

Abstract
Narcolepsy is characterized by chronic sleepiness and cataplexy, episodes of profound muscle weakness that are often triggered by strong, positive emotions. Narcolepsy with cataplexy is caused by a loss of orexin (also known as hypocretin) signaling, but almost nothing is known about the neural mechanisms through which positive emotions trigger cataplexy. Using orexin knock-out mice as a model of narcolepsy, we found that palatable foods, especially chocolate, markedly increased cataplexy and activated neurons in the medial prefrontal cortex (mPFC). Reversible suppression of mPFC activity using an engineered chloride channel substantially reduced cataplexy induced by chocolate but did not affect spontaneous cataplexy. In addition, neurons in the mPFC innervated parts of the amygdala and lateral hypothalamus that contain neurons active during cataplexy and that innervate brainstem regions known to regulate motor tone. These observations indicate that the mPFC is a critical site through which positive emotions trigger cataplexy.
Introduction
Cataplexy is a sudden loss of muscle tone that is frequently triggered by strong emotions, especially positive emotions such as those that occur with joking, laughter, or pleasant surprise (Overeem et al., 2011). Cataplexy occurs almost exclusively in narcolepsy, a sleep disorder that is caused by loss of the neurons producing the orexin neuropeptides and characterized by chronic sleepiness and unstable sleep/wake states (Peyron et al., 2000; Thannickal et al., 2000; Scammell, 2003). Although some episodes of cataplexy are partial, affecting only the face or neck, many episodes are severe, resulting in a collapse to the ground for up to 1–2 min with preserved consciousness. Even with current treatments, cataplexy can occur many times a day, hindering an individual's ability to socialize, work, and drive safely.
Similar to people with narcolepsy, mice lacking orexin signaling have episodes of cataplexy that are increased by stimuli that probably elicit positive emotions, including running wheels, palatable food, and group housing (Chemelli et al., 1999; Willie et al., 2003; España et al., 2007; Clark et al., 2009; Scammell et al., 2009). Similarly, in narcoleptic dogs, cataplexy is strongly elicited by palatable foods or play (Mitler et al., 1974; Babcock et al., 1976). This remarkable connection between emotional stimuli and cataplexy has been appreciated for >100 years (Westphal, 1877; Schenck et al., 2007), but remarkably little is known about the underlying neural mechanisms.
Using orexin knock-out (KO) mice, we examined the brain sites activated during cataplexy induced by chocolate, a highly palatable food. We then investigated whether reversible inhibition of one of the activated areas, the medial prefrontal cortex (mPFC), which has been implicated in the regulation of motivated behaviors and affect (Rushworth and Behrens, 2008; Etkin et al., 2011), could prevent chocolate-induced cataplexy and examined the downstream circuits that may mediate this effect.
Materials and Methods
Animals.
All procedures followed National Institutes of Health guidelines and were approved by the institutional animal care and use committee of Beth Israel Deaconess Medical Center. We backcrossed orexin KO mice (a generous gift from Dr. Takeshi Sakurai, Kanazawa University Kanazawa, Japan) to C57BL/6J mice for >10 generations. In these experiments, we used male orexin KO mice and wild-type littermates weighing 20–30 g and 10–20 weeks old. The numbers of mice used in each experiment were chosen based on expected variations between animals and variability of adeno-associated viral vector (AAV) microinjections. Mice were housed at an ambient temperature of ∼22°C on a 12:12 light/dark cycle (lights on at 07:00) with ad libitum access to food and water. Mice were housed individually after AAV injections.
Generation of AAV-glutamate-gated chloride channel.
For generation of the AAV-glutamate-gated chloride channel (GluCl), the pcDNA3.1-optGluClα-EYFP and pcDNA3.1-optGluClβY182F-ECFP plasmids, originally provided by Dr. Henry Lester (California Institute of Technology, Pasadena, CA), were obtained from Addgene. The genes encoding the optGluClα-EYFP and optGluClβY182F-ECFP fusion proteins were excised from the Addgene plasmids using SpeI and XhoI and ligated into the multiple cloning site of the pACP plasmid using XbaI and SalI restriction sites. The pACP plasmid contains AAV2 inverted terminal repeats flanking a CMV promoter, a multiple cloning site, and an intron and polyadenylation signal derived from SV40. The resulting plasmids were called pACP-optGluClα and pACP-optGluClβ. Packaging of AAVs was performed using a standard triple transfection protocol to generate helper-virus-free pseudotyped AAV10 virus. Briefly, AAV-293 cells were transfected via calcium phosphate precipitation with 1.33 pmol of pHelper and 1.15 pmol each of AAV2/10 and an AAV vector plasmid (either pACP-optGluClα or pACP-optGluClβ). The cells were harvested 72 h later and the pellets resuspended in DMEM, freeze-thawed three times, and centrifuged to produce a clarified viral lysate. The vector stocks were titered by real-time PCR using an Eppendorf Realplex machine. The titer of the preparations was ∼1 × 1013 vector genome copies/ml.
Injection of AAV into the anterior cingulate cortex and prelimbic cortex.
We anesthetized mice with ketamine/xylazine (100 and 10 mg/kg, i.p.) and microinjected littermates with either AAV-GFP (coding for green fluorescent protein [GFP] expressed under a CMV promoter; Lu et al., 2006) or AAV-GluCl into the anterior cingulate cortex (ACC) and prelimbic cortex (PLC) using a glass micropipette and air pressure injector system (Lu et al., 2006). Each mouse received two 150 nl injections into the ACC/PLC of each hemisphere (1.8 mm anterior to bregma; left–right 0.4 mm; 0.9 and 1.4 mm below the dural surface). The GluClα and GluClβ constructs exceeded the packaging limit for a single AAV, so they were injected as a 1:1 mixture of AAV-GluClα and AAV-GluClβ. For anterograde tracing, we injected similar volumes of AAV-GFP unilaterally into three wild-type mice.
Electroencephalogram and electromyogram recordings.
Immediately after injection of AAV, we implanted mice with epidural screw electrodes for recording electroencephalogram (EEG) activity and fine, stainless steel wires in the neck extensor muscles to record electromyogram (EMG) activity, as described previously (Clark et al., 2009). After 10 d of recovery, mice habituated to the EEG/EMG recording cables for 3 d before recordings. The EEG/EMG signals were amplified, filtered (EEG, 0.5–30 Hz; EMG, 5–50 Hz), digitized at a sampling rate of 128 Hz, and recorded with SleepSign software (Kissei Comtec).
Sleep–wake state analysis.
We used SleepSign for preliminary scoring of wake, rapid eye movement (REM), and non-REM (NREM) sleep in 10 s epochs and then examined all epochs and made corrections when necessary. We scored cataplexy according to previously published criteria using EEG, EMG, and simultaneous video recordings (Scammell et al., 2009). Specifically, we scored behavior as cataplexy if it met four criteria: (1) the behavior consisted of an abrupt episode of nuchal atonia lasting at least 10 s, (2) the mouse was immobile during the episode, (3) the EEG showed high theta activity and low delta activity during the episode, and (4) at least 40 s of wakefulness preceded the episode.
Feeding with palatable foods.
We recorded baseline behavior for 24 h starting at 19:00 in mice with ad libitum access to regular chow. At 19:00 on the next day, we gave mice ad libitum access to 10% (w/v) sucrose, peanut butter (Jif; Smucker), or milk chocolate (one Hershey's Kiss; Hershey) along with their regular chow. We measured food consumption by weighing the remaining food 24 h later. Mice ingested 30.5 ± 1.2 ml of 10% sucrose, 2.5 ± 0.3 g of peanut butter, or 3.3 ± 0.2 g of chocolate. For the Fos study, mice were killed at 24:00 after receiving chocolate at 19:00.
Ivermectin intraperitoneal injections and behavioral recordings.
Two weeks after AAV microinjection, we injected vehicle (propylene glycol, 1 μl/g) at 07:00 (light onset); 36 h later, we recorded baseline sleep/wake behavior and cataplexy for 1 d. On the next day, we gave mice chocolate at 19:00 (dark onset) and recorded behavior. Three days later, we administered ivermectin (IVM; 0.5% w/v in vehicle; Ivomec; Merial) at a dose of 5 mg/kg at 07:00 and recorded sleep/wake behavior for the next 2 d (without and with chocolate). As a control to establish that the effects of IVM can be completely reversed, we examined a second baseline period 2 weeks later without and with chocolate. We examined behavior beginning 36 h after injection of IVM because the effects of IVM last 3 d after injection based on our results and a prior study (Lerchner et al., 2007). In addition, in pilot experiments with four orexin KO mice that had not been injected with AAV-GluCl, we found that IVM administration reduced cataplexy for about 1 d after administration, suggesting that this initial, transient reduction in cataplexy may have been due to a nonspecific effect of the IVM injection itself (Davis et al., 1999). We recorded the second baseline period 2 weeks after the IVM injection because IVM has a long half-life, with behavioral effects lasting for several days but gone by 2 weeks (Lerchner et al., 2007).
Immunohistochemistry.
Under deep anesthesia with ketamine/xylazine (200 and 20 mg/kg, i.p., respectively), we perfused mice with PBS followed by 10% (v/v) formalin. We postfixed the brains in the same fixative overnight, cryoprotected them in 20% sucrose in PBS overnight, and then cut them into 40 μm coronal sections with a sliding microtome. For immunostaining, we placed sections in 0.3% (w/w) hydrogen peroxide for 30 min and then incubated them overnight in primary antibody at room temperature. Primary antibodies included rabbit anti-c-Fos (1:20,000; PC38; Oncogene Science) raised against amino acids 4–17 of human c-Fos; rabbit anti-GFP (1:5000; A6455; Invitrogen) raised against GFP isolated directly from Aequorea victoria; rabbit anti-melanin-concentrating hormone (MCH) (1:5000; a generous gift from Dr. Terry Maratos-Flier, Harvard University, Boston, MA) raised against the entire MCH peptide conjugated to keyhole limpet hemocyanin; and goat anti-orexin-A (1:5000; SC-8070; Santa Cruz Biotechnology) raised against the C terminus of human orexin-A. The anti-GFP antiserum recognizes both yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP) and produces no staining in uninjected mice. The MCH and orexin antisera produced no staining in brains of MCH and orexin KO mice, respectively. We then incubated sections with biotinylated secondary antibody (1:1000; Vector Labs) for 90 min, followed by horseradish peroxidase-conjugated avidin-biotin complex (1:1000; Vector Labs) for 1 h. We visualized immunoreactivity using 0.06% (w/v) DAB and 0.01% hydrogen peroxide. A black product was obtained by adding 0.01% (w/v) nickel ammonium sulfate and 0.005% (w/v) cobalt chloride to the DAB solution.
Quantitative analysis of histology.
To determine the number of Fos-immunoreactive (Fos-IR) cells, we counted nuclei using a counting box placed bilaterally over an area of interest. For the ACC, we placed a 500 × 350 μm box on sections 1.98 mm rostral to bregma, with the dorsal edge of the box 700 μm below the dorsal most point at which the hemispheres meet and the medial border along the midline. We counted cells in the PLC using an identical box placed just ventral to the ACC box. For the infralimbic cortex (ILC), we placed an identical box just ventral to the PLC box. For the piriform cortex, we placed a 1000 × 300 μm box with the lateral border along the rhinal fissure and the ventral border along the ventral brain surface 1.98 mm rostral to bregma. For the basolateral nucleus of the amygdala (BLA), we placed a 500 × 500 μm box with the ventral border along the ventral terminal of the external capsule and medial border along the lateral border of the central nucleus of the amygdala (CeA) 1.46 mm caudal from bregma. For the posterior basomedial nucleus of the amygdala, we placed a 500 × 400 μm box with the dorsal lateral edge at the terminal of the external capsule 1.94 mm caudal from bregma. For the ectorhinal cortex, we placed a 700 × 400 μm box with the ventral medial edge against the dorsal edge of the lateral nucleus of the amygdala 1.94 mm caudal from bregma. For the tuberomammillary nucleus, we used a 200 × 200 μm box with the dorsal border at the dorsal edge of third ventricle and the lateral border along the lateral surface of the hypothalamus 2.54 mm caudal to bregma. To count cells in the nucleus of the solitary tract, we used a trapezoidal box (dorsal side: 200 μm; lateral side: 300 μm; ventral side; 400 μm) with the medial edge adjacent to the area postrema 7.48 mm caudal to bregma. For each of these regions, the anterior–posterior coordinates are from the Paxinos mouse brain atlas (Paxinos and Franklin, 2001). We adjusted cell counts using the Abercrombie correction factor (Guillery, 2002).
To measure the extent of GluCl expression in the mPFC, we immunostained sections for YFP/CFP and measured the area of immunoreactivity in the ACC, PLC, and ILC using ImageJ software. We chose this method over counting somata because robust expression of YFP/CFP in dendrites and axons often obscured the cell bodies. Using a fixed grayscale level across all images, we calculated the area of GluCl expression that exceeded this threshold. All sections were scored blindly. An injection was defined as successful if >50% of the counting region in the right and left ACC/PLC was immunostained. Due to the variability of mouse stereotaxic injections, some injection sites were centered outside of the ACC/PLC and served as useful anatomic controls. Staining and imaging were done under identical conditions across groups.
To quantify the innervation of the orexin and MCH neurons by the mPFC, we injected AAV-GFP into the ACC/PLC of three mice and immunostained hypothalamic sections for GFP (black) and orexin or MCH (brown). We analyzed three sections (320 μm apart) through the rostrocaudal extent of the orexin or MCH fields at 1000× and counted axon terminals immediately adjacent to and in the same focal plane as labeled soma and proximal dendrites. Results are reported as the percentage of orexin or MCH neurons receiving at least one apposition.
In vitro electrophysiology.
Two weeks after injection of AAV-GluCl, we deeply anesthetized mice with 5% (v/v) isoflurane and rapidly removed the brains. We cut 250 μm coronal slices of the mPFC in ice-cold modified artificial CSF (ACSF) and allowed them to equilibrate for 15 min at 35°C in the same solution. We then transferred the slices to an incubating chamber containing standard ACSF at 22°C until ready for recording.
For brain slice preparation, we used modified ACSF containing the following (in mm): 100 NMDG, 2.5 KCl, 20 HEPES, 25 glucose, 30 NaHCO3, 1.24 NaH2PO4, 0.5 CaCl2, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, and 10 MgSO4, pH 7.3–7.4 after bubbling with 95% O2/5% CO2 and HCl (mOsm 310). We performed recordings at 22°C with standard ACSF containing the following (in mm): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1.24 NaH2PO4, 1.3 MgCl2, 10 glucose, and 2.5 CaCl2, pH 7.3–7.4 after bubbling with 95% O2, 5% CO2. The intracellular pipette solution contained the following (in mm): 128 K-gluconate, 2 KCl, 3 MgCl2, 10 HEPES, 2 MgATP, and 0.2 NaGTP, pH 7.3 with KOH.
For whole-cell voltage-clamp and current-clamp recordings, we visualized mPFC neurons expressing YFP/CFP using an Olympus BX51WI upright fluorescent microscope equipped with infrared differential interference contrast and an infrared-sensitive CCD camera (ORCA-ER; Hamamatsu Photonics). We made whole-cell recordings using a Multiclamp 700A amplifier, a Digidata 1322A interface, and Clampex 9 software (Molecular Devices) and analyzed the data with Clampfit software.
Statistical analysis.
All data are expressed as the mean ± SEM. Statistical analyses were performed using Prism software (GraphPad). We established the normality of each dataset using the Kolmogorov–Smirnov test and compared groups using paired two-tailed Student's t tests or one- or two-way repeated-measures ANOVA followed by Fisher's PLSD test. We used Pearson's correlation analysis to determine the relationship between chocolate intake and cataplexy and Fos-IR cells and cataplexy. In all cases, p < 0.05 was taken as the level of significance.
Results
Chocolate increases cataplexy in orexin KO mice
To examine the neural mechanisms of cataplexy, we first sought a stimulus that would produce high levels of cataplexy. We tested the response of orexin KO mice to 10% sucrose solution, peanut butter, or chocolate because palatable foods can increase cataplexy in animal models of narcolepsy (Babcock et al., 1976; Clark et al., 2009) and because these specific foods are often used as rewards in rodent studies. A 10% sucrose solution and peanut butter increased cataplexy 1.6- and 2.3-fold, respectively, but chocolate produced a striking 8.1-fold increase in cataplexy compared with regular chow alone (paired t tests: 10% sucrose: t(6) = 2.64, p = 0.038, n = 7; peanut butter: t(7) = 2.62, p = 0.040, n = 7; chocolate: t(10) = 6.58, p < 0.001, n = 11; Figure 1A). The mice also preferred chocolate over regular chow when presented with both (paired t test: t(8) = 4.01, p = 0.0035, n = 9; Fig. 1B). With chocolate, mice spent 6.3 ± 0.9% of the dark period in cataplexy and the high levels of cataplexy persisted for the entire dark period, with almost no cataplexy in the light period (two-way repeated-measures ANOVA with Fisher's PLSD test for feeding condition: F(1,20) = 29.0, p < 0.001, n = 11; Fig. 1D). The episodes of cataplexy all had the typical pattern of low EMG activity with increased EEG theta power and low delta power (Scammell et al., 2009). Chocolate also increased the amount of time spent awake by 1.4-fold in the dark period; however, even after normalizing for the increase in wake, chocolate still increased cataplexy 5.0-fold in the dark period. The amount of chocolate consumed was not correlated with the number of cataplexy bouts (r = 0.11, p = 0.75, n = 11; Fig. 1E). This lack of a dose–response relationship suggests that the increase in cataplexy was not due to a chemical constituent of chocolate causing the cataplexy, but rather to the hedonic characteristics of chocolate. Although it is difficult to infer the emotional state of mice, this striking increase in cataplexy with chocolate suggests that this highly palatable food may induce positive emotions that trigger cataplexy, as has been described in dogs and people with narcolepsy (Reid et al., 1994; Overeem et al., 2011).
Figure 1.
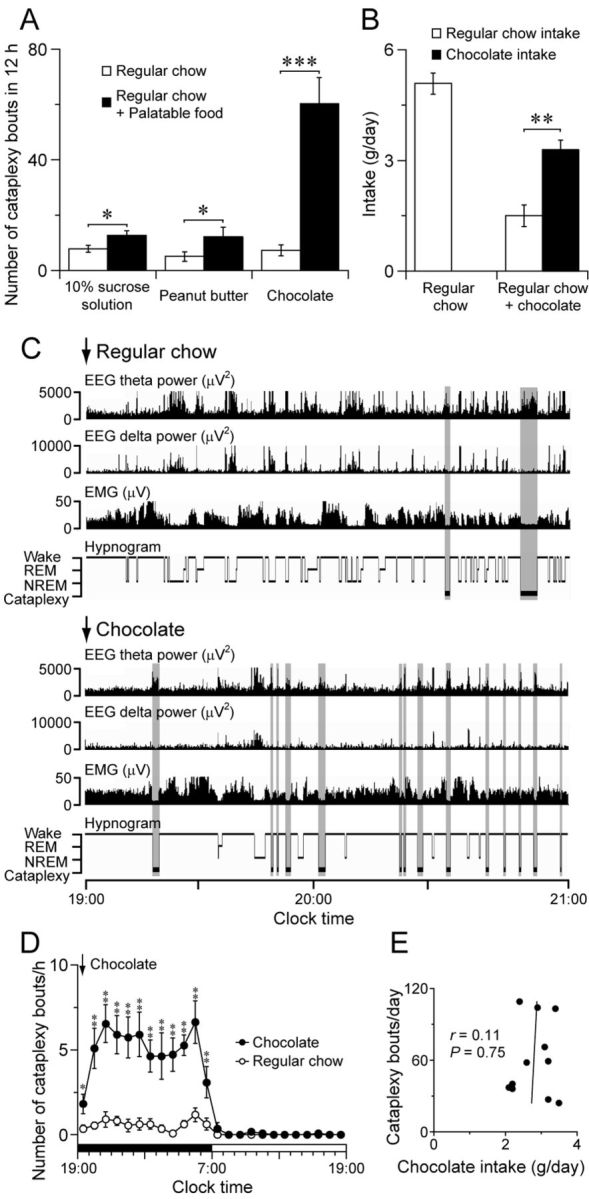
Chocolate increases cataplexy in orexin KO mice. A, Palatable foods increase cataplexy during the dark period; chocolate increases the number of cataplexy bouts 8.1-fold compared with regular chow alone (10% sucrose solution: n = 7; peanut butter: n = 7; chocolate: n = 11). B, Orexin KO mice prefer to eat chocolate over regular chow (n = 9). C, Typical examples of EEG theta power (4–9 Hz), EEG delta power (0.5–4 Hz), EMG, and hypnogram of an orexin KO mouse in the absence (top) or presence (bottom) of chocolate demonstrating the large increase in cataplexy with chocolate. Cataplexy bouts are highlighted in gray. D, Chocolate increases cataplexy across the dark period (n = 11). E, Chocolate consumption is not correlated with cataplexy (n = 11). *p < 0.05; **p < 0.01; ***p < 0.001.
Identifying brain regions active in association with cataplexy
We next used Fos immunostaining to identify brain regions activated in this cataplexy-promoting condition. We gave chocolate to orexin KO mice at dark onset and found increased Fos expression in 27 brain regions compared with mice fed only chow at the same time period (Table 1). Because some mice exhibited more cataplexy than others, we also examined the correlation between Fos expression in those 27 areas and the number of cataplexy bouts during the 3 h before killing of the mice given chocolate. Fos expression was correlated with cataplexy in brain regions that regulate olfaction, such as the piriform cortex (r = 0.91, p < 0.0001, n = 13), and areas involved in affect and motivation, such as the BLA (r = 0.90, p < 0.0001, n = 14), posterior basomedial nucleus of the amygdala (r = 0.90, p < 0.0001, n = 13), the ectorhinal cortex (r = 0.88, p < 0.0001, n = 13), and the mPFC (Figs. 2, 3). Within the mPFC, Fos expression was correlated with cataplexy in the ACC (r = 0.96, p < 0.0001, n = 12) and PLC (r = 0.94, p < 0.0001, n = 12), but not in the ILC (r = 0.34, p = 0.25, n = 12).
Table 1.
Brain regions with increased expression of Fos in orexin KO mice given chocolate
| Area | Chocolate | Regular chow |
|---|---|---|
| ACC | +++ | + |
| PLC | +++ | + |
| Agranular insular cortex | ++ | + |
| Orbitofrontal cortex | +++ | ++ |
| Piriform cortex | ++++ | ++ |
| Ectorhinal cortex | ++ | + |
| Dorsal tenia tecta | +++ | ++ |
| Claustrum | +++ | + |
| Nucleus accumbens | + | 0 |
| Bed nucleus of stria terminalis (lateral dorsal) | ++ | + |
| Anterior cortical nucleus of the amygdala | +++ | ++ |
| BLA | +++ | + |
| CeA | ++ | + |
| Posterior basomedial amygdala | +++ | + |
| Arcuate nucleus | +++ | + |
| Paraventricular hypothalamic nucleus | ++++ | ++ |
| Dorsomedial hypothalamic nucleus | +++ | ++ |
| Lateral hypothalamus | ++++ | +++ |
| Posterior hypothalamus | +++ | + |
| Paraventricular thalamic nucleus | ++++ | + |
| Rhomboid thalamic nucleus | +++ | + |
| Subparafascicular thalamic nucleus | ++++ | ++ |
| Parasubthalamic nucleus | ++++ | + |
| Prerubral field | +++ | + |
| Anterior pretectal nucleus | +++ | + |
| Edinger–Westphal nucleus | +++ | + |
| Nucleus of the solitary tract | ++++ | + |
Fos expression was scored as follows: 0, none; +, scarce; ++, low; +++, moderate; and ++++, high.
Figure 2.

Fos expression is positively correlated with cataplexy in several brain regions that regulate olfaction and affect. The number of cataplexy bouts during the 3 h before killing mice given chocolate is positively correlated with the number of Fos-IR cells in the piriform cortex, BLA, posterior basomedial nucleus of the amygdala, ectorhinal cortex, ACC, and PLC, but not in the ILC. Insets show the counting box for each region. Chocolate: n = 12–14; regular chow: n = 7–9.
Figure 3.

Fos expression in the mPFC is correlated with cataplexy. In the dorsal portion of the mPFC of orexin KO mice, Fos is heavily expressed in mice with frequent cataplexy and less so in mice with infrequent cataplexy in the 3 h before killing. Scale bar, 500 μm. fmi indicates forceps minor of the corpus callosum.
Other brain regions had increases in Fos unassociated with cataplexy. For example, Fos expression in the tuberomammillary nucleus was correlated with the amount of wakefulness (r = 0.53, p = 0.03, n = 14; data not shown), but not with the number of cataplexy bouts (r = 0.26, p = 0.38, n = 14). Chocolate also increased Fos in the nucleus of the solitary tract, which relays gastric information to the forebrain, but Fos expression was not correlated with the number of cataplexy bouts (r = 0.12, p = 0.70, n = 12).
The mPFC is required for chocolate-induced cataplexy
Although it is possible that this Fos expression could represent activity related to the consumption of chocolate or the emotional state, we hypothesized that the increases in Fos that were correlated with cataplexy might identify regions involved in triggering of cataplexy. Among the brain regions activated in cataplexy-inducing conditions, the mPFC may be especially critical for cataplexy, because prior research suggests that this area is activated in association with positive emotions (Damasio et al., 2000; Knutson et al., 2003; Rogers et al., 2004; Sabatinelli et al., 2007; Rushworth and Behrens, 2008; Etkin et al., 2011). To determine whether the mPFC is necessary for cataplexy, as opposed to covarying in activity with the emotional state that produces cataplexy, we reversibly inhibited neurons of the mPFC by focally expressing a modified chloride ion channel (GluCl) found in invertebrates that can be activated by the antiparasitic drug IVM. We chose this approach because IVM has a long half-life; prolonged, stable effects on behavior; is reversible; and at the doses used in this study, has little effect on mammalian neurons lacking the GluCl channel (Slimko et al., 2002; Lerchner et al., 2007).
To determine whether IVM could inhibit ACC/PLC neurons, we microinjected the mPFC of wild-type mice with AAV coding for the GluCl α and β subunits fused to YFP and CFP, respectively (AAV-GluCl). Two weeks later, we performed patch-clamp recordings on 12 fluorescently labeled neurons in frontal cortex slices (Fig. 4). ACC/PLC neurons had a mean resting membrane potential of −53.5 ± 1.8 mV (n = 12) and only two fired spontaneously, although all fired when injected with depolarizing currents. Six of these neurons were inhibited by IVM (50 nm), with membrane hyperpolarization and cessation of firing. In addition, voltage-clamp recordings in these same cells demonstrated an IVM-mediated outward current (Vh = −60 mV), which is consistent with an influx of Cl− (estimated ECl = −74 mV). The effect of IVM was reversible after a wash-out period of 45 min. Another six neurons displayed no response to IVM despite expressing YFP or CFP, indicating that these cells probably expressed only one of the subunits (α or β) required for the assembly of a functional GluCl channel, as described previously (Lerchner et al., 2007). In control mice, we microinjected AAV-GFP into the ACC/PLC, and IVM had no effect on any of the 12 GFP-labeled neurons, indicating that the response to IVM described above is mediated by the GluCl channel.
Figure 4.

IVM acts via GluCl channels to inhibit mPFC neurons. A, After an injection of AAV-GluCl, neurons in the ACC and PLC express marker proteins (YFP or CFP, middle) and can be visualized under infrared-differential interference contrast (right). B, C, In a mPFC neuron expressing GluCl, IVM (50 nm, black bar above traces) silences firing (B; current-clamp recording) and induces an outward current indicated by the bracket, hyperpolarizing the membrane (C; voltage-clamp recording; Vh = −60 mV). D, In a control mouse injected with AAV-GFP, IVM has no effect on a GFP-labeled neuron (voltage-clamp recording; Vh = −60 mV).
Next, to determine whether IVM inhibits mPFC neurons in vivo, we examined Fos expression in wild-type mice with unilateral injections of AAV-GluCl in the ACC and PLC. In mice given chocolate 1, 3, and 5 d after IVM injection (n = 3–4/group), IVM unilaterally reduced the number of Fos-IR cells by 39 ± 14%, 61 ± 14%, and 29 ± 8%, respectively, within the ACC/PLC region. In four control mice that did not receive IVM, unilateral expression of AAV-GluCl did not change the number of Fos-IR cells (−3 ± 7%). This result suggests that IVM produces long-lasting reductions in mPFC neuronal activity through GluCl, as was demonstrated in a prior study of striatal neurons (Lerchner et al., 2007).
We then investigated whether inactivation of the mPFC reduces cataplexy in orexin KO mice. We injected AAV-GluCl bilaterally into the ACC and PLC and then measured cataplexy and sleep/wake behavior after injection of vehicle (baseline 1) or IVM on nights with and without chocolate (Fig. 5). To determine whether the response to IVM is reversible, we recorded behavior after a second injection of vehicle 2 weeks later (baseline 2). We similarly tested a control group of orexin KO mice injected with AAV-GFP into the ACC/PLC.
Figure 5.

The ACC/PLC is necessary for chocolate-induced cataplexy. A, Typical injection site showing GluCl expression in the ACC and PLC. Scale bar, 500 μm. B, Experimental design to test the effect of reversible silencing of the ACC/PLC using IVM on cataplexy. C, Effect of IVM on cataplexy bouts, wakefulness, and chocolate intake of orexin KO mice injected with AAV-GluCl (n = 5, top) or AAV-GFP (controls, n = 5, bottom). In mice injected with AAV-GluCl, IVM reversibly reduces the number of cataplexy bouts with chocolate, but has no effect in the baseline condition. *p < 0.05; **p < 0.01. D, Superimposed GluCl injection sites in the ACC/PLC (n = 5). E, Mice with more GluCl expression in the ACC/PLC have greater reductions in cataplexy. This correlation was not apparent in the ILC (n = 17).
In five mice with the most robust expression of GluCl in the ACC/PLC (Fig. 5C), IVM had no effect on cataplexy on nights when mice had regular chow. In contrast, on nights with chocolate, IVM reduced the number of cataplexy bouts by 51% compared with vehicle (one-way repeated-measures ANOVA with Fisher's PLSD test: F(2,8) = 15.9, p < 0.01). IVM had no effects on wakefulness, REM, or NREM sleep amounts (data not shown) or on chocolate consumption. In 12 mice with injections of AAV-GluCl that were unilateral or that partially missed the mPFC, IVM reduced cataplexy only 14%. The response to IVM was mediated by the GluCl channel, because IVM had no effect on cataplexy in five control orexin KO mice expressing GFP in the ACC/PLC.
In the entire group of 17 mice with AAV-GluCl injections bilaterally involving the mPFC, the reduction in cataplexy was correlated with the GluCl-positive area in the ACC/PLC (r = 0.67, p < 0.005; Fig. 5E). This correlation was not apparent in the ILC. These data demonstrate that the ACC/PLC is necessary for chocolate-induced cataplexy in orexin KO mice.
Identifying ACC/PLC targets that may regulate cataplexy
To investigate the neural pathways through which the ACC/PLC may drive cataplexy, we microinjected AAV-GFP into the ACC/PLC of wild-type mice to anterogradely label the projections of this region (Fig. 6). ACC/PLC neurons innervated a variety of brain regions, such as the olfactory tubercle, lateral preoptic area, lateral hypothalamus, periaqueductal gray matter, and dorsal raphe nucleus, as well as areas involved in affect and motivated behaviors such as the BLA, nucleus accumbens, and ventral tegmental area, in a pattern very similar to that previously reported in rats (Sesack et al., 1989; Hurley et al., 1991; Vertes, 2004). Neurons of the BLA may signal emotional salience, especially with feeding, social play, and other rewarding conditions (Murray, 2007; Trezza et al., 2012), and we found that Fos expression in BLA neurons was positively correlated with cataplexy in orexin KO mice that were fed chocolate (Fig. 2), suggesting that the BLA may also regulate cataplexy.
Figure 6.

The ACC/PLC innervates brain regions that may regulate cataplexy. A, B, Many ACC/PLC neurons express GFP immunoreactivity after injection of the anterograde tracer AAV-GFP and they heavily innervate the BLA. C, GFP-labeled axons (black) also closely appose MCH neurons (brown). D, E, Double labeling for Fos (black) and MCH (brown) shows that Fos expression in the MCH neurons is positively correlated with the number of cataplexy bouts (chocolate: n = 12; regular chow: n = 8). F, GFP-labeled axons from the ACC/PLC (black) closely appose orexin neurons (brown). Scale bars, 200 μm (A, B,D) and 20 μm (C,F). BMA indicates anterior basomedial nucleus of the amygdala; opt, optic tract.
We also found that axon terminals from the ACC/PLC innervated a moderate number of neurons in the lateral hypothalamus that produce MCH. Specifically, GFP-IR nerve terminals closely apposed 61 ± 9% of the MCH neurons ipsilateral to the injection site (n = 3). Appositions were more common in the perifornical part of the MCH field. The MCH neurons are mainly active during REM sleep (Verret et al., 2003; Hassani et al., 2009) and may play an essential role in triggering muscle atonia and EEG theta activity in REM sleep and cataplexy. To determine whether the MCH neurons are active under cataplexy-inducing conditions, we examined Fos expression in MCH neurons of orexin KO mice given chocolate. We found a strong positive correlation between the number of cataplexy bouts and Fos expression in the MCH neurons (r = 0.93, p < 0.001, n = 12; Fig. 6E). The absolute number of MCH neurons expressing Fos was not large, but the correlation was strong, suggesting that the ACC/PLC may trigger cataplexy at least in part through a subset of MCH neurons.
Finally, we found that the ACC/PLC innervated approximately half of the orexin neurons. Ipsilateral to the injection site, GFP-IR nerve terminals closely apposed 47 ± 7% of the orexin neurons in the lateral half of the orexin field and 50 ± 14% in the medial half (n = 3). There were no obvious variations along the rostrocaudal extent of the orexin field and appositions were sparse contralaterally. Unlike the MCH and BLA neurons, the orexin neurons may play an opposite role, acting upon brainstem pathways and motor neurons to maintain muscle tone and prevent cataplexy in mice and humans with an intact orexin system (Mileykovskiy et al., 2005; Blouin et al., 2013). However, the loss of orexin neurons in animals or humans with narcolepsy may upset this balance so that when positive emotional stimuli activate the ACC/PLC, BLA, and MCH neurons, their inhibition of brainstem regions regulating muscle tone is unopposed, resulting in cataplexy.
Discussion
For >100 years, researchers have known that cataplexy is often triggered by positive emotional stimuli, but the neural circuitry linking emotions to cataplexy has remained unknown. We found here that chocolate, a rewarding stimulus, activated neurons in the ACC and PLC and markedly increased cataplexy in orexin KO mice. Reversible inhibition of the ACC/PLC with IVM strongly suppressed the increase in cataplexy with chocolate, suggesting that the ACC/PLC is necessary for the triggering of cataplexy by positive emotions. In addition, the ACC/PLC innervates MCH and BLA neurons and these target neurons were also activated with cataplexy. These findings shed light on specific neural pathways that may trigger cataplexy through positive emotions.
Technical considerations
These experiments highlight the importance of the ACC/PLC in cataplexy, but some methodological limitations should be kept in mind. In several brain regions, we observed increased expression of Fos that was correlated with cataplexy. Activation of these neurons suggests that they could be important for cataplexy, but this needs to be tested directly with additional experiments such as those using IVM. In addition, the expression of Fos most likely reflects patterns of neural activity extending over several hours and it may lack the temporal resolution to examine infrequent, brief states such as cataplexy. Even with chocolate, mice spent only approximately 6% of the dark period in cataplexy and we did not see increased Fos in brain regions that promote muscle atonia, such as the sublaterodorsal nucleus (SLD) (Lu et al., 2006; Luppi et al., 2012). Therefore, increased expression of Fos in regions such as the ACC, PLC, and BLA probably reflects the emotional tone that causes frequent cataplexy rather than the occurrence of cataplexy itself.
Second, we selected chocolate as a stimulus that should trigger positive affect because it is frequently used in studies of reward in mice (Hikida et al., 2010; Dragoi and Tonegawa, 2011). However, chocolate contains psychoactive compounds such as caffeine and theobromine that could potentially affect cataplexy. In our experiments, chocolate clearly increased wakefulness (and cataplexy only occurs during wakefulness), but even after normalizing for the amounts of wake time, chocolate still markedly increased cataplexy. Prior studies have shown that caffeine either has no effect or reduces cataplexy in narcoleptic mice (Willie et al., 2003; Okuro et al., 2010) and we found no correlation between cataplexy and the amount of chocolate consumed, suggesting that the effect is not caused directly by a chemical constituent of chocolate. We think it is most likely that the rewarding aspects of chocolate best explain the increase in cataplexy.
Finally, in mice with robust GluCl expression in the ACC and PLC, IVM reduced the frequency of cataplexy by 50% but did not prevent it entirely. The persistence of some cataplexy may indicate alternative pathways for promoting it, but it is likely that GluCl only partially inhibited the ACC/PLC in our experiment. In slice recordings, we found that half of the fluorescently labeled neurons did not respond to IVM, probably because these cells expressed insufficient amounts of the α or β subunit of GluCl and both are required to produce a functional channel. Although this inhibition was sufficient to reduce cataplexy by approximately 50%, it may be possible in future studies to reduce neuronal activity more thoroughly using a viral vector that enables inhibition of a larger percentage of ACC/PLC neurons.
mPFC and the neurobiology of positive affect
Several lines of evidence suggest that the mPFC mediates positive emotions in humans and animals. Activity in the mPFC, as measured by PET, is correlated with subjective feelings of happiness (Damasio et al., 2000) and functional MRI studies show that the mPFC is selectively activated by pleasurable visual stimuli (Sabatinelli et al., 2007) and reward (Knutson et al., 2003; Rogers et al., 2004). In addition, the mPFC is activated by positive outcomes in controls and in patients with narcolepsy (Ponz et al., 2010). Inspired by these clinical observations, researchers have begun to examine the neural mechanisms of positive affect in animals. In rats, chocolate and sucrose increase Fos expression in the mPFC (Schroeder et al., 2001; Mitra et al., 2011). Electrical stimulation of the mPFC in rats robustly elicits ultrasonic vocalizations that have been proposed to reflect pleasure or positive emotions (Burgdorf et al., 2007). Conversely, pharmacological inactivation of the mPFC reduces reward-seeking behaviors (Ishikawa et al., 2008a, b). These observations indicate that the mPFC plays a key role in positive emotional experience. We therefore focused on whether the mPFC is a key brain region for mediating cataplexy in response to positive emotional stimuli.
Model for forebrain regulation of cataplexy
Our findings not only indicate that the mPFC does indeed contribute to cataplexy produced by positive emotional stimuli, but also shed light on some of the forebrain circuitry through which this may occur (Fig. 7). Most likely, the brainstem pathways that generate atonia during cataplexy are the same as those active during REM sleep because the two states have many similarities (Scammell et al., 2009; Burgess and Scammell, 2012). The SLD produces atonia by exciting interneurons in the medulla and spinal cord that inhibit motoneurons (Lu et al., 2006; Krenzer et al., 2011; Luppi et al., 2012) and, during wakefulness, the SLD is inhibited by neurons in the ventrolateral periaqueductal gray matter and adjacent lateral pontine tegmentum (vlPAG/LPT).
Figure 7.

A model for the triggering of cataplexy by positive emotions. A, In wild-type mice, a positive stimulus such as chocolate activates the ACC and PLC that excite orexin, MCH, and amygdala neurons. The MCH and amygdala neurons inhibit the vlPAG/LPT neurons, but this inhibition is normally offset by excitation from the orexin neurons, resulting in sustained inhibition of the SLD by the vlPAG/LPT and no substantial loss of muscle tone. B, In orexin KO mice, the MCH and amygdala neurons are activated by the ACC and PLC, but in the absence of orexins, these systems can reduce activity in the vlPAG/LPT, leading to activation of the SLD and cataplexy. For simplicity, additional systems that regulate cataplexy and motor tone, including neurons producing norepinephrine, serotonin, and dopamine, are not shown in this model.
We found that the ACC/PLC of mice innervates the BLA, as was shown previously in rats (Vertes, 2004). This is probably an excitatory projection because the synapses appear asymmetrical (Brinley-Reed et al., 1995) and BLA neurons also express Fos in relation to cataplexy. The BLA may then relay signals to the CeA (Savander et al., 1995), because cataplexy-active neurons have been identified in both regions of narcoleptic dogs (Gulyani et al., 2002). The CeA sends inhibitory inputs to the vlPAG/LPT (Rizvi et al., 1991; Oka et al., 2008) and, in separate studies, we found that excitotoxic lesions of the amygdala reduce cataplexy by approximately 50% without affecting sleep, suggesting that it too is necessary for triggering cataplexy (Burgess et al., 2013). We also observed that the ACC/PLC innervates MCH and orexin neurons in the lateral hypothalamus. MCH neurons are active during REM sleep (Verret et al., 2003; Hassani et al., 2009) and send inhibitory projection to the vlPAG/LPT (Clément et al., 2012). We have found that at least some of the MCH neurons are active in proportion to the amount of cataplexy, suggesting that they may also help inhibit the vlPAG/LPT. Normally, during strong, positive emotional experiences, these inhibitory inputs to the vlPAG/LPT may be counterbalanced by excitatory inputs from the orexin neurons (Peyron et al., 1998; Blouin et al., 2013), but when orexin signaling is lost in narcolepsy, we propose that the inhibition is unopposed, resulting in cataplexy.
Our results demonstrate that the ACC and PLC, regions long known to play key roles in positive affect and motivation, are necessary for the triggering of cataplexy by chocolate. The findings of this study provide a rough framework for the mechanisms that mediate the triggering of cataplexy by positive emotions, but it is likely that additional brain regions are also important, including those implicated in reward and the hedonic aspects of feeding, such as the basal forebrain and mesolimbic pathways (Berridge et al., 2010). From a therapeutic perspective, as we learn more about the specific connections through which the forebrain regulates cataplexy, it may become possible to develop more effective and rational therapies to target the limbic pathways that trigger cataplexy without affecting other aspects of emotion.
Footnotes
This work was supported by the Japan Society for the Promotion of Science (Postdoctoral Fellowship for Research Abroad), the G. Harold and Leila Y. Mathers Foundation, and the National Institutes of Health (Grant #NS055367, Grant #HL095491, Grant #NS061863, and Grant #NS073613). We thank C. Bass for assistance in generating the adeno-associated viral vectors containing the GluCl subunits and M. Yamamoto for superb technical assistance.
The authors declare no competing financial interests.
References
- Babcock DA, Narver EL, Dement WC, Mitler MM. Effects of imipramine, chlorimipramine, and fluoxetine on cataplexy in dogs. Pharmacol Biochem Behav. 1976;5:599–602. doi: 10.1016/0091-3057(76)90298-7. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Ho CY, Richard JM, DiFeliceantonio AG. The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain Res. 2010;1350:43–64. doi: 10.1016/j.brainres.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin AM, Fried I, Wilson CL, Staba RJ, Behnke EJ, Lam HA, Maidment NT, Karlsson KÆ, Lapierre JL, Siegel JM. Human hypocretin and melanin-concentrating hormone levels are linked to emotion and social interaction. Nat Commun. 2013;4:1547. doi: 10.1038/ncomms2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinley-Reed M, Mascagni F, McDonald AJ. Synaptology of prefrontal cortical projections to the basolateral amygdala: an electron microscopic study in the rat. Neurosci Lett. 1995;202:45–48. doi: 10.1016/0304-3940(95)12212-5. [DOI] [PubMed] [Google Scholar]
- Burgdorf J, Wood PL, Kroes RA, Moskal JR, Panksepp J. Neurobiology of 50-kHz ultrasonic vocalizations in rats: electrode mapping, lesion, and pharmacology studies. Behav Brain Res. 2007;182:274–283. doi: 10.1016/j.bbr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Burgess CR, Scammell TE. Narcolepsy: neural mechanisms of sleepiness and cataplexy. J Neurosci. 2012;32:12305–12311. doi: 10.1523/JNEUROSCI.2630-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess CR, Oishi Y, Mochizuki T, Peever JH, Scammell TE. Amygdala lesions reduce cataplexy in Orexin KO mice. J Neurosci. 2013;33:9734–9742. doi: 10.1523/JNEUROSCI.5632-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/S0092-8674(00)81973-X. [DOI] [PubMed] [Google Scholar]
- Clark EL, Baumann CR, Cano G, Scammell TE, Mochizuki T. Feeding-elicited cataplexy in orexin knockout mice. Neuroscience. 2009;161:970–977. doi: 10.1016/j.neuroscience.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clément O, Sapin E, Libourel PA, Arthaud S, Brischoux F, Fort P, Luppi PH. The lateral hypothalamic area controls paradoxical (REM) sleep by means of descending projections to brainstem GABAergic neurons. J Neurosci. 2012;32:16763–16774. doi: 10.1523/JNEUROSCI.1885-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasio AR, Grabowski TJ, Bechara A, Damasio H, Ponto LL, Parvizi J, Hichwa RD. Subcortical and cortical brain activity during the feeling of self-generated emotions. Nat Neurosci. 2000;3:1049–1056. doi: 10.1038/79871. [DOI] [PubMed] [Google Scholar]
- Davis JA, Paylor R, McDonald MP, Libbey M, Ligler A, Bryant K, Crawley JN. Behavioral effects of ivermectin in mice. Lab Anim Sci. 1999;49:288–296. [PubMed] [Google Scholar]
- Dragoi G, Tonegawa S. Preplay of future place cell sequences by hippocampal cellular assemblies. Nature. 2011;469:397–401. doi: 10.1038/nature09633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- España RA, McCormack SL, Mochizuki T, Scammell TE. Running promotes wakefulness and increases cataplexy in orexin knockout mice. Sleep. 2007;30:1417–1425. doi: 10.1093/sleep/30.11.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Egner T, Kalisch R. Emotional processing in anterior cingulate and medial prefrontal cortex. Trends Cogn Sci. 2011;15:85–93. doi: 10.1016/j.tics.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW. On counting and counting errors. J Comp Neurol. 2002;447:1–7. doi: 10.1002/cne.10221. [DOI] [PubMed] [Google Scholar]
- Gulyani S, Wu MF, Nienhuis R, John J, Siegel JM. Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience. 2002;112:355–365. doi: 10.1016/S0306-4522(02)00089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci U S A. 2009;106:2418–2422. doi: 10.1073/pnas.0811400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikida T, Kimura K, Wada N, Funabiki K, Nakanishi S. Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron. 2010;66:896–907. doi: 10.1016/j.neuron.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Hurley KM, Herbert H, Moga MM, Saper CB. Efferent projections of the infralimbic cortex of the rat. J Comp Neurol. 1991;308:249–276. doi: 10.1002/cne.903080210. [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Ambroggi F, Nicola SM, Fields HL. Dorsomedial prefrontal cortex contribution to behavioral and nucleus accumbens neuronal responses to incentive cues. J Neurosci. 2008a;28:5088–5098. doi: 10.1523/JNEUROSCI.0253-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa A, Ambroggi F, Nicola SM, Fields HL. Contributions of the amygdala and medial prefrontal cortex to incentive cue responding. Neuroscience. 2008b;155:573–584. doi: 10.1016/j.neuroscience.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson B, Fong GW, Bennett SM, Adams CM, Hommer D. A region of mesial prefrontal cortex tracks monetarily rewarding outcomes: characterization with rapid event-related fMRI. Neuroimage. 2003;18:263–272. doi: 10.1016/S1053-8119(02)00057-5. [DOI] [PubMed] [Google Scholar]
- Krenzer M, Anaclet C, Vetrivelan R, Wang N, Vong L, Lowell BB, Fuller PM, Lu J. Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One. 2011;6:e24998. doi: 10.1371/journal.pone.0024998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerchner W, Xiao C, Nashmi R, Slimko EM, van Trigt L, Lester HA, Anderson DJ. Reversible silencing of neuronal excitability in behaving mice by a genetically targeted, ivermectin-gated Cl− channel. Neuron. 2007;54:35–49. doi: 10.1016/j.neuron.2007.02.030. [DOI] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clément O, Sapin E, Peyron C, Gervasoni D, Léger L, Fort P. Brainstem mechanisms of paradoxical (REM) sleep generation. Pflugers Arch. 2012;463:43–52. doi: 10.1007/s00424-011-1054-y. [DOI] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–798. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitler MM, Boysen BG, Campbell L, Dement WC. Narcolepsy-cataplexy in a female dog. Exp Neurol. 1974;45:332–340. doi: 10.1016/0014-4886(74)90122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Lenglos C, Martin J, Mbende N, Gagné A, Timofeeva E. Sucrose modifies c-fos mRNA expression in the brain of rats maintained on feeding schedules. Neuroscience. 2011;192:459–474. doi: 10.1016/j.neuroscience.2011.06.033. [DOI] [PubMed] [Google Scholar]
- Murray EA. The amygdala, reward and emotion. Trends Cogn Sci. 2007;11:489–497. doi: 10.1016/j.tics.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Oka T, Tsumori T, Yokota S, Yasui Y. Neuroanatomical and neurochemical organization of projections from the central amygdaloid nucleus to the nucleus retroambiguus via the periaqueductal gray in the rat. Neurosci Res. 2008;62:286–298. doi: 10.1016/j.neures.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Okuro M, Fujiki N, Kotorii N, Ishimaru Y, Sokoloff P, Nishino S. Effects of paraxanthine and caffeine on sleep, locomotor activity, and body temperature in orexin/ataxin-3 transgenic narcoleptic mice. Sleep. 2010;33:930–942. doi: 10.1093/sleep/33.7.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overeem S, van Nues SJ, van der Zande WL, Donjacour CE, van Mierlo P, Lammers GJ. The clinical features of cataplexy: a questionnaire study in narcolepsy patients with and without hypocretin-1 deficiency. Sleep Med. 2011;12:12–18. doi: 10.1016/j.sleep.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Bassetti CL, Schwartz S. Abnormal activity in reward brain circuits in human narcolepsy with cataplexy. Ann Neurol. 2010;67:190–200. doi: 10.1002/ana.21825. [DOI] [PubMed] [Google Scholar]
- Reid MS, Tafti M, Geary JN, Nishino S, Siegel JM, Dement WC, Mignot E. Cholinergic mechanisms in canine narcolepsy-I. Modulation of cataplexy via local drug administration into the pontine reticular formation. Neuroscience. 1994;59:511–522. doi: 10.1016/0306-4522(94)90173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi TA, Ennis M, Behbehani MM, Shipley MT. Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: topography and reciprocity. J Comp Neurol. 1991;303:121–131. doi: 10.1002/cne.903030111. [DOI] [PubMed] [Google Scholar]
- Rogers RD, Ramnani N, Mackay C, Wilson JL, Jezzard P, Carter CS, Smith SM. Distinct portions of anterior cingulate cortex and medial prefrontal cortex are activated by reward processing in separable phases of decision-making cognition. Biol Psychiatry. 2004;55:594–602. doi: 10.1016/j.biopsych.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Rushworth MF, Behrens TE. Choice, uncertainty and value in prefrontal and cingulate cortex. Nat Neurosci. 2008;11:389–397. doi: 10.1038/nn2066. [DOI] [PubMed] [Google Scholar]
- Sabatinelli D, Bradley MM, Lang PJ, Costa VD, Versace F. Pleasure rather than salience activates human nucleus accumbens and medial prefrontal cortex. J Neurophysiol. 2007;98:1374–1379. doi: 10.1152/jn.00230.2007. [DOI] [PubMed] [Google Scholar]
- Savander V, Go CG, LeDoux JE, Pitkänen A. Intrinsic connections of the rat amygdaloid complex: projections originating in the basal nucleus. J Comp Neurol. 1995;361:345–368. doi: 10.1002/cne.903610211. [DOI] [PubMed] [Google Scholar]
- Scammell TE. The neurobiology, diagnosis, and treatment of narcolepsy. Ann Neurol. 2003;53:154–166. doi: 10.1002/ana.10444. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Willie JT, Guilleminault C, Siegel JM, International Working Group on Rodent Models of Narcolepsy A consensus definition of cataplexy in mouse models of narcolepsy. Sleep. 2009;32:111–116. [PMC free article] [PubMed] [Google Scholar]
- Schenck CH, Bassetti CL, Arnulf I, Mignot E. English translations of the first clinical reports on narcolepsy and cataplexy by Westphal and Gelineau in the late 19th century, with commentary. J Clin Sleep Med. 2007;3:301–311. [PMC free article] [PubMed] [Google Scholar]
- Schroeder BE, Binzak JM, Kelley AE. A common profile of prefrontal cortical activation following exposure to nicotine- or chocolate-associated contextual cues. Neuroscience. 2001;105:535–545. doi: 10.1016/S0306-4522(01)00221-4. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol. 1989;290:213–242. doi: 10.1002/cne.902900205. [DOI] [PubMed] [Google Scholar]
- Slimko EM, McKinney S, Anderson DJ, Davidson N, Lester HA. Selective electrical silencing of mammalian neurons in vitro by the use of invertebrate ligand-gated chloride channels. J Neurosci. 2002;22:7373–7379. doi: 10.1523/JNEUROSCI.22-17-07373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/S0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trezza V, Damsteegt R, Manduca A, Petrosino S, Van Kerkhof LW, Pasterkamp RJ, Zhou Y, Campolongo P, Cuomo V, Di Marzo V, Vanderschuren LJ. Endocannabinoids in amygdala and nucleus accumbens mediate social play reward in adolescent rats. J Neurosci. 2012;32:14899–14908. doi: 10.1523/JNEUROSCI.0114-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Goutagny R, Fort P, Cagnon L, Salvert D, Léger L, Boissard R, Salin P, Peyron C, Luppi PH. A role of melanin-concentrating hormone producing neurons in the central regulation of paradoxical sleep. BMC Neurosci. 2003;4:19. doi: 10.1186/1471-2202-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- Westphal C. Eigentümliche mit Einschlafen verbundene Anfälle. Arch Psychiatr Nervenkr. 1877;7:631–635. [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE, Yanagisawa M. Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron. 2003;38:715–730. doi: 10.1016/S0896-6273(03)00330-1. [DOI] [PubMed] [Google Scholar]