERM proteins, two pharmacological inhibitors of SK were employed; SKX (17), an amidine-based SK inhibitor, and SKi-II (16), a nonlipid, small molecule inhibitor of SK. HeLa cells were pretreated with dimethylsulfoxide (DMSO) vehicle or SK inhibitor for 1 h, prior to addition of 10 ng/mL EGF for 2 min and ERM phosphorylation was examined (Fig. 2B, C). The EGF-mediated pERM response remained intact in cells subjected to DMSO vehicle treatment (Fig. 2B, C). In contrast to DMSO-treated cells, cells pretreated with 0.1 μM SKX or 10 μM SKi-II did not elicit pERM responses following EGF treatment (Fig. 2B, C). These findings, taken together, provide evidence solidifying a role for SK in EGF-induced ERM phosphorylation.
EGF activates the SK/S1P pathway
Based on the data above, it became important to determine whether EGF-induced ERM phosphorylation requires activation of SK and subsequent S1P signaling. With the purpose of determining whether EGF induces activation of SK and consequent S1P production, C17-Sph (sphingosine with a 17-carbon backbone) labeling was utilized (ref. 22 and Fig. 3A). HeLa cells were first exposed to C17-Sph, and then treated with 10 ng/ml EGF. Next, HPLC/MS was used to measure incorporation of C17-Sph into C17-S1P. Media from cells treated with EGF contained twice the levels of C17-S1P than did media from non-EGF-treated cells, supporting previous reports of EGF-mediated SK activation and S1P production (17). Next, to determine whether EGF-induced S1P generation was the result of SK activation, a pharmacological inhibitor of SK was employed. The presence of C17-S1P in the medium from cells treated with SKX alone was undetectable; furthermore, addition of EGF to SKX-treated cells did not induce C17-S1P generation. These data corroborate reports that EGF activates the SK/S1P pathway (11, 23, 24) in HeLa cells. With the aim of examining the physiological effect of EGF-generated extracellular S1P, S1PR internalization was used as a tool. Using confocal microscopy, GFP-tagged S1PR2 localization was observed primarily at the plasma membrane in untreated cells (Fig. 3B) or cells treated with SK inhibitor, SKi-II (Fig. 3C), or S1PR2 antagonist JTE-013 (Fig. 3D) alone. On the other hand, cells treated with 5 μM sphingosine (Fig. 3E) or 100 nM S1P (Fig. 3F), exhibited GFP-S1PR2 redistribution to punctate intracellular spots, which is indicative of receptor internalization occurring in response to activation. Treating HeLa cells with EGF also resulted in S1PR internalization (Fig. 3H), suggesting that S1P in the medium following EGF treatment is physiologically relevant and sufficient to activate S1P cell-surface receptors. Finally, to determine whether EGF-mediated S1PR internalization was SK dependent, and to rule out other possible EGF effects, cells were pretreated with SKi-II prior to EGF or sphingosine treatment, and GFP-tagged S1PR localization was assessed (Fig. 3G, I). Inhibition of SK obstructed sphingosine- and EGF-induced S1PR internalization (Fig. 3G, I), consolidating evidence that EGF-mediated S1PR internalization is SK dependent. In addition,, cells were pretreated with S1PR2 antagonist, and EGF-induced S1PR2 internalization was evaluated (Fig. 3J). As expected, JTE-013 was able to inhibit EGF-induced receptor internalization (Fig. 3J). These data demonstrate that EGF-induced extracellular S1P is biologically significant and capable of acting on cell surface S1PRrs, in particular S1PR2. Collectively these results suggest that EGF induces activation of the SK/S1P pathway, and indicates that activation of the SK/S1P pathway may be necessary for some EGF-mediated events.
Figure 3.
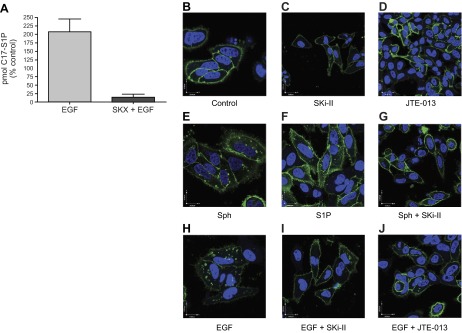
EGF generates S1P and transactivates S1PRs. A) HeLa cells were pretreated for 1 h with DMSO vehicle or 0.1 μM SKX, and 1 μM C17-Sph was added for 15 min. Following 15 min C17-Sph labeling, 10 ng/ml EGF was added for 2 min. Medium was analyzed via mass spectroscopy for C17-S1P levels; n ≥ 3. B–J) Hela cells were transfected with S1PR2 tagged with GFP for 24 h. Following transfection, cells were serum starved overnight, then pretreated for 6 h with 10 uM SKI (C, G, I) or for 1 h with 5 uM JTE-013 (D, J). Cells were treated treated with either 5 μM Sph (E, G), 100 nM S1P (F), or 100 ng/ml EGF (H–J) for 5 min. Cells were then fixed, and nuclei were stained with DAPI. Images were taken using SP8 Leica confocal microscope. Images are representative ≥4 independent experiments.
EGF induces ERM phosphorylation and translocation to lamellipodia via S1PR2
Having previously reported that S1P-mediated ERM phosphorylation occurs in a S1PR2-dependent manner (8), we were interested in determining the role of S1PRs in EGF-induced activation of ERM proteins. To investigate the role of S1PR signaling in EGF-stimulated ERM activation, gene-silencing techniques, as well as receptor-specific antagonists, were used. HeLa cells were treated with siRNA targeting S1PR2, and the effect on EGF-stimulated pERM was evaluated (Fig. 4). Cells exposed to scrambled control siRNA exhibited robust ERM phosphorylation in response to both S1P and EGF treatment (Fig. 4A, B). In contrast, S1PR2 knockdown significantly inhibited ERM phosphorylation following both S1P and EGF treatment, suggesting that S1PR2 plays a pivotal role in EGF-induced ERM phosphorylation.
Figure 4.
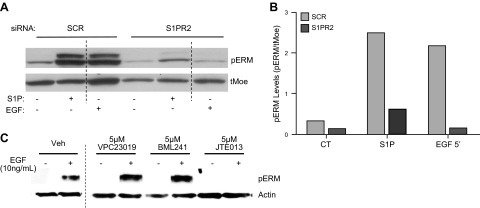
Effect of S1PR modulationon EGF-mediated pERM. A) HeLa cells were treated with 20 nM S1R2 siRNA for 48 h. After 48 h of siRNA, cells were serum starved, then treated with 10 ng/ml EGF for 5 min; n = 2. tMoe, total moesin. B) Quantification of pERM levels in A using ImageJ software. C) HeLa cells were serum starved for 16–18 h. Following 1 h pretreatment with DMSO vehicle or indicated S1PR antagonists, cells were treated with 10 ng/ml EGF for 5 min. pERM and actin levels were analyzed via Western blot; blots are representative, n = 5.
To further solidify the role of S1PR2 in EGF-mediated pERM, pharmacological inhibitors were used, and EGF-stimulated ERM phosphorylation was evaluated (Fig. 4B). Cells receiving vehicle treatment maintained the ability to activate ERM proteins following treatment with EGF (Fig. 4B); likewise, pretreatment with antagonist for S1PR1 (VPC23019) or S1PR3 (VPC23019, BML241) had no effect on EGF-induced ERM phosphorylation (Fig. 4B), indicating that S1PR1and S1PR3 are not involved in the activation of ERM proteins by EGF. Next, cells were pretreated with S1PR2-specific antagonist, JTE013, and the effects on EGF-mediated ERM phosphorylation were assessed. Pretreating HeLa cells for 1 h with 5 μM JTE013 completely inhibited the ability of EGF to induce phosphorylation of ERM proteins (Fig. 4C). These data provide genetic and pharmacological evidence supporting a major role for S1PR2in EGF-mediated C-terminal phosphorylation of ERM proteins.
Previous work from our laboratory showed that S1P-mediated cytoskeletal changes were dependent on ezrin T567 phosphorylation (8); moreover, others have shown that ezrin T567 phosphorylation is required for cytoskeletal rearrangement events brought about by EGF (7), such that C-terminal ezrin phosphorylation is required for EGF-induced lamellipodia formation (7). To determine the cytoskeletal changes that occur in response to EGF treatment, immunofluorescence microscopy was used to evaluate pERM localization, as well as morphological changes, following the addition of EGF. As visualized in Fig. 5A with pERM (green) and F-actin (red), HeLa cells that received no treatment exhibited low levels of phosphorylated ERM that exists intracellularly in a punctate pattern. Treatment with 100 nM S1P resulted in increased levels of pERM that localized to newly formed filopodial protrusions, consistent with our previous findings (Fig. 5A, B and ref. 8). In accordance with other reports, treating cells with EGF resulted in pERM-dependent translocation to newly formed lamellipodia which was accompanied by nuclear retraction (Fig. 5C and ref. 25). Having established that EGF induction of ERM phosphorylation occurs via SK and S1PR2, we were interested in determining the mechanism by which EGF-induces lamellipodia formation and cellular polarization occurs. Given the role SK and S1PR2 play in S1P-induced pERM-dependent filopodia formation (8) and their role in EGF-induced ERM phosphorylation, S1PR2-specific antagonist JTE013 was used and EGF-mediated pERM and lamellipodia formation evaluated (Fig. 5D). HeLa cells pretreated with S1PR2 inhibitor, followed by treatment with EGF, displayed no increase in pERM levels, and were unable to undergo cell polarization, evidenced by lack of lamellipodia formation and nuclear retraction (Fig. 5D). These findings suggest that S1PR2 signaling is necessary for EGF-induced cellular polarization. Next, and in order to evaluate the role of ezrin T567 phosphorylation in EGF-mediated cytoskeletal changes, VSV-G-tagged WT and nonphosphorylatable T567A (TA)-ezrin plasmid constructs were used, and cytoskeletal architecture was evaluated following EGF treatment (Fig. 6). Cells overexpressing WT ezrin display cytoskeletal protrusions, even in the absence of treatment, which is visualized with total ezrin (VSV-G) in green and F-actin in red (Fig. 6A). Treatment of WT-ezrin-expressing cells with S1P only slightly enhanced cytoskeletal protrusions compared to that of nontreated cells (Fig. 6B), while treatment with EGF induced lamellipodia formation, as expected (Fig. 6C). Cells overexpressing TA-ezrin displayed no cytoskeletal protrusions in the absence of treatment (Fig. 6D); moreover, S1P was unable to provoke filopodia formation in TA-ezrin-expressing cells (Fig. 6E), just as EGF was unable to induce lamellipodia formation and cellular polarization (Fig. 6F), indicating that T567 phosphorylation of ezrin is required for EGF-induced cytoskeletal rearrangement. Collectively, these data indicate that EGF induces pERM, as well as lamellipodia formation and cellular polarization, in a S1PR2-dependent manner.
Figure 5.
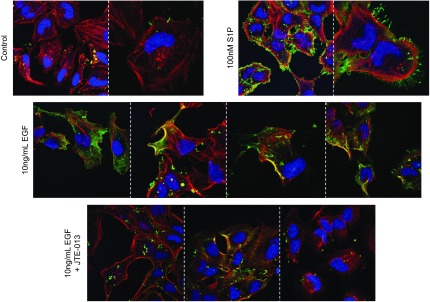
EGF-mediated pERM localization to lamellipodia occurs via S1PR2. HeLa cells were serum deprived and received no treatment (A) or were treated with 100 nM S1P for 5 min (B), 10 ng/ml EGF for 5 min (C), or 5 μM JTE-013 for 1 h, followed by 10 ng/ml EGF for 5 min (D). Cells were fixed and stained for pERM (green). pERM, nuclei (Draq5, blue), and F-actin (phalloidin, red) were visualized using confocal microscopy. Images are representative of ≥2 independent experiments.
Figure 6.
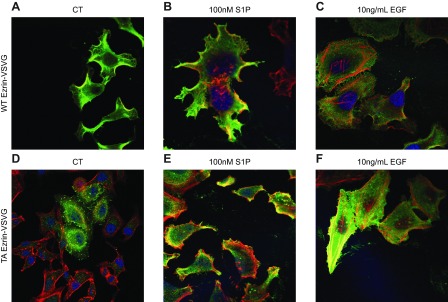
Ezrin T567 phosphorylation is required for EGF-mediated cytoskeletal changes. HeLa cells were transfected with VSV-G-tagged WT ezrin (A–C) or VSV-G-tagged nonphosphorylatable ezrin mutant T567A (D–F). Following transfection, cells were serum starved and received no treatment (A, D), 100 nM S1P treatment for 5 min (B, E), or 10 ng/ml EGF for 5 min (C, F). Cells were fixed, and VSV-G (green), nuclei (DRAQ5, blue), and F-actin (phalloidin, red) were visualized using laser-scanning confocal microscopy. Images are representative of ≥2 independent experiments.
EGF-mediated cell invasion occurs via S1PR2-dependent phosphorylation of ezrin T567
ERM protein activation and EGF have both been related to metastasis; thus, in order to assess the biological significance of EGF-mediated ERM phosphorylation and its potential role in metastasis, cell invasion assays were performed. Using trans-well chambers, invasion was measured as a readout of metastatic behavior. Nontransfected HeLa cells or HeLa cells expressing VSV-G-tagged WT ezrin or nonphosphorylatable ezrin mutant (TA) were assessed for their ability to invade through matrigel. In the presence or absence of S1PR2 antagonist, cells were allowed to invade toward complete medium (CM) or 50ng/ml EGF (EGF) for 48 h (Fig. 7). Nontransfected and WT-ezrin-expressing HeLa cells exhibited invasion toward CM with WT-ezrin-expressing cells invading more than nontransfected cells, while cells overexpressing the nonphosphorylatable TA-ezrin mutant invaded less than nontransfected HeLa cells (Fig. 7), suggesting that ezrin T567 phosphorylation plays a pivotal role in cellular invasion. Albeit at lower levels, cells invaded toward EGF in a pattern similar to that of CM, with WT-expressing cells invading the most, followed by normal HeLa cells, then TA-expressing cells, indicating that EGF-induced cell invasion is mediated by ERM proteins and specifically by phosphorylation of ezrin T567 (Fig. 7). To determine the role of S1PR2 in EGF-mediated cell invasion, cells were plated in the presence of S1PR2 inhibitor, and invasion toward EGF was evaluated. Inhibition of S1PR2 resulted in inhibition of EGF-stimulated invasion, inhibiting invasion by ≥50% under all 3 ezrin conditions (endogenous ezrin-expressing cells, P < 0.05; WT-ezrin-overexpressing cells, P < 0.05; and TA-ezrin-overexpressing cells, P=0.055; Fig. 7). In concert with the previously presented data, these findings suggest that EGF-mediated cell invasion occurs through S1PR2-dependent phosphorylation of ezrin T567 and may reveal novel targets for the development of therapeutics for pathologies that are driven by EGF, including many cancers.
Figure 7.

EGF induces invasion by S1PR2-dependent ezrin T567 phosphorylation. Nontransfected HeLa cells and cells overexpressing WT or TA ezrin were serum starved in serum-free (SF) medium for 4–6 h, plated in the presence or absence of 5 μM JTE-013 in the apical chamber of matrigel-coated trans-well inserts, and allowed to invade for 48 h toward CM or 50 ng/ml EGF. Invading cells were stained with fluorescent dye, photographed, and counted. Statistical analysis was performed using a Student's t test. *P < 0.05; aP = 0.055; n ≥ 3.
DISCUSSION
Here, a novel mechanism of EGF-induced cell invasion that requires S1P-mediated activation of ERM proteins has been identified. EGF led to stimulation of the SK/S1P pathway such that treatment with EGF resulted in elevated levels of extracellular S1P, activation of S1PR2, and ERM-dependent cell invasion. S1PR2-mediated C-terminal threonine phosphorylation of ezrin was required for EGF-induced lamellipodia formation and consequent cellular invasion. Taken together, these results have important implications for ERM regulation, for the role of the SK/S1P pathway and ERM in EGF-mediated events, particularly those involved in cancer promotion, and for the role of S1PR2 in cellular invasion, which has been unclear until now.
EGF regulates ERM proteins through the SK/S1P pathway
The effects of EGF on cell growth and proliferation have been well studied, and perhaps best studied are the roles of EGF in the initiation, maintenance, and progression of cancer (26, 27). EGF contributes to cancer development and metastasis by promoting cell motility and invasion, which both require specific, coordinated changes in the actin cytoskeleton (28). EGF promotes lamellipodia formation dependent on direct phosphorylation of ezrin by Nck-interacting kinase (NIK) in breast adenocarcinoma cells (7) and regulates invasion through ezrin tyrosine phosphorylation in ovarian cancer (29). In line with previous studies, we found that cells expressing a nonphosphorylatable ezrin mutant were unable to form lamellipodia or invade in response to EGF, suggesting that EGF-stimulated cytoskeletal rearrangement and invasion are dependent on ezrin phosphorylation at the C-terminal threonine.
The SK/S1P pathway is a secondary messaging system in response to numerous ligands, including EGF. EGF has been shown to induce acute activation, as well as late transcriptional up-regulation of SK1, both of which were required for EGF-mediated MCF7 breast cancer cell growth and motility (30). EGF has also been shown to activate the SK/S1P pathway and provoke migration of MB-MDA-453 via extracellular regulated kinase (ERK)-mediated phosphorylation and activation of SK2 (31). The latter work is in line with our current results showing that SK2 is predominantly involved in mediating EGF-induced ERM phosphorylation. Interestingly, SKX is thought to be specific for SK1 inhibition; however, it was still able to inhibit EGF-induced ERM phosphorylation, unlike SK1 siRNA. This can be due to several reasons. The inhibitor effectively removed all S1P from medium (Fig. 3A) thus leading to loss of EGF's ability to induce ERM phosphorylation; it is also possible that this inhibitor may lead to excessive ceramide accumulation, which our group previously showed is able to dephosphorylate ezrin by activating PP1α (5); and it is also possible that this inhibitor nonspecifically inhibits another kinase that phosphorylates ezrin independent of S1P. As for Ski-II, it is a pan-SK inhibitor and also inhibited EGF-induced ezrin phosphorylation, thus corroborating a role for SK2 in this pathway. In summary, the SK/S1P/S1PR2 pathway is required for EGF-induced ezrin phosphorylation, and details of the specific molecular mechanism of SK1/SK2 regulation are currently under investigation in our laboratory.
Several kinases have been reported to phosphorylate ezrin on T567, among which are several PKC isoforms (32), p38 MAP kinases (33), RhoA/ROCK (34), protein Rho kinase, G-protein-coupled receptor kinase 2 (GRK2; ref. 35), myotonic dystrophy kinase-related cell division cycle 42 (Cdc42)-binding kinase (36), and NIK (7). Interestingly, several of these kinases have also been shown to be activated by the SK/S1P pathway, such as PKCs (37), p38 MAPK (38), and RhoA (39). However, the specific kinase responsible for ezrin phosphorylation in response to SK/S1P pathway activation is also under investigation in our laboratory.
By studying EGF activation of ERM proteins, we were able to implicate the SK/S1P pathway in yet another EGF-mediated biology. In our study, EGF induced pERM through a mechanism involving the SK/S1P pathway, such that S1P generation and S1PR internalization following EGF treatment were found to be SK dependent. These findings are in agreement with previous work from our laboratory indicating that exogenously added S1P induced cytoskeletal rearrangement via activation of ERM proteins (8), as well as with work by Dudek et al. (40) implicating S1P-mediated phosphorylation of ERM proteins in lung endothelial barrier function. These data provide evidence that EGF leads to ERM phosphorylation via activation of the SK/S1P pathway and suggest that SK/S1P may participate in other EGF-mediated events, including changes in cellular architecture. Our results give merit to the idea of targeting the SK/S1P pathway in cancers with primary or acquired resistance to EGF/EGFR-targeted therapies, such as occurs in NSCLCs (15) and breast cancers (41).
EGF induces cellular invasion via S1PR2-dependent activation of ERM proteins
One of the many direct connections between EGF and S1P is highlighted in cancer cell migration and invasion. EGF induces invasion of breast cancer cells through CD44-mediated expression of matrix metalloproteinase 9 (MMP9); interestingly, CD44 is an ERM-binding protein also known to coordinate actin-based cytoskeletal reorganization (42). Like EGF, S1P induces expression of MMP9, leading to MCF10A breast cancer cell invasion (42). Also in support of a role for the SK/S1P signaling pathway in the invasive behavior of some cancers are reports of glioma cell invasion via S1PR2 and up-regulation of the metastatic inducer urokinase-type plasminogen activator (uPA; ref. 43). Supporting the aforementioned findings, the SK/S1P pathway was found to be downstream of EGF and upstream of ERM activation and invasion. Stimulation of cells with EGF led to cellular invasion through extracellular matrix, which may have occurred through a mechanism involving MMPs; however, while ezrin is intimately involved in metastasis, no correlations with MMPs have been made. It will be interesting to explore the potential connection between ezrin and MMPs. While S1PR1 and S1PR3 have already been related to cell migration and invasion, S1PR2 has opposing effects (44). In this work, a novel mechanism of EGF-stimulated invasion is unveiled, whereby S1P-mediated activation of S1PR2 and phosphorylation of ERM proteins are required. Notably, we do not discard the possibility that other S1PRs become activated following EGF treatment; however, we do propose that receptor localization is a key to determining the invasive or noninvasive result of S1PR2 activation.
Role of S1PR2 in cell migration and invasion
There is evidence supporting a major role for S1P and S1PRs in the promotion of cancer cell migration and invasion; however, many cancer-promoting functions of the SK/S1P pathway rely on S1PRs other than S1PR2; for example, S1PR1 mediates hepatocellular carcinoma metastasis (45), and S1PR3 promotes MCF10A breast cancer cell invasion (42). Contrary to the role of S1PR1 and S1PR3 in cell migration and invasion, S1PR2 has been shown to inhibit migration and invasion. S1PR2 has been reported to negatively regulate nephroblastoma tumor growth and progression (46), as well as prevent migration of hematopoietic stem cells (47) and invasion of cervical cancer cells (8). Despite evidence supporting inhibitory roles for S1PR2 in cancer, there is also evidence supporting a role for S1PR2in cancer promotion. While S1PR2 inhibits migration of glioma cells, it instead enhances invasion (48, 49). Our findings provide additional evidence supporting a positive role for S1PR2 in cancer, such that inhibition of S1PR2 impeded EGF-stimulated cell invasion. The mechanisms or circumstances encompassing the role of S1PR2in cancer prevention or promotion are not very well understood; therefore, we put forth the idea that S1PR2 cellular localization dictates its pro- or anti-invasive functions.
Induction and regulation of cell polarization, the differential cellular compartmentalization of proteins and signals (50), is an overlapping function shared by EGF and ezrin. While EGF signals for cells to undergo polarization, ERM proteins carry out this function directly. EGF induces front-rear polarity and directional migration in epidermal keratinocytes (51); moreover, EGF maintains an evolutionarily conserved role in organizing the dorsoventral axis of Drosophila by establishing ovum polarity during oogenesis (52–54). In our study, EGF induced lamellipodia formation and cell invasion, both of which were dependent on activation of ezrin, suggesting that EGF and ERM function coordinately to regulate cell polarity under these conditions. In support of our findings, ERM proteins are known to participate in the establishment of cell polarity such that mice devoid of ezrin exhibit improper development and homeostasis of the intestine, a result of disrupted apical integrity of the gut epithelium (55). While the role of the SK/S1P pathway in polarization is not well understood, there are reports of SK/S1PR2 localizing to the leading edge of migrating cells (56), reinforcing our observation that EGF-mediated cell polarization is S1PR2 dependent. Further investigation into ERM-mediated regulation of S1PR2 localization following EGF treatment is necessary; however, we suggest that S1PR2 may undergo specific cellular localization in response to EGF, leading to the asymmetrical activation of ERM proteins, which drives cell migration and invasion. Perhaps ligand-induced polarization of cells leads to localized S1P production and signaling, driving directional cell migration or invasion. In stark contrast, treating cells with exogenous S1P, in the absence of cellular polarization, inhibits migration and invasion due to global activation of ERM proteins around the entire periphery of the cell. Our data show that EGF induces cell polarity in the form of lamellipodia and mediates cell invasion via induction of ERM C-terminal threonine phosphorylation, through a mechanism involving SK and S1PR2.
Implications
The findings from this study have significant implications to cancer biology. The EGF and SK/S1P signaling pathways, along with ERM proteins, encompass important prognostic indicators of disease, especially for cancer. Components of the EGF signaling pathway currently function as markers of cancer grade and gauge treatment responsiveness and prognosis for numerous forms of cancer (57, 58). Likewise, SK/S1P pathway components are used as markers of disease, including cardiovascular disease, inflammatory disease, and cancer (59, 60). Not surprisingly, ERM proteins have great potential to be used as prognostic indicators offering information about cancer severity and metastatic potential. For example, up-regulation of ERM proteins, along with down-regulation of E-cadherin, correlates with invasion in vitro (61, 62) and is associated with lymph node metastasis in vivo (61, 63, 64). Given the role that S1PR2 plays in EGF-mediated, pERM-dependent cell invasion, it may serve as a potential prognosticator and/or biomarker of EGF and SK/S1P-mediated diseases, often occurring in cancer.
Supplementary Material
Acknowledgments
This article is based on work supported in part by Merit Award BX000156-01A1 (L.M.O.) from the Office of Research and Development, Department of Veterans Affairs, Ralph H. Johnson Veterans Affairs Medical Center (Charleston, SC, USA); U.S. Department of Education Graduate Assistance in Areas of National Need (GAANN) in Lipid Biology and New Technologies grant P200A070596 (K.A.O.G.); U.S. National Institutes of Health (NIH) grant HL-007260 (K.A.O.G.); National Cancer Institute grants R01-CA87584 (Y.A.H.) and PO1-CA97132 (Y.A.H. and L.M.O.), and NIH National Institute of General Medical Sciences grant R01-GM062887 (L.M.O.). The imaging facilities for this research were supported, in part, by Cancer Center Support Grant P30 CA138313 to the Hollings Cancer Center (Medical University of South Carolina). The content of this material does not represent the views of the U.S. Department of Veterans Affairs or the U.S. Government.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- BSA
- bovine serum albumin
- C17-Sph
- d-erythro-17-carbon sphingosine
- CM
- complete medium
- DMEM
- Dulbecco's modified Eagle's medium
- DMSO
- dimethylsulfoxide
- EGF
- epidermal growth factor
- ERM
- ezrin, radixin, and moesin
- FBS
- fetal bovine serum
- GFP
- green fluorescent protein
- MMP
- matrix metalloproteinase
- NIK
- Nck-interacting kinase
- NSCLC
- non-small cell lung cancer
- PDGF
- platelet-derived growth factor
- pERM
- phospho–ezrin, radixin, and moesin
- PIP2
- phosphatidylinositol-4,5-bisphosphate
- PP1α
- protein phosphatase 1α
- RT-PCR
- reverse transcription–polymerase chain reaction
- S1P
- d-erythro-sphingosine-1-phosphate
- S1PR1–5
- sphingosine-1-phosphate receptor 1–5
- SK
- sphingosine kinase
- SK1/2
- sphingosine kinase 1/2
- Sph
- d-erythro-sphingosine
- T558
- threonine 558
- T564
- threonine 564
- T567
- threonine 567
- T567A
- threonine 567 to alanine mutant (TA)
- TA
- threonine 567 to alanine mutant (T567A)
- WT
- wild type
REFERENCES
- 1. Neisch A. L., Fehon R. G. (2011) Ezrin, radixin and moesin: key regulators of membrane-cortex interactions and signaling. Curr. Opin. Cell Biol. 23, 377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kunda P., Rodrigues N. T., Moeendarbary E., Liu T., Ivetic A., Charras G., Baum B. (2012) PP1-mediated moesin dephosphorylation couples polar relaxation to mitotic exit. Curr. Biol. 22, 231–236 [DOI] [PubMed] [Google Scholar]
- 3. Li J., Yang H., Zhang S., Yu N., Zhou Q. (2007) [Expression and their significance of ezrin and E-cadherin in non-small cell lung cancer]. Zhongguo Fei Ai Za Zhi 10, 183–187 [DOI] [PubMed] [Google Scholar]
- 4. Louvet-Vallee S. (2000) ERM proteins: from cellular architecture to cell signaling. Biol. Cell 92, 305–316 [DOI] [PubMed] [Google Scholar]
- 5. Canals D., Roddy P., Hannun Y. A. (2012) Protein phosphatase 1alpha mediates ceramide-induced ERM Protein Dephosphorylation: a novel mechanism independent of phosphatidylinositol 4,5-biphosphate (PIP2) and myosin/erm phosphatase. J. Biol. Chem. 287, 10145–10155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fehon R. G., McClatchey A. I., Bretscher A. (2010) Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baumgartner M., Sillman A. L., Blackwood E. M., Srivastava J., Madson N., Schilling J. W., Wright J. H., Barber D. L. (2006) The Nck-interacting kinase phosphorylates ERM proteins for formation of lamellipodium by growth factors. Proc. Natl. Acad. Sci. U. S. A. 103, 13391–13396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Orr Gandy K. A., Canals D., Adada M. M., Wada M., Roddy P. L., Snider A., Hannun Y. A., Obeid L. M. (2012) Sphingosine 1-phosphate induces filopodia formation through S1P2R activation of ERM proteins. Biochem. J. 449, 661–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Canals D., Jenkins R. W., Roddy P., Hernandez-Corbacho M. J., Obeid L. M., Hannun Y. A. (2010) Differential effects of ceramide and sphingosine 1-phosphate on ERM phosphorylation: probing sphingolipid signaling at the outer plasma membrane. J. Biol. Chem. 285, 32476–32485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zeidan Y. H., Jenkins R. W., Hannun Y. A. (2008) Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J. Cell Biol. 181, 335–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paugh B. S., Paugh S. W., Bryan L., Kapitonov D., Wilczynska K. M., Gopalan S. M., Rokita H., Milstien S., Spiegel S., Kordula T. (2008) EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCdelta, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 22, 455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hait N. C., Sarkar S., Le Stunff H., Mikami A., Maceyka M., Milstien S., Spiegel S. (2005) Role of sphingosine kinase 2 in cell migration toward epidermal growth factor. J. Biol. Chem. 280, 29462–29469 [DOI] [PubMed] [Google Scholar]
- 13. Balaban N., Moni J., Shannon M., Dang L., Murphy E., Goldkorn T. (1996) The effect of ionizing radiation on signal transduction: antibodies to EGF receptor sensitize A431 cells to radiation. Biochim. Biophys. Acta 1314, 147–156 [DOI] [PubMed] [Google Scholar]
- 14. Cheng L., Zhang S., Alexander R., Yao Y., MacLennan G. T., Pan C. X., Huang J., Wang M., Montironi R., Lopez-Beltran A. (2011) The landscape of EGFR pathways and personalized management of non-small-cell lung cancer. Future Oncol. 7, 519–541 [DOI] [PubMed] [Google Scholar]
- 15. Batus M., Fidler M. J., Bonomi P. D. (2010) Primary and secondary therapeutic strategies for EGF receptor pathway inhibition in non-small-cell lung cancer. Expert Rev. Anticancer Ther. 10, 1589–1599 [DOI] [PubMed] [Google Scholar]
- 16. French K. J., Schrecengost R. S., Lee B. D., Zhuang Y., Smith S. N., Eberly J. L., Yun J. K., Smith C. D. (2003) Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 63, 5962–5969 [PubMed] [Google Scholar]
- 17. Kharel Y., Mathews T. P., Gellett A. M., Tomsig J. L., Kennedy P. C., Moyer M. L., Macdonald T. L., Lynch K. R. (2011) Sphingosine kinase type 1 inhibition reveals rapid turnover of circulating sphingosine 1-phosphate. Biochem. J. 440, 345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodgers A., Mormeneo D., Long J. S., Delgado A., Pyne N. J., Pyne S. (2009) Sphingosine 1-phosphate regulation of extracellular signal-regulated kinase-1/2 in embryonic stem cells. Stem Cells Dev. 18, 1319–1330 [DOI] [PubMed] [Google Scholar]
- 19. Bretscher A. (1989) Rapid phosphorylation and reorganization of ezrin and spectrin accompany morphological changes induced in A-431 cells by epidermal growth factor. J. Cell Biol. 108, 921–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gould K. L., Cooper J. A., Bretscher A., Hunter T. (1986) The protein-tyrosine kinase substrate, p81, is homologous to a chicken microvillar core protein. J. Cell Biol. 102, 660–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anelli V., Gault C. R., Cheng A. B., Obeid L. M. (2008) Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. Role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 283, 3365–3375 [DOI] [PubMed] [Google Scholar]
- 22. Spassieva S., Bielawski J., Anelli V., Obeid L. M. (2007) Combination of C(17) sphingoid base homologues and mass spectrometry analysis as a new approach to study sphingolipid metabolism. Methods Enzymol. 434, 233–241 [DOI] [PubMed] [Google Scholar]
- 23. Estrada-Bernal A., Lawler S. E., Nowicki M. O., Ray Chaudhury A., Van Brocklyn J. R. (2011) The role of sphingosine kinase-1 in EGFRvIII-regulated growth and survival of glioblastoma cells. J. Neurooncol. 102, 353–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sukocheva O., Wadham C., Holmes A., Albanese N., Verrier E., Feng F., Bernal A., Derian C. K., Ullrich A., Vadas M. A., Xia P. (2006) Estrogen transactivates EGFR via the sphingosine 1-phosphate receptor Edg-3: the role of sphingosine kinase-1. J. Cell Biol. 173, 301–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Libotte T., Kaiser H. W., Alt W., Bretschneider T. (2001) Polarity, protrusion-retraction dynamics and their interplay during keratinocyte cell migration. Exp. Cell Res. 270, 129–137 [DOI] [PubMed] [Google Scholar]
- 26. Takeuchi K., Ito F. (2010) EGF receptor in relation to tumor development: molecular basis of responsiveness of cancer cells to EGFR-targeting tyrosine kinase inhibitors. FEBS J. 277, 316–326 [DOI] [PubMed] [Google Scholar]
- 27. Yotsumoto F., Sanui A., Fukami T., Shirota K., Horiuchi S., Tsujioka H., Yoshizato T., Kuroki M., Miyamoto S. (2009) Efficacy of ligand-based targeting for the EGF system in cancer. Anticancer Res. 29, 4879–4885 [PubMed] [Google Scholar]
- 28. Lopez-Perez M., Salazar E. P. (2006) A role for the cytoskeleton in STAT5 activation in MCF7 human breast cancer cells stimulated with EGF. Int. J. Biochem. Cell Biol. 38, 1716–1728 [DOI] [PubMed] [Google Scholar]
- 29. Chen Z., Fadiel A., Feng Y., Ohtani K., Rutherford T., Naftolin F. (2001) Ovarian epithelial carcinoma tyrosine phosphorylation, cell proliferation, and ezrin translocation are stimulated by interleukin 1alpha and epidermal growth factor. Cancer 92, 3068–3075 [DOI] [PubMed] [Google Scholar]
- 30. Doll F., Pfeilschifter J., Huwiler A. (2005) The epidermal growth factor stimulates sphingosine kinase-1 expression and activity in the human mammary carcinoma cell line MCF7. Biochim. Biophys. Acta 1738, 72–81 [DOI] [PubMed] [Google Scholar]
- 31. Hait N. C., Bellamy A., Milstien S., Kordula T., Spiegel S. (2007) Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J. Biol. Chem. 282, 12058–12065 [DOI] [PubMed] [Google Scholar]
- 32. Ren L., Hong S. H., Cassavaugh J., Osborne T., Chou A. J., Kim S. Y., Gorlick R., Hewitt S. M., Khanna C. (2009) The actin-cytoskeleton linker protein ezrin is regulated during osteosarcoma metastasis by PKC. Oncogene 28, 792–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Antoine-Bertrand J., Ghogha A., Luangrath V., Bedford F. K., Lamarche-Vane N. (2011) The activation of ezrin-radixin-moesin proteins is regulated by netrin-1 through Src kinase and RhoA/Rho kinase activities and mediates netrin-1-induced axon outgrowth. Mol. Biol. Cell. 22, 3734–3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen Y., Wang D., Guo Z., Zhao J., Wu B., Deng H., Zhou T., Xiang H., Gao F., Yu X., Liao J., Ward T., Xia P., Emenari C., Ding X., Thompson W., Ma K., Zhu J., Aikhionbare F., Dou K., Cheng S. Y., Yao X. (2011) Rho kinase phosphorylation promotes ezrin-mediated metastasis in hepatocellular carcinoma. Cancer Res. 71, 1721–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kahsai A. W., Zhu S., Fenteany G. (2010) G protein-coupled receptor kinase 2 activates radixin, regulating membrane protrusion and motility in epithelial cells. Biochim. Biophys. Acta 1803, 300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakamura N., Oshiro N., Fukata Y., Amano M., Fukata M., Kuroda S., Matsuura Y., Leung T., Lim L., Kaibuchi K. (2000) Phosphorylation of ERM proteins at filopodia induced by Cdc42. Genes Cells 5, 571–581 [DOI] [PubMed] [Google Scholar]
- 37. Gorshkova I., He D., Berdyshev E., Usatuyk P., Burns M., Kalari S., Zhao Y., Pendyala S., Garcia J. G., Pyne N. J., Brindley D. N., Natarajan V. (2008) Protein kinase C-epsilon regulates sphingosine 1-phosphate-mediated migration of human lung endothelial cells through activation of phospholipase D2, protein kinase C-zeta, and Rac1. J. Biol. Chem. 283, 11794–11806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schenten V., Melchior C., Steinckwich N., Tschirhart E. J., Brechard S. (2011) Sphingosine kinases regulate NOX2 activity via p38 MAPK-dependent translocation of S100A8/A9. J. Leukoc. Biol. 89, 587–596 [DOI] [PubMed] [Google Scholar]
- 39. Medlin M. D., Staus D. P., Dubash A. D., Taylor J. M., Mack C. P. (2010) Sphingosine 1-phosphate receptor 2 signals through leukemia-associated RhoGEF (LARG), to promote smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 30, 1779–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adyshev D. M., Moldobaeva N. K., Elangovan V. R., Garcia J. G., Dudek S. M. (2011) Differential involvement of ezrin/radixin/moesin proteins in sphingosine 1-phosphate-induced human pulmonary endothelial cell barrier enhancement. Cell. Signal. 23, 2086–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kruser T. J., Wheeler D. L. (2010) Mechanisms of resistance to HER family targeting antibodies. Exp. Cell Res. 316, 1083–1100 [DOI] [PubMed] [Google Scholar]
- 42. Kim S., Han J., Kim J. S., Kim J. H., Choe J. H., Yang J. H., Nam S. J., Lee J. E. (2011) Silibinin suppresses EGFR ligand-induced CD44 expression through inhibition of EGFR activity in breast cancer cells. Anticancer Res. 31, 3767–3773 [PubMed] [Google Scholar]
- 43. Young N., Pearl D. K., Van Brocklyn J. R. (2009) Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol. Cancer Res. 7, 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamaguchi H., Kitayama J., Takuwa N., Arikawa K., Inoki I., Takehara K., Nagawa H., Takuwa Y. (2003) Sphingosine-1-phosphate receptor subtype-specific positive and negative regulation of Rac and haematogenous metastasis of melanoma cells. Biochem. J. 374, 715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bao M., Chen Z., Xu Y., Zhao Y., Zha R., Huang S., Liu L., Chen T., Li J., Tu H., He X. (2012) Sphingosine kinase 1 promotes tumour cell migration and invasion via the S1P/EDG1 axis in hepatocellular carcinoma. Liver Int. 32, 331–338 [DOI] [PubMed] [Google Scholar]
- 46. Li M. H., Sanchez T., Milne G. L., Morrow J. D., Hla T., Ferrer F. (2009) S1P/S1P2 signaling induces cyclooxygenase-2 expression in Wilms tumor. J. Urol. 181, 1347–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Donati C., Nincheri P., Cencetti F., Rapizzi E., Farnararo M., Bruni P. (2007) Tumor necrosis factor-alpha exerts pro-myogenic action in C2C12 myoblasts via sphingosine kinase/S1P2 signaling. FEBS Lett. 581, 4384–4388 [DOI] [PubMed] [Google Scholar]
- 48. Van Brocklyn J. R., Jackson C. A., Pearl D. K., Kotur M. S., Snyder P. J., Prior T. W. (2005) Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 64, 695–705 [DOI] [PubMed] [Google Scholar]
- 49. Van Brocklyn J., Letterle C., Snyder P., Prior T. (2002) Sphingosine-1-phosphate stimulates human glioma cell proliferation through Gi-coupled receptors: role of ERK MAP kinase and phosphatidylinositol 3-kinase beta. Cancer Lett. 181, 195–204 [DOI] [PubMed] [Google Scholar]
- 50. Stiess M., Bradke F. (2011) Neuronal polarization: the cytoskeleton leads the way. Dev. Neurobiol. 71, 430–444 [DOI] [PubMed] [Google Scholar]
- 51. Pullar C. E., Baier B. S., Kariya Y., Russell A. J., Horst B. A., Marinkovich M. P., Isseroff R. R. (2006) beta4 integrin and epidermal growth factor coordinately regulate electric field-mediated directional migration via Rac1. Mol. Biol. Cell 17, 4925–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lynch J. A., Peel A. D., Drechsler A., Averof M., Roth S. (2010) EGF signaling and the origin of axial polarity among the insects. Curr. Biol. 20, 1042–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kagesawa T., Nakamura Y., Nishikawa M., Akiyama Y., Kajiwara M., Matsuno K. (2008) Distinct activation patterns of EGF receptor signaling in the homoplastic evolution of eggshell morphology in genus Drosophila. Mech. Devel. 125, 1020–1032 [DOI] [PubMed] [Google Scholar]
- 54. Duchek P., Rorth P. (2001) Guidance of cell migration by EGF receptor signaling during Drosophila oogenesis. Science 291, 131–133 [DOI] [PubMed] [Google Scholar]
- 55. Saotome I., Curto M., McClatchey A. I. (2004) Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev. Cell 6, 855–864 [DOI] [PubMed] [Google Scholar]
- 56. Rosenfeldt H. M., Hobson J. P., Maceyka M., Olivera A., Nava V. E., Milstien S., Spiegel S. (2001) EDG-1 links the PDGF receptor to Src and focal adhesion kinase activation leading to lamellipodia formation and cell migration. FASEB J. 15, 2649–2659 [DOI] [PubMed] [Google Scholar]
- 57. Asghar U., Hawkes E., Cunningham D. (2010) Predictive and prognostic biomarkers for targeted therapy in metastatic colorectal cancer. Clin. Colorectal Cancer 9, 274–281 [DOI] [PubMed] [Google Scholar]
- 58. Domingo G., Perez C. A., Velez M., Cudris J., Raez L. E., Santos E. S. (2010) EGF receptor in lung cancer: a successful story of targeted therapy. Expert Rev. Anticancer Ther. 10, 1577–1587 [DOI] [PubMed] [Google Scholar]
- 59. Brakch N., Dormond O., Bekri S., Golshayan D., Correvon M., Mazzolai L., Steinmann B., Barbey F. (2010) Evidence for a role of sphingosine-1 phosphate in cardiovascular remodelling in Fabry disease. Eur. Heart J. 31, 67–76 [DOI] [PubMed] [Google Scholar]
- 60. Nunes J., Naymark M., Sauer L., Muhammad A., Keun H., Sturge J., Stebbing J., Waxman J., Pchejetski D. (2012) Circulating sphingosine-1-phosphate and erythrocyte sphingosine kinase-1 activity as novel biomarkers for early prostate cancer detection. Br. J. Cancer 106, 909–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhai J. W., Yang X. G., Yang F. S., Hu J. G., Hua W. X. (2010) Expression and clinical significance of Ezrin and E-cadherin in esophageal squamous cell carcinoma. Chin. J. Cancer 29, 317–320 [DOI] [PubMed] [Google Scholar]
- 62. Musial J., Sporny S., Nowicki A. (2007) Prognostic significance of E-cadherin and ezrin immunohistochemical expression in prostate cancer. Pol. J. Pathol. 58, 235–243 [PubMed] [Google Scholar]
- 63. Wang L., Gao Y., Tu Q., Hong J. (2010) [Expression of Ezrin and E-cadherin in nasopharyngeal carcinoma and its significance]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 35, 969–975 [DOI] [PubMed] [Google Scholar]
- 64. Cui Y., Li T., Zhang D., Han J. (2010) Expression of Ezrin and phosphorylated Ezrin (pEzrin) in pancreatic ductal adenocarcinoma. Cancer Invest. 28, 242–247 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.