

Abstract
Intracranial tumors are the most common solid tumors in children. The infratentorial compartment will be the primary site for 60% to 70% of these tumors, including astrocytomas, medulloblastomas, and ependymomas. Several technological advancements have increased our knowledge of the cell biology of pediatric brain tumors, facilitated earlier diagnosis, and improved neurosurgical resections while minimizing neurological deficits. These in turn have not only improved the survival of children with brain tumors but also their quality of life. Current management strategies in most cases rely on surgery coupled with adjuvant therapies, including radiation therapy and chemotherapy. The vulnerability of the immature brain to adjuvant therapies creates many challenges for the treating physician. We review current diagnostic and therapeutic approaches and outcome for children harboring the most common pediatric brain tumors: astrocytomas (low- and high-grade glioma), ependymoma, medulloblastoma, and craniopharyngioma. The emphasis will be on the neurosurgical management of children with these tumors.
Keywords: craniopharyngioma, ependymoma, glioma, medulloblastoma, pediatric brain tumors, neurosurgery
Introduction
Intracranial tumors are the most common solid neoplasms in children and the second most common malignancy of childhood.1 About 60% to 70% of all pediatric brain tumors, which include astrocytomas, medulloblastomas, and ependymomas, develop in the infratentorial compartment.2,3 The remaining 30% to 40% of tumors are supratentorial in origin and consist of optic pathway tumors, hypothalamic tumors, craniopharyngiomas, intraventricular tumors, and gliomas.4 The reason why pediatric brain tumors have a propensity to occur in the posterior fossa has not yet been elucidated.
The diagnosis of a brain tumor is often difficult to establish in a child, because many of the signs and symptoms may mimic those of more common childhood illnesses. Brain tumors produce neurologic symptoms that vary depending on the size, location, and invasiveness of the tumor. A list of the most common signs and symptoms of pediatric brain tumors is found in Table 1. Recurrent bouts of headache, nausea, or vomiting without focal deficits are observed in many children.5,6 Fundoscopy is imperative in any child with recurrent or progressive headache, as papilledema is commonly found.
Table 1.
Signs and Symptoms Associated with an Intracranial Tumor
| Signs and Symptoms |
|---|
|
An estimated 5% to 10% of brain tumors are due to a genetic predisposition. The occurrence of brain tumors in individuals with hereditary syndromes such as tuberous sclerosis, neurofibromatosis types 1 and 2, nevoid basal cell carcinoma syndrome, and familial adenomatous polyposis may be quite common. Some familial cancer syndromes such as the Li-Fraumeni syndrome, which is caused by germline mutations in the p53 gene, are associated with an increased risk of developing brain tumors. 7
In the past two decades, technological advances in many areas have provided us with a better appreciation of the biological behavior of pediatric brain tumors. In particular, the improvements in neuro-imaging have enabled us to make earlier diagnosis and to be more certain about tumor recurrence and dissemination. Magnetic resonance imaging (MRI) (Figure 1), positron emission tomography (PET), magnetoencephalography, diffusion tensor imaging, and functional MRI have enabled us to map brain tumors with extraordinary precision (Figure 2). For the neurosurgeon, the operating microscope, variable angled neuro-endoscopes, neuronavigation platforms, intraoperative MRI, cortical mapping, intraoperative evoked potentials, and high-resolution ultrasound have improved the safety and outcome of neurosurgical procedures, increasing the percentage of affected children who survive to adulthood.
Figure 1.

Sagittal MRIs of pediatric brain tumors in different brain regions. (A) Posterior fossa tumor in 14-year-old male with brief history of nausea, vomiting, and headache. At surgery, the lesion proved to be a medulloblastoma. (B) Midline suprasellar mass lesion with invagination into 3rd ventricle in 10-year-old female with progressive visual loss and headaches. This tumor was a craniopharyngioma. (C) Parasagittal tumor in 2-year-old female with brief history of seizures and leg weakness. This tumor was an ependymoblastoma. (D) Posterior third ventricular tumor in 2-year-old male with a history of vomiting and headaches. The lesion was a pineoblastoma.
Figure 2.
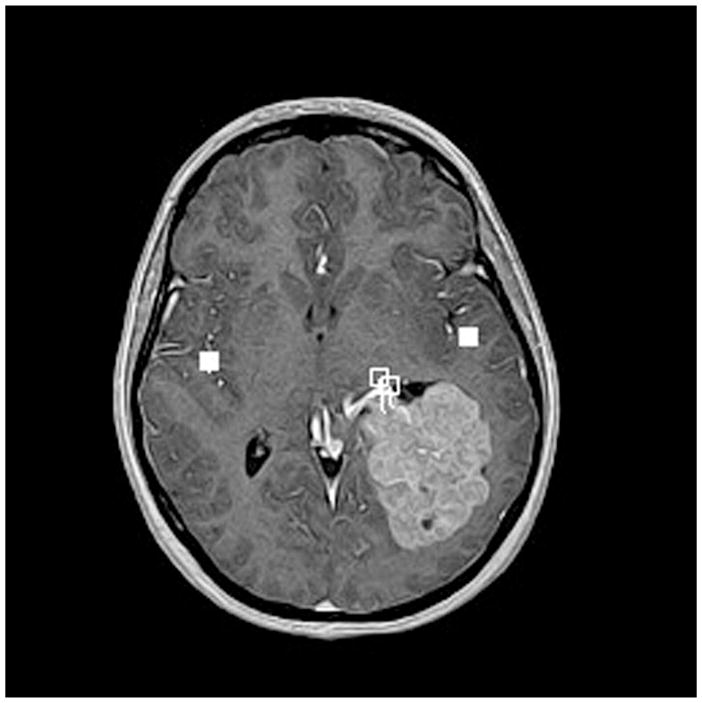
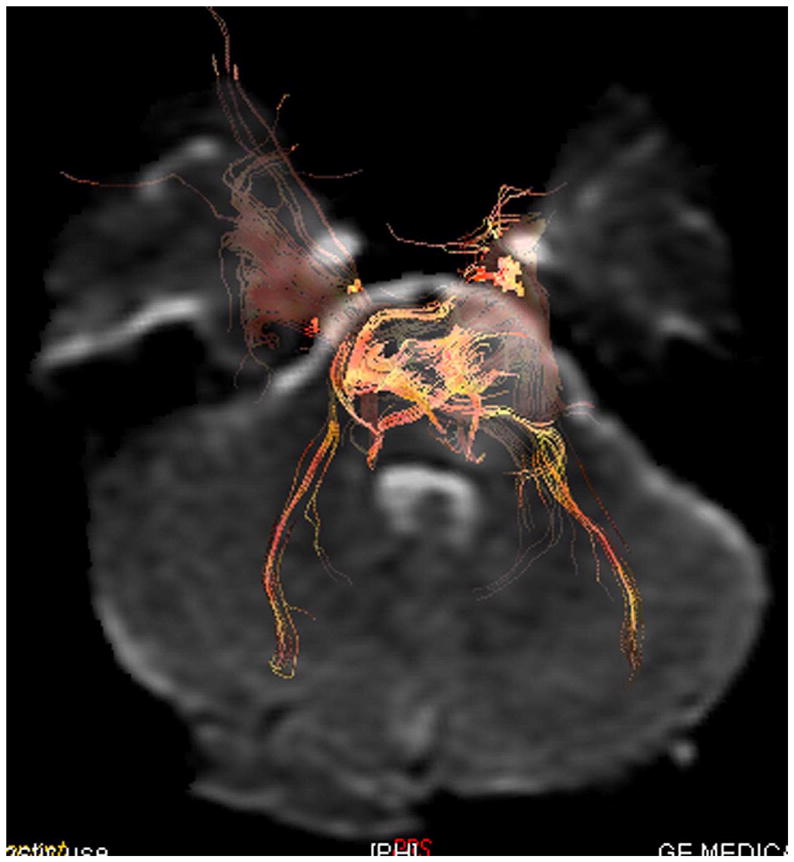
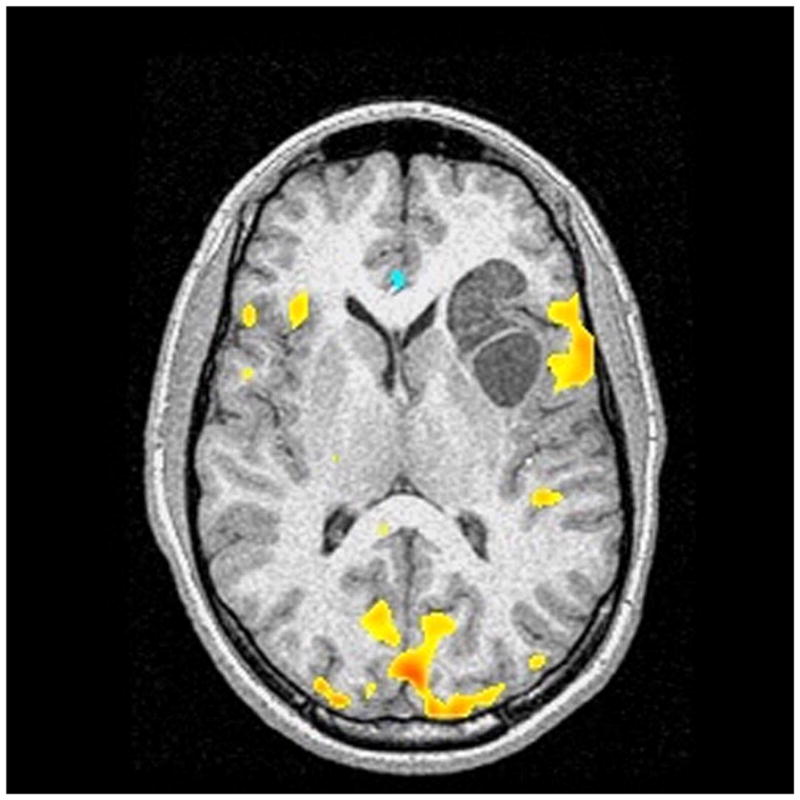
Advanced neuroimaging techniques in children with brain tumors. These techniques allow for mapping of eloquent regions of the child’s brain and aid the neurosurgeon in the approach to their removal so as to avoid injuring functional brain tissue. (A) Magnetoencephalography in 14-year-old female with left intraventricular tumor. The tumor was a choroid plexus papilloma. The white squares show language activation using typical magnetoencephalography paradigms. This patient has bilateral language activation. (B) Diffusion tensor imaging in 6-year-old male with left brainstem tumor showing slight compression of the corticospinal tract on the left side by the tumor mass. (C) Functional MRI in 10-year-old male with left frontal subcortical brain tumor showing primary language activation (yellow shading) in left inferior frontal region adjacent to the tumor.
In this review, we describe intracranial tumors for which neurosurgery clearly plays a role in patient outcome. These include gliomas (low-grade and high-grade), ependymoma, medulloblastoma, and craniopharyngioma.
Gliomas
More than half of all pediatric brain tumors are gliomas, which are usually supratentorial in location.8 Gliomas are classified into different grades from I through IV according to the World Health Organization. Grade I gliomas are benign tumors that include pilocytic astrocytoma and subependymal giant cell astrocytoma. The low-grade fibrillary astrocytoma roughly corresponds to the World Health Organization grade II. World Health Organization grade III and grade IV gliomas include anaplastic astrocytoma and glioblastoma multiforme, respectively. Glioma grades I and II are classified as low-grade gliomas and grades III and IV are considered high-grade gliomas.9
Low-Grade Gliomas
Low-grade gliomas comprise the most common type of brain tumor in children. Most are well-circumscribed tumors with varying histopathological features and include pilocytic astrocytomas, gangliogliomas, pleomorphic xanthoastrocytomas, and subependymal giant cell astrocytomas. Children with low-grade gliomatypically present with an insidious onset of symptoms suggestive of intracranial hypertension or a protracted history of seizures.10
Pilocytic astrocytomas are the most common type of low-grade glioma. They occur mostly in younger children (median, 4 years old) and form the majority of posterior fossa, optic apparatus, lobar, and dorsal exophytic brainstem tumors. They appear as a hypointense cystic mass on T1-weighted images with a solid part that enhances brightly.11
Less commonly, the neurosurgeon will face a child with a diffusely infiltrating low-grade glioma (World Health Organization grade II). These typically occur at a median age of 10 years, mostly in the cerebral hemispheric and pontine regions. These tumors may spread widely into regions of normal brain. Rarely, they can progress to higher-grade tumors. They are hypointense on T1-weighted images and hyperintense on T2-weighted images, and do not enhance after contrast.11
Management
The treatment of choice for low-grade gliomas that are in favorable locations is gross total resection (Figure 3). Pilocytic astrocytomas variants can be effectively treated in most cases by surgical resection, even when the lesions are located in eloquent locations such as the brainstem or thalamus. Functional brain mapping with functional MRI and magnetoencephalography, microneurosurgical techniques, intraoperative neuro-monitoring and imaging have enabled resection of previously inoperable tumors while minimizing surgical morbidity.11
Figure 3.

Intraoperative panel of images in 13-year-old male with left occipital pilocytic astrocytoma. This child was operated upon using frameless stereotaxy and intraoperative neuronavigation to ensure complete removal of his lesion.
While neurosurgical removal of a low-grade glioma, such as a pilocytic astrocytoma, is the optimal treatment, there are several locations in the brain where this cannot always be performed safely, such as the optic apparatus, hypothalamus, and certain regions of the brainstem. In these cases, chemotherapy or radiation therapy can be used as primary or adjuvant therapies.
The primary role of chemotherapy in the treatment of neurosurgically inaccessible low-grade gliomas is to delay radiation therapy or other treatments, particularly for young children. Most studies in the literature have focused on low-grade gliomas of the optic apparatus or hypothalamus. Here, chemotherapy can have a dramatic response or stabilizing influence without subjecting the child to the neurological sequelae of surgery or radiation therapy. Typically, radiation therapy is used when there are no further neurosurgical options or when chemotherapy has failed in a child who is old enough to receive radiation therapy, typically older than age 5.11 There have been recent advances in the delivery of radiation therapy using conformal, stereotactic fractionated radiation therapy and radiosurgery with 3-dimensional planning12 that have attempted to minimize some of the feared sequelae of radiation therapy to the developing brain.
Prognostic factors
Patient age, tumor grade, extent of resection, and tumor location are variables that have prognostic significance for patients with low-grade gliomas.13–16 The extent of resection may be the most important predictor of clinical outcome.17 In children where resection was greater than 95%, the 5- and 10-year survival rates ranged from 75% to 100%.11,13,16
High-Grade Glioma
High-grade gliomas are classically divided into anaplastic astrocytoma (grade III) and glioblastoma multiforme (grade IV) based on the presence of characteristic microscopic features. Childhood high-grade gliomas consistently display more frequent chromosomal aberrations and imbalance with multiple regions of abnormality.18 There is speculation that a high number of chromosomal aberrations may correlate with an aggressive biological phenotype.10,19
Symptoms of high-grade glioma are typically far more abrupt, with rapidly progressive symptoms or signs of elevated intracranial pressure or a focal neurological deficit.20 Seizures as a mode of presentation of high-grade glioma are less common. Nearly 5% to 10% of those with high-grade glioma deteriorate suddenly as a result of an intratumoral hemorrhage (Figure 4).10
Figure 4.
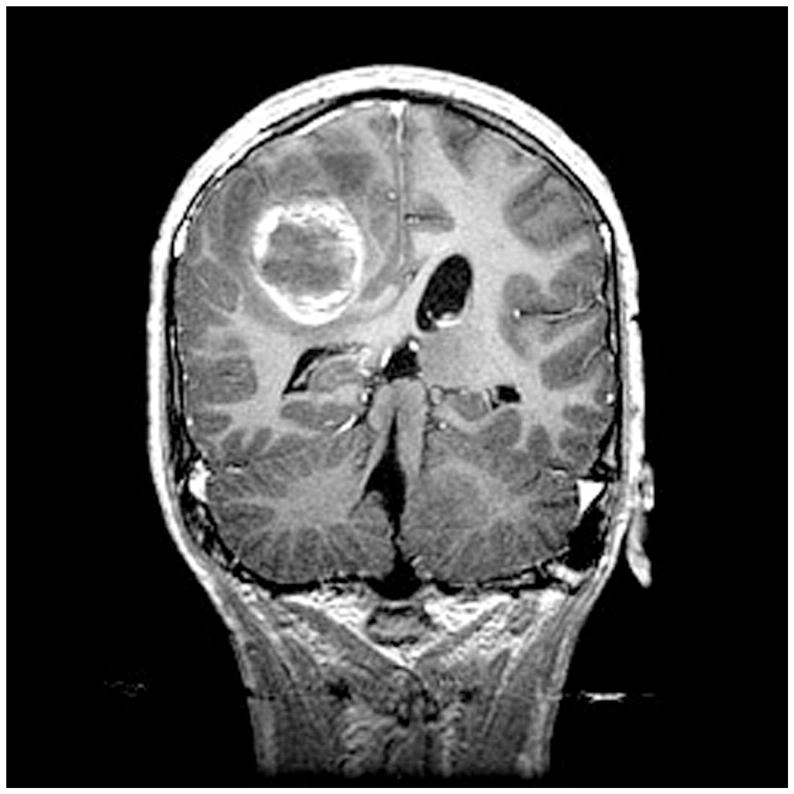
Coronal MRI scan in 14-year-old male with neurofibromatosis type 1 who presented with sudden onset of headache. The MRI shows both hemorrhage and tumor, and proved to be a glioblastoma multiformet surgery.
The characteristic MRI appearance of non-brainstem high-grade glioma is that of low signal intensity on T1-weighted sequences; cortical and white matter T2-signal hyperintensity, representing infiltrating tumor and reactive vasogenic edema, and an irregular pattern of contrast enhancement.10,21
Diffuse intrinsic pontine gliomas are the most commonly encountered tumor of the brainstem. Unfortunately, most are highly malignant neoplasms. Children with diffuse pontine gliomas will often present acutely with multiple cranial nerve signs, ataxia, long tract signs, and cerebellar signs.22,23 They appear hypointense on MRI with indistinct margins, reflecting the infiltrative nature of these high-grade lesions and indiscrete hyperintensity on T2-weighted imaging. Gadolinium enhancement can be variable and has no prognostic implication (Figure 5).24
Figure 5.
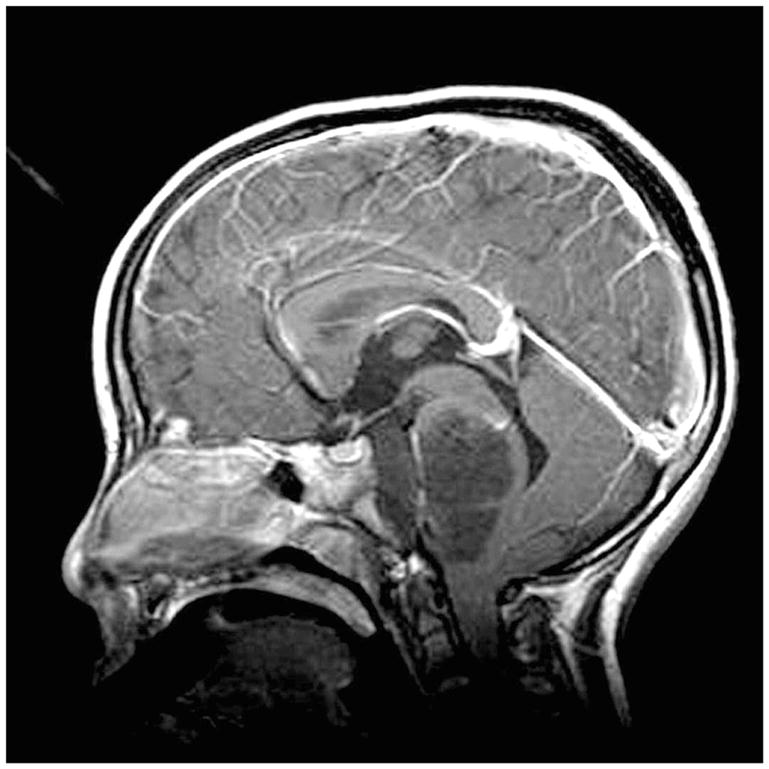
Sagittal gadolinium-enhanced MRI showing a diffuse intrinsic pontine glioma in an autistic child, one of identical twins. The tumor not only fills the pons, but encases the basilar artery. The prognosis for these tumors is extremely poor, with a 12- to 18-month survival noted in most cases.
Management
The general approach for children with non-brainstem high-grade gliomas is neurosurgical resection followed by adjuvant radiation therapy and chemotherapy. Complete tumor removal is the strongest prognostic factor.25,26 The outcome of children with high-grade gliomas is highly dependent on tumor histology. In the contemporary literature, 5-year survival rates ranged from 5% to 15% for children with glioblastoma multiforme and from 20% to 40% for those with anaplastic astrocytoma.27 Multi-institutional studies of pediatric patients with high-grade gliomas evaluated the significance of extent of resection and histology in children who were treated with aggressive multimodality therapies involving radiotherapy and chemotherapy.25,27–29 They demonstrated statistically significant different 5-year progression-free survival in patients with anaplastic astrocytoma who underwent a tumor resection of greater than 90% compared with children with glioblastoma multiforme who underwent less than 90% resection (44% and 4%, respectively).25
A significant association between overexpression of p53 and outcome in high-grade glioma has been described; 5-year progression free survival was 44 ± 6% in the group with low-level expression of p53 and 17 ± 6% in the group with overexpression of p53.30
Several studies have shown superiority of chemotherapy in high-grade glioma patients undergoing surgery and radiotherapy.27–29 Others have documented promising rates of long-term disease control in studies involving high-dose chemotherapy in conjunction with stem cell rescue. However, the toxicity of stem cell rescue must be taken into consideration.31
Most children with diffuse pontine gliomas die within 18 months of diagnosis, which is similar to the clinical course for glioblastoma multiforme.22,32,33 There is currently no role for radical surgery or biopsy since stereotactic biopsy does not change the management strategy.32 A biopsy should be reserved for indeterminate lesions on MRI accompanied by an unusual presentation or when mandated by a study protocol. In the context of neurofibromatosis type 1, however, diffuse brainstem tumors have a more favorable prognosis.34 Radiation therapy is the current mainstay of treatment for diffuse intrinsic pontine gliomas23,35,36 The role of chemotherapy remains to be determined.37
Often, a recurrence at the primary site of high-grade glioma may be amenable to additional local therapy, including resection or stereotactic radiosurgery. High-dose myeloablative chemotherapy and autologous stem cell rescue yielded a 4-year event-free survival rate of 22 ± 7% compared with 2 ± 1% who underwent conventional chemotherapy at the time of recurrence.10 Perhaps one of the most important aspects of the management of children with high-grade gliomas is the use of new chemotherapeutic agents in cooperative clinical trials. It is likely that convection-enhanced delivery of chemotherapeutics will be an option for future therapy in children with high-grade gliomas in upcoming clinical trials.
Ependymoma
Intracranial ependymomas are a common brain tumor in children, comprising 2% to 9% of all central nervous system tumors in this age category.38 They represent the third most common central nervous system neoplasms in this age range, following astrocytoma and medulloblastoma. 39 The peak incidence is between birth and 4 years of age, with little variation after ages 7 to 9. Males are 1.4 times more likely to develop ependymomas than females are. More than 70% of ependymomas occur in the posterior fossa,40,41 usually arising from neoplastic transformation of the ependymal cells in floor of 4th ventricle in the midline.42,43 The cause of ependymomas is unknown, but recent work has suggested a tumor suppressor gene for ependymoma resides on chromosome 22.44
According to the World Health Organization, four major subtypes occur, including ependymoma, anaplastic (malignant) ependymoma, myxopapillary ependymoma, and subependymoma.40,45 Children typically present with headache, vomiting, and lethargy subsequent to obstructive hydrocephalus. Later in the course of the disease, truncal ataxia, nystagmus, and VIth nerve palsy may develop. Ependymomas often extend through the foramen of Luschka into the cerebellopontine angle, and cranial nerve dysfunction may be noted.42,43,46
On computed tomography (CT) scanning, ependymomas are usually low-density masses that enhance strongly and cause hydrocephalus. Calcifications and cystic components may also be seen.47 MRI with and without contrast is the preferred neuroimaging modality for both diagnosis and surgical planning because it provides greater soft-tissue detail, especially for infratentorial lesions (Figure 6). On T1-weighted images, a hypo- to isointense contrast-enhancing mass is demonstrated that typically fills and expands 4th ventricle. Ependymomas are usually hyperintense on T2-weighted sequences.40,47
Figure 6.
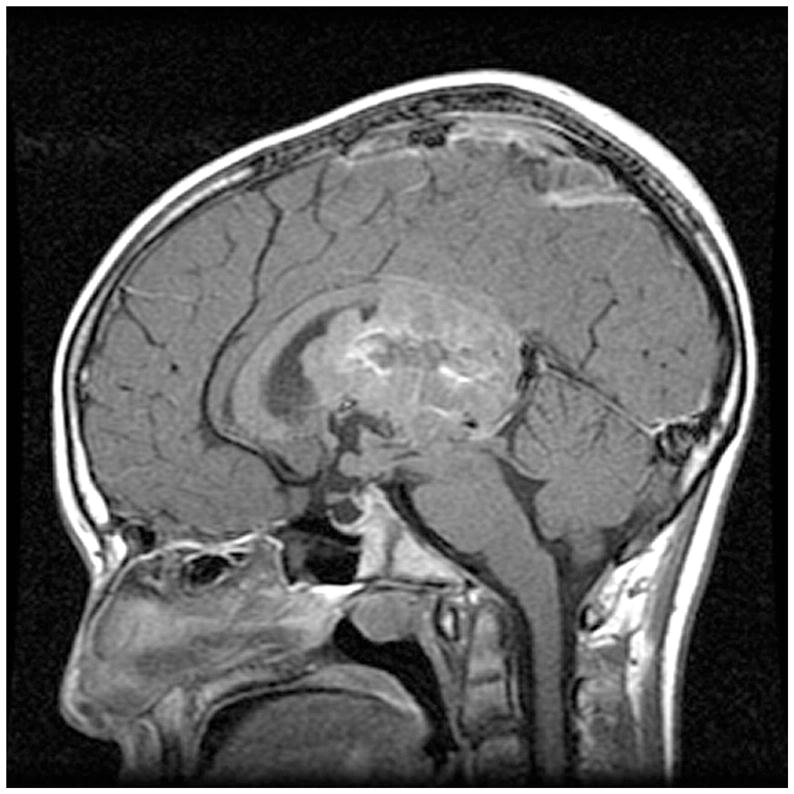
Sagittal MRI of intraventricular ependymoma in 14-year-old male who presented with brief history of headaches and memory loss.
The incidence of dissemination of ependymoma is only 11% to 17%.48,49 Nonetheless, it is important to demonstrate its presence or absence, because disseminated disease is a strong adverse prognostic factor. Accordingly, it is preferable to perform a staging spinal MRI preoperatively.40
Management
A number of studies have shown that complete surgical resection offers the best hope of cure.48–50 As such, gross total resection should be the intent of surgery when it can be accomplished safely (Figure 7).51 In reality, the actual percentage of cases where ependymomas of the posterior fossa can be completely resected is 30% to 50%.40 To enhance the safety of surgery for posterior fossa ependymoma, the neurosurgeon can take advantage of intraoperative monitoring of lower cranial nerves and brainstem pathways, and neuronavigation.
Figure 7.
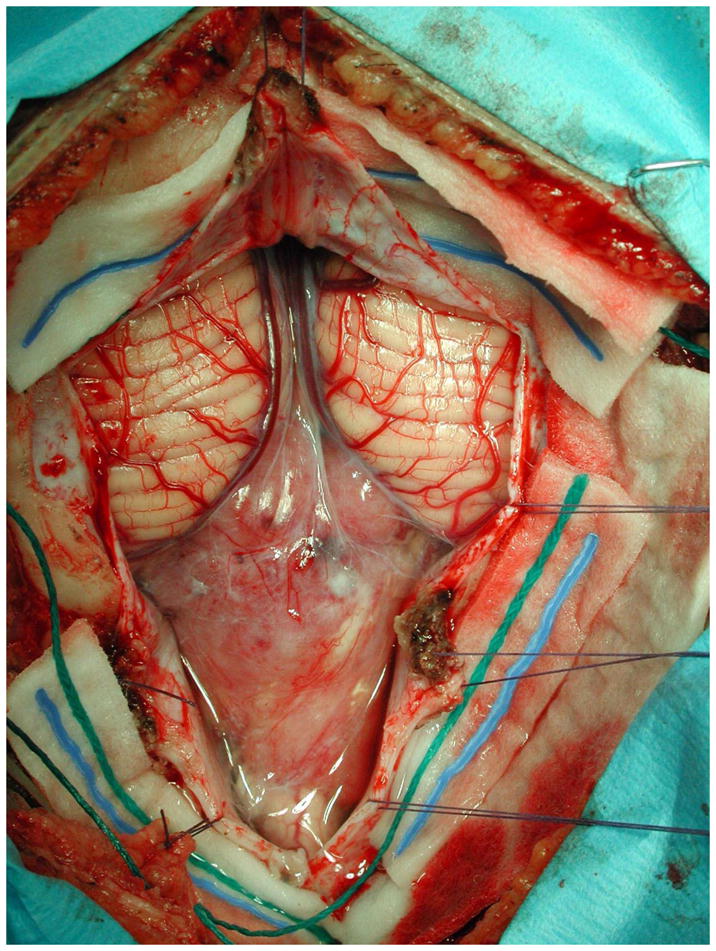
Intraoperative photomicrograph of posterior fossa ependymoma. The tumor descends below the cerebellar tonsils, and extends into the high cervical spinal canal. The goal with ependymoma is complete neurosurgical resection if possible.
Intracranial ependymomas are relatively radiosensitive. Craniospinal axis fields are used only when spinal seeding is radiologically or pathologically evident.52 In young children (less than 5 years of age) the provision of radiation therapy is usually avoided, if possible. Because of the severity of this disease, however, even young children with these tumors usually require chemotherapy and radiation therapy.40,53
Some authors recommend the use of postoperative radiotherapy, regardless of whether the resection is gross total or subtotal.51 The dose of radiation for the treatment of ependymoma has traditionally been in the range of 4500 cGy to 5600 cGy.41,46 Because the primary problem with ependymomas is local tumor control, brachytherapy with 125iodine has been used at recurrence.46 Radiosurgery also may be an option in patients with ependymomas.42,54
In comparison with medulloblastomas and even astrocytomas, ependymomas are quite chemoresistant. Chemotherapy has not yielded a significant improvement in survival for patients with ependymal tumors.46,55 High-dose chemotherapy followed by autologous bone marrow transplantation has been tried experimentally.40 At this point, the data are not sufficient to justify the use of chemotherapy in patients with ependymomas, except in younger children (to substitute for or delay radiation therapy56) or patients in whom surgery and radiation therapy have failed to control tumor growth.40
Prognostic factors
Unless a complete removal of the tumor can be accomplished, ependymoma almost always progresses. Generally, the 5-year survival rate for children with intracranial ependymomas is approximately 50% to 60% and the 10-year survival is approximately 40% to 50%.42 Controversial results have been published regarding the roles of age, tumor location and histological composition, radiotherapy, and chemotherapy on progression-free survival.38–40,42
Disease dissemination at diagnosis carries a poor prognosis. In fact, this may be the single most important prognostic factor.42,57–59 Total tumor excision was strongly related to patient prognosis in both uni- and multivariate analyses.39,40 Higher MIB-1 labeling index and subtotal resection were shown to be the strongest indicators of the tumor’s aggressive behavior and poor prognosis.60
Medulloblastoma
Medulloblastoma is the second most common posterior fossa tumor in children and represents 15% to 25% of pediatric brain tumors.61 Patients with this tumor usually present in the first decade of life,62 with peak incidences between 3 years and 4 years and 8 years and 9 years of age.63
Medulloblastomas are most frequently found in the 4th ventricle. They tend to be invasive of normal cerebellar tissue, and in 15% of cases they infiltrate the brainstem.64 The child with a medulloblastoma may present with obstructive hydrocephalus secondary to the posterior fossa mass followed by cerebellar dysfunction. Older children usually complain of morning headaches. Vomiting is frequent because of increased intracranial pressure. Later, patients develop ataxia caused by cerebellar compression and coexisting hydrocephalus.65 Development of a stiff neck or head tilt usually suggests tonsillar involvement by the tumor or signs of impending herniation.62,66
By CT, medulloblastoma is often seen as a midline hyperdense cerebellar vermian mass. Ventriculomegaly is noted in approximately 85% to 90% of cases.63,66 The best imaging modality for these tumors is MRI, which reveals a heterogeneous hypointense mass on T1-weighted imaging67,68 and a hyperintense mass on T2-weighted sequence which enhances heterogeneously after gadolinium infusion.62,63 MRI of the spine enables demonstration of spinal subarachnoid dissemination. Drop metastases occur in up to 40% of patients, most commonly in the lumbosacral and thoracic areas (Figure 8). Because of the significant incidence of dissemination at the time of diagnosis, neuroimaging of the entire neuraxis is required prior to initiating treatment.66,67 Magnetic resonance spectroscopy shows an elevated choline (a marker of biomembrane) and decreased N-acetyl aspartate (NAA, a neuronal cell marker) and other mobile lipids.69
Figure 8.
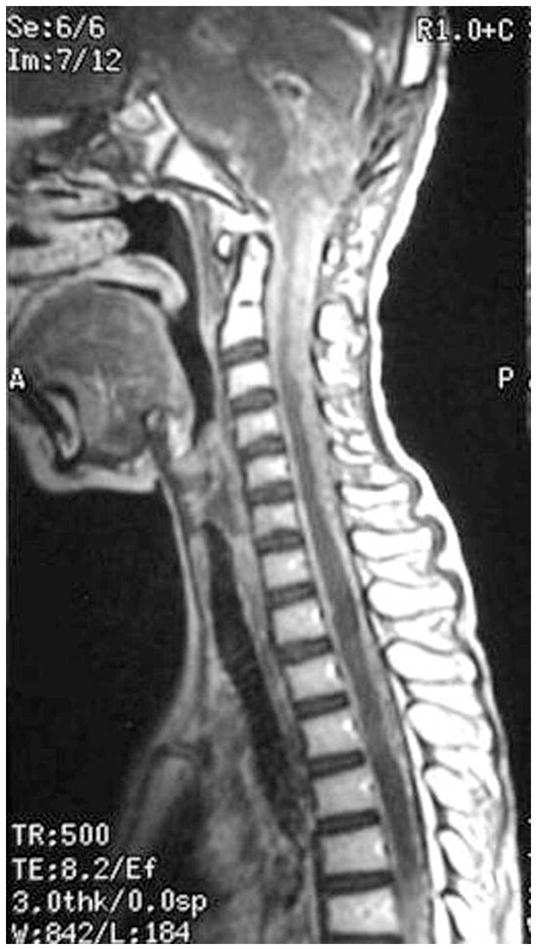
MRI of disseminated medulloblastoma. (A) Coating of the spinal cord with tumor is seen on this sagittal MRI. (B) Diffuse leptomeningeal seeding of the brain is shown.
Management
Surgical resection of medulloblastoma is an essential part of treatment that has led to improved survival, particularly in children with localized disease.70 Using micro-operative techniques, intraoperative imaging and monitoring and the ultrasonic aspirator facilitate gross total resection with minimal neurological deficits.71 When the tumor infiltrates the brainstem, the neurosurgeon should not be aggressive in this location.66
Postoperative cerebellar mutism is a relatively common complication. It develops over 48 hours to 72 hours after resection of the tumor and can persist from weeks to months, with associated findings of dysmetria, hypotonia, dysphagia, hemiparesis, and increased mood lability. Other common temporary postoperative complications are ataxia, hemiparesis, and VIth cranial nerve palsy.11, 71
Radiation therapy is an integral part of the treatment of patients with medulloblastoma that, compared with surgery alone, improves survival.11 Irradiated fields include the posterior fossa and the entire neuraxis, where there is potential spread of neoplastic cells. The standard dose to the primary site is 5400 rad. However, attempts are being made to identify the most appropriate dose.72 Radiation therapy is delayed or not given to children younger than 3 years of age.63 Proton beam irradiation has been proposed in the management of young patients, as it has a lower incidence of adverse effects than does conventional radiation toxicity.73 Some of the late complications of conventional radiation therapy include focal or diffuse demyelination, white-matter necrosis, focal calcifications, and microangiopathy.74,75
A range of chemotherapeutic drugs have been used in the treatment of medulloblastoma. One goal of chemotherapy is to augment, delay (especially in young children), or even avoid radiation; however, chemotherapy alone can be curative in patients with nonmetastatic medulloblastoma after gross total resection but not in cases of metastases.70 Using lower-dose craniospinal radiotherapy with chemotherapy in children older than 3 years with nondisseminated disease may lead to higher 5-year event-free survival rates.76,77
Despite impressive improvements in therapy, 20% to 30% of children with medulloblastoma still experience tumor progression,78 mostly within 2 years after diagnosis and in the primary tumor site of the posterior fossa.79 Once there is a recurrence, prognosis is very dismal and the patients usually die within a year.62 An increased probability of survival exists for patients whose tumors recur at only the primary tumor site.78 Salvage therapies include the use of second surgery, localized radiotherapy, chemotherapy, and high-dose chemotherapy with autologous stem cell rescue, which help overall survival rates of 0 to 22.6%.66,78
Prognostic factors
At the present time, children less than 3 years of age,72 residual tumor of more than 1.5 cm2, and/or any evidence of dissemination are associated with poor prognosis.66 It is expected that future treatment protocols will take advantage of the growing body of information on molecular pharmacology, neurogenetics, cell biology, and developmental biology to cure more children with medulloblastoma while minimizing the cognitive and neuropsychological effects of current therapies.63,66
Craniopharyngioma
Craniopharyngioma is the most common intracranial nonglial tumor of childhood and constitutes 2.5% to 9% of pediatric brain tumors.80,81 This tumor can occur at any age, with a peak at 5 years to 9 years.82 Males and females are affected in equal proportions.80 Clinical symptoms are usually related to endocrine dysfunction, even though these problems may not be the reason for the initial medical evaluation. Growth failure has been reported in 93% of children. Visual loss, headache, apathy, incontinence, depression, memory loss, and mental changes may also be observed. 83
Craniopharyngiomas typically arise in the suprasellar region from epithelial remnants of the Rathke’s pouch. Although the histological appearance of these lesions is benign, they are locally aggressive, with fingerlike attachments that invade adjacent critical structures such as the optic nerves and chiasm, pituitary gland, hypothalamus, 3rd ventricle, and blood vessels.81 Approximately 90% of craniopharyngiomas are primarily cystic, whereas in approximately 30% of the cases a small solid tumor with one or more cysts is found. Only about 10% of cases are wholly solid.80,81
Both CT and MRI are useful in the diagnosis. Brain CT often reveals a cystic mass with calcification; hydrocephalus is not an uncommon radiological finding. The tumor mass, its general configuration, and its relationship to the ventricular system and major cranial arteries are demonstrated precisely by MRI. Enhancement with contrast generally defines cyst walls and allows visualization of the solid portions of the tumor.83
Management
In 15% to 30% of patients the presenting symptoms are related exclusively to hydrocephalus. Often, there is bilateral ventricular dilation subsequent to obstruction of foramen of Monro, which can be managed with a single shunting system in conjunction with endoscopic fenestration of the septum pellucidum. Tumor surgery can be done after the ventricles have been decompressed and the patient has been stabilized.84
Cystic craniopharyngiomas frequently require therapies aimed at reducing cyst size. This can be achieved through various neurosurgical approaches, including stereotactic- or ultrasound-guided aspiration, direct shunting, fenestration into the ventricular system, and intracystic instillation of chemotherapeutic agents. An Ommaya reservoir system is commonly used for drainage of the cystic part; it is placed during open surgery or by stereotactic accesses, which typically involve the use of a neuroendoscope or intraoperative imaging.80,82
Surgical extirpation is an ideal goal for children with craniopharyngioma if it can be performed safely and with minimal morbidity. Cure can result following complete resection.81 However, the neurosurgeon must be fully cognizant of the critical location of the tumor adjacent to the optic apparatus, pituitary gland and stalk, hypothalamus, and vessels of the circle of Willis. A number of neurosurgical approaches have been used in the resection of craniopharyngiomas, including subfrontal, pterional, interhemispheric, transcallosal, and transsphenoidal. The choice of approach is based on intrinsic tumor features and neurosurgeon familiarity. Recently, the neuroendoscope has been used to resect craniopharyngiomas via a transnasal, transsphenoidal approach with promising early results. Despite the neurosurgeon’s best efforts at resection of craniopharyngioma, the recurrence of these tumors remains high at approximately 30%.82,85
To expand on the potential complications of craniopharyngioma surgery, impairment of anterior pituitary function necessitating replacement of two or more anterior pituitary hormones is seen in approximately 80% of cases.86 The non-endocrine morbidity (15%) includes hypothalamic damage resulting in morbid obesity, severe appetite disturbance, loss of diurnal rhythm, and cognitive impairment.87 Aneurysmal dilatation of the cerebral vessels of the circle of Willis has been reported postoperatively, and visual field alterations after surgery are commonly seen. For these reasons, some believe that children with craniopharyngioma should undergo limited resection followed by cyst-directed therapy or irradiation strategies (Figure 9). The change in approach from radical resection to limited resection followed by focal radiotherapy has afforded over 80% long-term tumor control without the cognitive and endocrine impairments.86,88
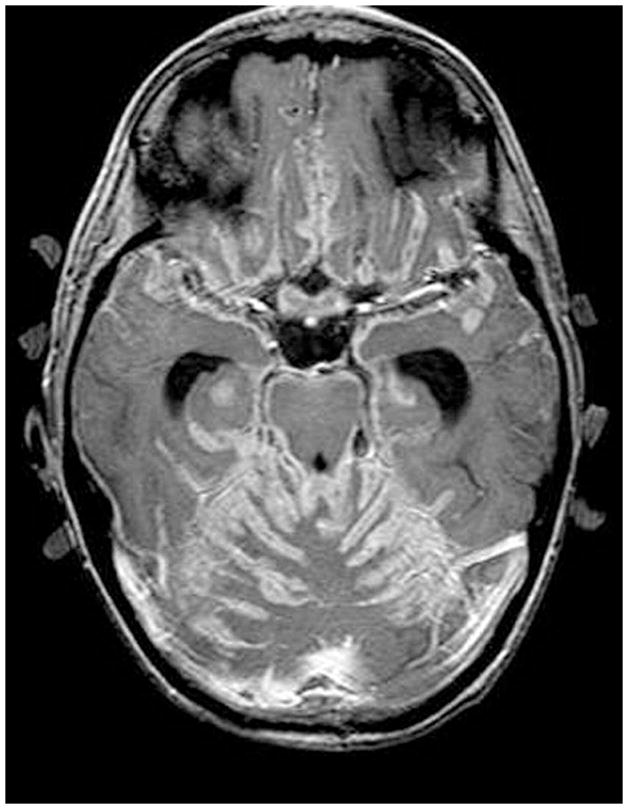
Figure 9.
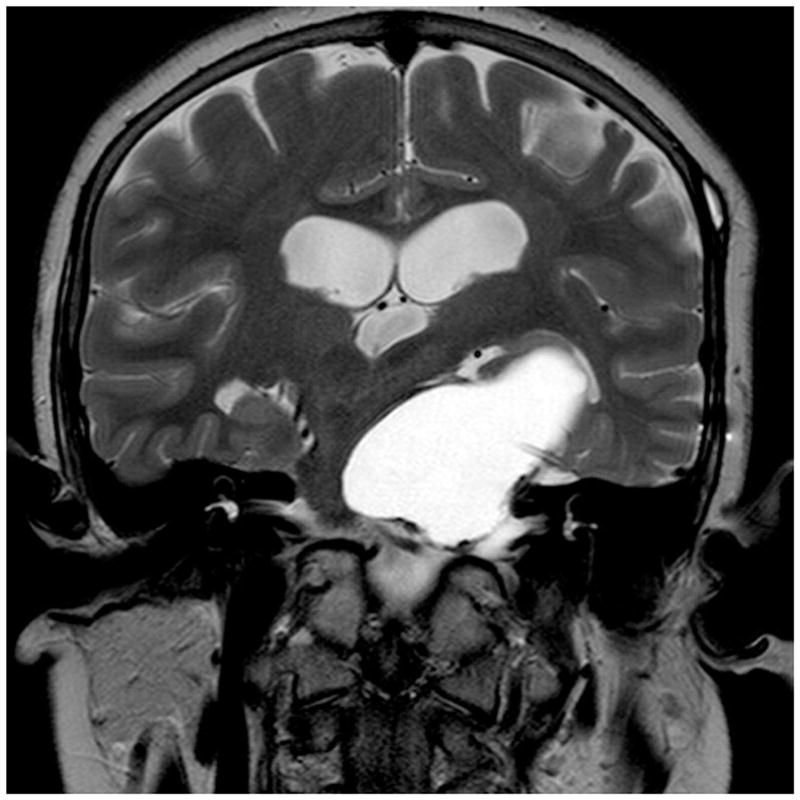
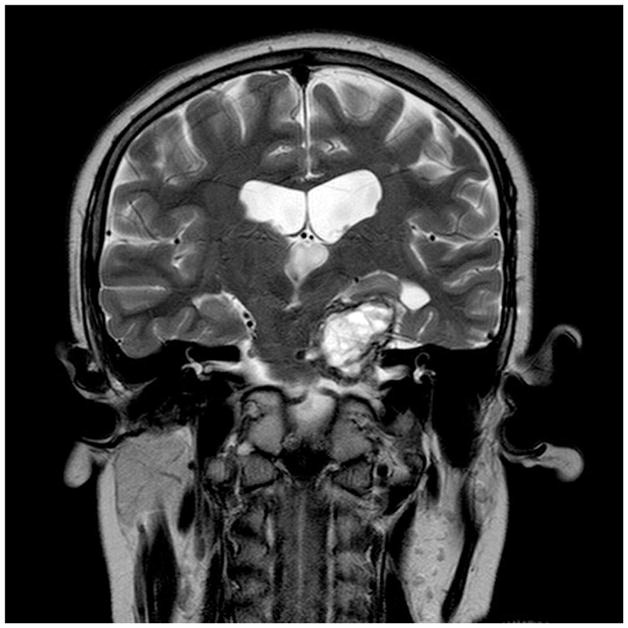
Cystic craniopharyngioma treated conservatively with Ommaya reservoir and repeated tapping. Coronal MRI scans are shown (A) prior to insertion of the Ommaya reservoir and (B) after insertion of the Ommaya reservoir and following repeated tapping; shows markedly diminished cyst size.
New 3-dimensional conformal radiation protocols using CT and MRI for target localization, dose-volume histograms, computer-driven multi-leaf collimators, and stereotactically guided delivery units are on the horizon for treatment of craniopharyngioma. These have all increased the potential for improving the therapeutic ratio of craniopharyngiomas.81,89,90 The 10-year survival in children treated with the aid of CT or MR imaging technology was 85% to 96% compared with 56% to 78% among children treated without using modern imaging.89
Stereotactic radiosurgery and fractionated stereotactic radiotherapy are increasingly being used in the management of pediatric craniopharyngioma, improving tumor control rates. Indications for radiosurgery are usually limited to craniopharyngiomas that are solid with a size less than 2.5 cm, and in tumors that are at least 3 mm away from the optic chiasm. Tumor control rates of 70% to 100% have been reported 1 year to 5 years after radiosurgery, with very few complications directly attributable to radiosurgery.81,89,91
Lastly, the use of intracavitary irradiation using yttrium-90 or phosphorus-32 is an option for cystic craniopharyngioma. Long-term control of cystic craniopharyngioma has been reported using this technique. 92
Conclusion
The neurosurgeon has traditionally played an integral role in the management of children with brain tumors. This trend will likely continue far into the future for many tumor types. Here, we have presented the data showing that neurosurgical extirpation of brain tumors can improve survival for children with low-grade glioma, high-grade glioma, medulloblastoma, ependymoma, and craniopharyngioma. In some instances, neurosurgery must be combined with expertly administered adjuvant therapies such as radiation therapy and chemotherapy, as in the case of medulloblastoma. In all instances, neurosurgery must be performed with the utmost skill and planning, and be bolstered by the latest technologies, such as intraoperative neuronavigation or MRI, functional mapping, and microneurosurgical techniques, to ensure that patients who survive the biology of their tumors are not held prisoner to devastating long-term neurological impairments. In the field of neurosurgery, we can expect to see minimally invasive techniques, such as neuro-endoscopy and stereotactic delivery of chemotherapeutics via convection-enhanced delivery, play an increasingly important role.
Acknowledgments
Presented in part at the Neurobiology of Disease in Children Symposium on Central Nervous System Tumors in conjunction with the 36th annual meeting of the Child Neurology Society, Quebec City, Quebec, October 10, 2007. This work was supported in part by grants from the National Cancer Institute of Canada/Canadian Cancer Society, the Pediatric Brain Tumor Foundation, Brainchild, and the Wiley Fund for Brain Tumour Research at the Hospital for Sick Children.
References
- 1.Young JL, Jr, Miller RW. Incidence of malignant tumors in U.S. children. J Pediatr. 1975;86:254–258. doi: 10.1016/s0022-3476(75)80484-7. [DOI] [PubMed] [Google Scholar]
- 2.Packer RJ, Bleyer WA, Pochedly C, editors. Pediatric neuro-oncology: new trends in clinical research. Chur, Switzerland: Harwood Academic; 1992. [Google Scholar]
- 3.Newman LA, Boop FA, Sanford RA, et al. Postoperative swallowing function after posterior fossa tumor resection in pediatric patients. Childs Nerv Syst. 2006;22:1296–1300. doi: 10.1007/s00381-006-0065-z. [DOI] [PubMed] [Google Scholar]
- 4.Albright AL. Brain tumors in neonates, infants, and toddlers. Contemp Neurosurg. 1985;7:1–8. [Google Scholar]
- 5.Kirk EA, Howard VC, Scott CA. Description of posterior fossa syndrome in children after posterior fossa brain tumor surgery. J Pediatr Oncol Nurs. 1995;12:181–187. doi: 10.1177/104345429501200402. [DOI] [PubMed] [Google Scholar]
- 6.Pollack IF. Brain tumors in children. N Engl J Med. 1994;331:1500–1507. doi: 10.1056/NEJM199412013312207. [DOI] [PubMed] [Google Scholar]
- 7.Bondy M, Wiencke J, Wrensch M, Kyritsis AP. Genetics of primary brain tumors: a review. J Neurooncol. 1994;18:69–81. doi: 10.1007/BF01324606. [DOI] [PubMed] [Google Scholar]
- 8.Davis FG, Freels S, Grutsch J, et al. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: An analysis based on surveillance, epidemiology, and end results (SEER) data 1973–1991. J Neurosurg. 1998;88:1–10. doi: 10.3171/jns.1998.88.1.0001. [DOI] [PubMed] [Google Scholar]
- 9.Cavenee WK, Furnari FB, Nagane M, et al. Diffusely nfiltrating astrocytomas. In: Kleihues P, Cavenee WK, editors. Tumours of the Nervous System. Lyon, France: IARC Press; 2000. pp. 10–21. [Google Scholar]
- 10.Tamber MS, Rutka JT. Pediatric supratentorial high-grade gliomas. Neurosurg Focus. 2003;14:Article 1. doi: 10.3171/foc.2003.14.2.2. [DOI] [PubMed] [Google Scholar]
- 11.Rutka JT, Kuo JS. Pediatric surgical neuro-oncology: current best care practices and strategies. J Neuro-Oncol. 2004;69:139–150. doi: 10.1023/b:neon.0000041877.14749.b6. [DOI] [PubMed] [Google Scholar]
- 12.Karim AB, Maat B, Hatlevoll R, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36:549–556. doi: 10.1016/s0360-3016(96)00352-5. [DOI] [PubMed] [Google Scholar]
- 13.Pollack IF, Claassen D, Al-Shboul Q, et al. Low-grade gliomas of the cerebral hemispheres in children: an analysis of 71 cases. J Neurosurg. 1995;82:536–547. doi: 10.3171/jns.1995.82.4.0536. [DOI] [PubMed] [Google Scholar]
- 14.Gajjar A, Sanford RA, Heideman R, et al. Low-grade astrocytoma: a decade of experience at St. Jude’s children’s research hospital. J Clin Oncol. 1997;15:2792–2799. doi: 10.1200/JCO.1997.15.8.2792. [DOI] [PubMed] [Google Scholar]
- 15.Butler D, Jose B, Summe R, et al. Pediatric astrocytomas. The Louisville experience: 1978–1988. Am J Clin Oncol. 1994;17:475–479. [PubMed] [Google Scholar]
- 16.Morreale VM, Ebersold MJ, Quast LM, Parisi JE. Cerebellar astrocytoma: experience with 54 cases surgically treated at the Mayo Clinic, Rochester, Minnesota from 1978 to 1990. J Neurosurg. 1997;87:257–261. doi: 10.3171/jns.1997.87.2.0257. [DOI] [PubMed] [Google Scholar]
- 17.Bowers DC, Gargan L, Kapur P, et al. Study of the MIB-1 labeling index as a predictor of tumor progression in pilocytic astrocytomas in children and adolescents. J Clin Oncol. 2003;21:2968–2973. doi: 10.1200/JCO.2003.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Kleihues P, Burger PC, Scheithauer BW, editors. Histological Typing of Tumors of the Central Nervous System. 2. Berlin, Germany: Springer-Verlag; 1993. [Google Scholar]
- 19.Rickert CH, Sträter R, Kaatsch P, et al. Pediatric high-grade astrocytomas show chromosomal imbalances distinct from adult cases. Am J Pathol. 2001;158:1525–1532. doi: 10.1016/S0002-9440(10)64103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Artico M, Cervoni L, Celli P, et al. Supratentorial glioblastoma in children: a series of 27 surgically treated cases. Childs Nerv Syst. 1993;9:7–9. doi: 10.1007/BF00301926. [DOI] [PubMed] [Google Scholar]
- 21.Zimmerman RA. Neuroimaging of primary brainstem gliomas: Diagnosis and course. Pediatr Neurosurg. 1996;25:45–53. doi: 10.1159/000121096. [DOI] [PubMed] [Google Scholar]
- 22.Wisoff JH, Boyett JM, Berger MS, et al. Current neurosurgical management and the impact of the extent of resection in the treatment of malignant gliomas of childhood: a report of the Children’s Cancer Group Trial No. CCG-945. J Neurosurg. 1998;89:52–59. doi: 10.3171/jns.1998.89.1.0052. [DOI] [PubMed] [Google Scholar]
- 23.Bucci MK, Maity A, Janss AJ, et al. Near complete surgical resection predicts a favorable outcome in pediatric patients with nonbrainstem, malignant gliomas. Cancer. 2004;101:817–824. doi: 10.1002/cncr.20422. [DOI] [PubMed] [Google Scholar]
- 24.Sposto R, Ertel IJ, Jenkin RD, et al. The effectiveness of chemotherapy for treatment of high grade astrocytoma in children: results of a randomized trial. A report from the Childrens Cancer Study Group. J Neurooncol. 1989;7:165–177. doi: 10.1007/BF00165101. [DOI] [PubMed] [Google Scholar]
- 25.Phuphanich S, Edwards MS, Levin VA, et al. Supratentorial malignant gliomas of childhood. Results of treatment with radiation therapy and chemotherapy. J Neurosurg. 1984;60:495–499. doi: 10.3171/jns.1984.60.3.0495. [DOI] [PubMed] [Google Scholar]
- 26.Finlay JL, Boyett JM, Yates AJ, et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Children’s Cancer Group. J Clin Oncol. 1995;13:112–123. doi: 10.1200/JCO.1995.13.1.112. [DOI] [PubMed] [Google Scholar]
- 27.Pollack IF, Finkelstein SD, Woods J, et al. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med. 2002;346:420–427. doi: 10.1056/NEJMoa012224. [DOI] [PubMed] [Google Scholar]
- 28.Mason WP, Grovas A, Halpern S, et al. Intensive chemotherapy and bone marrow rescue for young children with newly diagnosed malignant brain tumors. J Clin Oncol. 1998;16:210–221. doi: 10.1200/JCO.1998.16.1.210. [DOI] [PubMed] [Google Scholar]
- 29.Albright AL, Guthkelch AN, Packer RJ, et al. Prognostic factors in pediatric brain-stem gliomas. J Neurosurg. 1986;65:751–755. doi: 10.3171/jns.1986.65.6.0751. [DOI] [PubMed] [Google Scholar]
- 30.Jallo GI, Biser-Rohrbaugh A, Freed D. Brainstem gliomas. Childs Nerv Syst. 2004;20:143–153. doi: 10.1007/s00381-003-0870-6. [DOI] [PubMed] [Google Scholar]
- 31.Fischbein NJ, Prados MD, Wara W, et al. Radiologic classification of brain stem tumors: correlation of magnetic resonance imaging appearance with clinical outcome. Pediatr Neurosurg. 1996;24:9–23. doi: 10.1159/000121010. [DOI] [PubMed] [Google Scholar]
- 32.Albright AL, Packer RJ, Zimmerman R, et al. Magnetic resonance scans should replace biopsies for the diagnosis of diffuse brain stem gliomas: a report from the Children’s Cancer Group. Neurosurgery. 1993;33:1026–1030. doi: 10.1227/00006123-199312000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Epstein F, Wisoff JH. Intrinsic brainstem tumors in childhood: surgical indications. J Neurooncol. 1988;6:309–317. doi: 10.1007/BF00177425. [DOI] [PubMed] [Google Scholar]
- 34.Raffel C, McComb JG, Bodner S, Gilles FE. Benign brain stem lesions in pediatric patients with neurofibromatosis: case reports. Neurosurg. 1989;25:959–964. doi: 10.1097/00006123-198912000-00018. [DOI] [PubMed] [Google Scholar]
- 35.Allen JC, Siffert J. Contemporary chemotherapy issues for children with brainstem gliomas. Pediatr Neurosurg. 1996;24:98–102. doi: 10.1159/000121024. [DOI] [PubMed] [Google Scholar]
- 36.Pincus DW, Richter EO, Yachnis AT, et al. Brainstem stereotactic biopsy sampling in children. J Neurosurg. 2006;104(2 Suppl):108–114. doi: 10.3171/ped.2006.104.2.108. [DOI] [PubMed] [Google Scholar]
- 37.Broniscer A, Iacono L, Chintagumpala M, et al. Role of temozolomide after radiotherapy for newly diagnosed diffuse brainstem glioma in children: results of a multiinstitutional study (SJHG-98) Cancer. 2005;103:133–139. doi: 10.1002/cncr.20741. [DOI] [PubMed] [Google Scholar]
- 38.Shu HK, Sall WF, Maity A, et al. Childhood intracranial ependymoma: twenty-year experience from a single institution. Cancer. 2007;110:432–441. doi: 10.1002/cncr.22782. [DOI] [PubMed] [Google Scholar]
- 39.Figarella-Branger D, Civatte M, Bouvier-Labit C, et al. Prognostic factors in intracranial ependymomas in children. J Neurosurg. 2000;93:605–613. doi: 10.3171/jns.2000.93.4.0605. [DOI] [PubMed] [Google Scholar]
- 40.Maksoud YA, Hahn YS, Engelhard HH. Intracranial ependymoma. Neurosurg Focus. 2002;13:e4. doi: 10.3171/foc.2002.13.3.5. [DOI] [PubMed] [Google Scholar]
- 41.Salazar OM. A better understanding of CNS seeding and a brighter outlook for postoperatively irradiated patients with ependymomas. Int J Radiat Oncol Biol Phys. 1983;9:1231–1234. doi: 10.1016/0360-3016(83)90186-4. [DOI] [PubMed] [Google Scholar]
- 42.Sanford RA, Gajjar A. Ependymomas. Clin Neurosurg. 1997;44:559–570. [PubMed] [Google Scholar]
- 43.Lyons MK, Kelly PJ. Posterior fossa ependymomas: report of 30 cases and review of the literature. Neurosurg. 1991;28:659–665. [PubMed] [Google Scholar]
- 44.Dimopoulos VG, Fountas KN, Robinson JS. Familial intracranial ependymomas. Report of three cases in a family and review of the literature. Neurosurg Focus. 2006;15:20, E8. doi: 10.3171/foc.2006.20.1.9. [DOI] [PubMed] [Google Scholar]
- 45.Oppenheim JS, Strauss RC, Mormino J, et al. Ependymomas of the third ventricle. Neurosurgery. 1994;34:350–353. doi: 10.1227/00006123-199402000-00020. [DOI] [PubMed] [Google Scholar]
- 46.Sutton LN, Goldwein J. Intracranial ependymomas. In: Winn HR, editor. Youmans Neurological Surgery. 5. New York, NY: WB Saunders; 2004. pp. 3623–3637. [Google Scholar]
- 47.Peretti-Viton P, Perez-Castillo AM, Martini P, et al. Supratentorial ependymomas. Neuroradiological study. J Neuroradiol. 1991;18:201–209. [PubMed] [Google Scholar]
- 48.Rezai A, Woo H, Lee M, et al. Disseminated ependymomas of the central nervous system. J Neurosurg. 1996;85:618–624. doi: 10.3171/jns.1996.85.4.0618. [DOI] [PubMed] [Google Scholar]
- 49.Sutton L, Goldwein J, Perilongo G, et al. Prognostic factors in childhood ependymomas. Pediatr Neurosurg. 1990–1991;16:57–65. doi: 10.1159/000120509. [DOI] [PubMed] [Google Scholar]
- 50.Kovalic IT, Plaris N, Grigsby PW, et al. Intracranial ependymoma: Long term outcome, patterns of failure. J Neurooncol. 1993;15:125–131. doi: 10.1007/BF01053933. [DOI] [PubMed] [Google Scholar]
- 51.Rogers L, Pueschel J, Spetzler R, et al. Is gross-total resection sufficient treatment for posterior fossa ependymomas? J Neurosurg. 2005;102:629–636. doi: 10.3171/jns.2005.102.4.0629. [DOI] [PubMed] [Google Scholar]
- 52.McLaughlin MP, Marcus RB, Buatti JM, et al. Ependymoma: results, prognostic factors and treatment recommendations. Int J Radiat Oncol Biol Phys. 1998;40:845–850. doi: 10.1016/s0360-3016(97)00893-6. [DOI] [PubMed] [Google Scholar]
- 53.Duffner PK, Krischer JP, Sanford RA, et al. Prognostic factors in infants and very young children with intracranial ependymomas. Pediatr Neurosurg. 1998;28:215–222. doi: 10.1159/000028654. [DOI] [PubMed] [Google Scholar]
- 54.Grabb PA, Lunsford LD, Albright AL, et al. Stereotactic radiosurgery for glial neoplasms of childhood. Neurosurg. 1996;38:696–702. [PubMed] [Google Scholar]
- 55.Souweidane M, Bouffet E, Finlay I. The role of chemotherapy in newly diagnosed ependymoma of childhood. Pediatr Neurosurg. 1998;28:273–278. doi: 10.1159/000028664. [DOI] [PubMed] [Google Scholar]
- 56.Geyer JR, Zeltzer PM, Boyett JM, et al. Survival of infants with primitive neuroectodermal tumors and malignant ependymomas of the CNS treated with eight drugs in 1 day: A report from the Children’s Cancer Group. J Clin Oncol. 1994;12:1607–1615. doi: 10.1200/JCO.1994.12.8.1607. [DOI] [PubMed] [Google Scholar]
- 57.Nazar G, Hoffman H, Becker L, et al. Infratentorial ependymomas in childhood: Prognostic factors and treatment. Neurosurg. 1990;72:408–417. doi: 10.3171/jns.1990.72.3.0408. [DOI] [PubMed] [Google Scholar]
- 58.Healey E, Barnes P, Kupsky W, et al. The prognostic significance of postoperative residual tumor in ependymoma. Neurosurg. 1991;28:666–671. doi: 10.1097/00006123-199105000-00005. [DOI] [PubMed] [Google Scholar]
- 59.Chiu J, Woo S, Ater J, et al. Intracranial ependymoma in children: analysis of prognostic factors. J Neurooncol. 1992;13:283–290. doi: 10.1007/BF00172482. [DOI] [PubMed] [Google Scholar]
- 60.Zamecnik J, Snuderl M, Eckschlager T, et al. Pediatric intracranial ependymomas: prognostic relevance of histological, immunohistochemical, and flow cytometric factors. Mod Pathol. 2003;16:980–991. doi: 10.1097/01.MP.0000087420.34166.B6. [DOI] [PubMed] [Google Scholar]
- 61.Whelan HT, Krouwer HG, Schmidt MH, et al. Current therapy and perspectives in the treatment of medulloblastoma. Pediatr Neurol. 1998;18:103–115. doi: 10.1016/s0887-8994(97)00221-x. [DOI] [PubMed] [Google Scholar]
- 62.Modha A, Vassilyadi M, George A, et al. Medulloblastoma in children-the Ottawa experience. Childs Nerv Syst. 2000;16:341–350. doi: 10.1007/s003810050529. [DOI] [PubMed] [Google Scholar]
- 63.Crawford JR, MacDonald TJ, Packer RJ. Medulloblastoma in childhood: new biological advances. Lancet Neurol. 2007;6:1073–1085. doi: 10.1016/S1474-4422(07)70289-2. [DOI] [PubMed] [Google Scholar]
- 64.Jenkin O. The radiation treatment of medulloblastoma. J Neurooncol. 1996;29:45–54. doi: 10.1007/BF00165517. [DOI] [PubMed] [Google Scholar]
- 65.Raimondi AJ, Tomita T. Hydrocephalus and infratentorial tumors: incidence, clinical picture, and treatment. J Neurosurg. 1981;55:174–182. doi: 10.3171/jns.1981.55.2.0174. [DOI] [PubMed] [Google Scholar]
- 66.Packer RJ, Cogen P, Vezina G, Rorke LB. Medulloblastoma: clinical and biologic aspects. Neuro Oncol. 1999;1:232–250. doi: 10.1215/15228517-1-3-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar R, Achari G, Banerjee D, Chhabra DK. Uncommon presentation of medulloblastoma. Childs Nerv Syst. 2001;17:538–542. doi: 10.1007/s003810100446. [DOI] [PubMed] [Google Scholar]
- 68.Buhring U, Strayle-Batra M, Freudenstein D, et al. MRI features of primary, secondary and metastatic medulloblastoma. Eur Radiol. 2002;12:1342–1348. doi: 10.1007/s00330-001-1189-x. [DOI] [PubMed] [Google Scholar]
- 69.Peet AC, Davies NP, Ridley L, et al. Magnetic resonance spectroscopy suggests key diff erences in the metastatic behaviour of medulloblastoma. Eur J Cancer. 2007;43:1037–1044. doi: 10.1016/j.ejca.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 70.Grill J, Sainte-Rose C, Jouvet A, et al. Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol. 2005;6:573–580. doi: 10.1016/S1470-2045(05)70252-7. [DOI] [PubMed] [Google Scholar]
- 71.Tomita T. Surgical management of cerebellar peduncle lesions in children. Neurosurg. 1986;18:568–575. doi: 10.1227/00006123-198605000-00010. [DOI] [PubMed] [Google Scholar]
- 72.Kaufman BA. Medulloblastoma. In: Winn HR, editor. Youmans Neurological Surgery. 5. New York, NY: WB Saunders; 2004. pp. 3639–3654. [Google Scholar]
- 73.Kirsch DG, Tarbell NJ. New technologies in radiation therapy for pediatric brain tumors: the rationale for proton radiation therapy. Pediatr Blood Cancer. 2004;42:461–464. doi: 10.1002/pbc.10471. [DOI] [PubMed] [Google Scholar]
- 74.Riva D, Milani N, Pantaleoni C, et al. Combined treatment modality for medulloblastoma in childhood: effects on neuropsychological functioning. Neuropediatrics. 1991;22:36–42. doi: 10.1055/s-2008-1071413. [DOI] [PubMed] [Google Scholar]
- 75.Massimino M, Gandola L, Cefalo G, et al. Management of medulloblastoma and ependymoma in infants: a single-institution long-term retrospective report. Childs Nerv Syst. 2000;16:15–20. doi: 10.1007/PL00007279. [DOI] [PubMed] [Google Scholar]
- 76.Carrie C, Lasset C, Alapetite C, et al. Multivariate analysis of prognostic factors in adult patients with medulloblastoma. Cancer. 1994;74:2352–2360. doi: 10.1002/1097-0142(19941015)74:8<2352::aid-cncr2820740821>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 77.Taylor RE, Bailey CC, Robinson K, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for non-metastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21:1581–1591. doi: 10.1200/JCO.2003.05.116. [DOI] [PubMed] [Google Scholar]
- 78.Bowers DC, Gargan L, Weprin BE, et al. Impact of site of tumor recurrence upon survival for children with recurrent or progressive medulloblastoma. J Neurosurg. 2007;107(1 Suppl):5–10. doi: 10.3171/PED-07/07/005. [DOI] [PubMed] [Google Scholar]
- 79.Belza MG, Donaldson SS, Steinberg GK, et al. Medulloblastoma: freedom from relapse longer than 8 years a therapeutic cure? J Neurosurg. 1991;75:575–582. doi: 10.3171/jns.1991.75.4.0575. [DOI] [PubMed] [Google Scholar]
- 80.Aryan HE, Ozgur BM, Jandial R, Levy ML. Subfrontal transbasal approach and technique for resection of craniopharyngioma. Neurosurg Focus. 2005;18:E10. [PubMed] [Google Scholar]
- 81.Kalapurakal JA. Radiation therapy in the management of pediatric craniopharyngioma—a review. Childs Nerv Syst. 2005;21:808–816. doi: 10.1007/s00381-005-1188-3. [DOI] [PubMed] [Google Scholar]
- 82.Tomita T, Bowman RM. Craniopharyngiomas in children: surgical experience at Children’s Memorial Hospital. Childs Nerv Syst. 2005;21:729–746. doi: 10.1007/s00381-005-1202-9. [DOI] [PubMed] [Google Scholar]
- 83.Connally ES, Winfree Q, Carmel PW. Giant posterior fossa cystic craniopharyngiomas presenting with hearing loss. Report of three cases and review of the literature. Surg Neurol. 1997;47:291–299. doi: 10.1016/s0090-3019(96)00253-4. [DOI] [PubMed] [Google Scholar]
- 84.Petito CK, DeGirolami U, Earle KM. Craniopharyngiomas: a clinical and pathological review. Cancer. 1976;37:1944–1952. doi: 10.1002/1097-0142(197604)37:4<1944::aid-cncr2820370446>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 85.Tomita T. Editorial on Current surgical management of craniopharyngiomas. Childs Nerv Syst. 2005;21:604–605. doi: 10.1007/s00381-005-1229-y. [DOI] [PubMed] [Google Scholar]
- 86.Thompson D, Phipps K, Hayward R. Craniopharyngioma in childhood: our evidence-based approach to management. Childs Nerv Syst. 2005;21:660–668. doi: 10.1007/s00381-005-1210-9. [DOI] [PubMed] [Google Scholar]
- 87.De Vile CJ, Grant DB, Hayward RD, et al. Obesity in childhood craniopharyngioma: relation to post-operative hypothalamic damage shown by magnetic resonance imaging. J Clin Endocrinol Metab. 1996;81:2734–2737. doi: 10.1210/jcem.81.7.8675604. [DOI] [PubMed] [Google Scholar]
- 88.Boop FA. Craniopharyngioma. J Neurosurg. 2007;106(1 Suppl):1–2. doi: 10.3171/ped.2007.106.1.1. [DOI] [PubMed] [Google Scholar]
- 89.Habrand JL, Ganry O, Couanet D, et al. The role of radiation therapy in the management of craniopharyngioma: a 25-year experience and review of the literature. Int J Radiat Oncol Biol Phys. 1999;44:255–263. doi: 10.1016/s0360-3016(99)00030-9. [DOI] [PubMed] [Google Scholar]
- 90.Rajan B, Ashley S, Gorman C, et al. Craniopharyngioma—a long-term results following limited surgery and radiotherapy. Radiother Oncol. 1993;26:1–10. doi: 10.1016/0167-8140(93)90019-5. [DOI] [PubMed] [Google Scholar]
- 91.Amendola BE, Wolf A, Coy SR, et al. Role of radiosurgery in craniopharyngiomas: a preliminary report. Med Pediatr Oncol. 2003;41:123–127. doi: 10.1002/mpo.10364. [DOI] [PubMed] [Google Scholar]
- 92.Van den Berge JH, Blaauw G, Breeman WA, et al. Intracavitary brachytherapy of cystic craniopharyngiomas. J Neurosurg. 1992;77:S45–550. doi: 10.3171/jns.1992.77.4.0545. [DOI] [PubMed] [Google Scholar]