

Abstract
Secondary alkylcarbamic acid biphenyl-3-yl esters are a class of Fatty Acid Amide Hydrolase (FAAH) inhibitors, which include the reference compounds URB597 and URB694. Given the intrinsic reactivity of the carbamate group, the in vivo potency of these molecules in rats is strongly affected by their hydrolysis in plasma or hepatic metabolism. In the present study, in vitro chemical and metabolic stability assays (rat plasma and rat liver S9 fraction) were used to investigate the structure-property relationships (SPRs) for a focused series of title compounds, where lipophilicity and steric hindrance of the carbamate N-substituent had been modulated. The resulting degradation rates indicate that a secondary or tertiary alkyl group at the carbamate nitrogen atom increases hydrolytic stability towards rat plasma esterases. The calculated solvent accessible surface area (SASA) of the carbamate fragment was employed to describe the differences observed in rate constants of hydrolysis in rat plasma (log kplasma), suggesting that stability in plasma increases if the substituent exerts a shielding effect on the carbamate carbonyl. Stability in rat liver S9 fraction is increased when a tertiary carbon is bound to the carbamate nitrogen atom, while other steric effects showed complex relationships with degradation rates. The SPRs here described may be applied at the pharmacokinetic optimization of other classes of carbamate FAAH inhibitors.
Keywords: alkylcarbamates, FAAH inhibitors, liquid chromatography, SPR, stability, rat plasma
1. Introduction
Fatty Acid Amide Hydrolase [1,2,3,4] (FAAH, EC number 3.5.1.4), is, with the more lately discovered FAAH2 [5], a mammalian member of the ‘amidase signature’ family of enzymes. It catalyzes the hydrolytic cleavage of biologically active fatty acid ethanolamides, which include the endocannabinoid agonist arachidonoylethanolamide (anandamide) [6]. FAAH also catalyzes the hydrolysis of endogenous peroxisome proliferator-activated receptor type-α agonists such as oleoylethanolamide (OEA) [7,8] and palmitoylethanolamide (PEA) [9,10,11]. These natural substrates of FAAH activity are thought to regulate many physiological processes in the central nervous system and in peripheral tissues. Furthermore, deficits in the signaling activity of these molecules have been linked to a variety of pathologic conditions, including pain, inflammation, anxiety and depression [12].
The ability of selective FAAH inhibitors to upregulate anandamide signaling at cannabinoid receptors, thus causing a variety of potentially favorable effects in animal models, has fueled the discovery of various chemical scaffolds that result in FAAH inhibition [13,14,15,16,17]. Our work has focused on cyclohexylcarbamic acid biphenyl-3-yl esters [18,19] and has led to several interesting molecules such as URB597 [18,20,21], URB694 [22,23,24] and URB937 [25] (See Figure 1). In particular, URB597 has been intensely investigated with regard to its pharmacological properties [18,26,27,28] and mechanism of action [29,30,31,32].
Figure 1.
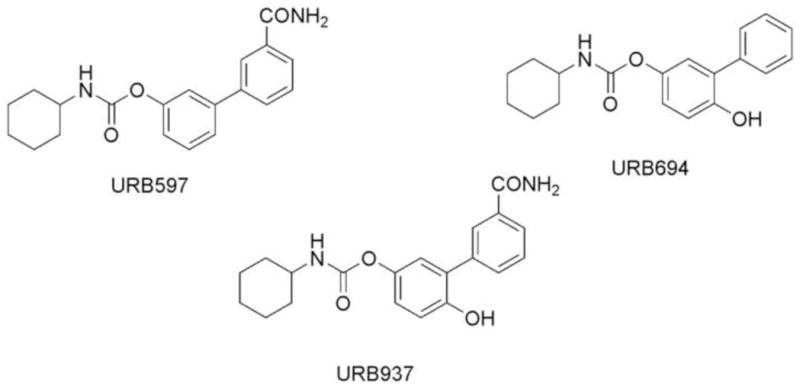
Chemical structures of URB597, URB694 and URB937.
In recent studies, we observed that intrinsic reactivity of the carbamate group influences reaction rate [22], inhibitory potency [22], off-target protein selectivity and affinity for metabolic enzymes [23,24]. In particular, small polar electrondonating groups (e.g. OH) at the para position of the proximal phenyl ring, as in URB694, are well tolerated with regard to FAAH inhibitory potency [22], and are also able to increase in vitro chemical and rat plasma hydrolytic stability [23]. This enhanced in vitro stability was also associated to improved in vivo [24] distribution and reduced inhibition of broad spectrum liver carboxylesterases, which had been reported as off-targets for URB597 when this compound is administrated at concentrations that exceed those needed to fully block FAAH activity [33].
The reactivity of the carbamate is, however, just one of the factors influencing the interaction with FAAH or with other possible targets, including different enzymes that may be carbamoylated by this group or catalyze its metabolic cleavage. Equally crucial for biological activity, in vivo selectivity and pharmacokinetic profile is molecular recognition at FAAH binding cavity, a process influenced by the overall size, shape and lipophilicity of the inhibitor. The recent publication of several co-crystal structures with FAAH and covalent [34,35,36] and non-covalent [37] inhibitors has shed new light on inhibitor-enzyme interactions within the catalytic core. The structure of humanized rat FAAH carbamoylated by URB597 confirmed the orientation of the cyclohexyl substituent within the acyl chain binding pocket and provided new information about the shape of the lipophilic channel allocating the N-substituent [35]. This structure also pointed out that carbamates monosubstituted at nitrogen are a peculiar class of FAAH inhibitors, as their amino group can make a relevant hydrogen bond in the carbamoylated enzyme. As also shown by structure-activity relationship (SAR) studies on these compounds, a primary or a secondary carbon, attached to the nitrogen atom, is well tolerated at FAAH binding site, while the tertiary carbon of an adamantyl derivative led to a drop of inhibitor potency [38]. On the other hand, as recently evidenced by comparison of in vivo potencies, carbamate stability is also affected by the size and shape of their N-substituent [24].
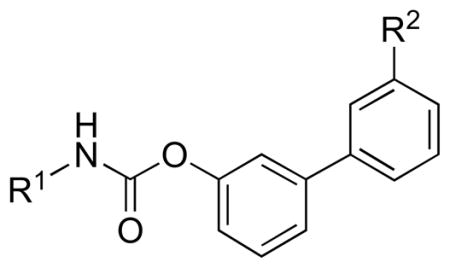
In the present study, we focused our attention on the relationship between the structure of this fragment and carbamate stability in different chemical environments, as well as in rat plasma and liver. To this aim, we employed a series of alkylcarbamic acid biphenyl-3-yl esters (compounds 1–20, Table 1), previously investigated for their SARs [38], in which the substituents at the carbamate nitrogen atom have different degrees of lipophilicity and steric hindrance. The compounds were selected in order to minimize the differences of intrinsic reactivity of the carbamate group and evaluate the sole effect produced by steric hindrance modulation at the N-portion on the molecular recognition process.
Table 1.
Alkylcarbamic acid biphenyl-3-yl esters: chemical and enzymatic stability and FAAH inhibitory potency.
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | pH 7.4a |
t1/2 (min) pH 9.0b |
t1/2 (min) 80% RPb,c |
t1/2 (min) Liver S9b |
pIC50d |
| 1 (URB524) |
|
H | 41.1±2.2%e | 40.7±2.3e | 42.7±3.7e | 6.0±1.8e | 7.20 |
| 2 (URB597) |
|
CONH2 | 40.2±1.2%e | 33.0±2.6e | 33.0±0.5e | 58.7±5.9e | 8.34 |
| 3 |
|
H | 33.9±2.3% | 43.8±0.4 | 3.8±0.6 | 7.4±0.7 | 4.86 |
| 4 |
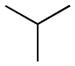
|
H | 34.6±1.1% | 61.8±0.5 | 20.0±2.0 | 1.9±0.4 | 6.28 |
| 5 |
|
H | 46.7±3.9% | 42.9±0.6 | 21.6±3.5 | 1.8±0.7 | 6.95 |
| 6 |
|
H | 46.7±1.2% | 68.3±5.9 | 7.5±2.0 | 8.0±2.0 | 7.24 |
| 7 |
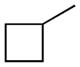
|
H | 22.6±0.2% | 27.5±0.8 | 19.9±1.2 | 2.5±0.5 | 7.27 |
| 8 |
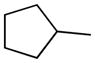
|
H | 34.4±3.6% | 45.1±3.6 | 40.0±6.0 | 2.5±0.5 | 7.47 |
| 9 |
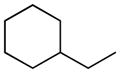
|
H | 65.4±6.2% | 104.8±12.2 | 15.3±0.2 | 9.8±2.0 | 7.18 |
| 10 |
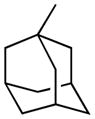
|
H | 66.7±3.8% | 260.4±14.1 | 183.3±30.9 | 99.2±1.0%f | 5.39 |
| 11 |
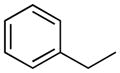
|
H | 22.7±2.3% | 26.5±3.2 | 14.3±0.7 | 10.8±0.5 | 6.86 |
| 12 |
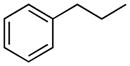
|
H | 34.9±1.8% | 40.0±1.6 | 22.1±1.6 | 2.7±0.1 | 6.32 |
| 13 |
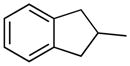
|
H | 16.2±3.3% | 22.4±1.3 | 16.5±1.1 | 63.1±1.8 | 6.67 |
| 14 |
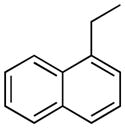
|
H | 24.8±4.1% | 38.7±5.7 | 7.3±0.3 | 46.8±5.9 | 7.23 |
| 15 |
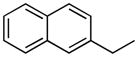
|
H | 18.4±4.1% | 23.9±1.8 | 1.0±0.1 | 27.5±0.9 | 8.27 |
| 16 |
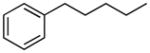
|
H | 35.9±5.4% | 42.8±2.6 | 6.6±0.7 | 19.3±2.4 | 8.03 |
| 17 |
|
H | 61.1±6.1% | 73.8±4.2 | 8.4±0.2 | 39.4±6.3 | 7.89 |
| 18 |
|
H | 30.9±8.0% | 33.4±4.1 | 8.0±0.9 | 13.0±3.1 | 7.40 |
| 19 |
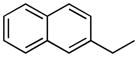
|
CONH2 | 22.8±5.4% | 13.9±0.02 | 5.6±0.9 | 39.0±0.5 | 9.20 |
| 20 |
|
CONH2 | 18.8±1.0% | 30.3±1.6 | 4.2±0.8 | 46.9±2.3 | 9.48 |
Percent of compound remaining after 24 h at 37 °C.
t1/2 values calculated from pseudo-first-order rate constants; mean ± SD, n = 3.
RP = Rat Plasma.
Published data. See reference [23].
Compound was stable for the tested time period (90 min, 37 °C).
Therefore, as reported in Table 1, the lipophilicity of substituents was increased from methyl (3) and isopropyl (4) to n-butyl (5) and n-hexyl (6). The size and length of N-alkyl substituent were varied with cycloalkyl groups such as cyclobutyl (7), cyclopentyl (8), and cyclohexylmethyl (9), arylalkyl ones such as benzyl (11) and phenethyl (12), and phenylalkyl ones of increasing lengths (16, 17). Highly bulky groups such as 1-adamantyl (10), a hydrophilic terminal group such as 6-morpholinohexyl (18), and conformationally constrained substituents, such as 2-indanyl (13), and α- and β-naphthylmethyl (14 and 15, respectively) were also included. Two derivatives bearing the hydrophilic 3′-carbamoyl group on the biphenyl-3-yl moiety (19, 20) were considered for their high in vitro FAAH inhibitory potency and to compare them with their more lipophilic unsubstituted analogues (15 and 16, respectively).
Rat plasma and rat liver S9 fraction were chosen as reference in vitro models for the hydrolytic and oxidative metabolic cleavage, respectively. Chemical stability was evaluated at physiological (7.4) and alkaline (9.0) pH values.
Experimental data were analyzed with the aim to disclose the most convenient set of physico-chemical properties (lipophilicity, steric hindrance), which would allow to keep in vitro inhibitory potency on FAAH combined with a diminished affinity for rat plasma hydrolases and liver oxidative enzymes, two critical elements for in vivo potency [24].
2. Results and discussion
The results of chemical and enzymatic stability assays on compounds 1–20 are reported in Table 1, which also lists pIC50 values for all tested compounds on rat brain membrane FAAH activity [38].
2.1. Effect of N-substitutions on chemical stability
The stability to chemical hydrolysis was evaluated by measuring residual concentrations of the compounds 1-20 at various time points in thermostated, buffered solutions at physiological (7.4) or alkaline (9.0) pH.
A 24 h cleavage of all carbamates at physiological pH yields percentages of remaining compound that range from 16.2% (13) to 66.7% (10). Hydrolysis at alkaline pH was significantly faster than that at pH 7.4, with half-lives (t1/2) ranging from 13.9 min (19) to 260.4 min (10). With the exception of the cyclohexylmethyl (9) and 1-adamantyl (10) derivatives, substitution of the N-cyclohexyl (1) group with an alkyl (3–6), cycloalkyl (7, 8) or arylalkyl (11, 12, 14-20) group did not significantly influence t1/2 values, which showed an averaged value of 39.2 min (n = 18). These results are consistent with the fact that the chemical stability of O-aryl carbamates is pH-dependent, with hydrolysis rates increasing with hydroxide ion concentration as a consequence of two concurring mechanisms, a common BAC2 and an elimination-addition one (E1cB) [39,40,41,42].
For three representative compounds (i.e. the lead compound URB524, the hindered adamantyl derivative 10 and the lipophilic phenylbutyl 16) we measured dependence of pseudo half-lives on concentration at pH 9.0. No significant difference was observed between pseudo half-lives at 1 μM, 300 nM, 100 nM (see Table 3 in Supplementary Material). This seems to rule out that variations in chemical stability can be ascribed to different solubility of the tested compounds, even if accurate solubility data should be needed to confirm this.
The intrinsic reactivity of the carbamate group was conserved along the series. The modulation of size, shape and lipophilicity of the N-substituent had, therefore, only little effect on the propensity of the carbamate to be chemically cleaved, in accordance to what we could expect from lipophilicity and steric hindrance change of the N-portion.
2.2. Effect of N-substitutions on rat plasma stability
In biological model systems endowed with high hydrolytic activity, e.g. rat plasma, the stability of carbamate-based compounds depends on their interaction with various non-specific hydrolases, where both carbamate reactivity and recognition processes may markedly influence hydrolysis rates [43].
For the present series of derivatives, no quantitative correlation between hydrolysis rate constants at alkaline pH (log kpH9) and in the presence of rat plasma (log kplasma, Table 2) could be found for compounds 1-20. No significative correlation was also found between log kplasma and calculated Lowest Unoccupied Molecular Orbital (LUMO) energy, previously employed by us as a descriptor of carbamate electrophilicity [23] (data not shown). Therefore, we may infer that additional factors other than the intrinsic reactivity of the hydrolyzable center could influence and control carbamate hydrolysis.
Table 2.
Data employed to model the stability of carbamates in biological media.
| Compound | log kplasma | log kS9 | SASAa | clogP | MR |
|---|---|---|---|---|---|
| 1 | −1.79 | −0.94 | 44.44 | 4.63 | 8.90 |
| 3 | −0.74 | −1.03 | 63.99 | 2.98 | 6.78 |
| 4 | −1.46 | −0.45 | 45.32 | 3.94 | 7.68 |
| 5 | −1.49 | −0.41 | 53.10 | 4.25 | 8.18 |
| 6 | −1.03 | −1.06 | 53.02 | 4.68 | 9.12 |
| 7 | −1.46 | −0.56 | 48.00 | 4.01 | 7.96 |
| 8 | −1.76 | −0.56 | 43.76 | 4.33 | 8.43 |
| 9 | −1.35 | −1.15 | 46.79 | 4.91 | 9.36 |
| 10 | −2.42 | b | 33.00 | 5.38 | 10.27 |
| 11 | −1.32 | −1.19 | 45.37 | 4.82 | 9.27 |
| 12 | −1.50 | −0.59 | 51.26 | 5.22 | 9.74 |
| 13 | −1.38 | −1.96 | 45.63 | 5.31 | 9.97 |
| 14 | −1.03 | −1.83 | 48.35 | 5.72 | 10.88 |
| 15 | −0.17 | −1.60 | 41.95 | 5.77 | 10.87 |
| 16 | −0.98 | −1.44 | 53.16 | 5.99 | 10.68 |
| 17 | −1.08 | −1.75 | 53.08 | 6.76 | 11.61 |
| 18 | −1.06 | −1.27 | 55.30 | 4.36 | 11.33 |
Solvent accessible surface area, Å2
Compound was stable for the tested time period (see also Table 1)
Replacing of N-cyclohexyl group of 1 (t1/2, plasma = 42.7 min) with a small alkyl group such as methyl (3) brought about in a 10-fold reduction in stability (t1/2 = 3.8 min), and such stability could not be recovered with the introduction of a long n-hexyl chain (6, t1/2 = 7.5 min). The presence of two additional alkyl substituents, i.e. isopropyl (4) and n-butyl (5), yielded t1/2 values of approximately 20 min. The cyclobutyl derivative (7, t1/2 = 19.9 min) showed a resistance to hydrolysis similar to that of its linear isomer (5), whereas the introduction of bulkier cycloalkyl groups such as cyclopentyl (8, t1/2 = 40.0 min) and, particularly, 1-adamantyl (10, t1/2 = 183.3 min) markedly increased the stability of the carbamate group to rat plasma hydrolases. Homologation, as exemplified by the methylcyclohexyl derivative (9), caused a drop in stability (t1/2 = 15.3 min).
The dependence of rat plasma stability on N-alkylaryl substitution (11–20) was more complex, as it depended on the chain length and the characteristics of the aromatic group. In general, the presence of short alkyl chains (11, 12) led to a moderate decrease in plasma stability (t1/2 = 14.3 and 22.1 min, respectively), which became critical when the phenyl ring was replaced by a naphthalene one (14, t1/2 = 7.3 min; 15, t1/2 = 1.0 min) or linked to an n-butyl (16, t1/2 = 6.6 min) or n-hexyl (17, t1/2 = 8.4 min) spacer. Finally, replacement of the aromatic portion of 17 with a ionizable morpholine group (18) had no effect on plasma stability (t1/2 = 8.0 min), suggesting that lipophilicity is not a major factor influencing interaction with plasma hydrolases. In principle, plasma protein binding can significantly affect stability of our compounds in different ways [44].
In fact, we had previously observed that albumin protects a class of prodrugs, having a carbamate fragment, from both enzymatic and chemical hydrolysis [45]; as all the present compounds resulted more stable in buffer at pH 7.4 than in plasma, protection from chemical hydrolysis looks negligible. On the other hand, different protein binding could affect the esterase-catalysed hydrolysis, thus explaining the different stabilities in rat plasma. In this regard, as it is reasonable to expect higher protein binding for more lipophilic compounds [46], the lack of correlation between lipophilicity and stability could be due to compound-specific competitions between hydrolytic enzymes and other proteins, or to the absence of a significant role for protein binding.
The introduction of a 3′-carbamoyl group on the biphenyl-3-yl moiety of compounds 15 and 16 led to compounds 19 (t1/2 = 13.9 min) and 20 (t1/2 = 30.3 min), which displayed t1/2 values that were comparable to those of their parent carbamates.
A QSPR analysis of rat plasma hydrolysis data was attempted to assess the role of different steric and lipophilic descriptors of the selected N-substituents on hydrolysis rate constants. Multiple regression analysis (MRA) employing traditional descriptors for lipophilicity (clogP) and steric hindrance (MR, L, B1, B5) [47], alone or in combination, did not yield any statistically significant model.
The 3D descriptor solvent accessible surface area (SASA) [48] for compounds 1 and 3-18 was therefore calculated for the carbamate fragment (SASAcarbamate), as a measure of the shielding effect provided by the substituents at the carbamate nitrogen, and used in the search of QSPR models (see Experimental section). In Figure 2, the SASAcarbamate (in blue colour) for the lead compound URB524 (1) is represented. Similar descriptors had been successfully applied to model kinetic data on the hydrolysis of esters by plasmatic carboxylesterases [49] or rat liver esterases [50]. While a general trend in the plot of log kplasma versus SASA can be discerned (See Figure 3), a single compound (15) behaves as an outlier, strongly affecting the correlation statistics (N=17, R2= 0.28, s=0.43, F=5.7). This structure, having a β-naphthylmethyl substituent, was less stable in rat plasma than expected, in particular if compared to the benzyl derivative 11 (t1/2= 1.0 min. vs 14.3 min, respectively) and to compounds of similar lipophilicity. Notably, the compounds 11 and 15 showed similar chemical stability at pH 9.0, suggesting that the peculiar unstability of compound 15 is likely due to specific interactions with some undefined rat plasma esterases. When 15 was excluded from the dataset, linear regression of the data represented in Figure 3 gave a statistically significant equation (Equation 1).
Figure 2.
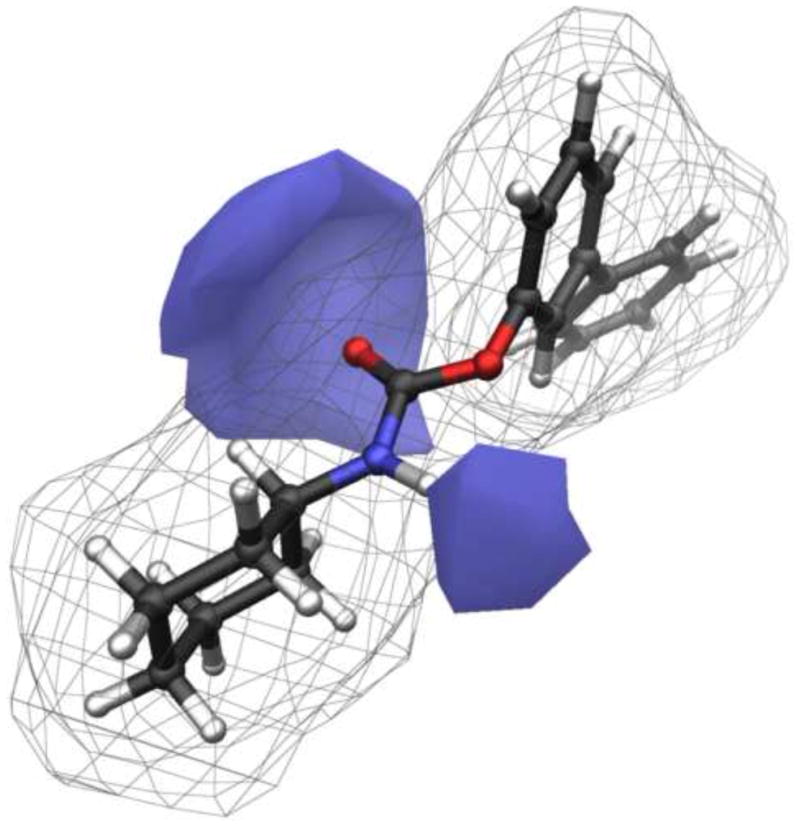
3D graphical representation of solvent accessible surface area for the carbamate group (SASAcarbamate) of URB524 (1).
Figure 3.

Plot of SASAcarbamate vs hydrolysis constants in rat plasma (log kplasma) for compounds 1 and 3-18.
| Eq. 1 |
This linear trend strongly depends on two data points, i.e. on compounds endowed with the lowest (10, adamantyl) and highest (3, methyl) SASA values. Their exclusion from the data set reduces the variation in SASA and stability. However, further exclusion of these two compounds gave an equation retaining similar regression coefficient (0.045±0.014) and standard error (0.21), even if R2 drops to 0.46, due to the reduced standard deviation of log kplasma (from 0.40 to 0.22). This is indicative of model robustness, even if anomalous behaviours, due to specific biomolecular processes, should be expected for plasma stability.
From a general point of view, from the analysis of the plot we can infer that the presence of a secondary (1, 8) or, better, tertiary (10) cycloalkyl group directly linked to the carbamate nitrogen atom is favourable to maintaining or enhancing rat plasma stability, because shielding of the carbamate carbonyl group might prevent cleavage by rat plasma enzymes. If a methylene group or even a longer alkyl chain is interposed between the bulky branched carbon atom and carbamate nitrogen, e.g. going from the N-cyclohexyl (1) to N-methylcyclohexyl (9) substituted carbamates, rat plasma stability drops.
All carbamates presenting SASA values over the 45.0 Å2 limit value (i.e. higher surface area of the carbamate fragment accessible to solvent, see also Table 2) showed lower stability in rat plasma, with pseudo half-lives (t1/2) spanning in the 4–20 min range.
An evidence on how a certain degree of in vitro liability to rat plasma hydrolytic enzymes could affect in vivo potency came from the comparison between the phenylbutyl derivative 16 and parent compound 1 (pIC50 = 8.03 vs 7.20, respectively). Despite being almost ten fold more potent in vitro than 1, the in vivo potency on FAAH inhibition of 16, expressed as ID50, was comparable (0.96 vs 0.81 mg kg−1, respectively) [24]. This result could be explained on the basis of an increased sensitivity of 16 to plasma esterases hydrolysis (t1/2, plasma = 6.6 min for 16 vs. 42.7 min for 1), also because a similar chemical stability was observed in aqueous buffer at physiological pH [23].
2.3. Effect of N-substitutions on metabolism by rat liver S9 fraction
Liver subcellular fractions, such as S9, cytosol and microsomes, obtained by differential centrifugation methods, have become reference in vitro models for drug stability studies in drug discovery setting [51]. To our aim, data of this kind could enable us to evaluate which modifications at the carbamate N-portion are related to a reduced clearance by rat liver metabolic enzymes and, therefore, are favourable to improve the in vivo half-life and oral bioavailability of the compounds [52]. All carbamates, with the sole exception of the 1-adamantyl derivative (10), which remained stable for the entire testing period, were susceptible to in vitro liver metabolism with half-lives ranging from less than 2 min for the isopropyl (4) and n-butyl (5) derivatives to over 1 hour for the 2-indanyl carbamate (13). A plot of log kS9 versus calculated molar refractivity (MR) showed a trend, which suggests that the increase in steric hindrance is inversely related to rat liver metabolic clearance (See Figure 4). A similar plot could also be drawn between log kS9 and calculated lipophilicity, expressed as clogP (clogP, see Table 2); however, since as clogP significantly correlated with MR (clogP = 0.68(± 0.02) MR −1.49(±0.28); n = 16; R2 = 0.974; s = 0.15; F = 533), it did not increase the obtained information. Among most stable compounds we can enumerate the N-alkylaryl substituted carbamates 13-15 for which conformational constraints could have reduced the overall affinity for rat liver enzymes. However, the observed trend remained qualitative; for instance, the methyl derivative (3) was much more stable than expected, if compared to the other short alkyl substituents isopropyl (4) and n-butyl (5), probably due to a poorer recognition by rat liver enzymes. No correlation was found, as expected, between rate constants in rat plasma (kplasma) and liver S9 fraction (kS9), the latter being a composite function of hydrolytic and oxidative reactions taking place within rat liver preparation [51].
Figure 4.

Plot of molar refractivity (MR) vs hydrolysis constants in rat liver S9 fraction (log kS9) for compounds 1, 3-9 and 11-18.
3. Conclusion
We have examined a series of alkylcarbamic acid biphenyl-3-yl esters to show that it is possible to regulate their rat plasma hydrolases recognition by varying the steric hindrance of the carbamate N-substituent. In particular, we have shown that the introduction of a secondary or bulkier tertiary carbon atom in close proximity to the carbamate nitrogen atom (8, 10) shields the hydrolytic attack by rat plasma esterases, if compared to what observed for linear substituents. This SPR also provides an explanation for the better in vivo/in vitro potency ratios previously observed in rats for cyclohexyl derivatives over the linear phenylalkyl ones [24]. The calculated area-based 3D descriptor SASA helped to rationalize these findings, with a trend where the hydrolysis rate constants were inversely related to the polar surface area of the carbamate fragment.
The results should be taken into consideration in the design of new carbamate-based FAAH inhibitors endowed with adequate stability toward rat plasma hydrolysis.
4. Experimental section
4.1. Chemistry
All chemicals were purchased from Sigma-Aldrich (Sigma-Aldrich srl, Cologno Monzese, Italy), and Analyticals Carlo Erba (Carlo Erba, Rodano, Italy) in the highest quality commercially available. Syntheses of compounds 1-20 were published elsewhere [24,38].
4.2. Biological Media
Rat plasma was obtained from male Wistar rats, 250–300 g (Charles River Laboratories, Milan, Italy). Animals were housed, handled, and cared for according to the European Community Council Directive 86 (609) EEC, and the experimental protocol was carried out in compliance with Italian regulations (DL 116/92) and with local ethical committee guidelines for animal research. Pooled plasma was obtained by cardiac puncture, collected into heparinized tubes, centrifuged (1,900 g, 4 °C, 10 min) using a ALC refrigerated centrifuge (ALC srl, Cologno Monzese, Italy) and stored at −70 °C until use.
Rat liver S9 fractions were obtained from the same rats, transcardially perfused with 60 mL ice-cold KCl 0.15 M. Livers were removed, weighted, sliced into small pieces, and homogenized on ice with an ice-cold phosphate-buffered saline (PBS) solution (0.01 M, pH 7.4, 20% w/v). The S9 fraction was obtained by centrifugation (9,000 g, 4 °C, 30 min) and stored at −70 °C until use. Protein content was quantified by the colorimetric Bradford method, employing bovine serum albumin (BSA) as internal standard [53].
4.3. In vitro chemical stability
Chemical stability was investigated, at fixed ionic strength (μ = 0.15 M), under physiological (0.01 M PBS, pH 7.4), and alkaline (0.01 M borate buffer, pH 9.0) pH conditions. Stock solutions of compounds were prepared in DMSO, and each sample was incubated at a final concentration of 100 nM-1 μM in pre-warmed (37 °C) buffer solution; the final DMSO concentration in the samples was kept at 1%. The samples were maintained at 37 °C in a temperature-controlled shaking water bath (60 rpm). At various time points, 100 μL aliquots were removed and analyzed by HPLC. Apparent half-lives (t1/2) for the disappearance of carbamate drugs were calculated from the pseudo-first-order rate constants obtained by linear regression of plots of log [drug] versus time plots, and are reported in Table 1 as mean values along with their standard deviations (n = 3).
4.4. In vitro enzymatic stability
Rat plasma was quickly thawed and diluted to 80% (v/v) with PBS (pH 7.4) to buffer the solution pH, which was checked during the course of experiments. Pooled rat plasma (400 μL) was incubated with PBS buffer (95 μL, pH 7.4) and compound stock solution (5 μL) in DMSO (final DMSO concentration in samples: 1%; final compound concentration: 1 μM).
In rat liver S9 fraction stability experiments, aliquots (50 μL) of liver S9 fraction were quickly thawed and incubated for 5 min at 37 °C with the NADPH-regenerating system (2 mM NADP+, 10 mM glucose-6-phosphate, 0.4 U mL−1 glucose-6-phosphate dehydrogenase, 5 mM MgCl2) in PBS (pH 7.4). At the end of the incubation period, compound stock solution (5 μL, 100 μM) in DMSO was added (final DMSO concentration in samples: 1%; final compound concentration: 1 μM). Final protein concentration in the liver S9 fraction, measured according to Bradford method with BSA as standard [53], was 2 mg mL−1.
Samples from rat plasma and rat liver S9 fraction for stability studies were maintained at 37 °C in a temperature-controlled shaking water bath (60 rpm) throughout the experiments. At regular time points, aliquots (50 μL) were withdrawn, added with two volumes of CH3CN, centrifuged at 8,000 g for 5 min at 4 °C, and analyzed by RP-HPLC. Apparent half-lives (t1/2) for the disappearance of test compounds were calculated from the pseudo-first-order rate constants obtained by linear regression of plots of log [drug] versus time; t1/2 values reported in Table 1 are the mean values of three experiments along with standard deviations.
4.5. Analytical methods
The disappearance of compounds 1, 2, 4-17, 19 and 20 was monitored by RP-HPLC employing a Shimadzu gradient system (Shimadzu Corp., Kyoto, Japan) consisting of two Shimadzu LC-10ADvp solvent delivery modules, a 20 μL Rheodyne sample injector (Rheodyne LLC, Rohnert Park, CA, USA), a SPD-10Avp UV-VIS and a fluorometric detector, equipped with a reversed-phase C18 column (LC-18-DB, 5 μm, 150 × 4.6 mm i.d.; Supelco, Bellefonte, PA, USA). The HPLC system was interfaced with PeakSimple 2.83 software for data acquisition. Mobile phases consisted of various percentages of CH3CN: 10 mM phosphate buffer, pH 7.0 (70:30→50:50, v/v) delivered at a flow rate of 1 mL min−1. Each compound was monitored at its relative absorbance maximum for UV detection and at an excitation/emission wavelength of 285/315 nm for fluorescence detection.
Compounds 3 and 18 were monitored by an API150EX single quadrupole LC-MS system (AB/Sciex, Toronto, Canada) equipped with an ESI (Electrospray ionization) Turbo IonSpray and an Atmospheric Pressure Chemical Ionization (APCI) ion sources working in positive ion mode (PIM) and coupled to an Agilent 1100 series HPLC system (Agilent Technologies, Waldbronn, Germany) constituted of a G1312A binary pump, a G1379A degasser and a 5 μL Rheodyne sample injector.
Compound-dependent parameters were optimized by flow-injection analysis (FIA) and ramping of the potentials. Final settings were: for compound 3, declustering potential (DP): 3.0 V, focusing potential (FP): 100 V, entrance potential (EP): 10 V; [M+H]+ ion was detected at m/z = 228.09. For compound 18, DP = 30 V; FP = 200 V; EP = 10 V and [M+H]+ was detected at m/z = 383.23. In both cases, higher voltages led to compound in-source fragmentation. The ion source temperature was set at 400 °C. In the ESI-MS-LC system (for 18) an Ascentis™ C18 reverse-phase column, 100 × 2.1 mm, 5 μm (Supelco, Bellefonte, PA, USA) was used; flow rate was kept at 250 μL min−1. In the APCI-MS-LC system (for 3) a Supelcosil C18-DB column, 150 × 4.6 mm, 5 μm (Supelco, Bellefonte, PA, USA) was used; flow rate was kept at 1 mL min−1. Mobile phases consisted of various percentages of 0.1% HCOOH:CH3CN (45:55→25:75, v/v). Data were acquired employing Analyst 1.4 software package (AB/Sciex, Toronto, Canada).
4.6. SPR analysis
Three-dimensional molecular models of compound 1 and 3–18 were built by applying standard tools in Macromodel [54] and their geometry optimised with OPLS2005 force field [55] until an energy gradient of 0.01 kcal/(mol · Å). The resulting structures were submitted to 20 ns of molecular dynamics simulation at 298 K, (time step for integration = 1 fs) applying OPLS2005 force field, in combination with the Surface Generalized Born continuum model (GB/SA) for water representation [56].
For each simulation, 5,000 snapshots were collected and, for all of them, SASA of the carbamate group (based on its N, H, and O atoms) was measured with Maestro software [57], using a probe-radius of 1.4 Å. Finally, for each compound, the mean value of the SASA was calculated and employed as steric descriptor in the QSPR models. The dependent variable was log kplasma, i.e. the logarithm of the apparent first-order kinetic constant observed in the presence of rat plasma, calculated as ln2/t1/2[min]. clogP values of compounds 1 and 3–18 were calculated employing Qikprop [58] as implemented in Macromodel, while MR values of the same compounds were calculated using MOE software [59].
Supplementary Material
Highlights.
Biphenyl-3-yl alkylcarbamates are a class of fatty acid amide hydrolase inhibitors
Steric effects of carbamate N-alkyl substituents can influence metabolic stability
Secondary or tertiary N-alkyl groups increase hydrolytic stability in rat plasma
Stability in rat liver S9 fraction is increased by bulky tertiary N-alkyl groups.
Solvent Accessible Surface Area (SASA) is a suitable descriptor of rat plasma hydrolysis.
Acknowledgments
This work was supported by MiUR (Ministero dell’Istruzione, dell’Università e della Ricerca), Universities of Parma and Urbino “Carlo Bo”. The S.I.T.I. (Settore Innovazione Tecnologie Informatiche) and C.I.M. (Centro Interdipartimentale Misure) of the University of Parma are gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Desarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 2.Ueda N, Kurahashi Y, Yamamoto S, Tokunaga T. Partial purification and characterization of the porcine brain enzyme hydrolyzing and synthesizing anandamide. J Biol Chem. 1995;270:23823–23827. doi: 10.1074/jbc.270.40.23823. [DOI] [PubMed] [Google Scholar]
- 3.Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta. 1995;1257:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- 4.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 5.Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem. 2006;281:36569–36578. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- 6.Devane WA, Hanuš L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 7.Rodríguez de Fonseca F, Navarro M, Gómez R, Escuredo L, Nava F, Fu J, Murillo-Rodríguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 8.Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 9.Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- 10.Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, Mor M, Russo R, Maccarrone M, Antonietti F, Duranti A, Tontini A, Cuzzocrea S, Tarzia G, Piomelli D. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci USA. 2009;106:20966–20971. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solorzano C, Antonietti F, Duranti A, Tontini A, Rivara S, Lodola A, Vacondio F, Tarzia G, Piomelli D, Mor M. Synthesis and structure-activity relationships of N-(2-oxo-3-oxetanyl)amides as N-acylethanolamine-hydrolyzing acid amidase inhibitors. J Med Chem. 2010;53:5770–5781. doi: 10.1021/jm100582w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discovery. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 13.Deng H. Recent advances in the discovery and evaluation of fatty acid amide hydrolase inhibitors. Exp Opin Drug Discovery. 2010;5:961–993. doi: 10.1517/17460441.2010.513378. [DOI] [PubMed] [Google Scholar]
- 14.Sit SY, Conway CM, Xie K, Bertekap R, Bourin C, Burris KD. Oxime carbamate–discovery of a series of novel FAAH inhibitors. Bioorg Med Chem Lett. 2010;20:1272–1277. doi: 10.1016/j.bmcl.2009.11.080. [DOI] [PubMed] [Google Scholar]
- 15.Gattinoni S, De Simone C, Dallavalle S, Fezza F, Nannei R, Battista N, Minetti P, Quattrociocchi G, Caprioli A, Borsini F, Cabri W, Penco S, Merlini L, Maccarrone M. A new group of oxime carbamates as reversible inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett. 2010;20:4406–4411. doi: 10.1016/j.bmcl.2010.06.050. [DOI] [PubMed] [Google Scholar]
- 16.Gustin DJ, Ma Z, Min X, Li Y, Hedberg C, Guimaraes C, Porter AC, Lindstrom M, Lester-Zeiner D, Xu G, Carlson TJ, Xiao S, Meleza C, Connors R, Wang Z, Kayser F. Identification of potent, noncovalent fatty acid amide hydrolase (FAAH) inhibitors. Bioorg Med Chem Lett. 2011;21:2492–2496. doi: 10.1016/j.bmcl.2011.02.052. [DOI] [PubMed] [Google Scholar]
- 17.Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta MB, Smith SE, Dudley DT, Sadagopan N, Bhattachar SN, Kesten SJ, Nomanbhoy TK, Cravatt BF, Ahn K. Discovery of PF-04457845: A highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med Chem Lett. 2011;2:91–96. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 19.Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- 20.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. Cyclohexylcarbamic acid 3′- or 4′-substituted biphenyl-3-yl esters as fatty acid amide hydrolase inhibitors: synthesis, quantitative structure-activity relationships, and molecular modeling studies. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 21.Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597) CNS Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tarzia G, Duranti A, Gatti G, Piersanti G, Tontini A, Rivara S, Lodola A, Plazzi PV, Mor M, Kathuria S, Piomelli D. Synthesis and structure-activity relationships of FAAH inhibitors: cyclohexylcarbamic acid biphenyl esters with chemical modulation at the proximal phenyl ring. ChemMedChem. 2006;1:130–139. doi: 10.1002/cmdc.200500017. [DOI] [PubMed] [Google Scholar]
- 23.Vacondio F, Silva C, Lodola A, Fioni A, Rivara S, Duranti A, Tontini A, Sanchini S, Clapper JR, Piomelli D, Mor M, Tarzia G. Structure-property relationships of a class of carbamate-based fatty acid amide hydrolase (FAAH) inhibitors: chemical and biological stability. ChemMedChem. 2009;4:1495–1504. doi: 10.1002/cmdc.200900120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, Sanchini S, Tarzia G, Mor M, Piomelli D. A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem. 2009;4:1505–1513. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clapper JR, Moreno-Sanz G, Russo R, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 2007;62:1103–1110. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Russo R, LoVerme J, La Rana G, Compton TR, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- 29.Basso E, Duranti A, Mor M, Piomelli D, Tontini A, Tarzia G, Traldi P. Tandem mass spectrometric data-FAAH inhibitory activity relationships of some carbamic acid O-aryl esters. J Mass Spectrom. 2004;39:1450–1455. doi: 10.1002/jms.729. [DOI] [PubMed] [Google Scholar]
- 30.Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lodola A, Mor M, Rivara S, Christov C, Tarzia G, Piomelli D, Mulholland AJ. Identification of productive inhibitor binding orientation in fatty acid amide hydrolase (FAAH) by QM/MM mechanistic modelling. Chem Commun. 2008:214–216. doi: 10.1039/b714136j. [DOI] [PubMed] [Google Scholar]
- 32.Lodola A, Capoferri L, Rivara S, Chudyk E, Sirirak J, Dyguda-Kazimierowicz E, Sokalski WA, Mileni M, Tarzia G, Piomelli D, Mor M, Mulholland AJ. Understanding the role of carbamate reactivity in fatty acid amide hydrolase inhibition by QM/MM mechanistic modelling. Chem Commun. 2011:2517–2519. doi: 10.1039/c0cc04937a. [DOI] [PubMed] [Google Scholar]
- 33.Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, Lannoye GS, Richardson P, Stewart A, Rogers JC, Brioni JD, Surowy CS. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology. 2007;52:1095–1105. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Mileni M, Garfunkle J, Demartino JK, Cravatt BF, Boger DL, Stevens RC. Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by α-ketoheterocycle inhibitors revealed from cocrystal structures. J Am Chem Soc. 2009;131:10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation. J Mol Biol. 2010;400:743–754. doi: 10.1016/j.jmb.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mileni M, Garfunkle J, Ezzili C, Kimball FS, Cravatt BF, Stevens RC, Boger DL. X-ray crystallographic analysis of α-ketoheterocycle inhibitors bound to a humanized variant of fatty acid amide hydrolase. J Med Chem. 2010;53:230–240. doi: 10.1021/jm9012196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Min X, Thibault ST, Porter AC, Gustin DJ, Carlson TJ, Xu H, Lindstrom M, Xu G, Uyeda C, Ma Z, Li Y, Kayser F, Walker NPC, Wang Z. Discovery and molecular basis of potent noncovalent inhibitors of fatty acid amide hydrolase (FAAH) Proc Natl Acad Sci U S A. 2011;108:7379–7384. doi: 10.1073/pnas.1016167108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mor M, Lodola A, Rivara S, Vacondio F, Duranti A, Tontini A, Sanchini S, Piersanti G, Clapper JR, King AR, Tarzia G, Piomelli D. Synthesis and quantitative structure-activity relationship of fatty acid amide hydrolase inhibitors: modulation at the N-portion of biphenyl-3-yl alkylcarbamates. J Med Chem. 2008;51:3487–3498. doi: 10.1021/jm701631z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams A, Douglas KT. Elimination-addition mechanisms of acyl transfer reactions. Chem Rev. 1975;75:627–649. [Google Scholar]
- 40.Adams P, Baron FA. Esters of carbamic acid. Chem Rev. 1965;65:567–602. [Google Scholar]
- 41.Christenson I. Alkaline hydrolysis of some carbamic acid esters. Acta Chem Scand. 1964;18:904–922. [Google Scholar]
- 42.Hegarty AF, Frost LN. Elimination–addition mechanism for the hydrolysis of carbamates. Trapping of an isocyanate intermediate by an o-amino-group. J Chem Soc Perkin Trans. 1973;2:1719–1728. [Google Scholar]
- 43.Testa B, Krämer SD. The Biochemistry of Drug Metabolism: Principles, Redox Reactions, Hydrolyses, Helvetica Chimica Acta. Zurich: 2008. pp. 201–248. [Google Scholar]
- 44.Vacondio F, Silva C, Mor M, Testa B. Qualitative structure-metabolism relationships in the hydrolysis of carbamates. Drug Metab Rev. 2010;42:551–589. doi: 10.3109/03602531003745960. [DOI] [PubMed] [Google Scholar]
- 45.Rivara M, Vacondio F, Silva C, Zuliani V, Fantini M, Bordi F, Plazzi PV, Bertoni S, Ballabeni V, Flammini L, Barocelli E, Mor M. Synthesis and stability in biological media of 1H-imidazole-1-carboxylates of ROS203, an antagonist of the histamine H3 receptor. Chem Biodivers. 2008;5:140–152. doi: 10.1002/cbdv.200890004. [DOI] [PubMed] [Google Scholar]
- 46.Fessey RE, Austin RP, Barton P, Davis AM, Wenlock MC. The role of plasma protein binding in drug discovery. In: Testa B, Krämer SD, Wunderli-Allenspach H, Folkers G, editors. Pharmacokinetic profiling in drug research. VHCA; Zurich: 2006. pp. 137–139. [Google Scholar]
- 47.Verloop A, Tipker J. Physical basis of sterimol and related steric constants. In: Hadzi D, Jerman-Blazic B, editors. Pharmacochemistry library, QSAR in Drug Design and Toxicology. Vol. 10. Elsevier; Amsterdam: 1987. pp. 97–102. [Google Scholar]
- 48.Buchwald P. Structure-metabolism relationships: steric effects and the enzymatic hydrolysis of carboxylic esters. Mini Rev Med Chem. 2001;1:101–111. doi: 10.2174/1389557013407403. [DOI] [PubMed] [Google Scholar]
- 49.Buchwald P, Bodor N. Quantitative structure-metabolism relationships: steric and nonsteric effects in the enzymatic hydrolysis of noncongener carboxylic esters. J Med Chem. 1999;42:5160–5168. doi: 10.1021/jm990145k. [DOI] [PubMed] [Google Scholar]
- 50.Hirota M, Sakakibara K, Yuzuri T, Kuroda S. Evaluation of the steric substituent effect by Ωs: reinvestigation of the reaction dependency of the steric substituent constant. J Phys Org Chem. 2001;14:788–793. [Google Scholar]
- 51.Testa B, van de Waterbeemd H, Folkers G, Guy R. Helvetica Chimica Acta. Zurich: 2001. Pharmacokinetic optimization in drug research; pp. 231–234. [Google Scholar]
- 52.Masimirembwa CM, Bredberg U, Andersson TB. Metabolic stability for drug discovery and development: pharmacokinetic and biochemical challenges. Clin Pharmacokinetics. 2003;42:515–528. doi: 10.2165/00003088-200342060-00002. [DOI] [PubMed] [Google Scholar]
- 53.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 54.MacroModel, version 9.7. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 55.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 56.Ghosh A, Sendrovic Rapp C, Friesner RA. Generalized born model based on a surface integral formulation. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 57.Maestro, version 9.0. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 58.Qikprop, version 3.2. Schrödinger, LLC; New York, NY: 2009. [Google Scholar]
- 59.Molecular Operating Environment (MOE 2008.10) C. C. G., Inc; 1255 University St. Montreal, Quebec, Canada H3B 3X3: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.