

Abstract
In 1991, treatment with low dose intramuscular desferrioxamine (DFO), a trivalent chelator that can remove excessive iron and/or aluminum from the body, was reported to slow the progression of Alzheimer’s disease (AD) by a factor of two. Twenty years later this promising trial has not been followed up and why this treatment worked still is not clear. In this critical interdisciplinary review, we provide an overview of the complexities of AD and involvement of metal ions, and revisit the neglected DFO trial. We discuss research done by us and others that is helping to explain involvement of metal ion catalyzed production of reactive oxygen species in the pathogenesis of AD, and emerging strategies for inhibition of metal-ion toxicity. Highlighted are insights to be considered in the quests to prevent potentially toxic effects of aluminum toxicity and prevention and intervention in AD.
Keywords: Aluminum toxicity, Alzheimer’s disease, Antioxidants, Chelators, Inflammation, Oxidative stress
1. Evidence
1.1. The complex multifactorial nature of Alzheimer’s disease
Alzheimer’s disease [AD] is the most common form of dementia, accounting for 60–70% of cases of neurological impairment in the elderly [1]. After the age of 65, its prevalence approximately doubles every five years [2]. In 2001, the prevalence of dementia in persons over age 60 was estimated by a Delphi consensus method to range from 1.6 to 6.6% [3,4]. By 2040, the total number of people in the world with dementia is expected to more than triple relative to the number affected in 2001 [3,4]. AD affects the brain’s ability to perform daily activities and to control thought, memory and language [5–8]. The disorder is associated with gross changes in the brain and changes at the microscopic levels [4–6,8,9]. At the macroscopic level, AD brains are smaller than normal, folding of the cortex is decreased, and ventricle size is increased. At the microscopic level, neuron count and synapse density are decreased, and the brain is riddled with amyloid plaques (diffuse and neuritic) containing amyloid-beta (Aβ) peptides, a secretase-mediated breakdown product of the amyloid precursor protein (APP), and neurofibrillary tangles containing excessive amounts of hyperphosphorylated tau protein [1,4,8,9].
The AD brain also is characterized by markers of oxidative stress, neuroinflammation and dysregulated inflammatory signaling (Section 1.3). The term oxidative stress refers to an imbalance between the body’s production of free radicals and their neutralization by a number of different means [10,11]. It may occur as the result of excessive ROS production, reduced antioxidant defense or a combination of both. All of these processes may be relevant in AD. It has been suggested that high energy demand from dependence on oxidative metabolism plus a high concentration of polyunsaturated fatty acids, and relatively low antioxidant enzyme activity, render the brain more vulnerable to oxidative insult than most organs [12]. The term neuroinflammation refers to the “integrated response of all cells within the central nervous system, including the neurons, macroglia, microglia and infiltrating leukocytes” to both acute and pathological insults (pg 237) [13]. Neuroinflammation can be a cause or a consequence of excessive oxidative stress [13–18]. Different types of central nervous system or systemic infections also have been implicated in AD (Table 2) [19–27]. As well, there is increasing recognition that epigenetic processes are involved in AD and other neurodegenerative diseases [28,29]. The term epigenetic literally means “above” genetics, and refers to processes that affect the expression of particular genes in DNA but do not change the linear sequence of its nucleotides. Epigenetic processes include methylation of DNA, alteration of the degree of chromosome compaction as the result of modification to the tails of DNA-binding histone proteins, and regulation of gene expression by non-coding RNAs such as micro RNAs (miRNAs) [28]. Certain brain-abundant miRNAs, such as miRNA-125b and miRNA-146a, found to be specifically elevated in AD brain, are also up-regulated in aluminum-stressed human brain cells in primary culture [30, JIB, in press (this current journal)].
Table 2.
Different infections implicated in Alzheimer’s disease. *
| Agent | Reference |
|---|---|
| Herpes simplex virus 1 | [19–21] |
| Chlamydia pneumoniae | [22,23] |
| Toxoplasma gondii | [24] |
| Spirochetes | [25] |
| Helicobacter pylori** | [26] |
| Other agents | [27] |
Involvement of these infections is considered controversial as of 2011.
Treating helicobacter for 5 years significantly deterred development of dementia in a population with a relatively low infection prevalence.
There is evidence for involvement of metal ions in AD pathogenesis, though this is complex [31,32]. AD brain is characterized by unusual distribution of metal ions, including iron, copper, zinc [31–33], and aluminum [34–36] in regions that are degenerating. However, there is controversy as to whether average concentrations of these metals are significantly altered in particular brain regions [37–39]. Furthermore, although complexes of Aβ and redox metals, especially iron, can be sources of reactive oxygen species (ROS) production in vitro [40], it has been argued that metal iron binding by Aβ may play a protective role in vivo [41]. Brain changes characteristic of advanced AD are likely not reversible [42].
Risk factors for the sporadic form of AD, which accounts for approximately 95% of cases [4,9], include the aging process in combination with interaction between various genetic, metabolic and environmental factors [4,9] (Table 1). The E4 allele of apolipoprotein E, the major lipid carrier in the central nervous system, has been consistently identified as the major genetic risk factor for AD, although it is not specific for this disorder [43–45]. Other genetic factors also are involved in AD [4,9]. The impact of E4 on disease risk may, in part, be due to its influence on oxidative and/or immuno-inflammatory status [46]. Interest in iron, copper and zinc in AD is topical [31–33,40]. Although aluminum involvement in AD is still considered controversial, studies in various model systems continue to reveal toxic effects from aluminum exposures equivalent to those experienced by humans in daily living [39,47–50]. See also various publications in this issue.
Table 1.
| Increasing age |
|---|
Predisposing genetic factors
|
Factors listed here are ones that might be controlled for in epidemiological studies of Alzheimer’s disease or in clinical treatment or prevention trials in the opinion of the authors. As indicated in Table 2, various infections also are implicated in AD, though this topic is controversial.
Efforts to develop an effective treatment for AD have remained elusive. Drugs currently licensed for use temporarily slow disease progression in some affected individuals but there is no evidence that they delay or prevent onset of AD [4,42,51]. The reduction of oxidative stress has been a therapy target in clinical trials, but results have largely been negative, or mild at best [9,52,53] (See other sources for approaches currently being considered in AD [4,9,54–60].). While it is clear that oxidative stress is playing some contributory role to the AD process [40], oxidative stress alone is likely not the only pathological insult that needs to be targetted. We have shown that chelator/antioxidant combinations are synergistically more effective than single chelators or antioxidants in antioxidant therapy approaches in metallo-based in vitro models of AD [61,62] (see Section 1.4). The rationale for this combined approach was based, in part, on results from a promising but neglected clinical trial of a trivalent metal ion chelator called desferrioxamine in AD [63,64] (Fig. 1), and on known properties of this chelator, which include not only metal sequestering but also generation of radicals [65–71] (Table 3). The clinical trial of DFO is discussed next.
Fig. 1.

Structures of desferrioxamine and Feralex-G. (A): DFO: deferrioxamine mesylate (USAN); alternatively desferrioxamine mesilate (BAN). DFO is a commercially available siderophore (molecule that sequesters iron from the environment) produced by Streptomyces pilosus. It is a hexadentate chelator, and binds trivalent iron or aluminum in a 1:1 ratio. It is used in many countries in the mesylate form to treat various forms of iron overload, and also aluminum overload. A major drawback to its use is that it is effective only when administered parenterally [65]. Image kindly provided by Andrzej Wilk, PhD, Senior Scientific Liaison, US Pharmacopeia; CAS Number 138–14-7; DFO has a molecular formula of C25H48N6O8·CH4O3S; MW 657. (B): Feralex-G. Feralex-G is an experimental oral chelator made from three natural substances—glucosamine (1), an amino acid, glycine (1″) and maltol (1′) [76]. It was designed to enter cells via complexing of the glucosamine “tail” with glucose transporters, and to be a safe replacement for DFO which is effective only parenterally (i.e., by intramuscular, subcutaneous, or intravenous routes). Feralex-G is bidentate and complexes with trivalent metal ions in the ratio of 3:1. Hydrophilicity of Feralex can be altered by substitution of the amino acid linker. Image reproduced from Kruck et al. [61] with permission. Feralex-G has a molecular formula of C13H13N2O7; MW 344 [61,62,76].
Table 3.
Properties attributed to desferrioxamine.
| Forms complexes with Fe3+ and Al3+; complex formation constants are 1032 and 1025 respectively | [65] |
| Forms complexes with Fe2+, Cu2+, Zn2+, and Ca2+ with complex formation constants of 1014 or below | [65] |
| Chelates free iron and iron bound to ferritin and hemosiderin but does not readily chelate iron in transferrin, hemoglobin, or other iron containing proteins | [65] |
| Has a suppressant effect on lymphocytes | [65] |
| Inhibits iron-dependent production of free radicals | [66] |
| Affects eicosanoid synthesis | [66] |
| Generates a reactive nitroxide radical | [66] |
| Acts as a substrate for peroxidases | [66] |
| Acts as a lipid chain breaking antioxidant, independently of its iron chelating properties | [67] |
| Can inhibit the oxidative chemistry of peroxynitrite by reaction of the hydroxamic acid moieties with trans-peroxynitrous acid | [68] |
| “A known activator of the hypoxia-inducible transcription factor 1 (HIF- 1) and the subsequent transcription of erythropoietin. In the brain, HIF-1 is a master switch of the transcriptional response to hypoxia, whereas erythropoietin is a potent neuroprotectant.” | [69] |
| Triggers inflammatory signal expression and production in certain types of human cells | [71,72] |
| Under conditions of non-iron overload, protects against oxidative stress by reducing ferryl heme (in ferryl myoglobin and hemoglobin), and preventing formation of cytotoxic derivatives of myoglobin such as heme to protein cross-linking. | [73] |
1.2. The successful but neglected 1991 trial of desferrioxamine in Alzheimer’s disease revisited
In 1991, the intramuscular injection of low dose desferrioxamine mesylate (DFO; desferoxamine; deferoxamine; Desferal) was found to reduce the rate of progression of AD by a factor of two within a two-year period [63]. Furthermore, among 48 patients in the study, there were no deaths in the DFO-treated group, but five deaths among those who were not treated. In this study, the progression of AD was measured by a specially developed videotaped behavior assessment instrument designed to obtain “a reliable and representative sample of an individual’s level of functioning in his or her home repeatedly over an extended time period” (pg 603) [64]. DFO is a licensed trivalent metal ion chelator that can effectively remove excessive trivalent iron and aluminum from the body (Fig. 1, Table 3).
Twenty years later, this promising result still has not been followed up. In 1993 a large, phase III trial approved by Health Canada was aborted because Ciba-Geigy reversed a commitment to cover the drug cost. The reasons given were that intramuscular injection of DFO was not well-tolerated and might interfere with patient compliance (though this was not an issue in the phase II trial), and that development of new drugs was needed. Other drawbacks to DFO are its high cost, toxicity and short plasma half life [65]. Furthermore, DFO is an orphan drug (i.e., it is produced with relatively little profit margin for the application to transfusional iron overload resulting from rare disorders such as thalassemias, and sickle cell anemia). Yet DFO continues to be used successfully in these latter disorders, alone or in combination with other oral chelators [72].
Twenty years later, we still do not know why the DFO trial worked. Effects of treatment with low dose intramuscular injection of DFO (125 mg twice per day, 5 days per week) were compared to treatment with oral lecithin, or to no treatment [63]. Because treatment administration involved significant personal interaction, and quality of interaction is reported to ward off deterioration in AD [74], the possibility of a placebo effect cannot be excluded.
Since the trial was completed, we know that iron and/or aluminum treatment of brain cells in culture induces the production of ROS and ROS-driven changes in gene expression characteristic of AD (see Sections 1.3 and 1.4 below). As already mentioned (Section 1.1) we also know that DFO and an experimental trivalent metal ion chelator called Feralex-G [75], especially in combination with other anti-oxidants, alleviates effects of metal ion stress in studies with in vitro model systems [61,62]. Thus it is likely that in the clinical trial, DFO inhibited metal-ion catalyzed oxidative stress.
Since the DFO trial was completed, ways in which metal ions amplify levels of ROS are better understood [11,76,77] (Fig. 2). Iron-catalyzed ROS production is exacerbated by the presence of other redox metals such as divalent copper, and by trivalent aluminum which is not [78]. Aluminum has no redox potential, nor is it powerfully attracted to sulfhydryl groups [79]. However, it has an extremely high and unchanging 3+ charge density, and this may be, in part, the basis for its promotion of Fenton chemistries in biological systems [29]. It has been suggested that aluminum on its own reacts with the superoxide anion oxidation to form a superoxide semireduced radical ion that has greater oxidative power than superoxide itself [77]. Further, aluminum also appears to impair mitochondrial bioenergetics leading to the progressive accumulation of oxidatively modified cellular proteins [80]. Aside from its role in ROS production, aluminum can affect cell functions in other ways [29,32]. It rapidly promotes compaction of biological molecules [29]. It can affect the function of hundreds of different proteins and enzymes [49], and the metabolism of calcium and magnesium [32]. Finally, it binds to A-T rich regions of DNA and has been postulated to play a role in gene silencing [29].
Fig. 2.
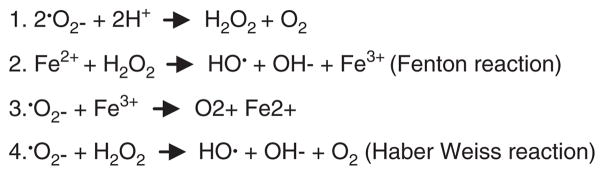
Metal-ion driven production of reactive oxygen species. Reactive oxygen species (ROS) include the superoxide radical (O2−), hydrogen peroxide (H2O2), and the hydroxyl radical (HO). HO is short-lived, reacting rapidly with lipids, DNA and proteins. Toxicity of O2− likely comes from reactions with H+, H2O2, iron, or other metals, to form more reactive ROS [11,77]. Al3+ may react with superoxide to form the superoxide semi-reduced radical ion , a more powerful pro-oxidant than superoxide [78]. Other redox metals, and non-redox metals can amplify iron-catalyzed ROS production.
Adapted from Kelly et al. [11].
DFO is a hexadentate chelator that avidly forms complexes with “free” trivalent iron or aluminum in a 1:1 ratio (Fig. 1). The term free refers to metal ions that are uncomplexed, or that can be readily removed from ligands such as ferritin or hemosiderin. DFO is a large molecule, but there is evidence that it crosses the blood–brain barrier [81]. Furthermore, in tracer experiments with rats, prolonged treatment with DFO was able to modestly reduce brain levels of aluminum [82].
Although DFO will remove excessive trivalent iron and/or aluminum from the body that is free or loosely bound, it has a number of other important properties (Table 3). In microenvironments containing excessive amounts of superoxide anion or peroxide, an important biological function of DFO must include metal ion complex formation per se. When fully sequestered by DFO, a trivalent metal ion is not able to catalyze production of ROS via Fenton-related reactions (Fig. 2). Another important property of DFO that has been demonstrated in rodents is its ability to induce hypoxia inducible transcription factor-1 and erythropoietin, a brain neuroprotective factor [69] (Table 3). That effects of iron and/or aluminum may have been altered by DFO in the clinical trial is strengthened by our studies of aluminum loaded nuclei [61] and cultured brain cells [62; Sections 1.3 and 1.4 below].
As of 2011 there is no information about DFO effects on relative levels of iron or aluminum in serum or urine of the AD patients in the clinical trial. However, in one unrelated study, AD patients were found to have elevated serum aluminum concentrations [83]. In a study of involvement of hemochromatosis mutations in sporadic AD, serum iron concentrations and related markers of body iron status were found, on average, to be within the normal range and not different from age and gender-matched healthy individuals [84]. To note is that measurement of aluminum in serum (and other tissues) is fraught with difficulties [85].
Analysis of brain autopsy samples from a series of AD patients who had received different total amounts of DFO prior to death indicated that extensive DFO treatment restored aluminum levels to nearly normal values, but had no effect on the brain iron levels [64]. These tantalizing observations, which suggest selective removal of aluminum from the brains of the treated patients, cannot be regarded as conclusive for two reasons. First, there is controversy about whether brain aluminum levels are elevated or not in AD patients [34–36,86]. Second, measurement of aluminum in brain tissue from patients treated with DFO was not done with reference to control samples to which DFO had been deliberately added to aid with interpretation of results.
The finding that side effects were neither serious nor frequent in the low dose DFO trial [63] suggests that the treatment protocol used did not seriously interfere with important iron-dependent processes in the AD brain, including normal functions of APP (Table 4) and/or Aβ (Table 5), or adaptive responses to infectious processes suspected of contributing to AD (e.g., iron-withholding [97,98]).
Table 4.
Some features and normal functions of APP.
| Is a classical heat shock protein | [87] |
| Has an iron-regulatory region in the 5′UTR | [88] |
| APP balance is critical for normal neuronal development, connection of synapses, and dendritic spine development | [89] |
| Has H-ferritin-like ferroxidase activity that is dependent upon copper binding but is inhibited by zinc | [90] |
| Interacts with ferroportin, facilitating iron export from neurons | [90] |
| Binds heparin | [91] |
| Its trafficking is regulated by copper | [92] |
Table 5.
Some normal functions of Abeta.
Studies with the metal-binding antibiotic clioquinol [99] (and with the related compound PBT2 [58]) support a role for metal catalyzed oxidative stress in AD as do experiments in a drosophila model of AD [100]. These underscore the potential importance of interventions based on metal chelation. Of relevance to the mechanism of action of clioquinol protection is the finding that in yeast cells, clioquinol sequesters iron, copper and zinc at the level of the plasma membrane [101]. This observation further emphasizes the potential importance of metal ion sequestering for inhibition of metal catalyzed Fenton reactions (Fig. 2).
1.3. Iron and/or aluminum induce ROS and changes in gene expression in model systems resembling those in AD
Abnormalities in metal-ion metabolism are implicated not only in AD but also in other neurodegenerative conditions [10,102–104]. Evidence from model systems is converging that metal ion exposure to aluminum or iron trigger changes in gene expression resembling those in the AD brain that are preceded by ROS production. The short-term exposure of mice to drinking water containing excessive aluminum induces pro-inflammatory processes in the brain [105,106]. Furthermore, effects have been demonstrated in rat brain that are aluminum specific [105]. Because mechanisms involved in such disturbances were not well-understood, we examined effects of physiological concentrations of various metal sulfates on ROS production and patterns of gene expression in cultured human brain cells [30,62,107–110]. The addition of iron and/or aluminum sulfates to cells in culture induces the production of ROS followed by changes in the expression of certain genes involved in stress sensing, inflammation, and in initiation of apoptosis that also have been observed to be upregulated in the brains of patients with moderate to late-stage AD [107]. Treatment with iron and aluminum together had greater genotoxic effects than either iron or aluminum alone. Evidence also was obtained for involvement of NF-kB (nuclear factor kappa-light chain-enhancer of activated B cells) and certain microRNAs (miRNAs). These regulate expression of specific sets of messenger RNA molecules (mRNAs) by binding to sequences in their 3′ untranslated regions. Furthermore, in cultures of brain glial cells (analogous to macrophages in the peripheral circulation), aluminum sulfate but not iron sulfate, appears to uniquely induce ROS-mediated up-regulation of an NF-kB-sensitive miRNA-146a that down-regulates the expression of complement factor H (CFH), an important repressor of inflammation [109]. This NF-kB-miRNA-146a-CFH signaling circuit is affected by Aβ42 peptides in the same way as by aluminum sulfate, and the circuit also is abnormal in the AD brain [110]. These findings underscore the concept that ROS induction by metal-catalyzed reactions is important in the regulation of genetic and epigenetic systems that define the essential mechanisms of AD pathogenesis.
1.4. Chelators and antioxidants in combination alleviate metal-induced oxidative damage and inflammation in model systems
Because neurotoxic metal-induced oxidative damage to nervous tissue has been implicated in several progressive neurodegenerative disorders as well as in AD (Section 1.3), and on the basis of positive findings in the DFO trial (Section 1.2), we have explored the ability of various agents to quench metal sulfate-induced production of reactive ROS and ROS-sensitive gene expression using cultured human brain cells [108]. Antioxidants included ascorbate, folic acid and phenyl butyl nitrone, a free radical trapping agent. Chelators included DFO and a new chelator designed for oral use called Feralex-G [75] (Fig. 1). Importantly, these reagents were found to quench ROS and also to inhibit the induction of genes whose expression was upregulated by treatment with metal ions alone to various degrees. Furthermore, when these reagents were used in certain combinations, synergistic effects were observed. These findings support the idea that specific anti-oxidants and metal ion chelators when used together can effectively and synergistically quench ROS-mediated induction of pathogenic gene expression induced by metal sulfates. Such effects may result, in part, from the fact that metal chelators can have antioxidant properties in addition to simply binding metals [73], and antioxidants (e.g., ascorbic acid) can have metal-chelating activity. As well, we have proposed that a mechanism called “molecular shuttle chelation” may be involved [61]. Low molecular weight chelators such as ascorbic acid may be able to quickly penetrate the nucleus of cells, bind to metals in this compartment, diffuse to regions accessible by larger chelators and transfer the metal to these molecules.
Subsequent studies have investigated the potential of naturally occurring antioxidants—docosahexaenoic acid (the major omega-3 fatty acid found in neurons, and abundant in cold-water oceanic fish oils), resveratrol (a polyphenol particularly abundant in red wine) and curcumin (a polyphenol derived from the curry spice turmeric) to inhibit metal sulfate induced genotoxic effects [30]. To note is that curcumin has been reported to counter aluminum-induced enhancement of certain aging related biochemical processes in young and old rats [111]. This is of potential relevance to AD since aging is a major risk factor for AD (Table 1).
2. Insights
2.1. Future research directions in aluminum toxicity
The review of information in Part 1 has identified a number of challenges to address in future research. First, because of aluminum’s unusual reactivity [29], and focal distribution in brain tissue [34,36,64], there is urgent need for standardized protocols and methods of analysis to use in the measurement of aluminum in fluids and tissues. Important considerations include the need for appropriate aluminum standards [37], specification of collection and storage containers, controlling for effects related to duration of sample storage, how to representatively sample brain tissue, and how best to compare data distributions with different patterns in test and control samples. When feasible in case/control studies, paired design and analysis can control for effects of “environmental” variables [112].
Second, ligand specific effects have been noted on aluminum incorporation and toxicity [113]. Our studies have been conducted with aluminum sulfate, because this is the usual contaminant in municipal drinking water. Others have used, for example, aluminum chloride [47,48], aluminum maltol [113], or aluminum lactate [114]. Reasons for ligand specific effects may be related to the hydrophilicity/hydrophobicity of the ligand, relative toxicity of the ligand and how it is metabolized, and the tendency of aluminum salts to hydrolyze and lower the pH. Relative effects of such factors need to be examined.
Third, studies of aluminum toxicity in cell culture systems and various animal models have tended to use aluminum exposures considered analogous to those experienced by humans [47,48]. Such exposures may not be appropriate for animals which have higher than human metabolic rates [115]. Other variables for consideration are apparent traits of aluminum resistance and aluminum sensitivity in mice. Earlier studies indicated that tissue distribution of aluminum in mouse brain and degree of toxicity following aluminum exposure depend markedly upon mouse strain. DBA/2 and CH3/2 strains are very sensitive to effects of dietary aluminum whereas A/J, BALB/c and C57BL/6 strains show no response [116]. To note is that CH3 mice have considerably higher serum levels of insulin-like growth factor-1 (IGF-1) than C57BL/6 mice and these strains also differ genetically at the IGF-1 locus [117]. Insulin and IGF-1 function deteriorate with progression of AD [118]. Furthermore, the IGF-1 pathway is known to be involved in neuronal protection, with dual effects on both Aβ and tau. Thus future studies might determine if the IGF-1 pathway is involved in defense against aluminum toxicity.
Fourth, evidence continues to accumulate that silicic acid may be nature’s antidote to aluminum toxicity, at least at low pH [119,120]. However, because aluminosilicates precipitate, and aluminosilicate deposits have been found within amyloid plaques and neurofibrillary tangles, it has been argued that AD and the amyotrophic lateral sclerosis/Parkinson disease (ALS/PD) complex of Guam are diseases of aluminosilicates in the brain [121]. Hence, consideration ought to be given to potentially toxic effects of silicic acid at least in certain circumstances.
Studies with plants have highlighted protective effects of magnesium against aluminum toxicity. A genome study of aluminum sensitive and aluminum resistant strains of soybean showed that when administered concomitantly with aluminum at low pH, magnesium ameliorated aluminum toxicity in the sensitive genotype by “(i) increasing the expression level of several genes that are upregulated in the aluminum-treated, aluminum-tolerant genotype in the absence of magnesium, and (ii) possibly saving energy by decreasing expression of most genes relative to expression under Al stress” [122]. Given that magnesium modulates APP trafficking and processing [123] and can counteract toxic effects of Ca2+ fluxes [124], there would be merit applying metabolomics (measurement of by products of metabolism in fluids and extracts) as well as genomics, to studies of aluminum sensitive and aluminum resistant strains of mice and in vitro model systems of aluminum toxicity and AD.
In addition to strategies highlighted in Section 1.4, other promising approaches to be further explored in prevention of aluminum ion toxicity include treatment with vitamins B1 and B6 after metal-ion exposure [125]. Finally, because metal ion-catalyzed ROS production is not only a problem per se, but likely also relevant to many different disorders and conditions as well as AD, the testing of different chelators [57] (including DFO, which now exists in pill form [126]), and the identification or development of new oral chelators is warranted. To note is that depending upon the dose and time of administration of an oral metal ion chelator relative to eating or drinking, consequences might include metal ion uptake or removal, or metal redistribution.
2.2. Strategies for reducing population exposures to aluminum
Because excessive exposure to aluminum continues to be implicated in the development and/or progression of AD and aging, as well as in other forms of human ill health [47,48,50,102,104,123], intervention activity should focus on the prevention of potential aluminum toxic effects. Persons with AD and adults with Down syndrome, who are at high risk of developing dementia resembling AD 20 to 30 years earlier than in the general population [9] may be particularly susceptible to oral exposures, since the ability of their gut to inhibit aluminum uptake may be compromised [127,128]. Workers in the aluminum industry and miners also are vulnerable as the result of breathing dust containing aluminum or being exposed to it. Excessive exposure in babies can result from overambitious immunization schedules [129] and from high levels of aluminum in prepared formula [130,131]. There is increasing recognition of aluminum contamination in products used in parenteral nutrition [132]. Jansson [133] previously indicated that regulatory agencies in Canada and the U.S. placed aluminum on priority lists for toxicity evaluation. In 2006, the Joint Food and Agricultural Organization/World Health Organization Expert Committee on Food Additives (JECFA) significantly lowered the tolerable weekly intake from 7 mg/kg body weight to 1 mg/kg body weight [134]. Followup to this report has not been consistent among different countries. For example, as of 2011 in the U.S. aluminum-containing food additives are “said” to be safe [135]. In Canada, the Bureau of Chemical Safety Branch of Health Canada’s Food Directorate initiated a review of aluminum in foods [136]. In response to this directive, an international group of authors have published a technical report about the toxicity of aluminum, aluminum oxide and aluminum hydroxide [115]. This document is available in printed form and on the web, but the content has not yet been prepared in a form that is meaningful to the average person. From this report it is clear that excessive aluminum exposures from foods and medicines are likely to be more of a hazard than from drinking water, which nevertheless is sometimes problematic.
On the basis of published and emerging scientific evidence, the neurotoxicity of environmental aluminum is still an issue [47–50]. Creative ways are needed to increase awareness of this problem and also to address it (Table 6). High on priority are labeling products for human consumption or use with the aluminum content and its contribution to the tolerable intake, as well as outreach and education at all levels. In addition, research activities should continue to explore ways to reduce the level of aluminum or its bioavailability in drinking water [120].
Table 6.
Some approaches for preventing excessive aluminum exposures.
Health policies
|
2.3. Future directions for Alzheimer’s treatment and prevention
There currently are no treatments for AD that slow down or prevent its development [4,9, Section 1.1]. There is currently difficulty in the early diagnosis of AD, and diagnostic techniques for AD need to be greatly improved [4,8]. As scientific publications continue to support the hypothesis that aluminum toxicity is involved in AD [39,50], it would be prudent to adopt strategies for preventing excessive aluminum exposures, and to continue testing approaches for alleviation of metal-related toxic effects in efforts to treat or prevent AD. With respect to clinical trials of AD, it has been argued that intervention efforts in AD should focus on prevention [60]. We suggest that data analysis in AD prevention or intervention trials include adjustment for known AD risk factors (Table 1). To consider as well is the collection of blood, urine, and if possible, cerebrospinal fluid (CSF) for “add-on” studies of biomarkers and mechanisms, with caution about the need for standardized procedures and methods for measurement of aluminum concentrations in particular (Section 2.1).
This review has drawn attention to the idea that chelators as well as antioxidants and other nutraceuticals (e.g., curcumin, dexahexaenoic acid, resveratrol) might not only help to counter effects of aluminum (and other metal) toxicity, but also be relevant to prevention or intervention in AD (Sections 1.4 and 2.2). As AD is an unequivocally complex neurological disorder, it may take some time to derive adequate combinations of chelators, antioxidants and nutraceuticals for use in clinical treatment of this insidious disease. Although excessive ROS production is problematic, to consider in prospective studies is the need for adequate ROS production to promote physiological adaptations to exercise [137] and also to maintain immune activation [138]. We question if one reason for apparent “failure” of antioxidant treatment for AD (see Section 1.1) is excessive dampening of hormetic levels of ROS that keep our antioxidant and anti-inflammatory defenses primed. Because of the multifactorial nature of AD (Section 1.1), interest continues to develop in the potential of lifestyle modulation [4,9,139], nutritional approaches [4,9,56], natural substances such as the hormone melatonin [140], and stage specific interventions that block causal events as well as those involved in damage at later stages [54].
3. Review highlights
3.1. Evidence
Alzheimer’s disease (AD) is a complex, multifactorial disorder involving oxidative stress and neuroinflammation. It may also involve CNS infection and epigenetic processes. There is no effective treatment for AD (Section 1.1). One promising but neglected approach published in 1991 involved the intramuscular injection of a trivalent metal ion chelator, desferrioxamine (DFO) (Section 1.2).
As of 2011, we still do not know why DFO treatment had a positive outcome in AD. The hypothesis that DFO was alleviating iron and/or aluminum toxicity remains viable (Section 1.2). Binding of metal ions by chelators in microenvironments, in addition to outright elimination of metal ion-chelator complexes from the body, and/or other properties attributed to DFO may be important in AD protective effects (Section 1.2, Table 2).
Generation of ROS and ROS-linked changes in gene expression resembling those in AD are induced by iron and aluminum sulfate in cultured brain cells and animal models, supporting the idea that these metals are primarily involved in AD brain pathogenesis (Section 1.3.).
Metal ion chelators and antioxidants (especially in combination) show promise for alleviation or prevention of iron and aluminum toxicity in model systems (Section 1.4).
3.2. Insights
Insights for consideration in future research in aluminum toxicity include: development of standard protocols for measuring aluminum in fluids and tissues; consideration of possible effects of aluminum ligands on outcome; studies of aluminum sensitive and aluminum resistant strains of animals, and co-application of genomics metabolomics in aluminum toxicity experiments; investigation of the potential toxicity of silicic acid in certain populations; use of Mg, and vitamins B1 and B6 in alleviation of aluminum toxicity (Section 2.1); testing of existing chelators and development of new ones.
Excessive exposures to aluminum should be avoided to prevent potentially toxic effects, especially in vulnerable populations. This will require the instigation of new health policy directives, as well as directed educational and research initiatives (Section 2.2; Table 6).
As of 2011, there is a general consensus that pleiotrophic intervention for AD should be started as early as possible. There is currently difficulty in the early diagnosis of AD, and diagnostic techniques for AD need to be greatly improved. The design and analysis of clinical trials themselves could be improved by adjustment for confirmed AD risk factors and use of add-on biomarker studies. If pathogenesis is resulting from redox metal and/or aluminum toxicity, at least in part, particular combinations of chelators, antioxidants and/or nutraceuticals may be effective. Excessive antioxidant use may interfere with some favorable, physiologically homeostatic, effects of ROS in maintaining the host activation of antioxidant defenses and the immune system (Section 2.3).
Acknowledgments
Helpful comments from the two anonymous reviewers are gratefully acknowledged. Because of space limitations, referencing of this paper was not exhaustive. We apologize to many authors whose valuable work was not cited.
References
- 1.Jalbert JJ, Daiello LA, Lapane KL. Epidemiol Rev. 2008;30:15–34. doi: 10.1093/epirev/mxn008. [DOI] [PubMed] [Google Scholar]
- 2.Lindsay J, Anderson L. BMC Womens Health. 2004;4:S20. doi: 10.1186/1472-6874-4-S1-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M Alzheimer’s Disease International. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Lancet. 2011;377:1019–1031. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- 5.Alzheimer A. Allgemeine Zeitschrift fur Psychiatrie und psychisch-Gerichtliche Medizin. Vol. 64. Berlin: 1907. pp. 146–148. [Google Scholar]
- 6.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 7.Association American Psychiatric. Diagnostic and Statistical Manual of Mental Disorders. 4. Author; Washington, DC: 2000. text rev. [Google Scholar]
- 8.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies T, Weintraub S, Phelps CH. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prasher V, Percy M, Jozsvai E, Berg JM. Developmental Disabilities in Ontario. In: Brown I, Percy M, editors. Ontario Association on Developmental Disabilities. 3. Toronto, ON: 2011. pp. 675–694. [Google Scholar]
- 10.Jomova K, Valko M. Toxicology. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Kelly KA, Havrilla CM, Brady TC, Abramo KH, Levin ED. Environ Health Perspect. 1998;106:375–384. doi: 10.1289/ehp.98106375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dringen R, Gutterer JM, Hirrlinger J. Eur J Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 13.Carson MJ, Thrash JC, Walter B. Clin Neurosci Res. 2006;6:237–245. doi: 10.1016/j.cnr.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mhatre M, Floyd RA, Hensley K. J Alzheimers Dis. 2004;6:147–157. doi: 10.3233/jad-2004-6206. [DOI] [PubMed] [Google Scholar]
- 15.Streit WJ, Mrak RE, Griffin WSJ. J Neuroinflammation. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGeer EG, McGeer PL. J Alzheimers Dis. 2010;19:355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 17.Galimberti D, Scarpini E. Front Biosci (Schol Ed) 2011;3:252–266. doi: 10.2741/s149. [DOI] [PubMed] [Google Scholar]
- 18.Pizza V, Agresta A, D’Acunto CW, Festa M, Capasso A. CNS Neurol Disord Drug Targets. 2011;10:621–634. doi: 10.2174/187152711796235014. [DOI] [PubMed] [Google Scholar]
- 19.Ball MJ, Lewis E, Haase AT. J Neural Transm Suppl. 1987;24:219–225. [PubMed] [Google Scholar]
- 20.Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. J Med Virol. 1991;33:224–227. doi: 10.1002/jmv.1890330403. [DOI] [PubMed] [Google Scholar]
- 21.Lukiw WJ, Cui JG, Yuan LY, Bhattacharjee PS, Corkern M, Clement C, Kammerman EM, Ball MJ, Zhao Y, Sullivan PM, Hill JM. Neuroreport. 2010;21:922–927. doi: 10.1097/WNR.0b013e32833da51a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreses-Werringloer U, Bhuiyan M, Zhao Y, Gérard HC, Whittum-Hudson JA, Hudson AP. Int J Med Microbiol. 2009;299:187–201. doi: 10.1016/j.ijmm.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammond CJ, Hallock LR, Howanski RJ, Appelt DM, Little CS, Balin BJ. BMC Neurosci. 2010;11:121. doi: 10.1186/1471-2202-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kusbeci OY, Miman O, Yaman M, Aktepe OC, Yazar S. Alzheimer Dis Assoc Disord. 2011;25:1–3. doi: 10.1097/WAD.0b013e3181f73bc2. [DOI] [PubMed] [Google Scholar]
- 25.Miklossy J. J Alzheimers Dis. 2008;13:381–391. doi: 10.3233/jad-2008-13404. [DOI] [PubMed] [Google Scholar]
- 26.Kountouras J, Boziki M, Gavalas E, Zavos C, Grigoriadis N, Deretzi G, Tzilves D, Katsinelos P, Tsolaki M, Chatzopoulos C, Venizelos I. J Neurol. 2009;256:758–767. doi: 10.1007/s00415-009-5011-z. [DOI] [PubMed] [Google Scholar]
- 27.Honjo K, van Reekum R, Verhoeff NP. Alzheimers Dement. 2009;5:348–360. doi: 10.1016/j.jalz.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Qureshi IA, Mehler MF. Curr Neurol Neurosci Rep. 2011 Jun 15; doi: 10.1007/s11910-011-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukiw WJ. J Inorg Biochem. 2010;104:1010–1012. [PubMed] [Google Scholar]
- 30.Pogue AI, Percy ME, Cui JG, Li YY, Bhattacharjee S, Hill JM, Kruck TPA, Zhao Y, Lukiw WJ, Inorg J. Biochem. 2011;105:1433–1436. doi: 10.1016/j.jinorgbio.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolognin S, Messori L, Zatta P. Neuromolecular Med. 2009;11:223–238. doi: 10.1007/s12017-009-8102-1. [DOI] [PubMed] [Google Scholar]
- 32.Bonda DJ, Lee HG, Blair JA, Zhu X, Perry G, Smith MA. Metallomics. 2011;3:267–270. doi: 10.1039/c0mt00074d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Todorich BM, Connor JR. Ann N Y Acad Sci. 2004;1012:171–178. doi: 10.1196/annals.1306.014. [DOI] [PubMed] [Google Scholar]
- 34.Crapper DR, Krishnan SS, Dalton AJ. Trans Am Neurol Assoc. 1973;AZ98:17–20. [PubMed] [Google Scholar]
- 35.Perl DP, Moalem S. J Alzheimers Dis. 2006;9:291–300. doi: 10.3233/jad-2006-9s332. [DOI] [PubMed] [Google Scholar]
- 36.Rusina R, Matěj R, Kašparová L, Kukal J, Urban P. Neurotox Res. 2011 May 13; doi: 10.1007/s12640-011-9246-y. [DOI] [PubMed] [Google Scholar]
- 37.Lovell MA, Ehmann WD, Markesbery WR, Melethil S, Swyt CR, Zatta PF. J Toxicol Environ Health. 1996;48:637–648. doi: 10.1080/009841096161113. [DOI] [PubMed] [Google Scholar]
- 38.Schrag M, Mueller C, Oyoyo U, Kirsch WM. Prog Neurobiol. 2011;94:296–306. doi: 10.1016/j.pneurobio.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomljenovic L. J Alzheimers Dis. 2011;23:567–598. doi: 10.3233/JAD-2010-101494. [DOI] [PubMed] [Google Scholar]
- 40.Smith MA, Zhu X, Tabaton M, Liu G, McKeel DW, Jr, Cohen ML, Wang X, Siedlak SL, Dwyer BE, Hayashi T, Nakamura M, Nunomura A, Perry G. J Alzheimers Dis. 2010;19:363–372. doi: 10.3233/JAD-2010-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castellani RJ, Lee HG, Siedlak SL, Nunomura A, Hayashi T, Nakamura M, Zhu X, Perry G, Smith MA. J Alzheimers Dis. 2009;18:447–452. doi: 10.3233/JAD-2009-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maarouf CL, Daugs ID, Kokjohn TA, Kalback WM, Patton RL, Luehrs DC, Masliah E, Nicoll JA, Sabbagh MN, Beach TG, Castaño EM, Roher AE. Mol Neurodegener. 2010;5:39. doi: 10.1186/1750-1326-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Lancet. 1993;342:697–699. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 44.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leduc V, Domenger D, De Beaumont L, Lalonde D, Bélanger-Jasmin S, Poirier J. Int J Alzheimers Dis. 2011 Apr 5; doi: 10.4061/2011/974361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jofre-Monseny L, Minihane AM, Rimbach G. Mol Nutr Food Res. 2008;52:131–145. doi: 10.1002/mnfr.200700322. [DOI] [PubMed] [Google Scholar]
- 47.Walton JR. Neurosci Lett. 2007;412:29–33. doi: 10.1016/j.neulet.2006.08.093. [DOI] [PubMed] [Google Scholar]
- 48.Walton JR. Neurotoxicology. 2009;30:182–193. doi: 10.1016/j.neuro.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 49.Bernardo JA, Edwards MR, Barnett B. http://emedicine.medscape.com/article/165315-overview#a0101.
- 50.Bondy SC. Neurotoxicology. 2010;31:575–581. doi: 10.1016/j.neuro.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Brien JT, Burns A. J Psychopharmacol. 2010 Nov 18; doi: 10.1177/0269881110387547. [DOI] [Google Scholar]
- 52.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 53.Lee HP, Zhu X, Casadesus G, Castellani RJ, Nunomura A, Smith MA, Lee HG, Perry G. Expert Rev Neurother. 2010;10:1201–1208. doi: 10.1586/ern.10.74. [DOI] [PubMed] [Google Scholar]
- 54.Frautschy SA, Cole GM. Mol Neurobiol. 2010;41:392–409. doi: 10.1007/s12035-010-8137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stone JG, Casadesus G, Gustaw-Rothenberg K, Siedlak SL, Wang X, Zhu X, Perry G, Castellani RJ, Smith MA. Ther Adv Chronic Dis. 2011;2:9–23. doi: 10.1177/2040622310382817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pocernich CB, Bader Lange ML, Sultana R, Butterfield DA. Curr Alzheimer Res. 2011;8:452–469. doi: 10.2174/156720511796391908. [DOI] [PubMed] [Google Scholar]
- 57.Bandyopadhyay S, Huang X, Lahiri DK, Rogers JT. Expert Opin Ther Targets. 2010;14:1177–1197. doi: 10.1517/14728222.2010.525352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H, Ingelsson M, Masters CL, Tanzi RF, Cummings JL, Herd CM, Bush AI. J Alzheimers Dis. 2010;20:509–516. doi: 10.3233/JAD-2010-1390. [DOI] [PubMed] [Google Scholar]
- 59.Menéndez-González M, Pérez-Piñera P, Martínez-Rivera M, Muñiz AL, Vega JA. Curr Pharm Des. 2011;17:508–520. doi: 10.2174/138161211795164112. [DOI] [PubMed] [Google Scholar]
- 60.Emery VO. J Neural Transm. 2011;118:1361–1378. doi: 10.1007/s00702-011-0663-0. [DOI] [PubMed] [Google Scholar]
- 61.Kruck TP, Cui JG, Percy ME, Lukiw WJ. Cell Mol Neurobiol. 2004;24:443–459. doi: 10.1023/B:CEMN.0000022773.70722.b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kruck TP, Percy ME, Lukiw WJ. Neuroreport. 2008;19:245–249. doi: 10.1097/WNR.0b013e3282f4cb7e. [DOI] [PubMed] [Google Scholar]
- 63.Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. Erratum in Lancet 1991 337 (1991) 1618 Comment in Lancet 338 (1991) 324–326. [DOI] [PubMed] [Google Scholar]
- 64.McLachlan DR, Smith WL, Kruck TP. Ther Drug Monit. 1993;15:602–607. [PubMed] [Google Scholar]
- 65.Desferal(r), Name of the Drug. 2010 Feb; http://www.novartis.com.au/PI_PDF/dsf.pdf19.
- 66.Halliwell B. Free Radic Biol Med. 1989;7:645–651. doi: 10.1016/0891-5849(89)90145-7. [DOI] [PubMed] [Google Scholar]
- 67.Hartley A, Davies M, Rice-Evans C. FEBS Lett. 1990;264:145–148. doi: 10.1016/0014-5793(90)80786-i. [DOI] [PubMed] [Google Scholar]
- 68.Denicola A, Souza JM, Reynaldo M, Gatti M, Augusto O, Radi R. Free Radic Biol Med. 1995;19:11–19. doi: 10.1016/0891-5849(94)00239-g. [DOI] [PubMed] [Google Scholar]
- 69.Prass K, Ruscher K, Karsch M, Isaev N, Megow D, Priller J, Scharff A, Dirnagl U, Meise A. J Cereb Blood Flow Metab. 2002;22:520–555. doi: 10.1097/00004647-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 70.Choi EY, Kim EC, Oh HM, Kim S, Lee HJ, Cho EY, Yoon KH, Kim EA, Han WC, Choi SC, Hwang JY, Park C, Oh BS, Kim Y, Kimm KC, Park KI, Chung HT, Jun CD. J Immunol. 2004;172:7069–7077. doi: 10.4049/jimmunol.172.11.7069. [DOI] [PubMed] [Google Scholar]
- 71.Lee HJ, Lee J, Lee SK, Lee SK, Kim EC. BMC Cancer. 2007;7:176. doi: 10.1186/1471-2407-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neufeld EJ. Hematol Am Soc Hematol Educ Program. 2010;2010:451–455. doi: 10.1182/asheducation-2010.1.451. [DOI] [PubMed] [Google Scholar]
- 73.Reeder BJ, Hider RC, Wilson MT. Free Radic Biol Med. 2008;44:264–273. doi: 10.1016/j.freeradbiomed.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 74.Amieva H, Stoykova R, Matharan F, Helmer C, Antonucci TC, Dartigues JF. Psychosom Med. 2010;72:905–911. doi: 10.1097/PSY.0b013e3181f5e121. [DOI] [PubMed] [Google Scholar]
- 75.Kruck TP, Burrow J. J Inorg Biochem. 2002;88:9–24. doi: 10.1016/s0162-0134(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 76.Dumont M, Beal MF. Free Radic Biol Med. 2010. Dec 1, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Exley C. Free Radic Biol Med. 2004;36:380–387. doi: 10.1016/j.freeradbiomed.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 78.Bondy SC, Guo-Ross SX, Pien J. Neurotoxicology. 1998;19:65–71. [PubMed] [Google Scholar]
- 79.Nesbitt SL, Emerson JA, Bell JP. Int J Adhes Adhes. 2000;20:429–436. [Google Scholar]
- 80.Kumar V, Gill KD. Arch Toxicol. 2009;83:965–978. doi: 10.1007/s00204-009-0455-6. [DOI] [PubMed] [Google Scholar]
- 81.Hua Y, Keep RF, Hoff JT, Xi G. Acta Neurochir Suppl. 2008;105:3–6. doi: 10.1007/978-3-211-09469-3_1. [DOI] [PubMed] [Google Scholar]
- 82.Yokel RA, Rhineheimer SS, Sharma P, Elmore D, McNamara PJ. Toxicol Sci. 2001;64:77–82. doi: 10.1093/toxsci/64.1.77. [DOI] [PubMed] [Google Scholar]
- 83.Zapatero MD, Garcia de Jalon A, Pascual F, Calvo ML, Escanero J, Marro A. Biol Trace Elem Res. 1995;47:235–240. doi: 10.1007/BF02790122. [DOI] [PubMed] [Google Scholar]
- 84.Percy M, Moalem S, Garcia A, Somerville MJ, Hicks M, Andrews D, Azad A, Schwarz P, Beheshti Zavareh R, Birkan R, Choo C, Chow V, Dhaliwal S, Duda V, Kupferschmidt AL, Lam K, Lightman D, Machalek K, Mar W, Nguyen F, Rytwinski PJ, Svara E, Tran M, Wheeler K, Yeung L, Zanibbi K, Zener R, Ziraldo M, Freedman M. J Alzheimers Dis. 2008;14:69–84. doi: 10.3233/jad-2008-14107. [DOI] [PubMed] [Google Scholar]
- 85.Milacic R, Murko S, Scancar J. J Inorg Biochem. 2009;103:1504–1513. doi: 10.1016/j.jinorgbio.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 86.Bjertness E, Candy JM, Torvik A, Ince P, McArthur F, Taylor GA, Johansen SW, Alexander J, Grønnesby JK, Bakketeig LS, Edwardson JA. Alzheimer Dis Assoc Disord. 1996;10:171–174. doi: 10.1097/00002093-199601030-00006. [DOI] [PubMed] [Google Scholar]
- 87.Dewji NN, Do C. Brain Res Mol Brain Res. 1996;35:325–328. doi: 10.1016/0169-328x(95)00214-d. [DOI] [PubMed] [Google Scholar]
- 88.Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR. J Biol Chem. 2002;277:45518–455128. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 89.Hoe HS, Lee HK, Pak TD. CNS Neurosci Ther. 2010 Dec 27; doi: 10.1111/j.1755-5949.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho HH, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI. Cell. 2010;142:857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Corrigan F, Pham CL, Vink R, Blumbergs PC, Masters CL, van den Heuvel C, Cappai R. Brain Res. 2011;1378:137–143. doi: 10.1016/j.brainres.2010.12.077. [DOI] [PubMed] [Google Scholar]
- 92.Acevedo KM, Hung YH, Dalziel AH, Li QX, Laughton K, Wikhe K, Rembach A, Roberts B, Masters CL, Bush AI, Camakaris J. J Biol Chem. 2011;286:8252–8862. doi: 10.1074/jbc.M110.128512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L. J Alzheimers Dis. 2010;19:441–449. doi: 10.3233/JAD-2009-1230. [DOI] [PubMed] [Google Scholar]
- 94.Cenini G, Maccarinelli G, Lanni C, Bonini SA, Ferrari-Toninelli G, Govoni S, Racchi M, Butterfield DA, Memo M, Uberti D. Amino Acids. 2010;39:271–283. doi: 10.1007/s00726-009-0438-1. [DOI] [PubMed] [Google Scholar]
- 95.Nair NG, Perry G, Smith MA, Reddy VP. J Alzheimers Dis. 2010;20:57–66. doi: 10.3233/JAD-2010-1346. [DOI] [PubMed] [Google Scholar]
- 96.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. PLoS One. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moalem S, Weinberg ED, Percy ME. Biometals. 2004;17:135–139. doi: 10.1023/b:biom.0000018375.20026.b3. [DOI] [PubMed] [Google Scholar]
- 98.Weinberg ED. Metallomics. 2010;2:732–740. doi: 10.1039/c0mt00023j. [DOI] [PubMed] [Google Scholar]
- 99.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Arch Neurol. 2003;60:1685. doi: 10.1001/archneur.60.12.1685. Erratum in Arch Neurol 61 (2004) 776. [DOI] [PubMed] [Google Scholar]
- 100.Rival T, Page RM, Chandraratna DS, Sendall TJ, Ryder E, Liu B, Lewis H, Rosahl T, Hider R, Camargo LM, Shearman MS, Crowther DC, Lomas DA. Eur J Neurosci. 2009;29:1335–1347. doi: 10.1111/j.1460-9568.2009.06701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li C, Wang J, Zhou B. J Alzheimers Dis. 2010;21:1249–1262. doi: 10.3233/jad-2010-100024. [DOI] [PubMed] [Google Scholar]
- 102.Exley C, Mamutse G, Korchazhkina O, Pye E, Strekopytov S, Polwart A, Hawkins C. Mult Scler. 2006;12:533–540. doi: 10.1177/1352458506071323. [DOI] [PubMed] [Google Scholar]
- 103.Jiang F, Sun ZZ, Tang YT, Xu C, Jiao XY. Diabetes Res Clin Pract. 2011;93:43–48. doi: 10.1016/j.diabres.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 104.Da Costa R, Szyper-Kravitz M, Szekanecz Z, Csépány T, Dankó K, Shapira Y, Zandman-Goddard G, Orbach H, Agmon-Levin N, Shoenfeld Y. Isr Med Assoc J. 2011;13:91–95. [PubMed] [Google Scholar]
- 105.Bondy SC, Ali SF, Guo-Ross S. Mol Chem Neuropathol. 1998;34:219–232. doi: 10.1007/BF02815081. [DOI] [PubMed] [Google Scholar]
- 106.Campbell A, Bondy SC. Cell Mol Biol (Noisyle-Grand) 2000;46:721–730. [PubMed] [Google Scholar]
- 107.Alexandrov PN, Zhao Y, Pogue AI, Tarr MA, Kruck TP, Percy ME, Cui JG, Lukiw WJ. J Alzheimers Dis. 2005;8:117–127. doi: 10.3233/jad-2005-8204. Discussion 209–215. [DOI] [PubMed] [Google Scholar]
- 108.Lukiw WJ, Pogue AI. J Inorg Biochem. 2007;101:1265–1269. doi: 10.1016/j.jinorgbio.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pogue AI, Li YY, Cui JG, Zhao Y, Kruck TP, Percy ME, Tarr MA, Lukiw WJ. J Inorg Biochem. 2009;103:1591–1595. doi: 10.1016/j.jinorgbio.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 110.Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ. J Biol Chem. 2010;285:38951–38960. doi: 10.1074/jbc.M110.178848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sharma D, Sethi P, Hussain E, Singh R. Biogerontology. 2009;10:489–502. doi: 10.1007/s10522-008-9195-x. [DOI] [PubMed] [Google Scholar]
- 112.Percy ME, Andrews DF. Can J Public Health. 1986;77:174–183. [PubMed] [Google Scholar]
- 113.Lévesque L, Mizzen CA, McLachlan DR, Fraser PE. Brain Res. 2000;877:191–202. doi: 10.1016/s0006-8993(00)02637-8. [DOI] [PubMed] [Google Scholar]
- 114.Becaria A, Lahiri DK, Bondy SC, Chen D, Hamadeh A, Li H, Taylor R, Campbell A. J Neuroimmunol. 2006;176:16–23. doi: 10.1016/j.jneuroim.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 115.Krewski D, Yokel RA, Nieboer E, Borchelt D, Cohen J, Harry J, Kacew S, Lindsay J, Mahfouz AM, Rondeau V, Toxicol J. Environ Health B Crit Rev. 2007;10:1–269. doi: 10.1080/10937400701597766. Erratum in J. Toxicol. Environ. Health B Crit. Rev. 11 (2008) 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fosmire GJ, Focht SJ, McClearn GE. Biol Trace Elem Res. 1993;37:115–121. doi: 10.1007/BF02783787. [DOI] [PubMed] [Google Scholar]
- 117.Iida K, Rosen CJ, Ackert-Bicknell C, Thorner MO. J Endocrinol. 2005;186:481–489. doi: 10.1677/joe.1.06200. [DOI] [PubMed] [Google Scholar]
- 118.Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. J Alzheimers Dis. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- 119.Exley C, Pinnegar JL, Taylor H. J Theor Biol. 1997;189:133–139. doi: 10.1006/jtbi.1997.0501. [DOI] [PubMed] [Google Scholar]
- 120.Exley C, Korchazhkina O, Job D, Strekopytov S, Polwart A, Crome P. J Alzheimers Dis. 2006;10:17–24. doi: 10.3233/jad-2006-10103. Discussion 29–31. [DOI] [PubMed] [Google Scholar]
- 121.Meyer C. Current Hypotheses—Alzforum. Updated 14 July 2005, http://www.alzforum.org/res/adh/cur/meyer/default.asp.
- 122.Duressa D, Soliman KM, Chen D. Genome. 2010;53:787–797. doi: 10.1139/g10-069. [DOI] [PubMed] [Google Scholar]
- 123.Yu J, Sun M, Chen Z, Lu J, Liu Y, Zhou L, Xu X, Fan D, Chui D. J Alzheimers Dis. 2010;20:1091–1106. doi: 10.3233/JAD-2010-091444. [DOI] [PubMed] [Google Scholar]
- 124.Lee M, Jantaratnotai N, McGeer E, McLarnon JG, McGeer PL. Brain Res. 2011;1369:21–35. doi: 10.1016/j.brainres.2010.10.084. [DOI] [PubMed] [Google Scholar]
- 125.Mehta R, Dedina L, O’Brien PJ. Toxicol In Vitro. 2011;25:1114–1122. doi: 10.1016/j.tiv.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 126.PRESS TV. Iran produces first desferal pills. http://edition.presstv.ir/detail/94172.html.
- 127.Moore PB, Edwardson JA, Ferrier IN, Taylor GA, Lett D, Tyrer SP, Day JP, King SJ, Lilley JS. Biol Psychiatry. 1997;1:488–492. doi: 10.1016/S0006-3223(96)00045-5. [DOI] [PubMed] [Google Scholar]
- 128.Moore PB, Day JP, Taylor GA, Ferrier IN, Fifield LK, Edwardson JA. Dement Geriatr Cogn Disord. 2000;11:66–69. doi: 10.1159/000017216. [DOI] [PubMed] [Google Scholar]
- 129.Tomljenovic L, Shaw CA. Curr Med Chem. 2011;18:2630–2637. doi: 10.2174/092986711795933740. [DOI] [PubMed] [Google Scholar]
- 130.Burrell SA, Exley C. BMC Pediatr. 2010;10:63. doi: 10.1186/1471-2431-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dabeka R, Fouquet A, Belisle S, Turcotte S. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2011;28:744–753. doi: 10.1080/19393210.2011.571795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gura GM. Nutrition. 2010;26:585–594. doi: 10.1016/j.nut.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 133.Jansson ET. J Alzheimers Dis. 2001;3:541–549. doi: 10.3233/jad-2001-3604. [DOI] [PubMed] [Google Scholar]
- 134.The International Program on Chemical Safety (IPCS) Evaluations of the Joint FAO/WHO Expert Committee on Food Additives (JECFA) Report TRS 940-JECFA 67/35, http://apps.who.int/ipsc/database/evaluations/chemical.aspx?chemID=298.
- 135.Agency for Toxic Substances & Disease Registry (ATSDR) Public Health Statement for Aluminum. http://www.atsdr.cdc.gov/phs/phs.asp?id=1076&tid=34.
- 136.Health Canada. Health Canada Review of Dietary Exposure to Aluminum. www.hc-sc.gc.ca/fn-an/securit/addit/aluminum-eng.php.
- 137.Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Blüher M. Proc Natl Acad Sci USA. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Los M, Dröge W, Stricker K, Baeuerle PA, Schulze-Osthoff K. Eur J Immunol. 1995;25:159–165. doi: 10.1002/eji.1830250127. [DOI] [PubMed] [Google Scholar]
- 139.Foster PP, Rosenblatt KP, Kuljiš RO. Front Neurol. 2011;2:28. doi: 10.3389/fneur.2011.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cardinali DP, Furio AM, Brusco LI. Curr Neuropharmacol. 2010;8:218–227. doi: 10.2174/157015910792246209. [DOI] [PMC free article] [PubMed] [Google Scholar]